
Biomineralization-inspired sandwich dentin desensitization strategy based on multifunctional nanocomposite with yolk–shell structure†
Luyao
Yi
,
Hongling
Wu
,
Yue
Xu
,
Jian
Yu
,
Yaning
Zhao
,
Hongye
Yang
 * and
Cui
Huang
*
* and
Cui
Huang
*
The State Key Laboratory Breeding Base of Basic Science of Stomatology (Hubei-MOST) & Key Laboratory for Oral Biomedical Ministry of Education, School & Hospital of Stomatology, Wuhan University, Wuhan, China. E-mail: yanghongye@whu.edu.cn; huangcui@whu.edu.cn
First published on 8th November 2022
Abstract
Dentin hypersensitivity (DH) treatment is far from being unequivocal in providing a superior strategy that combines immediate and long-term efficiency of dentinal tubule (DT) occlusion and clinical applicability. In order to achieve this aim, a type of multifunctional yolk–shell nanocomposite with acid resistance, mechanical resistance and biomineralization properties was developed in this study, which consists of a silica/mesoporous titanium–zirconium nanocarrier (STZ) and poly(allylamine hydrochloride) (PAH)-stabilized amorphous calcium phosphate (ACP) liquid precursor. First, the nanocomposite, named as PSTZ, immediately occluded DTs and demonstrated outstanding acid and mechanical resistance. Second, the PSTZ nanocomposite induced intrafibrillar mineralization of single-layer collagen fibrils and remineralization of demineralized dentin matrix. Finally, PSTZ promoted the odontogenic differentiation of dental pulp stem cells by releasing ACP and silicon ions. The reconstruction of the dentin-mimicking hierarchical structure and the introduction of newly formed minerals in the upper, middle and lower segments of DTs, defined as sandwich-like structures, markedly reduced the permeability and achieved superior long-term sealing effects. The nanocomposite material based on mesoporous yolk–shell carriers and liquid-phase mineralized precursors developed in this study represents a versatile biomimetic sandwich desensitization strategy and offers fresh insight into the clinical management of DH.
Introduction
Wonderful works of nature have provided humans with surprising inspiration, which exhibit remarkable biostructures and performances through ingenious bioprocesses.1 Biomineralization is a typical dynamic bioprocess by which organisms generate and remodel hierarchically mineralized tissues (e.g., bone and dentin). Dentin is composed of 70% inorganic minerals (35% intrafibrillar minerals and 65% extrafibrillar minerals) and 30% organic substances.2,3 When attacked by acid, its mineral homeostasis will be broken, accompanied by the dissolution of intrafibrillar and extrafibrillar minerals, the collapse of collagen structures, a considerable loss in mechanical strength and even disturbances in function.4 Dentin hypersensitivity (DH) can be regarded as a type of disease induced by an imbalance between demineralization and remineralization. Due to its high prevalence and irritability, DH is acknowledged to severely affect the quality of life of patients.5,6 Characteristic transient and sharp pain results from fluid movement within exposed dentinal tubules (DTs), which is generally induced by thermal, chemical, mechanical or osmotic stimuli. According to the hydrodynamic hypothesis, the widely accepted theory, DT occlusion is representative of an overwhelming majority of treatment strategies to avoid fluid flow and reduce dentin permeability, thereby alleviating DH symptoms.7,8Many types of materials with DT-occluding properties have been developed, including DT blockers (e.g., ions, mineral salts, proteins) and dentin sealers (e.g., glass ionomers, composites and dentin adhesives).7 Among them, biomimetic mineralization strategies have become a promising approach to the management of DH.9–13 Some studies induced hydroxyapatite (HAp) deposition in situ within DTs and proposed rapid and efficient therapy for treating DH with long-term effects but failed to restore the natural hierarchical structure of dentin and ignored the critical role played by collagen fibrils.11,12 Some studies have tried to occlude DTs via polymer-induced liquid precursor (PILP) mediating biomimetic collagen mineralization, yet the periodic replenishment of the mineralizing solution is infeasible and limits its clinical application.9 Above all, no immediate DT-occluding effect was presented in these studies, the mineralization time of which was at least 7 days. To date, it remains far from being unequivocal in providing a superior strategy exhibiting both immediate and long-term efficiency of DT occlusion and clinical applicability.7
Actually, dentin itself provides important clues for the design of newly desensitizing biomaterials. Honed by millions of years of evolution, dentin has already a set of formation and self-repair mechanisms. Dentinogenesis primarily includes two steps: secretion of proteins (e.g., collagen scaffold) and mineralization within the proteins. These components respectively act as steel and cement to provide the major mechanical strength of dentin.2 When dentin suffers small amounts of damage, odontoblasts, located in the dentin pulp cavity with their cytoplasmic extensions reaching to DTs, are stimulated and then produce restorative dentin. If a more serious injury occurs, dental pulp stem cells (DPSCs) in the pulp cavity will differentiate into odontoblasts to improve the efficiency of restorative dentin formation.14,15 Thus, with regard to DH and the repair of damaged dentin, both biomimetic dentin mineralization and restorative mineralization of DPSCs should be considered together.16 Inspired by the bottom-up biomineralization process, we propose to develop a novel sandwich desensitization strategy for DH treatment.
Intrafibrillar mineralization determining the mechanical properties of dentin is the key to the sandwich strategy. Extensive experiments demonstrated the effectiveness of PILP in mediating collagen mineralization, which is defined as an amorphous liquid-phase mineral precursor sequestered by biomimetic analogs of noncollagenous proteins, such as polyaspartic acid (PAsp), polyacrylic acid (PAA), poly(allylamine hydrochloride) (PAH) and poly(amidoamine) (PAMAM).17–20 Amorphous mineralization precursors with fluidic characteristics could infiltrate into the gap zone of collagen fibrils and then transform into needle-like HAp.21 Calcium phosphate is the primary mineral in vertebrates; therefore, as the first-formed mineral phase in biomineralization, amorphous calcium phosphate (ACP) is widely used in biomimetic mineralization.22 However, due to thermodynamic instability and rapid phase-transition properties, ACP usually ceases to be effective during the progress of mineralization and must be applied at high frequencies.23–25 To solve this problem, mesoporous silica nanoparticles (MSNs) were proposed in previous studies to load ACP and protect ACP from phase transition, and intrafibrillar collagen mineralization was verified.26,27 In addition, desensitizing material based on MSNs showed great propensity for acid resistance. Unfortunately, due to the hydrolysis of MSNs and the lack of new substances to fill the voids left after degradation, the long-term effect of DT occlusion showed a downward trend.28,29 Thus, more superior carriers for mineralization precursors are still required. As inert elements, titanium and zirconium are stable in biological environments. Many studies have shown that titanium–zirconium alloys have higher biomechanical strength, fracture toughness and corrosion resistance, similar to the related mesoporous materials.30–32 However, no related reports are available about titanium–zirconium-based delivery systems for precursor transportation.
To our knowledge, biomimetic strategies remain limited today. One target for biomimicry is the hierarchical structure of native dentin, which is primarily comprised of both intrafibrillar and extrafibrillar mineralization.2 Given the acid-resistance of silica, the chemical stability of titanium–zirconium and the biomimetic mineralization ability of PILP, we developed a PAH-ACP-loaded silica/mesoporous titanium–zirconium (STZ) yolk–shell nanocomposite (PSTZ) with acid and mechanical resistance. We comprehensively investigated its efficacy in mediating intrafibrillar mineralization of single-layer collagen fibrils, inducing remineralization of demineralized dentin matrix, occluding DTs and stimulating odontogenesis of human DPSCs (hDPSCs). On this basis, a sandwich dentin desensitization strategy with great potential for clinical application was proposed, which showed notable immediate and long-term desensitization effect by occluding the upper, middle and lower sections of the DTs.
Results and discussion
Characterization of the STZ and PSTZ nanocomposites
The synthesis process and typical morphology of STZ and PSTZ nanocomposites with yolk–shell structures are shown in Fig. 1A and Fig. S1.† Specifically, monodispersed SiO2 nanospheres with a diameter of 200 nm were used as a core, and then a hybrid shell with a thickness of 25 nm consisting of TiO2 and ZrO2 was formed. After ammonia etching, the SiO2 core was partially dissolved, and a cavity with a diameter of 50 nm was obtained. The final products were made up of a solid SiO2 core and mesoporous TiO2/ZrO2 shells (Fig. 1B). As shown in Fig. 1C, Ca and P elements were homogeneously distributed in the internal cavity of STZ nanospheres after PAH-ACP loading, illustrating that PAH-ACP precursors successfully infiltrated the PSTZ nanocomposites.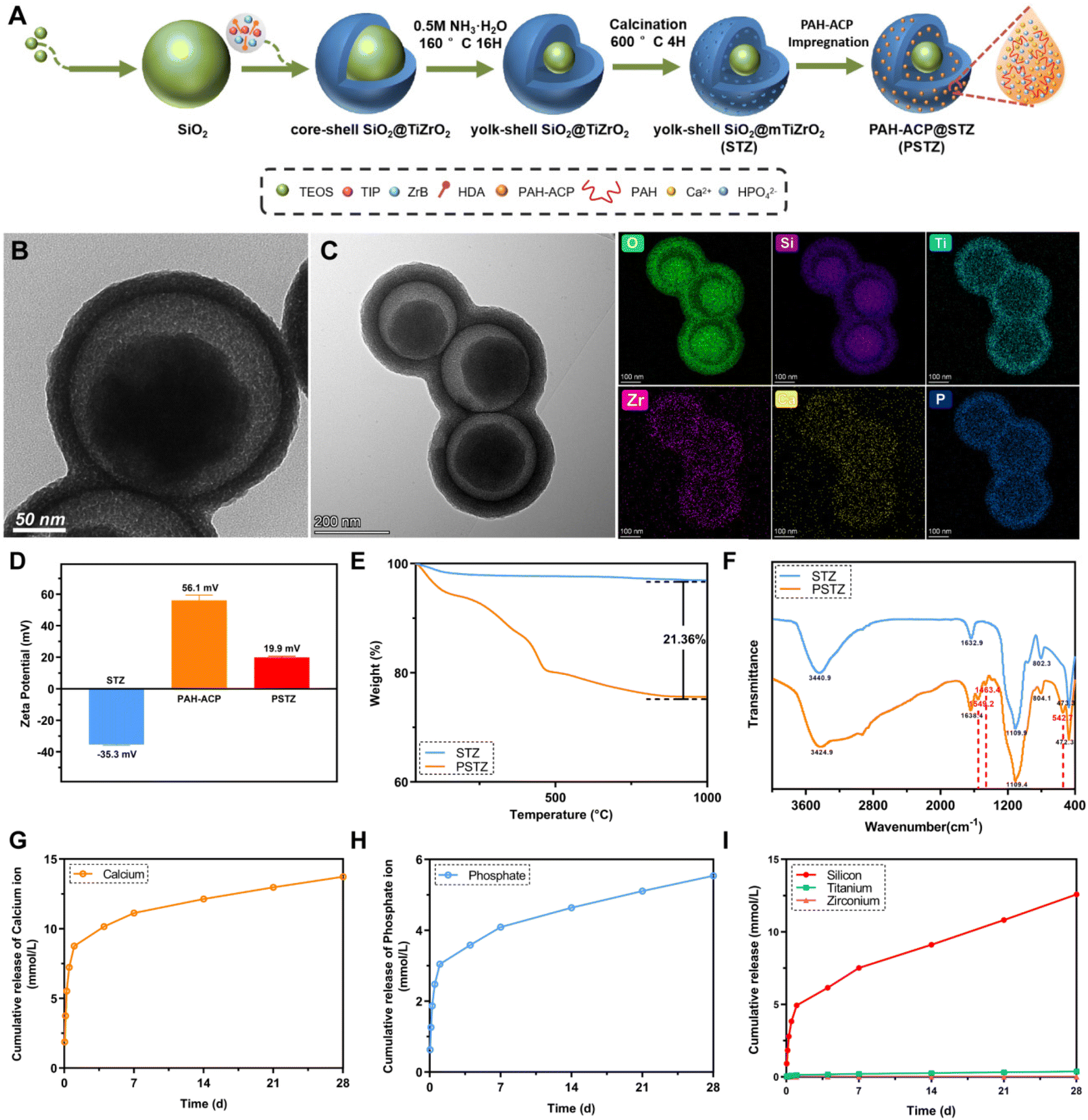 | ||
| Fig. 1 Characterization of STZ and PSTZ. (A) Schematic illustration of the synthesis process of STZ and PSTZ. (B) TEM microphotographs and corresponding elemental mapping of STZ and (C) PSTZ. (D) Zeta potential of STZ, PAH-ACP, and PSTZ. (E) TGA curves, (F) FTIR spectra of STZ and PSTZ. (G–I) Cumulative release profiles of Ca, P, Si, Ti, Zr from PSTZ in Tris-buffered saline (TBS) buffer for 28 days. | ||
The zeta potentials of STZ, PAH-ACP and PSTZ were −35.3 mV, 56.1 mV and 19.9 mV, respectively (Fig. 1D). The improved zeta potentials from STZ to PSTZ indicated that electrostatic action might be the primary mechanism for PAH-ACP precursor loading.26
Thermogravimetric analysis (TGA) showed that the weight loss of PSTZ was 21.36 wt% more than that of STZ (Fig. 1E), corresponding to the loading amount of PAH, suggesting that the amount of PAH-ACP adsorbed in STZ was higher than 21.36 wt%, outclassing previous materials.26,27,33 Also, the three-stage thermal degradation process curve for PSTZ represented the loss of loosely bound water below 25–150 °C, decomposition of organic polymer PAH in the range of 150–450 °C, and crystallization or phase transition beyond 450 °C.33
The molecular structure and chemical bonds of STZ and PSTZ were investigated using Fourier transform infrared (FTIR) spectra (Fig. 1F). For STZ, the broad absorption peak at 3440 cm−1 corresponded to the –OH stretching vibration, indicating water molecules associated with the nanocomposites.34 The peak at 1109 cm−1 was attributed to the Si–O–Si asymmetric stretching vibration, while the peaks at 802 cm−1 and 470 cm−1 were attributed to the symmetrical stretching vibration of Si–O.35 The peak at 1632 cm−1 was attributed to the bending vibration of the Ti–OH bond.36 Due to the integration of Zr ions with Ti, the peak of Zr–O stretching was shifted and an intense peak at 470 cm−1 was detected.37 Compared with STZ, PSTZ exhibited a broader peak at 1109 cm−1 (phosphate ν3 stretching vibration), and three additional peaks at 1549 cm−1 (N–H bending vibration), 1463 cm−1 (CH3 asymmetric deformation) and 543 cm−1 (phosphate ν4 bending vibration), indicating PAH-ACP loading.11,27 A single peak without splitting at ∼543 cm−1 was also observed, which shifted from 580 cm−1, due to the incorporation of PAH into ACP. The X-ray diffraction (XRD) pattern (Fig. S2†) of PSTZ showed two typical peaks: one was a broad peak at approximately 22°, suggesting that doping ZrO2 in TiO2 decreased the crystallinity and formed an amorphous titania–zirconia hybrid;36,38 and the other peak was a hump at 25–30°, confirming that PAH-ACP remained in an amorphous phase in agreement with the FTIR findings.19,39
The release profiles of Ca and P ions exhibited burst release within 24 h, followed by a slow sustained release for 28 days (Fig. 1G–I). In addition, the cumulative release amount was markedly higher than that in a previous study,27 reaching up to 13.7 mM and 5.5 mM for Ca and P, respectively, which might be attributed to the cavity of the yolk–shell structure providing sufficient capacity for PAH-ACP. In addition, the release of Si rose with time, which was reported to be advantageous for stimulating the mineralization process of stem cells.40,41 Conversely, the release of Ti and Zr was nearly undetectable, which implied that the shell of STZ does not dissolve in common solution, thereby preserving the long-term efficacy of DT occlusion.
PSTZ-mediated intrafibrillar mineralization of reconstructed collagen fibrils
Collagen fibrils, particularly type-I collagen, are the most abundant organic substances in dentin. Biomineralized collagen fibrils significantly enhance mechanical strength by combining with minerals to form an organic–inorganic hybrid.2 In this study, the capacity of the yolk–shell PSTZ nanocomposite to mediate intrafibrillar mineralization of collagen fibrils was evaluated.A single-layer type-I collagen model was constructed to investigate the mineralization process. As shown in Fig. 2A1 and Fig. S3A,† the reconstituted collagen fibrils stained by osmium tetroxide displayed a characteristic D-band pattern with an axial length of 67 nm as a result of the staggered arrangement of gap and overlap regions.42 In addition, the crystal diffraction ring was not present in SAED (inset in Fig. 2A1), and there was no Ca or P in the element mapping (Fig. 2A3 and A4), indicating that there were no minerals within the reconstituted collagen fibrils before mineralization.
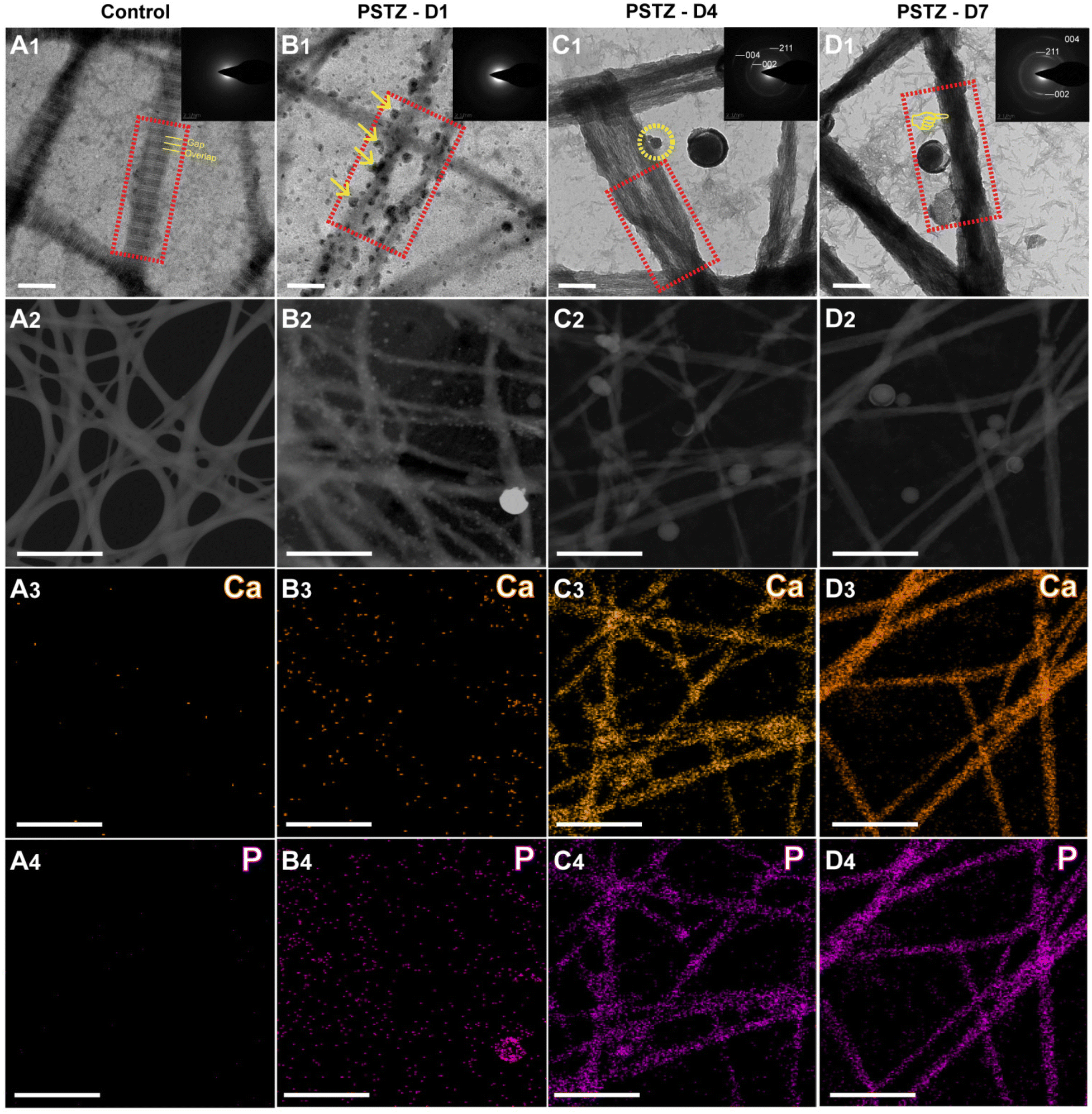 | ||
| Fig. 2 PSTZ-mediated intrafibrillar mineralization of reconstructed collagen fibrils. (A1–D1) High magnification of representative TEM images and SAED patterns of stained unmineralized collagen fibrils (A1) and PSTZ-mediated intrafibrillar mineralization after 1 d (B1), 4 d (C1), and 7 d (D1) incubation (scale bar, 200 nm). (A1) The periodic D-band pattern related to gap zone and overlap zone (yellow dotted line) was evident in the control group. (B1) At 1 d, mineralization precursors (yellow arrow) were released from the PSTZ and attached to the surface of collagen fibrils. (C1) At 4 d, collagen fibrils were partially mineralized and mineralization precursors (yellow circle) continued to infiltrate into the fibrils. (D1) At 7 d, complete intrafibrillar mineralization was observed. Red rectangles present the morphological changes of collagen fibrils during mineralization process. Inset: Corresponding SAED pattern. (A2–4–D2–4) Electronic images and corresponding elemental distribution of Ca and P (scale bar, 1 μm). | ||
After incubation in PSTZ-containing solution for 1 d, a lot of spherical liquid precursor PAH-ACP was released from the PSTZ nanocomposite, which proceeded to the surface of the collagen fibrils and arranged along the c-axis (Fig. 2B1 and Fig. S3B†). The electrostatic attraction of positively charged nucleation sites along the collagen fibrils or the osmotic pressure generated by the polyelectrolyte might be the driving force.43,44 SAED (inset in Fig. 2B1) showed a broad diffraction band, and element mapping (Fig. 2B3 and B4) revealed that Ca and P were dispersed outside of collagen fibrils. Both results confirmed that the liquid precursor was ACP clusters and that collagen fibrils were undergoing pre-mineralization.
When the mineralization period was prolonged to 4 days, discontinuous partial mineralization occurred (Fig. S3C†). From the image at high magnification (Fig. 2C1), the majority of the amorphous precursors were transformed into oriented needle-like crystals within collagen fibrils. Insufficiently, the overall density of mineralized collagen fibrils was relatively low, and some non-mineralized regions were still observed.
Complete intrafibrillar mineralization occurred after 7 days of incubation. The electron density of mineralized collagen fibrils demonstrated in Fig. 2D1 and Fig. S3D† was denser than that at 4 days (Fig. 2C1), indicating an increased mineralization degree. SAED analysis (inset in Fig. 2C1 and D1) of mineralized collagen fibrils showed three characteristic polycrystalline diffraction rings at surfaces (002), (211) and (004), which verified that the formed crystals were hydroxyapatite.45 According to the element mapping (Fig. 2C3, C4 and D3, D4), Ca and P were evenly distributed within collagen fibrils, confirming intrafibrillar mineralization. One interesting finding was that the electron density of Ca and P loaded in the PSTZ nanocomposite at 7 days also markedly decreased compared to that at 1 day, which was observed in the element mapping (Fig. 2B3, B4 and D3, D4). This phenomenon illustrated that the ACP precursors triggering intrafibrillar collagen were released from the PSTZ nanocomposite.
In conclusion, we demonstrated the capacity of the PSTZ nanocomposite to mediate intrafibrillar mineralization. Although a delivery system for mineralization precursors has been proposed to prompt intrafibrillar mineralization,26,27,33 this was the first time that the specific mineralization process from the release of precursors to the completion of mineralization, including amorphous-to-crystal transition, has been observed and described in detail. PSTZ-mediated intrafibrillar mineralization of reconstructed collagen fibrils consisted of the following processes: self-assembly of type-I collagen fibrils, delivery of PAH-ACP precursors from PSTZ nanocomposite to collagen fibrils, and partial and complete intrafibrillar mineralization.
PSTZ-mediated remineralization of demineralized dentin matrix
Dentin is an abundant collagenous structure, primarily consisting of type-I collagen fibrils mineralized intrafibrillarly/extrafibrillarly with carbonated HAp.2 In view of the complexity of the oral environment, dentin demineralization is ubiquitous in daily life. The demineralized collagen network is a weak layer that affects the mechanical properties or dentin bonding durability.46 Therefore, remineralization of demineralized dentin matrix (DDM) has emerged as a novel and successful strategy for dentin interface management.47 DDM is a suitable biomimetic mineralization model,2 but is rarely used in research on intrafibrillar mineralization, perhaps due to the high technical sensitivity of creating a three-dimensional demineralized collagen network and difficulty in observing the mineralization process. Based on the success of PSTZ-mediated intrafibrillar mineralization of single-layer collagen fibrils, we investigated the mineralization efficiency of PSTZ on DDM, a three-dimensional model.DDM was created by etching with 37% phosphoric acid for 20 s, fully flushing and gently blowing before immersion in mineralizing solution. As shown in Fig. 3A1–A4, both intrafibrillar and extrafibrillar minerals of the dentin surface were removed, and a typical periodic pattern of collagen fibrils was visible, ensuring mineral dissolution within collagen. In addition, collagen fibrils were generally fractured in the absence of inorganic mineral support, of which elastic modulus and hardness were decreased to 0.73 GPa and 0.03 GPa, respectively (Fig. 3E–G).48 The results of energy dispersive X-ray spectroscopy (EDX) analysis (Fig. S5A1–A4†) demonstrated that Ca and P were scattered over the dentin surface at relatively low concentrations.
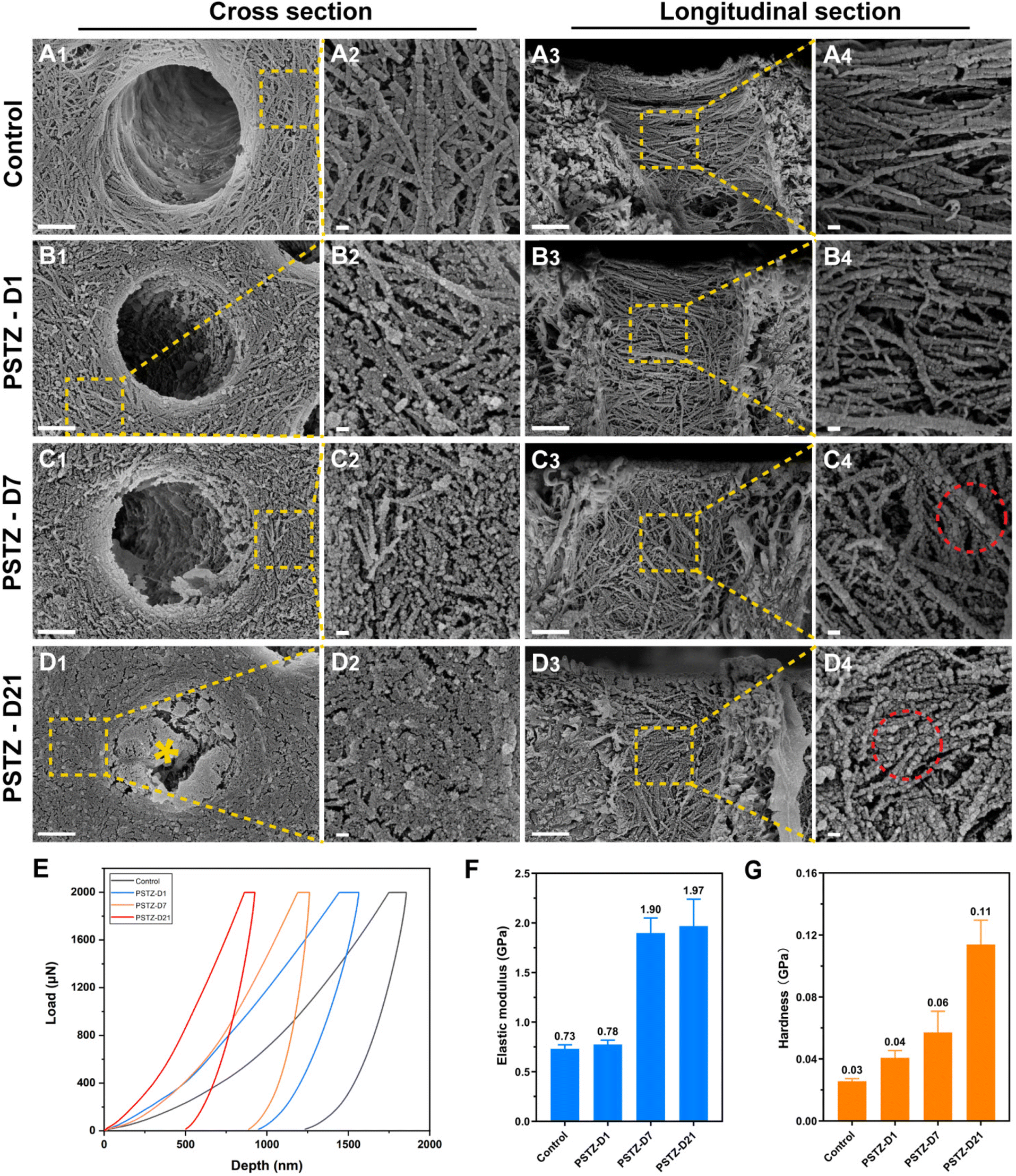 | ||
| Fig. 3 PSTZ-mediated remineralization of demineralized dentin matrix (DDM). (A–D) Representative FESEM images of DDM prepared by 37% phosphoric acid etching (A). DDM incubated in PSTZ-containing solution for 1 d (B), 7 d (C), and 21 d (D). Cross and longitudinal section images are shown in A1–D1, A2–D2 and A3–D3, A4–D4, respectively. High-magnification images (A2–D2 and A4–D4) were magnified from the selected area (yellow square) in the low-power image (A1–D1 and A3–D3). (Yellow asterisk: deposited mineral; red circle: extrafibrillar remineralization.) Scale bar in (A1–D1 and A3–D3), 1 μm; in (A2–D2 and A4–D4), 100 nm. (E) Typical loading–unloading curves extracted from nanoindentation tests at remineralization time points. (F) Elastic modulus of DDM and remineralized dentin matrix. (G) Hardness of DDM and remineralized dentin matrix. | ||
After immersion in PSTZ-containing solution for 1 day, the dentin collagen fibrils were attached by a large number of spherical particles, which likewise appeared in the single-layer collagen fibril model (Fig. 3B1–B4). However, different from the control group, the periodic pattern of collagen fibrils was no longer apparent in the PSTZ-D1 group, indicating that PAH-ACP precursors had penetrated into collagen fibrils. The electron densities of Ca and P significantly increased, particularly in the areas within the DTs (Fig. S5B1–B4†). This result could be related to the fact that the three-dimensional structure of DTs is suitable for PAH-ACP deposition and difficult to wash away during sample preparation. On the contrary, PAH-ACP on the surface of dentin specimens might be washed away in the aforesaid process, thus the modulus and hardness were similar to those of the control group (Fig. 3E–G).
After remineralization for 7 days, dentin collagen fibrils with increased width were found in FESEM images (Fig. 3C1–C4), confirming intrafibrillar mineralization. Furthermore, remineralized collagen fibrils presented a continuous state, indicating that the internal minerals provided support for the collagen network. Additionally, numerous spherical PAH-ACP precursors were attached to the collagen surface and even agglomerated into larger spheres, owing to the long-term delivery from PSTZ nanocomposite. EDX results (Fig. S5D1–D4†) showed that Ca and P contents in the PSTZ-D7 group dramatically increased when compared to the PSTZ-D1 group, and the hardness and elastic modulus (Fig. 3E–G) of remineralized dentin matrix were significantly higher than those of the control, implying a higher mineralization degree.
With a prolonged remineralization time of 21 days, extrafibrillar mineralization was observed in conjunction with intrafibrillar mineralization from a cross-sectional view (Fig. 3D1 and D2). DDM was coated by crystals to form a homogeneous surface. In addition, the elastic modulus was no different from that of the PSTZ-D7 group, but hardness was increased from 0.06 GPa to 0.11 GPa (Fig. 3E–G), which could be attributed to the synergistic action of intra- and extra-mineralization. Similarly, the EDX results (Fig. S5F1–F3†) also showed that Ca and P were distributed uniformly over peritubular and intratubular dentin. Compared with the PSTZ-D7 group, there was no variation in electron density within the DTs. Thus, we hypothesized that the marked increase in Ca and P contents were primarily caused by extrafibrillar mineralization (Fig. S5F4†). From the longitudinal view (Fig. 3D3 and D4), the diameter of spherical particles adhering to collagen fibrils increased, as a consequence of the further deposition of supersaturated Ca and P ions.
In conclusion, the 3D model of DDM and its remineralization with the PSTZ nanocomposite was successfully constructed. DDM was remineralized in the following order: intrafibrillar mineralization, then extrafibrillar mineralization. This process followed the bottom-up mineralization approach,49 and allowed the reconstruction of the complex hierarchical structure of dentin. The early process of collagen mineralization was the same as that in the single-layer model. The PSTZ nanocomposite released PAH-ACP, which then stimulated intrafibrillar mineralization. However, extrafibrillar mineralization occurred later, which was attributed to the specific confined environment provided by DDM.50 The degree of external mineralization improved as the mineralization time increased, which was corroborated by the FESEM images and EDX analysis of the remineralized samples on day 4 and day 14 (Fig. S4 and S5†). Therefore, a bottom-up mineralization strategy based on biomimetic remineralization has promising applications in dentin interface management.
PSTZ-mediated occlusion of dentinal tubules
According to hydrodynamic theory, the occlusion of DTs is regarded as an effective strategy to alleviate DH. In the past decade, the physically occlusion technique has received the most attention, yet the long-term effect diminishes because few minerals have been developed over time.7 After verifying the outstanding mineralization capability of the PSTZ nanocomposite through the above experiments, we considered whether this nanomaterial, which contains both a yolk–shell carrier and a liquid-phase mineralization precursor, can provide a time-independent effect on DT occlusion.FESEM observation and dentin permeability assessment were performed to achieve qualitative and quantitative examination of DT occlusion. Considering the complex and dynamic oral environment, an acid challenge subgroup and mechanical brushing subgroup were used to simulate food-induced pH changes and chewing- or brushing-induced friction, respectively. A modified fluid permeation device (schematic diagram is shown in Fig. 4A) was used to test dentin permeability. The permeability (Fig. 4B) was expressed as a percentage (Lp%), with the average Lp of etched dentin disks being determined to be 100%. The morphology of dentin specimens was analyzed from cross sections and longitudinal sections (Fig. 5 and 6), which highlighted the occluding efficiency and depth, respectively.29
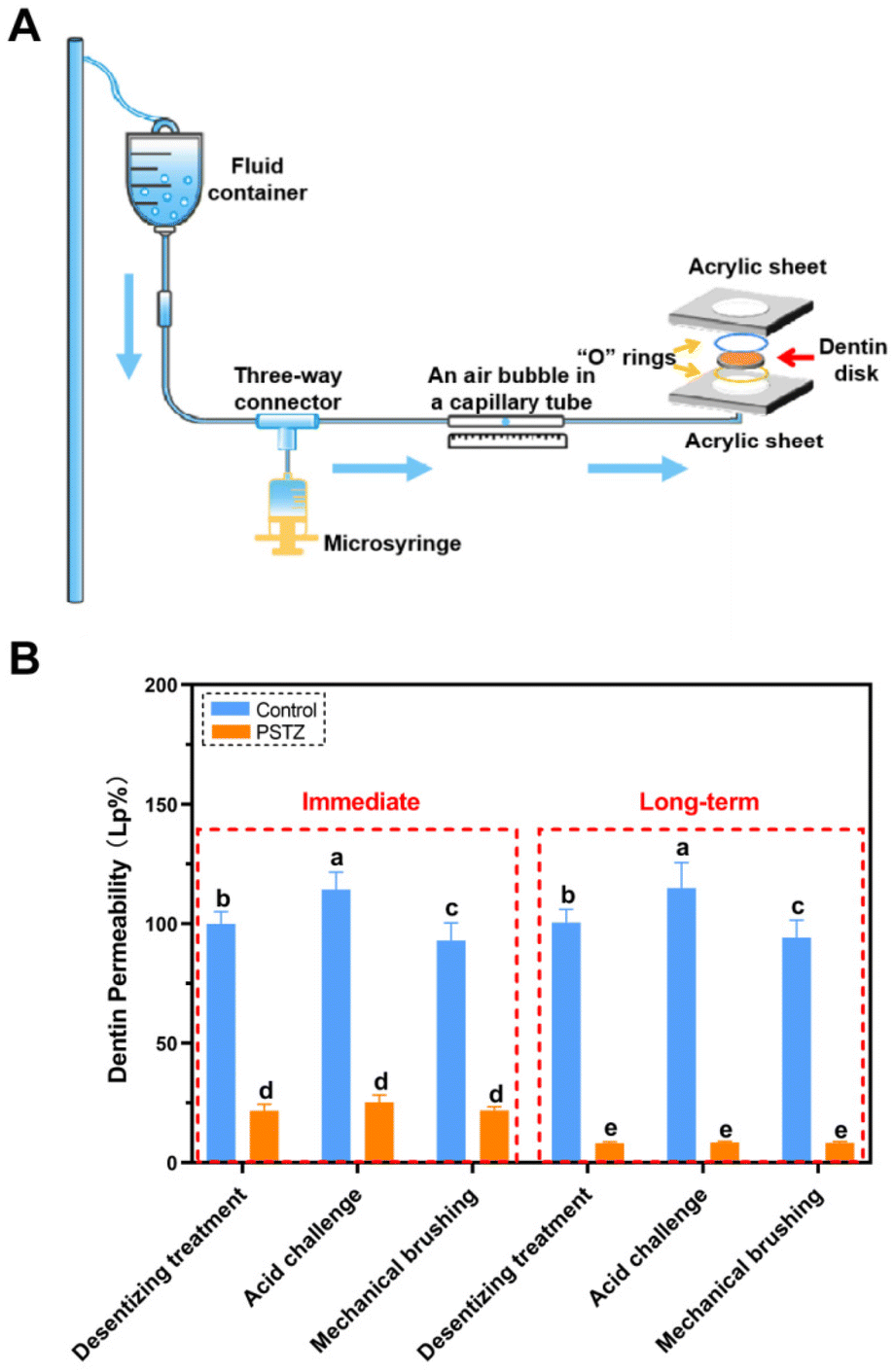 | ||
| Fig. 4 Evaluation of dentin permeability. (A) Schematic diagram of modified fluid permeation device for dentin permeability assessment. (B) Dentin permeability values obtained from different subgroups, after incubation for 0 or 28 days. The average Lp value of etched dentin disks (N = 12) was determined as 100% dentin permeability. Different letters (a–e) indicate significant differences (p < 0.05). | ||
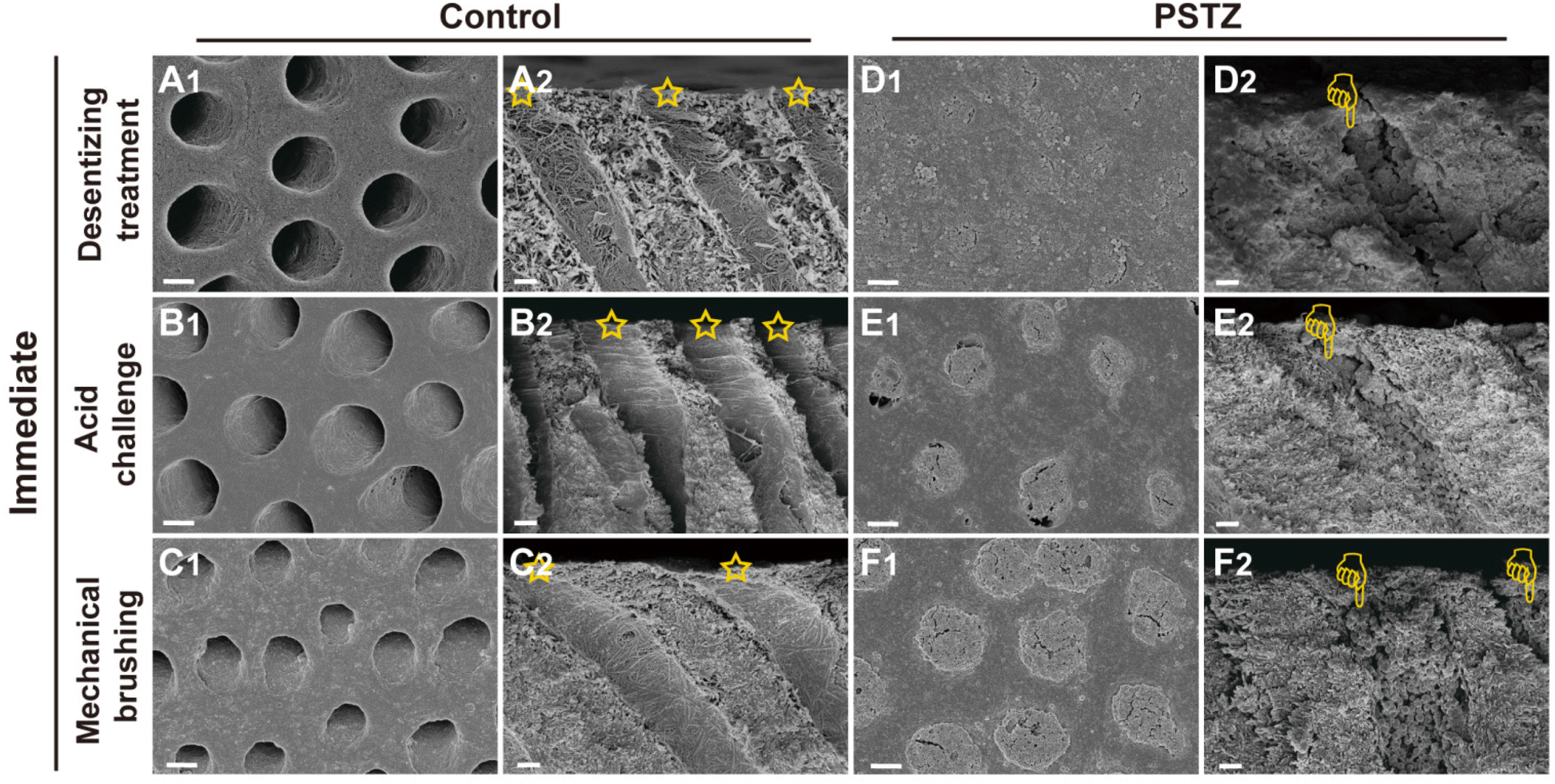 | ||
| Fig. 5 Representative FESEM images of PSTZ-mediated immediate DT occlusion from cross and longitudinal sections. FESEM images of (A and D) desensitizing treatment subgroup, (B and E) acid challenge subgroup and (C and F) mechanical brushing subgroup of the control and PSTZ group. (Yellow pentagram: patent tubules in the control group; yellow finger: tubules blocked with PSTZ nanocomposites.) Scale bar in (A1–F1), 2 μm; and in (A2–F2), 1 μm. | ||
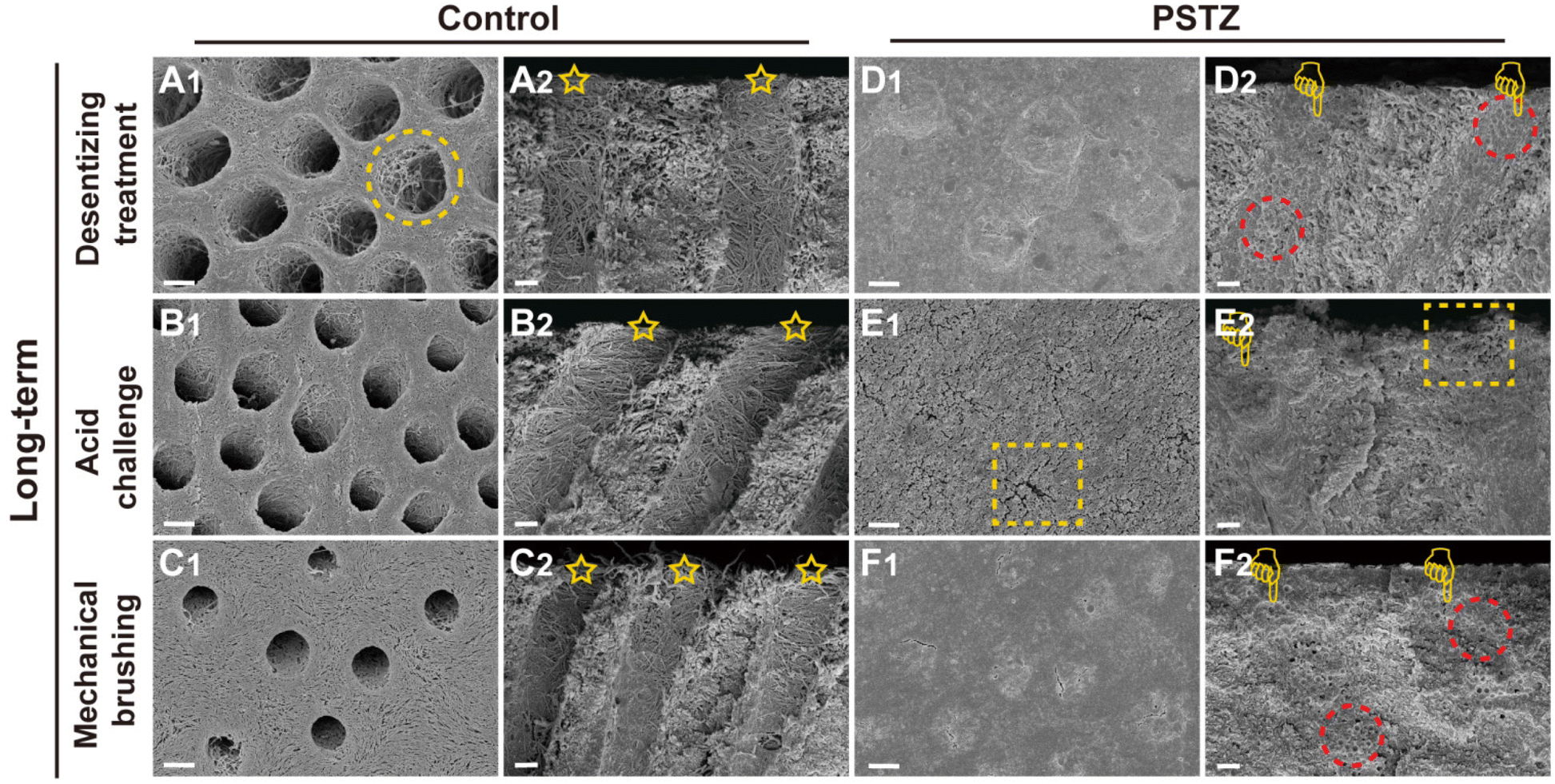 | ||
| Fig. 6 Representative FESEM images of PSTZ-mediated long-term DT occlusion from cross and longitudinal sections. FESEM images of (A and D) desensitizing treatment subgroup, (B and E) acid challenge subgroup and (C and F) mechanical brushing subgroup of the control and PSTZ group. (Yellow circle: demineralized dentin collagen fibrils; yellow pentagram: patent tubules in the control group; yellow finger: tubules blocked with PSTZ nanocomposites; red circle: PSTZ nanocomposites embedded in hydroxyapatite; yellow rectangle: a little exposure of blocked tubules.) Scale bar in (A1–F1), 2 μm; and in (A2–F2), 1 μm. | ||
As shown in Fig. 5A–C, all DTs of the control group were exposed, and collagen fibrils were demineralized. After citric acid challenge for 1 min, the diameter of tubule orifices was enlarged (Fig. 5B1 and B2). Simultaneously, increased permeability was detected (Fig. 4B). After mechanical brushing for 3 min, the surface of the dentin disks became rough, and the collagen fibrils were deformed (Fig. 5C1 and C2). Particularly in Fig. 5C2, the tubule orifices were even covered by collapsed collagen fibrils, indicating that the mechanical characteristics of demineralized dentin had markedly diminished. In addition, although the internal diameter of tubules remained unchanged, the dentin permeability increased due to the smaller orifice diameter (Fig. 4B).
In contrast, the PSTZ-treated group showed that DTs were primarily filled with materials, and the permeability was markedly reduced (Fig. 5D1 and D2). After acid etching, blockages were marginally dissolved, and the corresponding permeability increased to some extent (Fig. 5E1, E2 and 4B). However, plugging in this study was noticeably stronger than that in previous studies.28,29 This result was primarily attributable to the core of the PSTZ nanocomposite, which was composed of silica with strong acid resistance. Also, after mechanical brushing challenge, tubules were blocked more tightly owing to the vertical force (Fig. 5F1 and F2). This result was also supported by the lower permeability (Fig. 4B).
In general, the immediate effect of PSTZ-mediated occlusion of DTs exceeded the proposed expectations. Encouraged by this, a long-term experiment was conducted to validate whether the PSTZ nanocomposite can introduce newly formed minerals by releasing PAH-ACP precursors, thereby improving the long-term tubule plugging effect. Thus, half of the dentin specimens were immersed in artificial saliva for 28 days, and then the long-term occluding effects of PSTZ were evaluated by the same methods.
FESEM images (Fig. 6A–C) revealed that a demineralized collagen network with more evident pores existed in the control group because collagen fibrils were hydrolyzed during long-term storage. Similarly, the diameter of tubules increased in the acid challenge subgroup (Fig. 6B1 and B2) while being decreased in the mechanical brushing subgroup (Fig. 6C1 and C2). The surface collagen network of specimens in the long-term group was flattened after mechanical brushing for 3 min, implying that mechanical properties might be impaired. The permeability of the above three subgroups was marginally higher than that of the immediate group, but there was no statistically significant difference (Fig. 4B).
In contrast, the long-term PSTZ-treated group demonstrated excellent plugging. In the desensitizing treatment subgroup (Fig. 6D1 and D2), we discovered that spherical PSTZ nanocomposite particles were surrounded with newly generated crystals, forming a solid structure similar to intertubular dentin. In the acid challenge subgroup (Fig. 6E1 and E2), only a few minerals at the orifice were dissolved. In the mechanical brushing subgroup (Fig. 6F1 and F2), tiny cracks induced by mechanical stress were detected at the edge of the DT orifices. We inferred that the above crystals were formed under the induction of PAH-ACP released from the PSTZ nanocomposite. The blockages in the lower segment of tubules were not affected due to the high acid resistance of silica and the excellent mechanical strength of titania and zirconia.31,32,51,52 In addition, the permeability data (Fig. 4B) were consistent with the FESEM observations, which were lower than those of the immediate group, indicating that the participation of remineralization promoted DT occlusion.
A kind of composite material with great stability and long-term effectiveness was constructed and applied to dentin desensitization treatment. The proposed immediate and long-term experimental results jointly showed that the combined application of physical desensitization and biomimetic mineralization was a feasible strategy to improve the plugging effect of DTs.
Biocompatibility of STZ and PSTZ nanocomposites
Biocompatibility assessment of nanocomposites is required before biomedical application, of which hemocompatibility and cytocompatibility are two important evaluation indices.53 First, the hemolytic behaviors of STZ and PSTZ nanocomposites towards red blood cells (RBCs) were evaluated using a hemolysis assay, which is considered to be a reliable way to estimate the blood compatibility of bioactive nanomaterials.54 Deionized water and PBS buffer acted as the positive control and negative control, respectively. As shown in Fig. S6,† the hemolytic activity of STZ and PSTZ nanocomposites at representative gradient concentrations was markedly lower than 5% hemolysis, which is the minimum permissible value for biomaterials.55 The high blood compatibility was mainly due to the spatial placement of silica, which was located inside STZ nanoparticles. The silanol groups did not contact RBCs, thereby RBC rupture induced by the binding of silanol groups to cell membranes was avoided. In addition, the role played by the chemical inertness of titania and zirconia as well as the large size of STZ and PSTZ nanoparticles could not be ignored.56,57Second, the cytotoxicity of STZ and PSTZ nanocomposites on hDPSCs was investigated by CCK-8 assay. After incubation in STZ-containing culture medium at various doses for 1, 4, and 7 days, a very small decrease in cell activity was only detected at a concentration of 1280 μg mL−1 (Fig. 7A). PSTZ-containing culture medium at all concentrations showed no cytotoxicity on the 1st and 4th days (Fig. 7B). Despite the decrease in the high concentration group, the relative cell viability was still higher than 85% (Fig. 7A and B). Due to the great biocompatibility and bioactivity of silica, the cell activity of both the STZ and PSTZ groups at medium concentrations was higher than that of the control group, demonstrating that the dissolved Si ion promoted cell proliferation as well.58 In addition, previous studies have shown that Ca ions also have a positive regulatory effect on cell proliferation.59
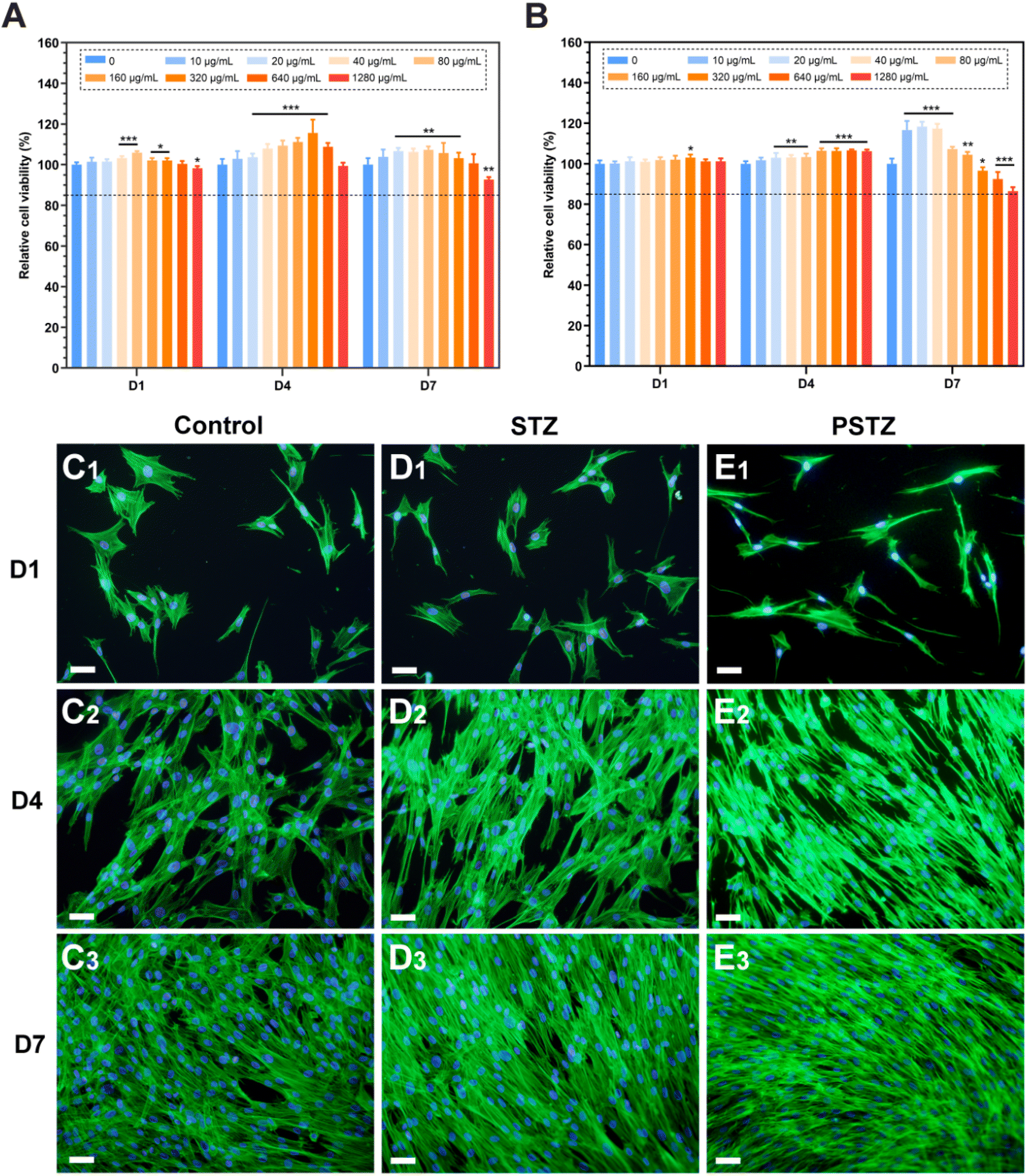 | ||
| Fig. 7 Biocompatibility of STZ and PSTZ. (A and B) CCK-8 assay of different concentrations of STZ and PSTZ on hDPSCs (N = 5). Data are presented as relative cell viability (%) compared with the control group (0 μg mL−1) at the same time point. Statistical analysis was performed against the corresponding control group (0 μg mL−1). *p < 0.05, **p < 0.01, ***p < 0.001. Dotted line: 85% relative cell viability. (C–E) Representative CLSM pictures from the control, STZ and PSTZ groups, after incubation for 1 d (C1–E1), 4 d (C2–E2) and 7 d (C3–E3). Scale bar = 100 μm. | ||
Third, FITC-phalloidin staining was applied for morphological analysis. Green and blue fluorescence represent F-actin stained with FITC-phalloidin and nuclei stained with DAPI, respectively. The STZ and PSTZ groups at 80 μg mL−1 were observed as representatives (Fig. 7D and E), and this concentration was then used to induce odontogenic differentiation. The hDPSCs in the STZ group were polygonal with apparent filamentous pseudopodia, which was consistent with the control group (Fig. 7C). Compared with the other two groups, some cells in the PSTZ group shrank marginally, and their pseudopodia decreased (Fig. 7E1 and E2), owing to the ion concentration.60 However, as culture time was increased, there was no marked variation in cell density, echoing the results of the CCK-8 assay. The STZ and PSTZ nanocomposites were generally biocompatible, which provided the required elements for cell proliferation by dissolving Ca, P and Si ions.
In vitro odontogenesis of hDPSCs induced by STZ and PSTZ nanocomposites
As aforementioned, because of the physiological structure of the pulp–dentin complex, DPSCs differentiate into odontoblasts and form reparative dentin when suffering outside incitement from tubules.14–16 Inspired by the natural biological defense mechanism, we inferred that the formation of reparative dentin was an entry point for DH treatment. However, there are a limited number of relevant studies and insufficient evidence. In view of the excellent plugging depth exhibited by the PSTZ nanocomposite, it or its exudate had the opportunity to contact hDPSCs. If the PSTZ nanocomposite could promote the odontogenic differentiation of hDPSCs, it is expected to achieve the proposed envisaged sandwich desensitization strategy. Calcium, phosphorus and silicon are critical regulatory elements in the formation and calcification of biomineralized tissues. Bioactive materials containing the above three elements are widely used in hard tissue engineering.61,62 To verify the possibility of reparative dentin formation induced by tubule-occluding materials, nanocomposites with liquid mineral precursor and continuously dissolved Si ions were tested for their ability to synergistically induce the odontogenesis of hDPSCs.The ions released by the occluding materials penetrate into the pulp cavity through the DTs, and inevitably, their concentration decreases as the distance increases.63 Considering the transport efficiency of released ions, mineralization medium containing STZ and PSTZ nanocomposites at a concentration of 80 μg mL−1 was used for biomineralization of hDPSCs.
ALP staining, ALP activity assays and Alizarin Red S staining were conducted to detect the ALP activity and the mineralization degree of the extracellular matrix. According to the ALP staining results (Fig. 8A), the PSTZ group showed the highest ALP activity after 7 days of mineralization induction. The ALP activity of the STZ group was marginally higher than that of the control group. These results were supported by the quantitative results of the ALP activity assay (Fig. 8C). After 21 days of cultivation, red-stained calcium nodules were observed in all groups (Fig. 8B), which were formed by small calcium phosphate crystal precursors and lipids secreted by odontoblasts/osteoblasts attracting calcium during mineralization.64 Significantly increased calcium salt deposition in hDPSCs was found in the PSTZ group, which then formed hydroxyapatite crystals, exhibiting the best odontogenic induction ability. Similar to findings from previous studies,61,62 Si ions released from STZ nanocomposites act individually; thus, the degree of mineralization of the STZ group was higher than that of the control group.
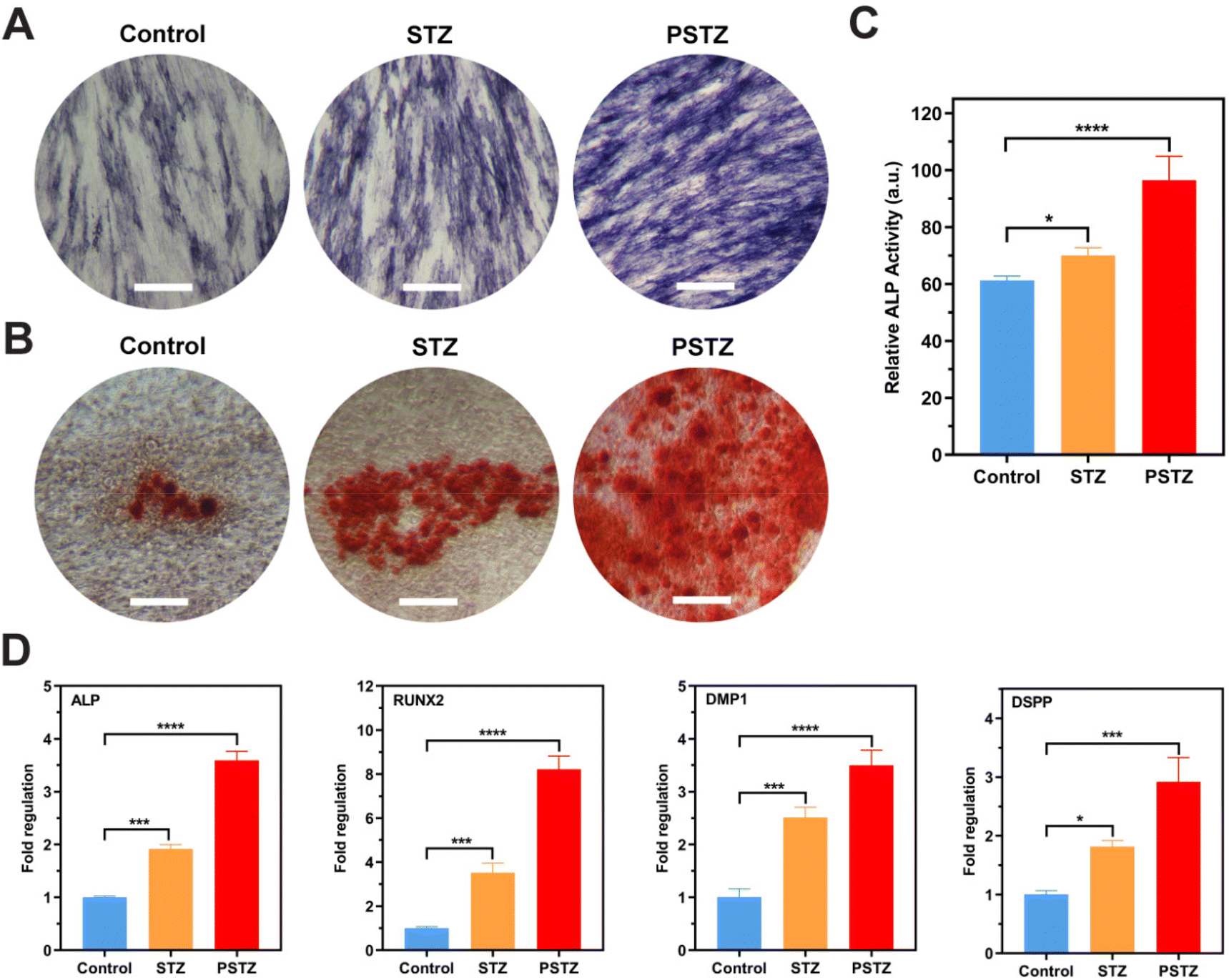 | ||
| Fig. 8 In vitro odontogenesis of hDPSCs induced by STZ and PSTZ. (A) ALP staining after hDPSCs cultured in odontogenic media for 7 d. Scale bar, 500 μm. (B) Alizarin Red S staining of the control, STZ, and PSTZ groups after 21 days of induction. Scale bar, 500 μm. (C) ALP activity (N = 3) after hDPSCs cultured in odontogenic media for 7 d. (D) RT-qPCR analysis of odontogenic-related gene (ALP, RUNX2, DMP1, DSPP) expression of hDPSCs after 7 days incubation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
The expression of odontogenesis-related genes was measured by RT-qPCR on the 7th day to demonstrate odontogenic differentiation potential (Fig. 8D). Among the genes, ALP and RUNX2 are critical and necessary in the early stage of odontogenic and osteogenic differentiation.65 DMP1 and DSPP are classic odontoblastic markers that are expressed in odontoblasts, and their presence is necessary during dentin matrix mineralization.66 DSPP induces mineralization in the extracellular matrix, while DMP1 directly regulates cytodifferentiation of hDPSCs into odontoblasts in both the early and advanced stages of the odontogenesis process.67Fig. 8D shows a statistically significant upregulation of the ALP, RUNX2, DMP1 and DSPP genes in the STZ and PSTZ groups compared to the control group. Consistent with the above results, the PSTZ group was the best performer, indicating that Ca and P ions played the most critical roles in biomineralization as the two critical determinants for bone mineralization.68,69 According to the literature,60,70 as a nonnegligible factor for cell fate determination, Ca ions at an optimal concentration could stimulate and maintain hDPSC-mediated mineralization. Ca ions were reported to regulate the odontogenic differentiation of hDPSCs by producing a local microenvironment with high concentrations that could activate stem cells and induce cell migration, or binding to specific receptors bound to the plasma membrane and then upregulating the gene expression of odontogenesis-related proteins, which could change cellular functions, or decreasing capillary permeability, which could lead to an increase in the concentration of calcium-dependent pyrophosphatase that inhibits metabolic mineralization.63 Also, some studies reported that P ions have a more prominent role in the regulation of bone mineralization. The lack of P ions could lead to osteomalacia regardless of the level of Ca ions.71 P ions participated in the mineralization process by affecting the rate of HAp formation, regulating the function of extracellular matrix proteins in mineralization and the expression of genes involved in osteogenic/odontogenic differentiation and the mineralization process, which was observed in experimental results.68 In addition, the odontogenic induction effect of the STZ group was also enhanced compared with that of the control; thus, we speculate that Si ions induced biomineralization via a synergistic effect with Ca and P ions.72
In general, the PSTZ nanocomposite greatly increased the activity of ALP, the amount of mineralized nodules and the expression of mineralization-related genes, thereby promoting odontogenesis of hDPSCs. According to previous research, newly differentiated cells activated by biological molecules could synthesize dentin matrix, stimulate and expedite reparative dentin formation and form a calcific barrier between the occluding materials and underlying pulp tissue.73 Proteins associated with odontogenic differentiation were shown to induce odontogenesis differentiation, promote tertiary dentin formation in vivo and reduce the symptoms of DH.74 By analogy, the PSTZ nanocomposite has a lot of potential for inducing the formation of tertiary dentin, for which direct evidence should be thoroughly investigated in future work. In this study, we validated the potential of the PSTZ nanocomposite to promote biomineralization and introduced a biological strategy for DH therapy, including odontogenic differentiation of stem cells.
Conclusions
In summary, we successfully constructed a PAH-ACP-loaded silica/mesoporous titanium–zirconium yolk–shell nanocomposite with acid and mechanical resistance and developed a sandwich desensitization strategy based on a bioinspired concept (Fig. 9). The sandwich strategy refers to physical occlusion of DTs by the PSTZ nanocomposite, collagen mineralization mediated by PAH-ACP released from the PSTZ, and restorative odontogenesis induced by Si, Ca and P ions dissolved from the PSTZ in the upper, middle and lower sections of DTs, respectively. Results showed that the PSTZ nanocomposite with biomimetic mineralization properties applied to the DH model achieved both excellent immediate and long-term DT-occluding effects. Accompanied by the reprinted hierarchical structure of dentin, we propose that this strategy based on biomimetic mineralization is innovative, of significance and has stability and shows great promise for clinical DH management.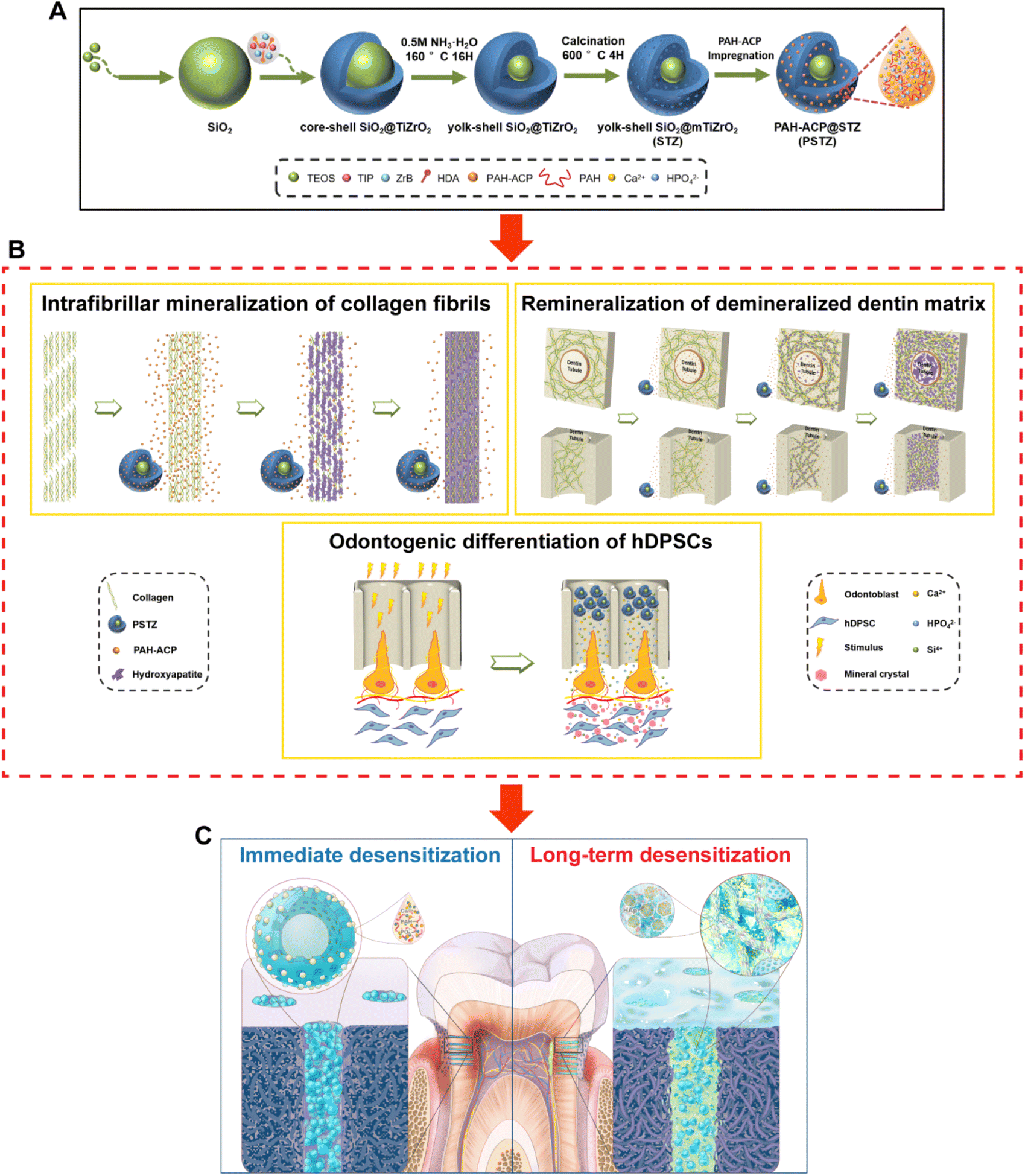 | ||
| Fig. 9 Graphical illustration for the biomineralization-inspired sandwich dentin desensitization strategy based on PSTZ multifunctional nanocomposite. (A) Schematic illustration of the synthesis process and PSTZ. (B) Schematic diagram of PSTZ-mediated intrafibrillar mineralization of reconstructed collagen fibrils, remineralization of DDM and odontogenesis. (C) Schematic illustrating the mechanism of the biomineralization-inspired sandwich dentin desensitization strategy. Left: Immediate desensitization induced by physical occlusion of DTs with the PSTZ nanocomposite. Right: Long-term desensitization induced by intrafibrillar and extrafibrillar mineralization, and restorative odontogenesis. | ||
Experimental section
Materials
Calcium chloride dihydrate (CaCl2·2H2O), disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O), calcium chloride hexahydrate (MgCl2·6H2O), potassium chloride (KCl), potassium dihydrogen phosphate (KH2PO4), ethanol, ammonia and 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Poly(allylamine hydrochloride) (PAH), titanium(IV) isopropoxide and zirconium butoxide were obtained from Aladdin Co., Ltd (Shanghai, China). Tetraethyl orthosilicate (TEOS), hexamethyldisilazane (HMDS), 0.5% Triton X-100, and 4% paraformaldehyde were purchased from Sigma-Aldrich (St Louis, MO, USA). Bovine serum albumin (BSA), 4′,6-diamidino-2-phenylindole (DAPI), BCIP/NBT Alkaline Phosphatase Color Development Kit and ALP activity assay kit were purchased from the Beyotime Institute of Biotechnology (Shanghai, China). Other chemicals and solvents without special mention were obtained from Thermo Fisher Scientific Inc. (USA). All materials were used as received.Synthesis of STZ nanospheres
STZ nanospheres were fabricated via a previously described technique and slightly improved for higher loading efficiency.38 Firstly, a modified Stöber method was adopted to prepare 200 nm SiO2 nanospheres as the core of STZ.75 In brief, 110 mL of ethanol, 40 mL of deionized water and 22.6 mL of concentrated ammonia were mixed and stirred at 35 °C for 30 min. Then, 1 mL TEOS was added rapidly to this mixture and allowed to react for 30 min. After that, 8.68 mL TEOS was added dropwise and reacted for 2 h. The precipitate was centrifuged, then rinsed with deionized water and ethanol before drying at room temperature. 0.8 g of SiO2 nanospheres was mixed with absolute ethanol by sonication, and 1.0 g of HDA and 2 mL of concentrated ammonia were added and stirred at 35 °C for 30 min. Then, 660 μL of titanium(IV) isopropoxide and 340 μL of zirconium butoxide solution were concurrently added dropwise and reacted for 2 h. The product was collected in the same way as SiO2 nanospheres. Yolk–shell STZ nanospheres were created using the ammonia etching method. In short, as-prepared materials were dispersed in 0.5 M ammonia solution, heated in a reactor at 160 °C for 16 h, and collected as mentioned above. Obtained powder was calcined in a muffle furnace at 600 °C for 4 h to remove leftover HDA templates.Preparation of PSTZ nanocomposite
In advance, 18 mM CaCl2·2H2O and 8.4 mM Na2HPO4·12H2O in TBS buffer (pH = 7.40) were produced. After dissolving 50 mg of PAH in 50 mL of CaCl2·H2O solution and adding equal volumes of Na2HPO4 solution through a peristaltic pump (BT101L, NanBei Instruments Ltd, China) at a rate of 2.5 mL min−1, PAH-ACP mineralizing solution was obtained. 0.1 g STZ nanospheres were suspended with 100 mL of PAH-ACP solution and agitated overnight to promote ACP infiltration into the channels of STZ. The PSTZ precipitate was retrieved, washed, freeze-dried and stored at −20 °C for further characterization and application.Characterization
The ultrastructural features and elemental mapping were examined by transmission electron microscopy (TEM, Talos F200S, Thermo Fisher Scientific Inc., Waltham, MA, USA). Zeta potential was immediately determined after ultrasonic dispersion with ethanol by a zetasizer (Nano ZS, Malvern Instruments Ltd, Malvern, UK). A thermogravimetric analyzer (TGA, NETZSCH STA 449 F3, Germany) was used to analyze the loading amount of PAH-ACP at a heating rate of 10 °C min−1 in nitrogen atmosphere. The molecular structure, chemical bonds and composition of nanospheres were evaluated by a FTIR spectrometer (Nicolet 5700, Thermo Fisher, USA). Phase analysis was performed using XRD (X'Pert Pro, Panalytical BV, Netherlands). The concentrations and rates of Ca, P, Si, Ti, Zr ions released from PSTZ were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, ICAP-6300, Thermo Fisher, USA). Briefly, 50 mg PSTZ was submerged in 10 mL of TBS buffer without Ca and P ions. 1 mL of the supernatant was removed and analyzed after centrifugation at 1, 3, 6, and 12 h, and 1, 4, 7, 14, 21 and 28 days; then equal volume of fresh solution was supplemented. The measurements were performed in triplicate for each group.Preparation of dentin specimens
The Ethics Committee of School and Hospital of Stomatology, Wuhan University authorized this research project. After achieving the informed consents of donors, sound human third molars were obtained, cleaned and stored in thymol solution at 4 °C. Dentin disks of around 1.0 mm in thickness were prepared using a water-cooled low-speed diamond saw (Isomet; Buehler, Evanston, IL, USA) and polished sequentially with 600-, 800-, 1200-, 2000- and 3000-grit SiC sandpaper. For remineralization, dentin disks were ultrasonically cleaned and etched with 37% phosphoric acid for 20 s to completely demineralize the dentin matrix. For tubule occlusion, dentin disks were treated with 0.5 M EDTA (pH = 7.4) for 2 min to simulate a DH model. Before usage, all specimens were flushed with deionized water twice.Mineralization of collagen fibrils
A single-layer collagen fibril model, reconstituted according to the literature,76 was used for examining the mineralization ability of PSTZ. Assembling solution consisted of 200 mM KCl and 50 mM glycine and the pH was adjusted to 9.20 with 1.0 M NaOH solution. 15 μL of collagen-I stock solution (5 mg mL−1, Coring Biotech, China) was mixed with 785 μL of assembling solution and then placed at room temperature. After 20 min, 2.5 μL of collagen-I solution was dripped on 200-mesh Ni TEM grids and wet-incubated in a sealed Petri-dish at 37 °C for 12 h. Collagen-covered TEM grids were cross-linked by floating in 0.05 wt% glutaraldehyde solution for 1 h and then washed with deionized water and air dried. 50 mg of PSTZ was immersed in 10 mL of TBS buffer (pH = 7.4) and gently shaken for 20 min. After that, PSTZ-containing mineralization solution was transferred into a 6 cm Petri-dish. Collagen-covered TEM grids were floated on the above suspension and incubated at 37 °C. After 1, 4, and 7 d, grids were taken out, rinsed with deionized water, air dried and then observed by TEM (HT7700, Hitachi, Japan). Subsequently, characteristic samples were analyzed by TEM (Tecnai G2 F20, FEI, USA) with selected area electron diffraction (SAED) and element mapping.Remineralization of demineralized dentin matrix
Remineralization solution was similarly generated by dispersing 50 mg of PSTZ into 10 mL TBS buffer (pH = 7.4) and shaking for 20 min to allow ACP release. The demineralized dentin specimens (N = 6) were then submerged in the remineralization solution and stored in a 37 °C incubator. The specimens were retrieved at predetermined intervals, segmented longitudinally from the reverse side, rinsed with deionized water and then fixed with 2.5% glutaraldehyde for 24 h. After that, disks were rinsed with deionized water again, dehydrated with graded ethanol, immersed into HMDS for 30 min and vacuum dried for 1 h. Specimens (N = 3) were sprayed with gold before field-emission scanning electron microscopy (FESEM, Sigma, Zeiss, Germany) observation and energy dispersive X-ray spectroscopy (EDX) analysis. To assess the mechanical properties of remineralized dentin matrix at the microscale, nanoindentation tests were conducted using a Hysitron TI-980 Triboindenter. The surfaces of specimens (N = 3) were polished with 600-, 800-, 1200-, 2000- and 3000-grit SiC sandpaper, followed by aqueous diamond suspensions from 1 μm to 0.25 μm. The maximum force of 2000 μN and a standard trapezoidal load function of 5–2–5 s were set in advance. Measurements (N = 10) were randomly selected from each group for statistical analysis of average modulus and hardness.Assessment of tubule-occluding effects
Dentin disks were thoroughly washed with deionized water before pretreatment and then the PSTZ-containing suspension was applied to the occlusal surface of dentin disks using a brush for 1 min, with a concentration of 25 mg mL−1. TBS buffer (pH = 7.4) was utilized to treat disks in the same way as the control. Pretreated dentin disks were randomly assigned into two groups: immediate and long-term groups. Samples from the latter group were stored in artificial saliva (pH = 7.4), which consisted of 0.2 mM MgCl2·6H2O, 0.7 mM CaCl2·2H2O, 30 mM KCl, 4.0 mM KH2PO4 and 20 mM HEPES buffer and was renewed every three days. At the designed time points (0 and 28 days), disks were retrieved and classified into three subgroups (n = 15) before FESEM observation and dentin permeability assessment. The desensitizing treatment subgroup was treated with deionized water for 1 min, the acid challenge subgroup was immersed into 6% citric acid solution for 1 min and the mechanical brushing subgroup received 3 min of gentle brushing with a soft-bristled toothbrush (Colgate-Palmolive Co., China). After treatment, disks were rinsed twice with deionized water and blown lightly parallel to the surface. For evaluating both tubule-occluding efficiency and depth, disks were further split into two sections, cross-section and longitudinal-section, respectively. Then, disks were dehydrated using graded ethanol, HMDS, and a vacuum drying oven successively and gold-sprayed before being analyzed with FESEM.Dentin permeability assessment was conducted by a modified fluid permeation device, as described in prior work.28 To replicate pulp pressure, a fluid container filled with deionized water was positioned 50 cm above the test bench and connected to a capillary tube through a three-way connector. A 10 mL syringe was linked to the three-way connector via a polyethylene hose, which was used to make air bubbles indicating liquid movement. Dentin disks were sandwiched between two acrylic sheets and sealed by rubber bands with inner diameter of 7 mm. The time and distance of an air bubble passing through a capillary tube were recorded and translated to hydraulic conductance (Lp). The average Lp value of etched dentin disks was determined as 100% dentin permeability. The dentin permeability of each subgroup was tested and expressed as a percentage (Lp%).
Isolation and culture of hDPSCs
Caries-free third molars extracted from 18- to 25-year-olds were immediately immersed in phosphate buffered saline (PBS) buffer (pH = 7.4) containing 2% penicillin–streptomycin, preserved at low temperature and employed as soon as possible to derive hDPSCs. Teeth were stripped of the associated soft tissues, disinfected with iodophor, and then the pulp cavity was exposed by smashing it with a hammer under the protection of double-layer sterile gauze. Dental pulp tissue was taken out with tweezers, rinsed several times and transferred to an Eppendorf tube containing 1 ml of α-modified minimal essential medium (α-MEM, Hyclone, Logan, UT, USA). In the Eppendorf tube, dental pulp tissue was cut into pieces and centrifuged at 1000 rpm for 5 min. Then, α-MEM was discarded, 500 μl of type I collagenase was added before storing in a 37 °C incubator for 45 min. After digestion, the supernatant was removed by centrifugation and washed twice with PBS buffer containing 2% penicillin–streptomycin (Amresco LLC, Solon, OH, USA). Finally, the dental pulp tissue was resuspended with α-MEM medium containing 20% FBS and 1% penicillin–streptomycin, transferred to a 25 cm2 cell culture flask and incubated in 5% CO2 at 37 °C. When hDPSCs were generated, the culture medium was changed every two days. When the fusion degree reached 80–90%, hDPSCs were digested from the culture flask and cultured for serial passage or cryopreservation at −80 °C. Passages 3–6 hDPSCs were used for biocompatibility tests and odontogenic differentiation experiments.Evaluation of biocompatibility
The biocompatibility of STZ and PSTZ nanocomposites was assessed by hemolysis assay, cytoskeleton observation and CCK-8 assay. Hemolysis assay was used for determining the toxicity of STZ and PSTZ to animal erythrocyte membrane. The experimental solution was produced by immersing STZ and PSTZ in PBS buffer at concentrations of 80, 320, and 1280 μg mL−1. The positive and negative controls were deionized water and PBS buffer, respectively. 4 mL of blood obtained from specific pathogen free (SPF) mice was diluted in PBS buffer at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dilutions. Then, each sample (N = 3) was prepared by mixing 1 mL of testing solution and 200 μL of diluted blood, and then incubated at 37 °C for 4 h. Subsequently, samples were centrifuged at 1690g for 5 min and the OD value at 545 nm of the supernatant was determined. The hemolysis rate was calculated using the following formula:
:5 dilutions. Then, each sample (N = 3) was prepared by mixing 1 mL of testing solution and 200 μL of diluted blood, and then incubated at 37 °C for 4 h. Subsequently, samples were centrifuged at 1690g for 5 min and the OD value at 545 nm of the supernatant was determined. The hemolysis rate was calculated using the following formula: 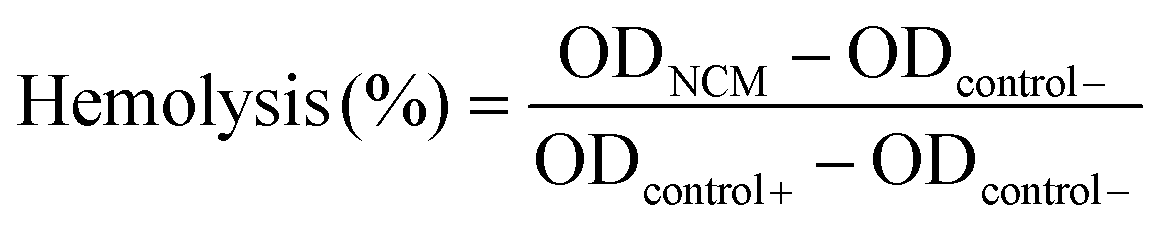 .
.
Cell counting kit-8 (CCK-8, Dojindo, Japan) and FITC-phalloidin staining kit (Yeasen, China) were used for measuring the effects of STZ and PSTZ nanocomposites on the proliferation of hDPSCs. The hDPSCs were seeded in a 96-well plate at 3000 cells per well (N = 5) for CCK-8 assay, or a 24-well plate with sterile cell climbing slice at 40000 cells for cytoskeleton observation per well (N = 3). Following 24 h of incubation, cells were adhered to walls and then subjected to extract solutions of gradient concentration of STZ and PSTZ, comprising 0, 10, 20, 40, 80, 160, 320, 640, 1280 μg mL−1. After incubation for 1, 4, and 7 days, these cells were processed for CCK-8 assay and FITC-phalloidin staining in accordance with the specification. With regard to CCK-8 assay, 110 μL of CCK-8 solution was placed into each well (N = 5) and incubated for 2 h in the absence of light. Absorbance was measured at 450 nm by a spectrophotometer in a microplate reader. The 0 μg mL−1 group at each time point was used as the control to calculate relative cell activity (%). The CCK-8 assay was implemented in triplicate. FITC-phalloidin was used to label the structure of the cytoskeleton. The cells were washed with PBS buffer twice, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100. Then, FITC-phalloidin supplemented with 1% BSA and DAPI were used for staining the cytoskeleton and nuclei, respectively. The morphology of the cells was observed using a fluorescence microscope (DM 4000B LED, Leica, Germany).
In vitro odontogenesis of hDPSCs
To verify the odontogenic efficiency, hDPSCs were seeded at a density of 105 and cultured in corresponding extract-containing odontogenic induction solution (α-MEM complete medium, 10 mM β-glycerophosphate, 50 μg mL−1 ascorbic acid, 10 nM dexamethasone), which was divided into 3 groups as follows: (1) control, (2) STZ, (3) PSTZ. The ultimate concentration of nanocomposites was 80 μg mL−1 which was changed every 3 days. Subsequently, ALP staining, ALP activity assay, real-time quantitative PCR (RT-qPCR), and Alizarin Red S staining were performed to assess the odontogenic differentiation degree of hDPSCs.After 7 days of culture, ALP staining and ALP activity measurement were conducted. For ALP staining, after removing the culture medium, the cells were gently washed with PBS, fixed with 4% paraformaldehyde solution, rinsed with PBS again and then assayed with a BCIP/NBT Alkaline Phosphatase Color Development Kit. The results of ALP staining were recorded by a microscope after washing with deionized water. ALP activity was measured using an ALP activity assay kit and a BCA protein assay kit. Cells (N = 3) were washed with PBS, and then lysed by 1% Triton X-100. The obtained samples were processed in accordance with the instructions. Absorbance was measured at 405 nm and 562 nm for the quantification of ALP activity and total protein contents respectively, and the ALP activity was calculated.
The expression levels of odontogenic differentiation-related genes were determined by RT-qPCR, including ALP, RUNX2, DMP1, and DSPP. Total cellular RNA was extracted from hDPSCs (N = 3) cultured in odontogenic induction solution for 7 days with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Then, cDNA was synthesized using a PrimeScript RT Reagent Kit (Takara, Tokyo, Japan) and RT-qPCR was performed with SYBR Green Reagent using an ABI QuantStudio 6 PCR system. GAPDH was utilized as the reference gene for normalizing gene expression.
Alizarin Red S staining (Servicebio, China) was employed to observe the formation of calcium nodules after 21 days of culture. Briefly, after removing the culture medium, the cells were washed with PBS, fixed with 4% paraformaldehyde solution for 10 min and washed with deionized water twice. Then, Alizarin Red S staining solution was applied to the cells and incubated for 20 min. The results were recorded using a microscope after washing with deionized water.
Statistical analysis
All of the quantitative data were presented as the mean ± standard deviation. SPSS software (IBM SPSS Statistics 26.0, Armonk, NY, USA) was applied for statistical analysis. One-way analysis of variance (ANOVA) and subsequent post-hoc Tukey's test were conducted for analyzing differences among the groups. P-Values < 0.05 were considered statistically significant.Author contributions
Luyao Yi carried out the experiments, analyzed the data and wrote the manuscript. Hongling Wu performed in vitro experiments and analyzed the data. Yue Xu, Jian Yu and Yaning Zhao participated in sample preparation and discussion. Hongye Yang and Cui Huang conceived the project, designed the experiments, analyzed the data, and revised the manuscript with contributions from all authors. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (grant numbers 81701012 and 81970918), the Large-Scale Instrument and Equipment Sharing Foundation of Wuhan University and Research Center for Medicine and Structural Biology of Wuhan University. We also want to offer special thanks to Prof. Buyuan Guan (Jilin University, Changchun, China) for his guidance on material synthesis, Prof. Ruikang Tang and Prof. Haihua Pan (Zhejiang University, Hangzhou, China) for their technical assistance in the self-assembly and mineralization of single-layer collagen fibril model, and Prof. Zhaoyong Zou (Wuhan University of Technology, Wuhan, China) for his detailed review of the manuscript.References
- J. J. Xie, H. Ping, T. N. Tan, L. W. Lei, H. Xie, X. Y. Yang and Z. Y. Fu, Prog. Mater. Sci., 2019, 105, 100571 CrossRef.
- B. M. Oosterlaken, M. P. Vena and G. With, Adv. Mater., 2021, 33, 2004418 CrossRef CAS.
- S. S. Yao, B. Jin, Z. M. Liu, C. Y. Shao, R. B. Zhao, X. Y. Wang and R. K. Tang, Adv. Mater., 2017, 29, 1605903 CrossRef PubMed.
- Y. Kuboki, K. Ohgushi and T. Fusayama, J. Dent. Res., 1977, 56, 1233–1237 CrossRef CAS PubMed.
- L. Favaro Zeola, P. V. Soares and J. Cunha-Cruz, J. Dent., 2019, 81, 1–6 CrossRef.
- D. W. Douglas-de-Oliveira, G. P. Vitor, J. O. Silveira, C. C. Martins, F. O. Costa and L. O. M. Cota, J. Dent., 2018, 71, 1–8 CrossRef PubMed.
- H. J. Shiau, J. Evid. Based Dent. Pract., 2012, 12(3), 220–228 CrossRef.
- A. B. O. D. Canadian, J. Can. Dent. Assoc., 2003, 69, 221–226 Search PubMed.
- Z. Chen, Y. Y. Duan, S. Z. Shan, K. D. Sun, G. Wang, C. Y. Shao, Z. H. Tang, Z. K. Xu, Y. Y. Zhou, Z. Chen, R. K. Tang, H. H. Pan and Z. J. Xie, Nanoscale, 2022, 14(3), 642–652 RSC.
- Y. Shi, D. N. Shen, H. Zheng, Z. F. Wu, C. Y. Shao, L. Q. Zhang, H. H. Pan, R. K. Tang and B. P. Fu, ACS Biomater. Sci. Eng., 2019, 5, 5481–5488 CrossRef CAS.
- J. K. He, J. H. Yang, M. Li, Y. C. Li, Y. Y. Pang, J. Y. Deng, X. Zhang and W. G. Liu, ACS Nano, 2022, 16, 3119–3134 CrossRef CAS PubMed.
- C. Li, D. Y. Lu, J. J. Deng, X. Zhang and P. Yang, Adv. Mater., 2019, 31, 1903973 CrossRef CAS.
- C. Berg, E. Unosson, H. Engqvist and W. Xia, ACS Biomater. Sci. Eng., 2020, 6, 3599–3607 CrossRef CAS.
- S. Dimitrova-Nakov, A. Baudry, Y. Harichane, O. Kellermann and M. Goldberg, J. Endod., 2014, 40, S13–S18 CrossRef PubMed.
- P. Hollands, D. Aboyeji and M. Orcharton, Br. Dent. J., 2018, 224, 747–750 CrossRef.
- N. Kawashima, Arch. Oral Biol., 2012, 57, 1439–1458 CrossRef.
- W. J. Fang, H. Ping, W. Wagermaier, S. B. Jin, S. Amini, P. Fratzl, G. Sha, F. J. Xia, J. S. Wu, H. Xie, P. C. Zhai, W. M. Wang and Z. Y. Fu, Nanoscale, 2021, 13, 8293–8303 RSC.
- K. Jiao, L. N. Niu, C. F. Ma, X. Q. Huang, D. D. Pei, T. Luo, Q. Huang, J. H. Chen and F. R. Tay, Adv. Funct. Mater., 2016, 26, 6858–6875 CrossRef CAS.
- M. J. Olszta, X. G. Cheng, S. S. Jee, R. Kumar, Y. Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- J. H. Li, J. J. Yang, J. Y. Li, L. Chen, K. N. Liang, W. Wu, X. Y. Chen and J. S. Li, Biomaterials, 2013, 34, 6738–6747 CrossRef CAS PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748–12753 CrossRef CAS.
- A. Lotsari, A. K. Rajasekharan, M. Halvarsson and M. Andersson, Nat. Commun., 2018, 9, 4170 CrossRef PubMed.
- C. Combes and C. Rey, Acta Biomater., 2010, 6, 3362–3378 CrossRef CAS PubMed.
- J. Zhao, Y. Liu, W. B. Sun and H. Zhang, Chem. Cent. J., 2011, 5, 40–46 CrossRef CAS.
- X. J. Luo, H. Y. Yang, L. N. Niu, J. Mao and F. R. Tay, Acta Biomater., 2016, 31, 378–387 CrossRef CAS.
- H. Y. Yang, L. N. Niu, J. L. Sun, X. Q. Huang, D. D. Pei, C. Huang and F. R. Tay, Int. J. Nanomed., 2017, 12, 839–854 CrossRef CAS.
- J. Yu, L. Y. Yi, R. Guo, J. M. Guo, H. Y. Yang and C. Huang, Int. J. Nanomed., 2021, 16, 3041–3057 CrossRef PubMed.
- J. Yu, H. Y. Yang, K. Li, H. Y. Ren, J. M. Lei and C. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 25796–25807 CrossRef CAS PubMed.
- S. Galli, R. Jimbo, Y. Naito, S. Berner, M. Dard and A. Wennerberg, Clin. Oral Implants Res., 2017, 28, 1234–1240 CrossRef PubMed.
- F. G. Qi, N. Wang, Z. W. Yang, Y. Wang and H. J. Li, Adv. Eng. Mater., 2022, 2200105 CrossRef CAS.
- L. M. Kang and C. Yang, Adv. Eng. Mater., 2019, 21, 1801359 CrossRef.
- X. Q. Huang, H. Y. Yang, T. Luo, C. Huang, F. R. Tay and L. N. Niu, Acta Biomater., 2018, 67, 366–377 CrossRef CAS.
- A. Guha, N. Biswas, K. Bhattacharjee, N. Sahoo and K. Kuotsu, Drug Delivery, 2016, 23, 3552–3561 CrossRef CAS.
- C. M. Li, J. S. Zhao and Y. S. Zhang, Nanomaterials, 2021, 11, 2314 CrossRef CAS.
- J. K. Zhao, S. S. Ge, D. Pan, Q. Shao, J. Lin, Z. K. Wang, Z. Hu, T. T. Wu and Z. H. Guo, J. Colloid Interface Sci., 2018, 529, 111–121 CrossRef CAS.
- Z. N. Kayani, A. Kamran, Z. Saddiqe, S. Riaz and S. Naseem, J. Photochem. Photobiol., B, 2018, 183, 357–366 CrossRef CAS PubMed.
- B. Y. Guan, T. Wang, S. J. Zeng, X. Wang, D. An, D. M. Wang, Y. Cao, D. X. Ma, Y. L. Liu and Q. S. Huo, Nano Res., 2014, 7, 246–262 CrossRef CAS.
- C. Y. Shao, B. Jin, Z. Mu, H. Lu, Y. Q. Zhao, Z. F. Wu, L. M. Yan, Z. S. Zhang, Y. C. Zhou, H. H. Pan, Z. M. Liu and R. K. Tang, Sci. Adv., 2019, 5, w9569 CrossRef.
- M. C. Shi, Y. H. Zhou, J. Shao, Z. T. Chen, B. T. Song, J. Chang, C. T. Wu and Y. Xiao, Acta Biomater., 2015, 21, 178–189 CrossRef CAS PubMed.
- J. L. Sun, K. Jiao, L. N. Niu, Y. Jiao, Q. Song, L. J. Shen, F. R. Tay and J. H. Chen, Biomaterials, 2017, 113, 203–216 CrossRef CAS PubMed.
- P. Fratzl, Collagen: Structure and Mechanics, an Introduction, Springer US, Boston, MA, 2008 Search PubMed.
- F. Nudelman, K. Pieterse, A. George, P. Bomans, H. Friedrich, L. J. Brylka, P. Hilbers, G. D. With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS.
- L. N. Niu, S. E. Jee, K. Jiao, L. Tonggu, M. Li, L. Wang, Y. D. Yang, J. H. Bian, L. Breschi and S. S. Jang, Nat. Mater., 2017, 16, 370–378 CrossRef CAS PubMed.
- S. S. Yao, X. F. Lin, Y. F. Xu, Y. W. Chen, P. C. Qiu, C. Y. Shao, B. Jin, Z. Mu, N. A. J. M. Sommerdijk and R. K. Tang, Adv. Sci., 2019, 6, 1900683 CrossRef CAS.
- M. Neshatian, J. Holcroft, A. Kishen, G. D. Souza and B. Ganss, Mater. Today Bio, 2022, 14, 100255 CrossRef CAS PubMed.
- M. K. Arifa, R. Ephraim and T. Rajamani, Int. J. Clin. Pediatr. Dent., 2019, 12, 139–144 CrossRef.
- R. Chen, R. Jin, X. Li, X. Fang and Z. Chen, Dent. Mater., 2020, 36, 1397–1406 CrossRef CAS PubMed.
- Y. Liu, S. Mai, N. Li, C. K. Y. Yiu, J. Mao, D. H. Pashley and F. R. Tay, Acta Biomater., 2010, 7, 1742–1751 CrossRef PubMed.
- J. Silvent, M. Robin, C. B. Tovani, Y. Wang and N. Nassif, Biomacromolecules, 2021, 22, 2802–2814 CrossRef CAS.
- Y. Feng, L. Zhong, Y. Hou, S. Jia and J. Cui, J. Biotechnol., 2019, 306, 54–61 CrossRef CAS PubMed.
- S. Galli, R. Jimbo, Y. Naito, S. Berner and M. Dard, Clin. Oral Implants Res., 2017, 28, 1234–1240 CrossRef PubMed.
- Y. W. Pan, X. H. Ma, C. Liu, J. Xing, S. Q. Zhou, B. Parshad, T. Schwerdtle, W. Z. Li, A. G. Wu and R. Haag, ACS Nano, 2021, 15, 15069–15084 CrossRef CAS PubMed.
- V. Agarwal, V. Gupta, V. K. Bhardwaj, K. Singh, P. Khullar and M. S. Bakshi, ACS Appl. Mater. Interfaces, 2022, 14, 6428–6441 CrossRef CAS.
- J. Autian, Biological Model Systems for the Testing of the Toxicity of Biomaterials, Springer, Boston, MA, 1975 Search PubMed.
- Y. Zhao, X. Sun, G. Zhang, B. G. Trewyn, I. I. Slowing and S. Y. Lin, ACS Nano, 2011, 5, 1366–1375 CrossRef CAS PubMed.
- J. D. Finan and F. Guilak, J. Cell. Biochem., 2010, 109, 460–467 CAS.
- M. Y. Shie, S. J. Ding and H. C. Chang, Acta Biomater., 2011, 7, 2604–2614 CrossRef CAS PubMed.
- M. M. Dvorak, A. Siddiqua, D. T. Ward, D. H. Carter, S. L. Dallas and E. F. Nemeth, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5140–5145 CrossRef CAS PubMed.
- S. An, G. Yan, J. Ling, W. Xi and X. Yin, J. Mater. Sci. Mater. Med., 2012, 23, 789–795 CrossRef CAS PubMed.
- E. O'Neill, G. Awale, L. Daneshmandi, O. Umerah and W. H. Lo, Drug Discovery Today, 2018, 23, 879–890 CrossRef PubMed.
- J. A. Seth, G. Boivin and O. Andersen, J. Trace Elem. Med. Biol., 2012, 26, 149–152 CrossRef.
- P. Sangwan, A. Sangwan, J. Duhan and A. Rohilla, Int. Endod. J., 2013, 46, 3–19 CrossRef CAS PubMed.
- E. E. Golub, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1592–1598 CrossRef CAS PubMed.
- A. V. Kotova, A. A. Lobov, J. A. Dombrovskaya, V. Y. Sannikova, N. A. Ryumina, P. Klausen, A. L. Shavarda, A. B. Malashicheva and N. I. Enukashvily, Biomedicines, 2021, 9, 1606 CrossRef CAS PubMed.
- B. Li, T. Ouchi, Y. B. Cao, Z. H. Zhao and Y. Men, Front. Cell Dev. Biol., 2021, 9, 654559 CrossRef PubMed.
- A. Almushayt, K. Narayanan, A. E. Zaki and A. George, Gene Ther., 2006, 13, 611–620 CrossRef CAS PubMed.
- S. C. Chaudhary, M. Kuzynski, M. Bottini, E. Beniash, T. Dokland, C. G. Mobley, M. C. Yadav, A. Poliard, O. Kellerman and J. L. Millán, Matrix Biol., 2016, 284–300 CrossRef CAS PubMed.
- V. P. Galván-Chacón, D. M. Pereira, S. Vermeulen, H. Yuan, J. Li and P. Habibović, Bioact. Mater., 2023, 19, 127–138 CrossRef PubMed.
- O. Grninger, S. Hess, D. Mohn, E. Schneider and J. Buschmann, Int. J. Mol. Sci., 2020, 21, 2627 CrossRef PubMed.
- N. Vidavsky, J. Kunitake and L. A. Estroff, Adv. Healthcare Mater., 2021, 10, 2001271 CrossRef CAS PubMed.
- E. Bosch-Rué, L. Diez-Tercero, B. Giordano-Kelhoffer, L. M. Delgado and R. A. Perez, Front. Cell Dev. Biol., 2021, 8, 613545 Search PubMed.
- S. Phung, C. Lee, C. Hong, M. Song, J. K. Yi, R. G. Stevenson, M. K. Kang, K. H. Shin, N. H. Park and R. H. Kim, J. Dent. Res., 2016, 96, 107–115 CrossRef PubMed.
- S. H. Park, Y. S. Lee, D. S. Lee, J. C. Park and W. J. Shon, J. Dent. Res., 2019, 98, 1239–1244 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- C. Y. Shao, R. B. Zhao, S. Q. Jiang, S. S. Yao, Z. F. Wu, B. Jin, Y. L. Yang, H. H. Pan and R. K. Tang, Adv. Mater., 2018, 30, 1704876 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04993g |
| This journal is © The Royal Society of Chemistry 2023 |