
Sulfur and phosphorus co-doping optimized electronic structure and modulated intermediate affinity on PdSP metallene for ethanol-assisted energy-saving H2 production†
Hongjing
Wang
 ,
Yanan
Guo
,
Qiqi
Mao
,
Hongjie
Yu
,
Kai
Deng
,
Ziqiang
Wang
,
Xiaonian
Li
,
You
Xu
* and
Liang
Wang
*
,
Yanan
Guo
,
Qiqi
Mao
,
Hongjie
Yu
,
Kai
Deng
,
Ziqiang
Wang
,
Xiaonian
Li
,
You
Xu
* and
Liang
Wang
*
State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: yxu@zjut.edu.cn; wangliang@zjut.edu.cn
First published on 5th April 2023
Abstract
Coupling cathodic hydrogen evolution reaction (HER) and anodic electrochemical oxidation of organic small molecules in a co-electrolysis system could simultaneously realize high-value chemical generation and energy-saving hydrogen production, which, however, require high-performance electrocatalysts. In this work, we developed a one-step solvothermal method to synthesize S, P-co-doped Pd metallene (PdSP metallene) and employed it as a bifunctional electrocatalyst for both the HER and ethanol oxidation reaction (EOR). The co-doping of S and P atoms into Pd metallene could introduce multiple active sites and increase the electrochemically-active surface area. Moreover, the electronic interactions between Pd, S, and P atoms could regulate the electronic structure of the active sites and modulate the intermediate affinity on the resultant PdSP metallene, thus boosting the electrocatalytic HER and EOR performance. In the HER-EOR co-electrolysis system with bifunctional PdSP metallene electrocatalysts, only a 0.88 V of electrolysis voltage was required to fulfill 10 mA cm−2 current density, much lower than that of pure water electrolysis (1.41 V) using the same electrocatalysts.
Introduction
Hydrogen has attracted attention due to its advantages of ecological friendliness and high energy density, which makes it widely applied.1–3 Electrochemical water splitting is a promising way to produce highly purified hydrogen. In a typical water splitting process, high voltages are indispensable for catalysts to drive the reaction mainly because of the slow reaction kinetics of the anodic oxygen evolution reaction (OER), which greatly confines the industrial application.4–7 To overcome the obstacles for electrolytic hydrogen production via the conventional water splitting process, one attractive strategy is replacing OER with thermodynamically more favorable small molecule oxidation reactions. In this regard, alcohol oxidation reaction shows great potential as an alternative to OER to achieve highly efficient hydrogen production.8–11 Among various alcohol substrates, ethanol endows itself with higher energy densities and is less toxic than methanol, comes from a wide range of renewable sources (including straw, sugarcane, and corn cob), and breaks through the bottleneck of storage and transportation.12–15 The construction of hybrid co-electrolysis systems by coupling the cathodic hydrogen evolution reaction (HER) and anodic ethanol oxidation reaction (EOR) could potentially reduce the electrolysis voltage for hydrogen production and simultaneously generate value-added chemicals at the anode; however, its realization requires high-performance electrocatalysts.16–19It has been proved that Pd and Pd-based materials as catalysts for EOR and HER are favorable for the improvement of activities and kinetics. Particularly, Pd has been recognized as an alternative material for Pt owing to its similar properties to Pt and better CO tolerance.20–23 As for the aspect of catalyst design, the way to construct and manipulate well-defined morphologies and components of nanostructure has a major contribution to enhancing the catalytic performances.24–27 Two-dimensional (2D) nanomaterials stand out from many nanomaterials owing to their accessible active sites, high specific surface area, and maximized atomic utility.28–32 Among them, metallene with ultrathin layer and maximally loaded active sites confers the unique charm and favourable protection from poisoning species, which has induced researchers to carry out extensive research.33–38 For metal nanocrystal catalysts, doping with nonmental elements (e.g., N, S, P, B, etc.) is an effective strategy to boost their catalytic performance by utilizing electronic and/or synergistic effects between metal and nonmetal elements.39,40 In particular, dual-doping of metal nanocrystals with suitable two different kinds of nonmetals was demonstrated to be powerful to enhance the catalytic performance due to the resulting synergy between heteroatoms can alter the surface electronic structure and moderate the affinity between intermediates and active sites, then acquiring much lower kinetic energy barrier and more pronounced catalytic activity than un-doping and mono-doping counterparts.41–44 Despite recent advances for Pd-based metallene and doped metal nanocrystals, few studies have been reported on the construction of dual-nonmetal doped Pd-based metallene for HER and/or EOR electrocatalysis applications.
Herein, we developed a one-step solvothermal method to synthesize S and P dual-doped Pd metallene (PdSP metallene) by using triphenylphosphine sulfide (TPS) as S and P sources, and Pd(acac)2 as the Pd source. In such a ternary PdSP metallene system, the co-incorporation of S and P atoms could endow the resultant PdSP metallene with multiple active sites and increased electrochemically-active surface area (ECSA). Moreover, the electronic interaction between metal (Pd) and non-metal (S and P) could optimize the electronic structure of active sites and modulate the intermediates’ affinity on active sites. As a result, the obtained PdSP metallene exhibited enhanced bifunctional HER and EOR electrocatalytic activities in alkaline solutions, better than the undoped Pd metallene counterparts. Impressively, the HER-EOR hybrid co-electrolysis system with bifunctional PdSP metallene as both the cathode and anode could drive a current density of 10 mA cm−2 for H2 production at a low cell voltage of 0.88 V, much lower than that of the conventional overall water electrolysis (1.41 V) using the same electrocatalysts.
Experimental
Synthesis of PdSP metallene
PdSP metallene was fabricated through a one-step solvothermal method. Typically, 1.1 g of potassium hydroxide (KOH) was added into a 30 mL bottle containing 5.7 mL of N,N-dimethylformamide (DMF), and 4 mL of ethylene glycol (EG) under ultrasonication for 30 min. Afterward, 10 mg of palladium acetylacetone (Pd(acac)2) was dropped into the above mixture solution. Next, 2 mg of triphenylphosphine sulfide (TPS) was dissolved in the above solution. Finally, 10 mL of diethylenetriamine (DETA) was added to the above solution. The obtained solution was transferred to a 25 mL Teflon-lined autoclave and incessantly heated at 200 °C for 8 h. After cooling down to room temperature, the acquired sample was washed several times with H2O and ethanol in turns. In the end, the resultant products were dried in a vacuum oven at 60 °C for further characterization and electrochemical tests. As a contrastive sample, the Pd metallene counterpart was synthesized by a similar procedure but without TPS addition.Electrochemical measurements
HER and EOR electrocatalytic performances were investigated on a CHI 760E instrument using a three-electrode configuration, with a Hg/HgO electrode (1.0 M KOH), catalyst-decorated glassy carbon electrode (GCE) (3 mm diameter, 0.071 cm2 area) and graphite rod as reference electrode, working electrode, and counter electrode, respectively. The electrocatalyst ink was obtained via a homogeneous distribution of 2 mg of PdSP metallene catalyst in 1 mL of mixed solution containing water (800 μL), isopropanol (100 μL), and Nafion (0.5 wt%, 100 μL). Then, 5 μL of the electrocatalyst ink was deposited on a polished GCE surface and dried (catalyst loading: 0.14 mg cm−2). HER and EOR measurements were independently carried out in 1.0 M KOH, and 1.0 M KOH with 1.0 M C2H5OH. The linear sweep voltammetry (LSV) curves with 95% iR-compensation were achieved under a scan rate of 5 mV s−1. Other electrochemistry data, such as chronopotentiometric (V–t) curves, chronoamperometric (i–t) curves, and electrochemical impedance spectroscopy (EIS) were also recorded in detail. The ECSAs of the catalysts were measured by underpotentially deposited Pd oxide reduction. The desorption peaks of Pd oxide reduction were recorded by cyclic voltammetry (CV) with a scan rate of 50 mV s−1 to calculate the ECSAs. Obtained potentials in this work have been transferred into the reversible hydrogen electrode (RHE) according to the formula E (vs. RHE) = Eθ (Hg/HgO) + E (vs. Hg/HgO) + 0.0592 × pH.As for the two-electrode HER-EOR hybrid co-electrolysis system, the PdSP metallene catalyst was loaded on a 1 × 1 cm−2 carbon paper (catalyst loading: 1 mg cm−2) and served as both the cathode and anode. Carbon paper was cleaned with 3 M HCl solution, ethanol, and deionized water, alternately, and dried before use. The HER-EOR co-electrolysis performance was tested in 1.0 M KOH with 1.0 M C2H5OH. To determine the Faradaic efficiency (FE) for HER, the produced H2 was collected by the water drainage method, and FE for H2 production was calculated using the following equation:
| FE = (N(H2) × Z × F/Q) × 100% |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), Z is the electron number transferred by one mole of H2 (Z = 2), Q is the recorded charge during the electrolysis, and N(H2) is the molar amount for hydrogen production.
485 C mol−1), Z is the electron number transferred by one mole of H2 (Z = 2), Q is the recorded charge during the electrolysis, and N(H2) is the molar amount for hydrogen production.
Results and discussion
Fig. 1 shows the synthetic process for typical wrinkled PdSP metallene under solvothermal conditions using TPS as S and P sources and Pd(acac)2 as the Pd precursor. In this synthetic system, DETA contributes to directing the growth of 2D nanosheets.45 Additionally, KOH is favourable for breaking down DMF into dimethylamine (DMA), which conduces to the generation of ultrathin nanosheets.29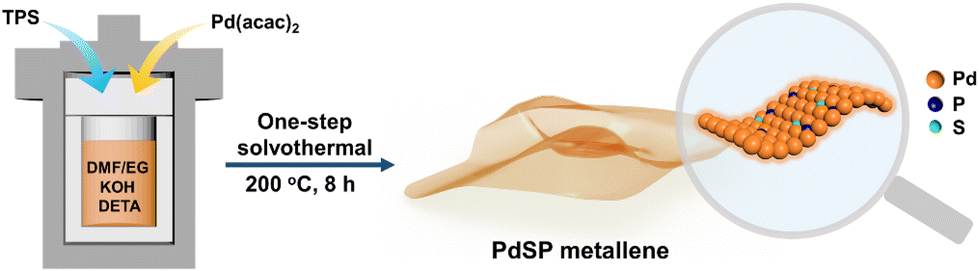 | ||
| Fig. 1 Scheme for the preparation of PdSP metallene. | ||
As revealed by high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) (Fig. 2a and b), the as-fabricated PdSP metallene is composed of crimped nanosheets. Transmission electron microscopy (TEM) image further certified that the sample is characteristic with creased ultrathin 2D nanosheets (Fig. 2c). In comparison with pure Pd metallene (Fig. S1†), the TEM image of PdSP metallene has no apparent changes, suggesting that the incorporation of S and P atoms into the Pd lattice has no damage to the existing structure and inherits the morphology and structure of Pd metallene. It was noted that adding amounts of TPS in the synthetic process of PdSP metallene has an influence on its final morphology. Increasing the TPS from 0 mg to 2 mg, the final samples display well-fined metallene morphology, that is of the pure Pd metallene (Fig. S1†) and typical PdSP metallene (Fig. 2c), respectively. However, further increasing the TPS content to 5 mg, agglomeration of samples is observed (Fig. S2a†), which is more serious in the presence of 10 mg of TPS (Fig. S2b†). The selected area electron diffraction (SAED) pattern for typical PdSP metallene in Fig. S3† reveals its polycrystallinity feature. Furthermore, the thickness of the PdSP metallene was tested using atomic force microscopy (AFM) technology and the corresponding thickness is approximately 1.22 nm (Fig. 2d), confirming the ultrathin 2D structure of PdSP metallene, ulteriorly. The high-resolution TEM (HRTEM) image reveals clear lattice stripes of PdSP metallene (Fig. 2e). Concurrently, integrated pixel intensities were gained according to the tagged areas and the mean lattice spacing of PdSP metallene is approximately 0.227 nm (Fig. 2e), corresponding to the Pd (111) crystal plane. The energy dispersive X-ray spectroscopy (EDX) mapping analysis clarifies the presence and homogeneous distribution of Pd, S, and P elements on the entire nanosheet (Fig. 2f). The atomic ratio of Pd, S, and P elements was approximately 92.10:3.51:4.39 as further analyzed by the EDX spectrum (Fig. S4†).
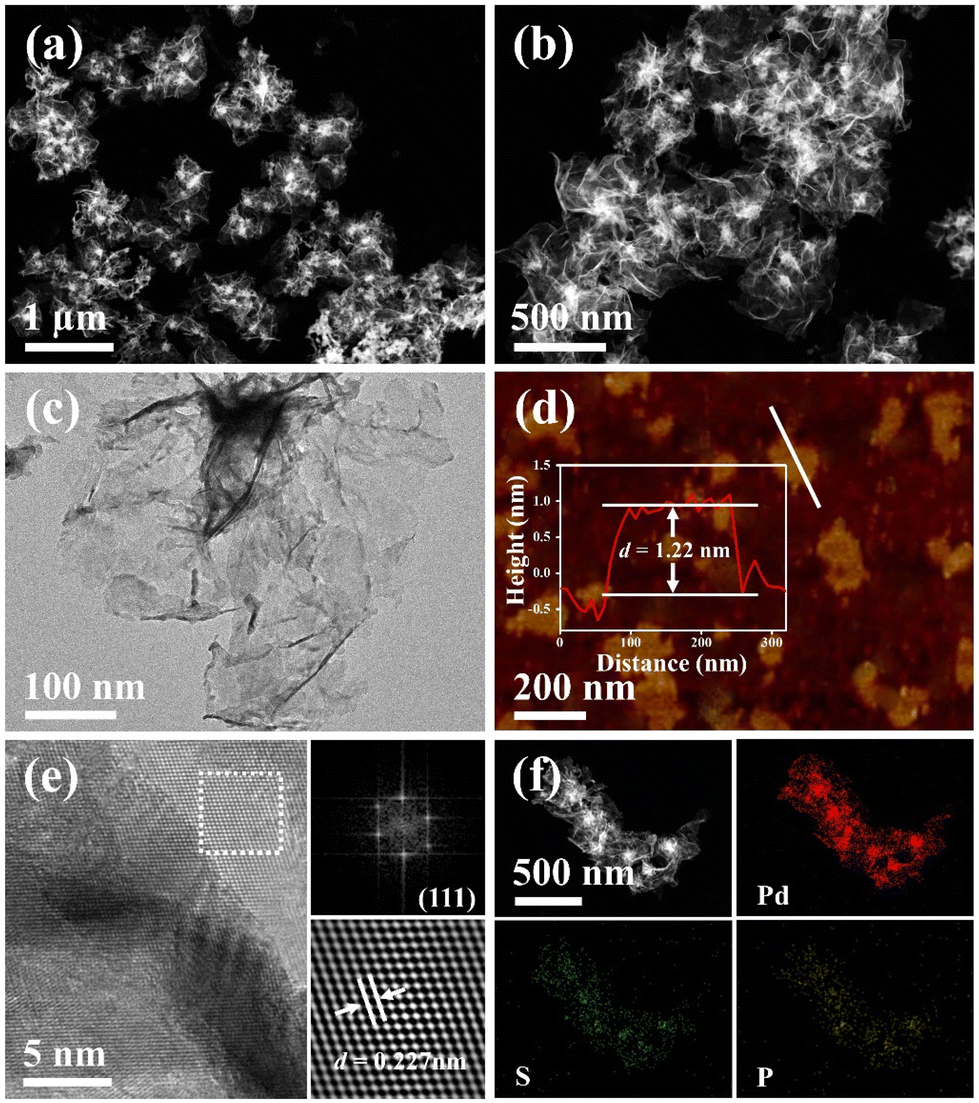 | ||
| Fig. 2 (a and b) HAADF-STEM images, (c) TEM image, (d) AFM image and corresponding height profiles, (e) HRTEM image, and (f) HAADF-STEM and element mapping images of the PdSP metallene. The inset in (e) displays the corresponding FFT pattern. | ||
To obtain more crystalline information on PdSP metallene, XRD analysis was conducted (Fig. 3a). Five characteristic diffraction peaks for PdSP metallene at 40.09°, 46.62°, 68.06°, 81.99°, and 86.60° could be indexed to the (111), (200), (220), (311), and (222) facets of the face-centered-cubic (fcc) crystal, respectively. Compared with pure Pd metallene, the XRD diffraction peaks for PdSP metallene shift to lower 2θ values indicative of the regulated Pd–Pd inter-lattice spacings because of the insertion of S and P atoms into Pd metallene.43,46 The chemical states, compositional and electronic interactions of Pd and PdSP metallene were further explored by XPS. The Pd 3d regions of the PdSP and Pd metallene are displayed in Fig. 3b. The peaks of PdSP metallene at 334.78 and 340.10 eV are fitted to Pd (0) 3d5/2 and Pd (0) 3d3/2,respectively, and the other peaks at 336.17 and 341.81 eV are matched with Pd(II).47 It can be apparently observed that the Pd (0) 3d5/2 peak for PdSP metallene shifts to a lower binding energy compared to that in Pd metallene, which testifies the modulation of electrophilic property in the PdSP system and consequently downshifts the d-band center of Pd because of the electronic synergy between S, P, and Pd.43,48–50 As for XPS spectra of S 2p in PdSP metallene (Fig. 3c), two peaks at 162.58 and 161.52 eV match with 2p1/2 and 2p3/2 peaks, respectively, of S species.40,42,51 Furthermore, the characteristic peaks at 129.9 eV is assigned to P (0), and the other peak at 133.27 eV can be attributable to the P (V) (Fig. 3d).41,42 Considering the above results, there is no doubt that S and P species have been successfully introduced into Pd metallene, forming ternary PdSP matellene.
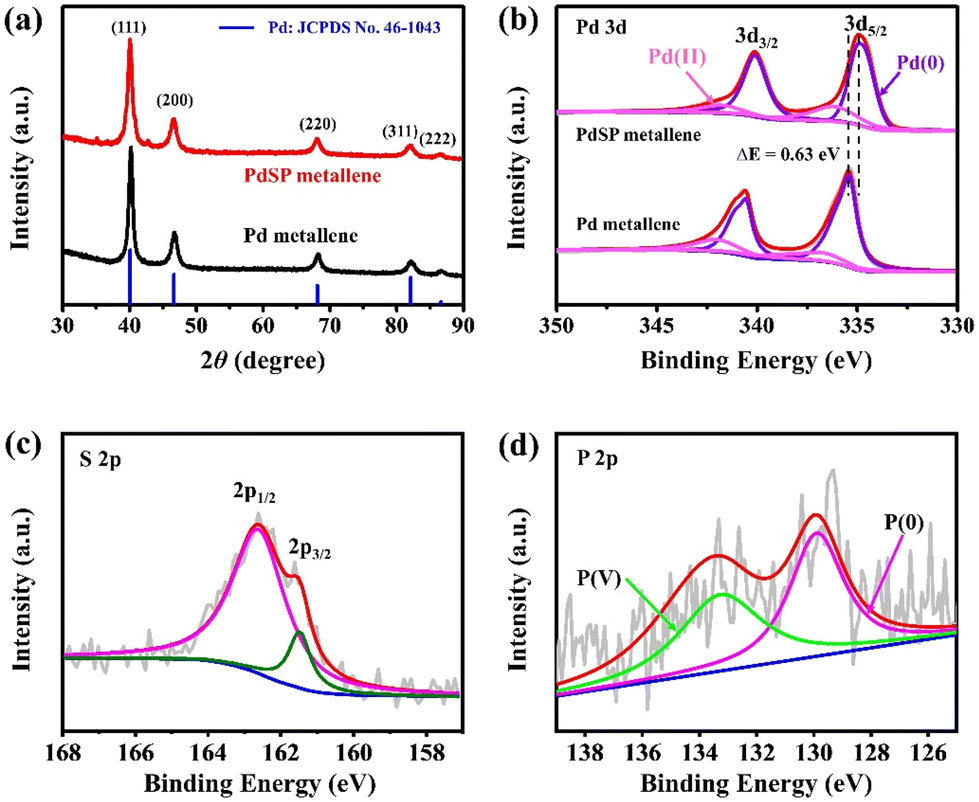 | ||
| Fig. 3 (a) XRD patterns for PdSP metallene and Pd metallene. (b) Pd 3d XPS spectra for PdSP metallene and Pd metallene. (c) S 2p and (d) P 2p XPS spectra for PdSP metallene. | ||
Pd metallene based on the preponderance of non-metallic doping and nanosheet architecture has a certain catalytic activity for ethanol oxidation.52,53 Inspired by this, the catalytic performance of PdSP metallene for EOR was explored. For studying the ECSA of the catalysts, CV was conducted over a potential range from 0.12 V to 1.43 V in 0.1 M KOH electrolyte (Fig. S5a–c†). Accordingly, the ECSA is calculated to be 56.06 m2 g−1 for PdSP metallene, which is larger than that for Pd metallene (46.95 m2 g−1) and Pd black (27.86 m2 g−1) (Fig. S5d†), indicative of increased ECSA for PdSP metallene thanks to the metallene structure and S and P atoms co-incorporation.
The EOR electrocatalytic activity of PdSP metallene, Pd metallene, and Pd black was investigated in a 1.0 M KOH containing 1.0 M ethanol solution. Fig. 4a and b show the EOR CV curves for various catalysts, with current densities normalized by Pd mass (mass activity) and ECSA (specific activity), respectively. As presented in Fig. 4a and c, the mass peak current for PdSP metallene reaches 1.52 mA μg−1Pd, which is 1.57 and 2.92 times higher than that at Pd metallene (0.97 mA μg−1Pd) and Pd black (0.52 mA μg−1Pd), respectively. According to Fig. 4b and c, ECSA-normalized CVs display that the specific activity at the peak potential of EOR on PdSP metallene (2.75 mA cm−2) is 1.33 and 1.50 times higher than that on Pd metallene (2.06 mA cm−2) and Pd black (1.83 mA cm−2), respectively, indicating that the catalytic activity of PdSP metallene for EOR precedes the initial Pd metallene and Pd black. Considering these results, as P and S atoms are doped into the structure of Pd metallene, it created synergistic effects that made it easier for the catalyst to modulate the electron distribution and improve the active sites.53 Concurrently, the optimization of the electronic structures of Pd metallene by S and P co-doping has a relatively weakened adsorption affinity of poisoning intermediates and accelerates the generation of O-related species during ethanol oxidation, which can facilitate the whole oxidation process.54 In addition, as displayed by the chronoamperometric (i–t) curves (Fig. 4d), the EOR current density for PdSP metallene outperforms that for Pd metallene and Pd black in the whole electrolysis, revealing the stability enhancement of PdSP metallene, thanks to the structure effect and the electronic effect through the inserted-S, P atoms.
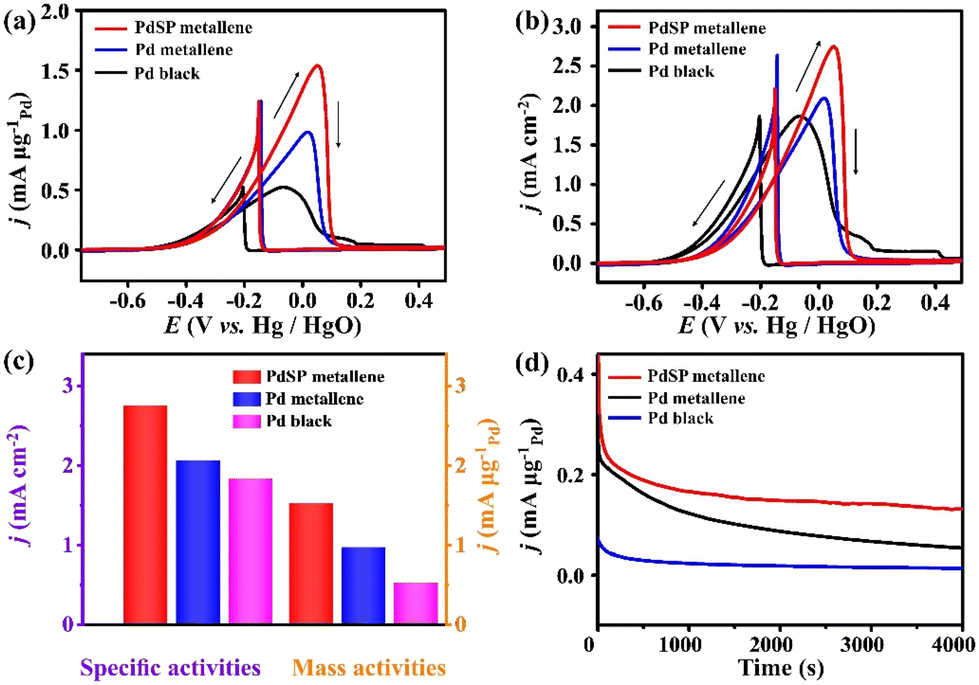 | ||
| Fig. 4 (a) Mass activities and (b) specific activities of EOR for various catalysts recorded at a scan rate of 50 mV s−1 with 1.0 M KOH solution containing 1.0 M CH3CH2OH. (c) Comparative peak mass activities and specific activities for various catalysts. (d) Chronoamperometric curves for various catalysts recorded at −0.2 V in 1.0 M KOH electrolyte containing 1.0 M CH3CH2OH. The arrows in (a) and (b) indicate the potential scan direction. | ||
The electrocatalytic activity of PdSP metallene towards HER was also investigated in 1.0 M KOH, with Pd metallene and Pd black as contrastive catalysts. According to the HER polarization curves for various catalysts (Fig. 5a), the overpotential of PdSP metallene at 10 mA cm−2 is 124 mV, which is relatively lower than that of Pd metallene (310 mV) and Pd black (496 mV) (Fig. 5b). A comparison of the achieved current densities of various catalysts at a cathodic potential of -0.18 V (vs. RHE) is also exhibited in Fig. 5c. Apparently, the PdSP metallene reached a higher current density (25.96 mA cm−2) than those of Pd metallene (2.36 mA cm−2) and Pd black (1.49 mA cm−2), further implying the ameliorative HER property of PdSP metallene (Fig. 5c). It seems that a synergic effect between P and S within catalysts significantly enhanced the catalytic activity of PdSP metallene. Because of the strong electronegativity of S and P elements, the acquired enriched active sites contributed to the adsorption of H2O under alkaline media and the relatively positive current density, which further makes it easier to achieve higher HER activity.55 Thereafter, the corresponding Tafel slopes were computed according to the LSV curves. As displayed in Fig. 5d, the PdSP metallene catalyst exhibits a lower Tafel slope of 138 mV dec−1 than that of Pd metallene (175 mV dec−1) and Pd black (178 mV dec−1), displaying the acceleration of kinetics for the HER process, thanks to incorporated S and P atoms on PdSP metallene. Besides, the EIS diagram for various catalysts under the same applied potential of−0.47 V (vs. RHE) illustrates that PdSP metallene possesses a lower charge transfer resistance than Pd metallene and Pd black, implying a much faster electron transfer rate and meliorative electrocatalytic activity for the HER as well (Fig. S6†). These can be ascribed to the fact that regulated surface electrons adjust to the affinity of Pd metallene to hydrogen by S and P co-doping, which agrees with the XPS results. To verify the durability and long-term stability of the PdSP metallene in 1.0 M KOH, the conducted LSV polarization curve after 1000 cycles nearly has no change compared to the initial curve (Fig. 5e). Additionally, the durability of PdSP metallene for HER was conducted by chronopotentiometry (V–t) measurements under a consecutive cathodic current density of 10 mA cm−2 and the applied potential did not change apparently through a 20 h of operating (Fig. 5f). Thereafter, the detached sample of PdSP metallene was collected to characterize again by TEM (Fig. S7†), which confirmed the exceptional architecture and compositional stability after undergoing a long testing period of HER electrocatalysis.
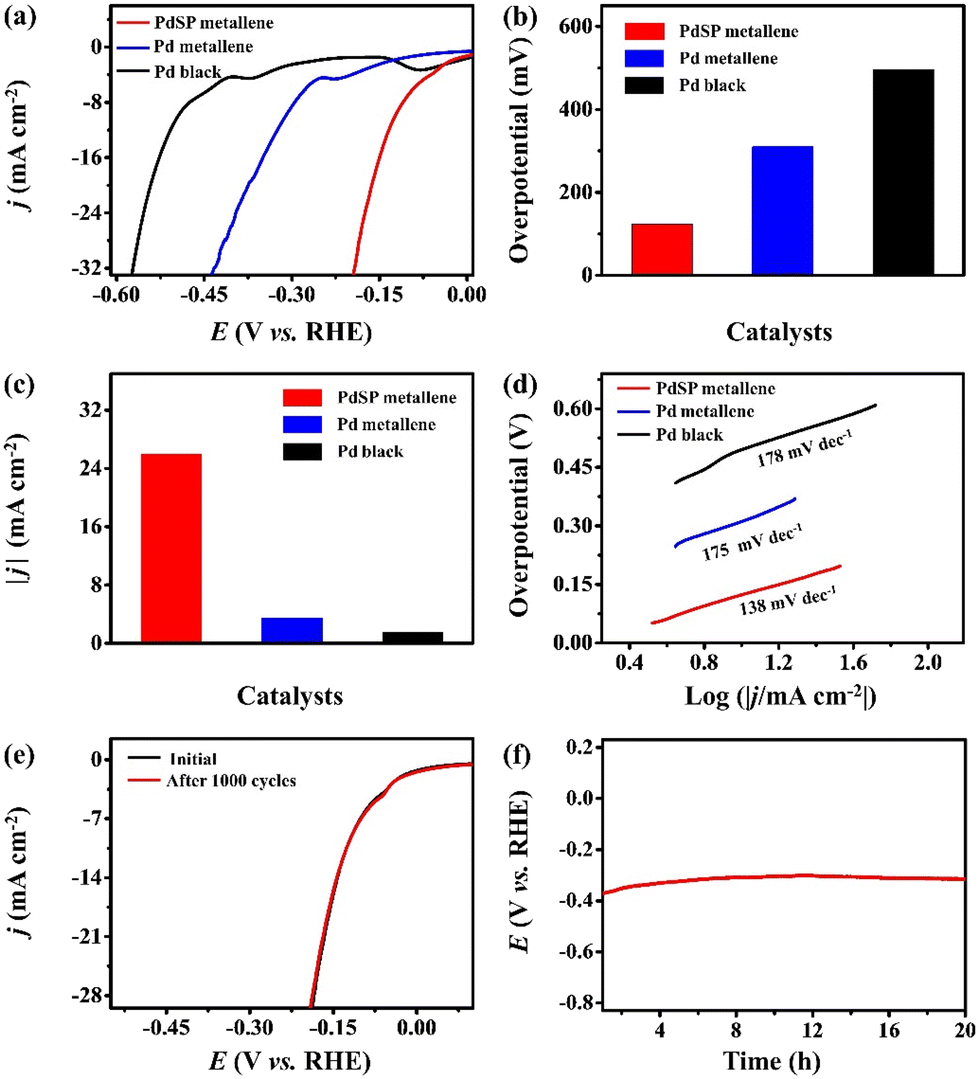 | ||
| Fig. 5 (a) HER polarization curves for PdSP metallene, Pd metallene, and Pd black in 1.0 M KOH electrolyte. (b) Comparison of the potentials at 10 mA cm−2 and (c) the current densities at -0.18 V (vs. RHE) for various catalysts. (d) Tafel slope plots for various electrocatalysts. (e) HER polarization curves for the PdSP metallene before and after 1000 cycles in 1.0 M KOH electrolyte. (f) Chronopotentiometry curves for the PdSP metallene at a constant cathodic current density of 10 mA cm−2 in 1.0 M KOH electrolyte for 20 h. Current density data in (a) and (e) were obtained by normalizing the current to the geometric area of the GC working electrode (0.071 cm2). | ||
Since PdSP metallene has a favourable performance for electrocatalyzing EOR and HER in alkaline media, an HER-EOR hybrid co-electrolysis system under 1.0 M KOH containing 1.0 M ethanol was constructed by applying the PdSP metallene as both cathode and anode electrode materials (PdSP metallene||PdSP metallene) and the relative device scheme is exhibited in Fig. 6a. As investigated in Fig. 6b, the PdSP metallene||PdSP metallene two-electrode system needs a cell voltage of 0.88 V to fulfill a current density of 10 mA cm−2 in 1.0 M KOH containing 1.0 M ethanol, which is largely lower than that in 1.0 M KOH without ethanol (1.41 V) using the same electrocatalysts, suggesting that replacing OER with EOR could reduce the electrolysis voltage for hydrogen production. Particularly, EOR-assisted water electrolysis only consumes smaller energy compared to pure water electrolysis, which is comparable to many other reported small organic molecules oxidation-assisted H2 generation systems (Table S1†). Combining the required higher voltage of Pd metallene couple (0.97 V) and Pt/C||RuO2 (1.66 V) at 10 mA cm−2, it appears that the incorporation of S and P into Pd metallene subtly moderates the catalytic properties because of its optimized structure and components (Fig. 6c and Fig. S8†). Additionally, the LSV curves of the PdSP metallene couple in 1.0 M KOH solution with various ethanol concentrations are illustrated in Fig. 6d. The corresponding data evidently demonstrate that the electrode obtained a high EOR response with the addition of ethanol. The slight degradation in ethanol-assisted H2 production performance in the presence of high ethanol concentrations is due to the adsorption of pre-existing oxidation products on the electrode surface preventing further oxidation reactions.56,57
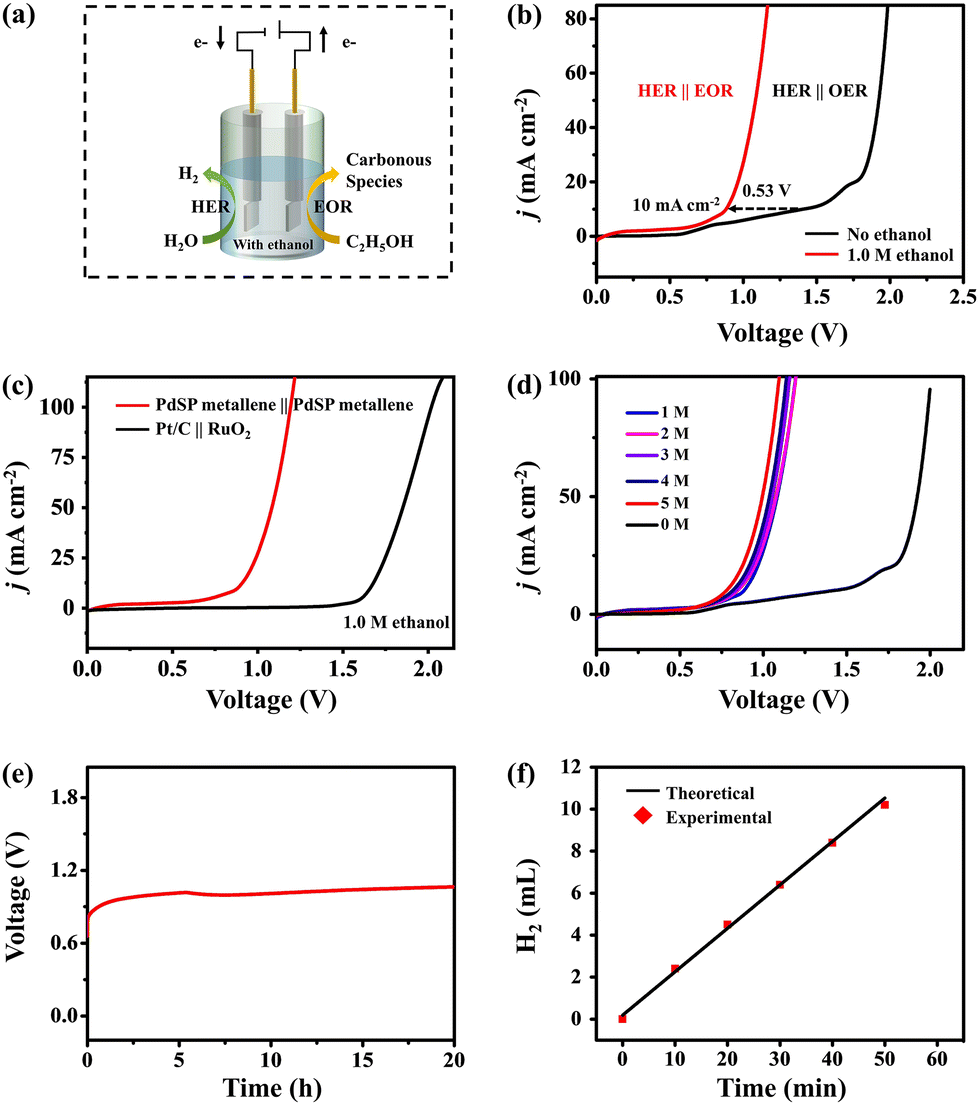 | ||
| Fig. 6 (a) Schematic illustration for two-electrode ethanol-assisted water splitting system. (b) LSV curves of the PdSP metallene as anode and cathode in 1.0 M KOH electrolyte with and without 1.0 M ethanol. (c) LSV curves of different electrocatalysts in 1.0 M KOH electrolyte containing 1.0 M ethanol in a two-electrode system. (d) LSV curves of PdSP metallene||PdSP metallene in 1.0 M KOH electrolyte with various ethanol concentrations. (e) Chronopotentiometry curves of PdSP metallene||PdSP metallene at a constant current density of 10 mA cm−2 in 1.0 M KOH electrolyte containing 1.0 M ethanol for 20 h. (f) The measured H2 quantity and theoretical values at the cathode. Current density data in (b–d) were obtained by normalizing the current to the geometric area of the carbon paper working electrode (1 cm2). | ||
To distinctively confirm the catalytic durability of HER-EOR co-electrolysis by the PdSP metallene||PdSP metallene two-electrode system, the chronopotentiometry (V–t) measurement at a constant current density of 10 mA cm−2 was conducted. As shown in Fig. 6e, apparently, there is no obvious change in the applied voltage during 20 h of ethanol-assisted water splitting process, demonstrating the remarkable catalytic durability. For estimating the FE of the co-electrolysis system for H2 generation, a bulk electrolysis experiment was conducted at a current density of 10 mA cm−2 for 50 min. As shown in Fig. 6f, the experimentally as-generated H2 amount is in conjunction with the theoretical computed values and the FE for H2 generation is nearly 98.21%. Post-electrolysis electrolyte was further qualitatively analyzed using 13C NMR spectroscopy. The results in Fig. S9† verified the presence of acetic acid and carbonate in the post-electrolysis electrolyte.
The distinguished HER and EOR electrocatalytic properties over the PdSP metallene could be ascribed to its unique metallene structure and ternary PdSP composition (Fig. 7). Firstly, the ultrathin nature of 2D nanosheets of metallene can supply plenty of active sites and enhance the stability of nanosheets benefitting from their high surface area and highly wrinkled property.29 With that, the co-incorporation of S and P atoms could further endow the resultant PdSP metallene with multiple active sites and increase the ECSA. Secondly, doping of S and P atoms effectively modifies the Pd electronic structure, which makes PdSP metallene acquire the relatively moderate surface adsorption capability of H* by modulating the affinity of Pd metallene to hydrogen, and consequently facilitating HER electrocatalysis.43,45 Thirdly, as for EOR electrocatalysis, the presence of P–O species on the surface of PdSP metallene could kinetically enlarge the adsorption numbers of OHads, which is formed from H2O dissociation, and thus facilitates the oxidation of C2H5OHads at a low potential.42,54,58
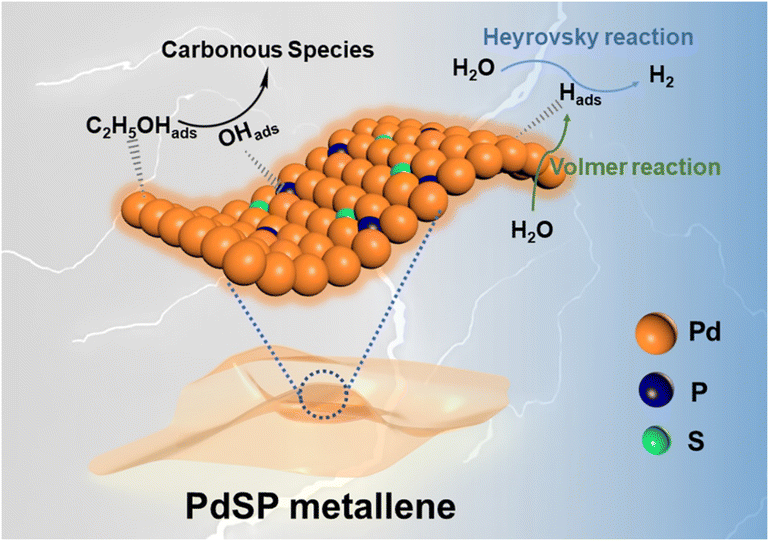 | ||
| Fig. 7 Schematic illustration for the ethanol-assisted HER process on the PdSP metallene. | ||
Conclusions
In summary, PdSP metallene was synthesized via a one-step solvothermal method and functioned as a bifunctional electrocatalyst for both HER and EOR in an alkaline medium. The experimental results evidenced that the co-doping of S and P increases the active sites and alternatively affects the electronic structure of PdSP metallene, which subtly makes the effective modulation of the affinity between intermediates and PdSP metallene come true and accordingly optimizes the electrochemical catalysis toward HER and EOR. More significantly, the constructed PdSP metallene||PdSP metallene two-electrode system just needs a lower voltage of 0.88 V than that of pure water splitting (1.41 V) to drive a current density of 10 mA cm−2 in ethanol-water electrolyte, implementing the concept of energy-efficient H2 production methods and enriching the application of value-added products.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21972126, 21978264, 21905250, and 22278369), Natural Science Foundation of Zhejiang Province (No. LQ22B030012 and LQ23B030010), and China Postdoctoral Science Foundation (2021M702889).References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- J. Yang, Y. J. Ji, Q. Shao, N. Zhang, Y. Y. Li and X. Q. Huang, Adv. Funct. Mater., 2018, 28, 1803722 CrossRef.
- Q. Zhou, R. Sun, Y. Ren, R. Tian, J. Yang, H. Pang, K. Huang, X. Tian, L. Xu and Y. Tang, Carbon Energy, 2023, 5, e273 CAS.
- H. Shi, Y. T. Zhou, R. Q. Yao, W. B. Wan, X. Ge, W. Zhang, Z. Wen, X. Y. Lang, W. T. Zheng and Q. Jiang, Nat. Commun., 2020, 11, 2940 CrossRef CAS PubMed.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef CAS PubMed.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS PubMed.
- D. Wu, K. Kusada, S. Yoshioka, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Chen, O. Seo, J. Kim, C. Song, S. Hiroi, O. Sakata, T. Ina, S. Kawaguchi, Y. Kubota, H. Kobayashi and H. Kitagawa, Nat. Commun., 2021, 12, 1145 CrossRef CAS PubMed.
- Y. Xu, T. Liu, K. Shi, H. Yu, K. Deng, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Commun., 2023, 59, 1817–1820 RSC.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian and L. Feng, Nano Res., 2022, 15, 8936–8945 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- J. Bai, D. Liu, J. Yang and Y. Chen, ChemSusChem, 2019, 12, 2117–2132 CrossRef CAS PubMed.
- J. Bai, X. Xiao, Y. Y. Xue, J. X. Jiang, J. H. Zeng, X. F. Li and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 19755–19763 CrossRef CAS PubMed.
- R. Rizo, R. M. Aran-Ais, E. Padgett, D. A. Muller, M. J. Lazaro, J. Solla-Gullon, J. M. Feliu, E. Pastor and H. D. Abruna, J. Am. Chem. Soc., 2018, 140, 3791–3797 CrossRef CAS PubMed.
- H. T. Zheng, Y. L. Li, S. X. Chen and P. K. Shen, J. Power Sources, 2006, 163, 371–375 CrossRef CAS.
- T. J. Wang, G. R. Xu, H. Y. Sun, H. Huang, F. M. Li, P. Chen and Y. Chen, Nanoscale, 2020, 12, 11526–11535 RSC.
- H. Y. Sun, Y. Ding, Y. Q. Yue, Q. Xue, F. M. Li, J. X. Jiang, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 13149–13157 CrossRef CAS PubMed.
- S. Sheng, K. Ye, L. N. Sha, K. Zhu, Y. Y. Gao, J. Yan, G. L. Wang and D. X. Cao, Inorg. Chem. Front., 2020, 7, 4498–4506 RSC.
- R. Du, J. Wang, Y. Wang, R. Hubner, X. Fan, I. Senkovska, Y. Hu, S. Kaskel and A. Eychmuller, Nat. Commun., 2020, 11, 1590 CrossRef CAS PubMed.
- J. Qi, N. Benipal, C. Liang and W. Li, Appl. Catal., B, 2016, 199, 494–503 CrossRef CAS.
- A. Mondal, A. De and J. Datta, Appl. Catal., A, 2018, 561, 87–95 CrossRef CAS.
- J. S. Guo, R. R. Chen, F. H. Zhu, S. G. Sun and H. M. Villullas, Appl. Catal., B, 2018, 224, 602–611 CrossRef CAS.
- H. An, L. N. Pan, H. Cui, B. J. Li, D. D. Zhou, J. P. Zhai and Q. Li, Electrochim. Acta, 2013, 102, 79–87 CrossRef CAS.
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen, G. Lu, D. Su and X. Huang, Angew. Chem., 2016, 128, 9176–9181 CrossRef.
- S.-B. Wang, W. Zhu, J. Ke, M. Lin and Y.-W. Zhang, ACS Catal., 2014, 4, 2298–2306 CrossRef CAS.
- J. Li, M. Hou and Z. Zhang, Nanoscale, 2022, 14, 8096–8102 RSC.
- D. Yi, C. Marcelot, I. Romana, M. Tasse, P. F. Fazzini, L. Peres, N. Ratel-Ramond, P. Decorse, B. Warot-Fonrose, G. Viau, P. Serp and K. Soulantica, Nanoscale, 2023, 15, 1739–1753 RSC.
- H. Wang, W. Wang, H. Yu, Q. Mao, Y. Xu, X. Li, Z. Wang and L. Wang, Appl. Catal., B, 2022, 307, 121172 CrossRef CAS.
- Q. Yang, L. Shi, B. Yu, J. Xu, C. Wei, Y. Wang and H. Chen, J. Mater. Chem. A, 2019, 7, 18846–18851 RSC.
- H. Cheng, N. Yang, X. Liu, Q. Yun, M. H. Goh, B. Chen, X. Qi, Q. Lu, X. Chen, W. Liu, L. Gu and H. Zhang, Natl. Sci. Rev., 2019, 6, 955–961 CrossRef CAS PubMed.
- J. Ge, P. Wei, G. Wu, Y. Liu, T. Yuan, Z. Li, Y. Qu, Y. Wu, H. Li, Z. Zhuang, X. Hong and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 3435–3438 CrossRef CAS PubMed.
- H. Wang, Q. Mao, T. Ren, T. Zhou, K. Deng, Z. Wang, X. Li, Y. Xu and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 44733–44741 CrossRef CAS PubMed.
- C. Cao, Q. Xu and Q.-L. Zhu, Chem Catal., 2022, 2, 693–723 CrossRef CAS.
- P. Prabhu and J. M. Lee, Chem. Soc. Rev., 2021, 50, 6700–6719 RSC.
- F. Lv, B. Huang, J. Feng, W. Zhang, K. Wang, N. Li, J. Zhou, P. Zhou, W. Yang, Y. Du, D. Su and S. Guo, Natl. Sci. Rev., 2021, 8, nwab019 CrossRef CAS PubMed.
- K. Deng, T. Zhou, Q. Mao, S. Wang, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Adv. Mater., 2022, 34, 2110680 CrossRef CAS PubMed.
- Q. Hong, B. Miao, T. Wang, F. Li and Y. Chen, Energy Lab., 2023, 1, 220022 Search PubMed.
- Y.-N. Li, Q.-L. Hong, B.-Q. Miao, T.-J. Wang, Y. Ding and Y. Chen, Renewables, 2023, 1, 90–99 CrossRef.
- H. Xia, Z. D. Shi, C. S. Gong and Y. M. He, J. Mater. Chem. A, 2022, 10, 19067–19089 RSC.
- C. Ouyang, X. Wang and S. Wang, Chem. Commun., 2015, 51, 14160–14163 RSC.
- Y. Xu, M. Z. Wang, S. S. Yu, T. L. Ren, K. L. Ren, Z. Q. Wang, X. N. Li, L. Wang and H. J. Wang, Chem. Eng. J., 2021, 421, 127751 CrossRef CAS.
- S. Yin, Y. Xu, S. Liu, H. Yu, Z. Wang, X. Li, L. Wang and H. Wang, Nanoscale, 2020, 12, 14863–14869 RSC.
- T. Jiang, L. Y. Yu, Z. J. Zhao, W. Wu, Z. C. Wang and N. C. Cheng, Chem. Eng. J., 2022, 433, 133525 CrossRef CAS.
- H. Lv, L. Z. Sun, D. D. Xu and B. Liu, Sci. Bull., 2020, 65, 1823–1831 CrossRef CAS PubMed.
- S. L. Yin, S. L. Liu, Z. Q. Wang, Y. Xu, X. N. Li, H. J. Wang and L. Wang, Chem. Eng. J., 2022, 435, 134711 CrossRef CAS.
- H. Lv, D. Xu, L. Sun, J. Henzie, S. L. Suib, Y. Yamauchi and B. Liu, ACS Nano, 2019, 13, 12052–12061 CrossRef CAS PubMed.
- H. Yu, T. Zhou, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 12027–12031 CrossRef CAS PubMed.
- A. L. Wang, X. J. He, X. F. Lu, H. Xu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2015, 54, 3669–3673 CrossRef CAS PubMed.
- S. Y. Shen, T. S. Zhao, J. B. Xu and Y. S. Li, J. Power Sources, 2010, 195, 1001–1006 CrossRef CAS.
- L. Chen, L. R. Zhang, L. Y. Yao, Y. H. Fang, L. He, G. F. Wei and Z. P. Lu, Energy Environ. Sci., 2019, 12, 3099–3105 RSC.
- Y. Wang, K. Xu, Z. Zhu, W. Guo, T. Yu, M. He, W. Wei and T. Yang, Chem. Commun., 2021, 57, 1368–1371 RSC.
- Z. R. Yang, S. Q. Wang, J. Wang, A. J. Zhou and C. W. Xu, Sci. Rep., 2017, 7, 15479 CrossRef PubMed.
- K. Yu, Y. Lin, J. C. Fan, Q. X. Li, P. H. Shi, Q. J. Xu and Y. L. Min, Catalysts, 2019, 9, 114 CrossRef.
- L. Sun, H. Lv, Y. Wang, D. Xu and B. Liu, J. Phys. Chem. Lett., 2020, 11, 6632–6639 CrossRef CAS PubMed.
- K. Liang, S. Pakhira, Z. Yang, A. Nijamudheen, L. Ju, M. Wang, C. I. Aguirre-Velez, G. E. Sterbinsky, Y. Du, Z. Feng, J. L. Mendoza-Cortes and Y. Yang, ACS Catal., 2018, 9, 651–659 CrossRef.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS.
- H. L. Yang, Z. N. Yu, S. W. Li, Q. L. Zhang, J. Jin and J. T. Ma, J. Catal., 2017, 353, 256–264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01112g |
| This journal is © The Royal Society of Chemistry 2023 |