
Structure engineering of CeO2 for boosting the Au/CeO2 nanocatalyst in the green and selective hydrogenation of nitrobenzene†
Junqing
Ye
ab,
Meizan
Jing
c,
Yu
Liang
a,
Wenjin
Li
a,
Wanting
Zhao
a,
Jianying
Huang
 de,
Yuekun
Lai
*de,
Weiyu
Song
c,
Jian
Liu
c and
Jian
Sun
*af
de,
Yuekun
Lai
*de,
Weiyu
Song
c,
Jian
Liu
c and
Jian
Sun
*af
aSchool of Life Science, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: jiansun@bit.edu.cn
bJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, School of Petrochemical Engineering, Changzhou University, Changzhou 213164, P. R. China
cState Key Laboratory of Heavy Oil Processing, College of Science, China University of Petroleum-Beijing, Beijing 102249, P. R. China
dCollege of Chemical Engineering, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: yklai@fzu.edu.cn
eQingyuan Innovation Laboratory, Quanzhou 362801, P. R. China
fAdvanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, P. R. China
First published on 23rd March 2023
Abstract
Exploring eco-friendly and cost-effective strategies for structure engineering at the nanoscale is important for boosting heterogeneous catalysis but still under a long-standing challenge. Herein, multifunctional polyphenol tannic acid, a low-cost natural biomass containing catechol and galloyl species, was employed as a green reducing agent, chelating agent, and stabilizer to prepare Au nanoparticles, which were dispersed on different-shaped CeO2 supports (e.g., rod, flower, cube, and octahedral). Systematic characterizations revealed that Au/CeO2-rod had the highest oxygen vacancy density and Ce(III) proportion, favoring the dispersion and stabilization of the metal active sites. Using isopropanol as a hydrogen-transfer reagent, deep insights into the structure–activity relationship of the Au/CeO2 catalysts with various morphologies of CeO2 in the catalytic nitrobenzene transfer hydrogenation reaction were gained. Notably, the catalytic performance followed the order: Au/CeO2-rod (110), (100), (111) > Au/CeO2-flower (100), (111) > Au/CeO2-cube (100) > Au/CeO2-octa (111). Au/CeO2-rod displayed the highest conversion of 100% nitrobenzene and excellent stability under optimal conditions. Moreover, DFT calculations indicated that nitrobenzene molecules had a suitable adsorption energy and better isopropanol dehydrogenation capacity on the Au/CeO2 (110) surface. A reaction pathway and the synergistic catalytic mechanism for catalytic nitrobenzene transfer hydrogenation are proposed based on the results. This work demonstrates that CeO2 structure engineering is an efficient strategy for fabricating advanced and environmentally benign materials for nitrobenzene hydrogenation.
New conceptsNano Au/CeO2 has attracted much attention in various fields, including fuel cells, CO2 hydrogenation, and advanced oxidation, due to its versatile morphologies, stable structure, and good redox ability. Traditionally, the fabrication of Au/CeO2 and its subsequent catalytic hydrogenation are performed in the presence of NaBH4 and hydrogen gas, but this raises environmental and safety issues. In this work, we developed and reported a new concept to employ hydroxyl compounds as green reductants for the synthesis of Au/CeO2 and for the nitrobenzene transfer hydrogenation. First, tannic acid is used as a natural reducing agent, chelator, and stabilizing agent to achieve the green formation and deposition of gold nanoparticles on CeO2, where CeO2 with different morphologies are obtained by a solvothermal method through ion-induced and confine-induced nucleation. Second, isopropanol is used as a safe hydrogen source instead of the more dangerous H2 from a cylinder in the nitrobenzene transfer hydrogenation reaction. Among four kinds of catalysts, Au/CeO2-rod exhibits the highest catalytic activity. Deep insights are gained into the structure–activity relationship of Au/CeO2, demonstrating that the higher the oxygen vacancy concentration and Ce(III) proportion, the higher the activity of Au/CeO2. This environmentally benign and safe method will be helpful to researchers engaged or interested in the nanoscience fields and in developing cutting-edge advanced nanomaterials and green catalysts. |
Introduction
Aniline and its derivatives (such as azoxybenzene and azobenzene) are a class of compounds with high added value and important industrial application value, whereby they are widely used in the production of dyes, pigments, plastics, and agricultural chemicals.1–4 As a classic way to produce aniline and its derivatives, the hydrogenation reaction routes of –NO2-containing compounds have been studied by researchers for around a century.5–7 However, the current challenge is to develop efficient and stable catalysts to enable them to carry out catalytic reactions under milder, greener, and safer conditions. Moreover, nitrobenzene hydrogenation is the most commonly used probe reaction to investigate the relationship between the structure and properties of hydrogenation catalysts. The hydrogen used in the hydrogenation of nitrobenzene can come from molecular hydrogen (H2), and so high-pressure gas cylinder H2 is still mainly used in industry, which is also very easy to use. However, explosion accidents caused by high-pressure hydrogen cylinders during transportation and usage occur every year. Therefore, to protect people's lives and the safety of equipment and devices, it is very necessary to find a hydrogen source that can replace high-pressure hydrogen. Fortunately, catalytic transfer hydrogenation can produce hydrogen directly during a reaction by adding a hydrogen donor reagent, and also offers the advantages of mild reaction conditions, easy operation, and more safety. However, the common hydrogen-transfer reagents, e.g., hydrazine hydrate (N2H4·H2O), sodium borohydride (NaBH4), formic acid, and silyl hydride, are expensive and environmentally unfriendly.8–12 Isopropanol is often chosen as a hydrogen donor due to its low price and environmental friendliness.13–15 In addition to the selection of the hydrogen-transfer reagents, the choice of efficient catalysts is also very important for the catalytic transfer hydrogenation reaction. Thus, researchers have devoted a lot of effort to the design and fabrication of catalysts for the catalytic transfer hydrogenation of nitrobenzene, as well as to the exploration of the reaction mechanism.CeO2 is an attractive heterogeneous catalyst support because of its stable structure and good redox ability. In particular, the mutual transformation between Ce4+/Ce3+ in CeO2 makes CeO2 capable of storing and releasing oxygen and forming sufficient oxygen vacancies on the surface.16–19 Li et al. reported that the 8Ni/CeO2 catalyst can be used in the selective transformation of C–O bonds in biomass-derived furans for the production of fuels and chemicals with excellent selectivity (>97%) and stability (>2400 h).20 The existence of O vacancies enables CeO2 to disperse and stabilize metal nanoparticles well. Therefore, CeO2-based nanocatalysts can be used in many oxidation reactions or in the hydrogenation of –NO2 functional groups.20–30 CeO2 has different morphologies with the exposure of different crystal planes. The crystal plane effect is an influential factor in many reactions;31,32 for example, CO oxidation,33–37 and the water–gas shift reaction,38–40 and so on. Generally speaking, CeO2 exposure to different crystal planes affects the formation energy of O vacancies, which can further regulate the interaction between the active metal and the CeO2 support, as well as the electronic properties of the active metal centers. Thus, the activity of the catalyst can also be regulated.41 Among the CeO2 forms loaded with different metal catalysts, environmentally benign Au/CeO2 shows extraordinarily high catalytic activity in various reactions.42 It has been reported that the Au nanoparticles deposited on the supports can be employed as adsorption sites for reactant molecules, thus showing excellent catalytic performance.43,44 In fact, Au nanoparticles with an unfilled orbital electronic structure can combine with –NO2 functional groups by occupying the unfilled orbital with lone-pair electrons. Au/CeO2 nanocatalyst, as a low-temperature water–gas shift catalyst, was first reported by Fu et al., who also showed there was a strong interaction between the CeO2 and dispersed Au nanoparticles.45 Bu et al. compared the catalytic activity of CeO2 with different structures for formaldehyde oxidation.46 Their results indicated that the rod-like Au/CeO2 (with mainly exposed (110) and (100) crystal planes) had a much higher catalytic performance and was more stable than the commercial CeO2 (with mainly exposed (111) crystal planes). The interface interaction between the nanorod-like CeO2 and Au facilitated oxygen vacancy generation, thereby promoting formate production. The catalytic performance improved when Au was loaded on to the CeO2 support. For example, Si et al. found that the Au/CeO2 catalyst exhibited a strong CeO2 morphology/crystal effect in the water–gas shift reaction.47 The catalytic activity of nanorods with (110) and (100) exposed crystal planes was higher than that of nanopolyhedra with (111) and (100) exposed crystal planes, and nanocubes with the (100) exposed crystal plane. Inspired by these studies, we believe that the design of a dual-sites catalyst by dispersing Au nanoparticles on CeO2 surface would be an effective strategy for the catalytic transfer hydrogenation of nitrobenzene. In this context, we aimed to address some outstanding questions: when Au/CeO2 catalysts are applied in nitrobenzene hydrogenation, will CeO2 have a similar morphology/crystal effect? How do Au/CeO2 catalysts with different morphologies of CeO2 work when adsorbing and hydrogenating nitrobenzene and the hydrogen-transfer reagent? What are the facts and mechanisms behind them? The current research on this subject is scarce and these have not been definitively answered yet.
In this work, to explore the morphology-dependent properties of CeO2-supported Au for the hydrogenation of nitrobenzene and the reaction mechanisms, we prepared four kinds of CeO2 supports with different morphologies: CeO2-rod, CeO2-flower, CeO2-cube, and CeO2-octa structures. Then, the corresponding Au/CeO2 catalysts were formed by loading these CeO2 supports with Au nanoparticles using tannic acid as the reductant, chelator, and stabilizer. Through comprehensive structural characterization techniques, experiments, and combined with DFT calculations, we studied the mechanism of the nitrobenzene transfer hydrogenation catalyzed by the Au/CeO2 catalysts with different morphologies of CeO2, using isopropanol as a hydrogen-transfer reagent. The results suggested that Au/CeO2-rod with (110) + (100) + (111) exposed crystal faces showed the best catalytic performance and stability among the Au/CeO2 catalysts, and the descending order was: Au/CeO2-rod (110), (100), (111) > Au/CeO2-flower (100), (111) > Au/CeO2-cube (100) > Au/CeO2-octa (111). Furthermore, DFT calculation results revealed that nitrobenzene could more easily adsorb on Au in Au/CeO2 (110), and the Au/CeO2 (110) crystal plane was more likely to dissociate the H from –OH and α-C–H from isopropanol to form active H* species, which were then used for the hydrogenation of nitrobenzene. The synergistic interaction of the Au/CeO2 interface contributed to the catalytic activity for nitrobenzene transfer hydrogenation.
Results and discussion
Characterizations of the various CeO2-based materials
The morphology and structural properties of the CeO2-based materials were investigated by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM). In Fig. S1a (ESI†), the well-defined rod-shaped morphology of CeO2 can be observed, and CeO2-rod exhibited a length of 30–100 nm and a width of 5–7 nm. As shown in Fig. S1b–d (ESI†), the d-spacings derived from the HRTEM results were 0.31, 0.27, and 0.19 nm for CeO2-rod, which were assigned to the (111), (100), and (110) orientations, respectively. The TEM and HRTEM images of CeO2-cube (Fig. S1e and f, ESI†), CeO2-flower (Fig. S1g and h, ESI†), and CeO2-octa (Fig. S1i and j, ESI†) indicated that CeO2-cube preferentially had exposed (100) crystal facets, CeO2-flower exposed (100) and (111), and CeO2-octa predominately exposed (111) crystal facets. The SEM images in Fig. S1k and l (ESI†) show that the morphologies of CeO2-octa and CeO2-flower were well-defined.As shown in Fig. 1(a)–(h), the TEM and HRTEM images show that the Au nanoparticles were dispersed on the different shapes of CeO2. The fast Fourier transform (FFT) pattern in the inset of Fig. 1(b) shows 10 peaks, corresponding to the 10 lattice fringes of Au nanoparticles with 2.3 nm spacings, and suggesting that the Au nanoparticles had preferentially exposed (111) facets with a face-centered cubic (fcc) structure. For the Au/CeO2-cube (Fig. 1(a) and (b)), Au/CeO2-rod (Fig. 1(c) and (d)), and Au/CeO2-flower (Fig. 1(e) and (f)), the average sizes of Au nanoparticles were 18, 28, and 39 nm, respectively. However, for the Au/CeO2-octa catalyst (Fig. 1(g) and (h)), the Au nanoparticles were aggregated with a size of 8 nm. The EDS elemental mapping results for Au/CeO2 (Fig. S2, ESI†) further confirmed the well-dispersed Au nanoparticles on the surface of CeO2, except for the CeO2-octa support. Table 1 presents the weight loadings of Au in the Au/CeO2-cube, Au/CeO2-rod, Au/CeO2-flower, and Au/CeO2-octa determined by ICP-OES as 2.2, 2.2, 2.1, and 2.1 wt%, respectively. These indicate that the Au/CeO2 catalysts were synthesized successfully using tannic acid as a reductant, chelating agent, and stabilizer.
 | ||
| Fig. 1 TEM and HRTEM images of Au/CeO2-cube (a and b), Au/CeO2-rod (c and d), Au/CeO2-flower (e and f), and Au/CeO2-octa (g and h). Interplanar crystal spacings of 0.19, 0.27, and 0.31 nm corresponded to the crystal planes of CeO2 (110), CeO2 (100), and CeO2 (111) (JCPDS 34-0394), respectively. | ||
| Catalyst | S BET (m2 g−1) | Total pore volume (cm3 g−1) | Average pore size (nm) | Actual content of Au (wt%) | Oads atomic% | Olatt atomic% | Ce(III) atomic% | Ce(IV) atomic% |
|---|---|---|---|---|---|---|---|---|
| Au/CeO2-cube | 106.5 | 0.33 | 11.1 | 2.2 | 26.84 | 73.16 | 19.62 | 80.38 |
| Au/CeO2-rod | 108.9 | 0.27 | 9.9 | 2.2 | 33.86 | 66.14 | 21.73 | 78.27 |
| Au/CeO2-flower | 126.6 | 0.16 | 5.6 | 2.1 | 31.26 | 68.74 | 20.47 | 79.53 |
| Au/CeO2-octa | 3.6 | 0.02 | 34.1 | 2.1 | 4.25 | 95.75 | 12.65 | 87.35 |
| CeO2-cube | 41.0 | 0.20 | 14.3 | — | 9.37 | 90.63 | 8.60 | 91.40 |
| CeO2-rod | 119.7 | 0.40 | 12.0 | — | 17.30 | 82.70 | 11.50 | 88.50 |
| CeO2-flower | 134.5 | 0.16 | 5.7 | — | 12.55 | 87.45 | 10.39 | 89.61 |
| CeO2-octa | 3.1 | 0.01 | 23.3 | — | 2.12 | 97.88 | 3.51 | 96.49 |
In Fig. S3a (ESI†), the crystal structures of CeO2 are shown in the XRD plot in the 2-theta range 10°–85°. All the observed diffraction peaks of the (111), (200), (220), (311), (222), (400), (331), and (420) facets could be attributed to the typical cubic fluorite structure of CeO2 (JCPDS 34-0394). As shown in Fig. 2(a), the peaks at 38.3°, 44.5°, and 65.4° could be ascribed to the characteristic diffraction peaks of Au, and the position of CeO2 remained unchanged, whereas the intensity of the diffraction peaks for Au/CeO2 changed. This suggests that the grain sizes of the Au/CeO2 catalysts were different, and such results are consistent with the TEM results.
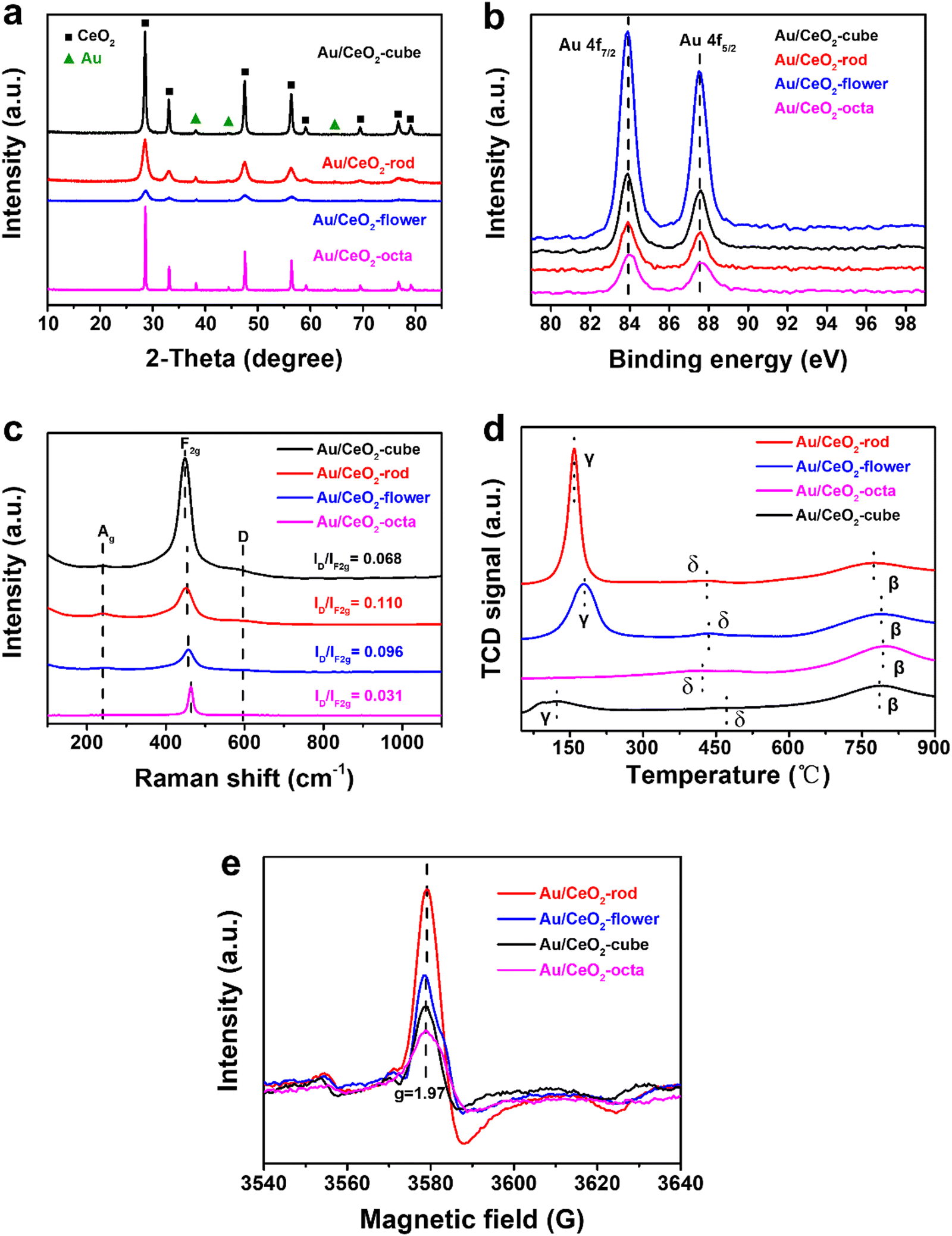 | ||
| Fig. 2 XRD patterns (a), Au 4f XPS results (b), Raman spectra (c), H2-TPR results (d), and ESR spectra (e) of the various Au/CeO2 catalysts. | ||
The BET surface area and pore volume were acquired from the nitrogen adsorption and desorption technique. As illustrated in Fig. S3b (ESI†), with the increase in relative pressure, the nitrogen adsorption and desorption isotherms suddenly increased, meaning they can be classified as IV isotherms according to the IUPAC classification.48 The formation of H3 hysteresis loops in the various Au/CeO2 structures confirmed the presence of mesopores. The BJH pore-size distribution curves and N2 adsorption–desorption isotherms of CeO2 with different shapes were similar to those of Au/CeO2 catalysts (Fig. S3c and d, ESI†). As can be seen from the BJH pore-size distribution curves in Fig. S4a (ESI†), there were few pores in Au/CeO2-octa with the size of 27 nm (Fig. S4a, ESI†), and the BET surface area was only 3.6 m2 g−1, and the total pore volume only 0.02 cm3 g−1 (Table 1). The order of the BET surface area of CeO2 followed the order: CeO2-flower (134.5 m2 g−1) > CeO2-rod (119.7 m2 g−1) > CeO2-cube (41 m2 g−1) > CeO2-octa (3.1 m2 g−1). Au/CeO2-flower had the highest specific surface area (126.6 m2 g−1) in the four different Au/CeO2 catalysts, but the pore volume of Au/CeO2-flower (0.16 cm3 g−1) was smaller than that of Au/CeO2-rod (0.27 cm3 g−1) and Au/CeO2-cube (0.33 cm3 g−1). Similarly, the specific surface area of the CeO2 supports in the four different Au/CeO2 catalysts followed the order: Au/CeO2-flower (126.6 m2 g−1) > Au/CeO2-rod (108.9 m2 g−1) > Au/CeO2-cube (106.5 m2 g−1) > Au/CeO2-octa (3.6 m2 g−1), where the surface areas of Au/CeO2-rod and Au/CeO2-cube were very close. However, when comparing the surface areas of CeO2 and Au/CeO2, it was found that the surface areas of the flower- and rod-shaped CeO2 decreased slightly after loading the nano Au particles. The reason for this may be that the interaction between nano Au particles and CeO2-flower and CeO2-rod was stronger than that for CeO2 with other morphologies, so a small part of the pores of CeO2 was blocked by the nano Au particles, resulting in the surface area of Au/CeO2 decreasing. However, the interaction between Au nanoparticles and CeO2-octa was weak, and the surface areas of Au/CeO2-octa and CeO2-octa were almost equal. The cubic CeO2 was different from the others. The specific surface area of Au/CeO2-cube loaded with nano Au particles was larger than that of CeO2-cube. We speculate that this may be due to the surface effect of the nanoparticles. As the pore size of CeO2 in Au/CeO2-cube decreased, the pore volume increased, and the number of particles increased, resulting in an increase in the particle surface area. Guided by these observations, we can say that the BET surface area was not directly correlated with the total pore volume between CeO2 and Au/CeO2, and there was no regular order to follow between the BET surface area of Au/CeO2 and CeO2.
To study the surface chemical environment of the different Au/CeO2 catalysts and the chemical valence states of the elements, XPS experiments were carried out. Fig. S4b (ESI†) shows the full XPS spectra of the various Au/CeO2, including the XPS results for Au 4f, O 1s, and Ce 3d. The XPS results of Au 4f are shown in Fig. 2(b), where the two sharp peaks at 83.8 and 87.5 eV in Au/CeO2 were consistent with the dominant existence of Au(0) species.49,50 These findings show that the valence states of Au nanoparticles loaded on CeO2 supports with different morphologies via the impregnation method were the same, all of which were Au(0). Fig. S4c (ESI†) presents the XPS results for Ce 3d, wherein the peaks could be divided into Ce(III) for the red line and Ce(IV) for the blue line. Table 1 lists the calculated atomic percentages of Ce(III) and Ce(IV), showing that for various CeO2 and Au/CeO2, the surface Ce(III) proportions decreased in the order: rod > flower > cube > octa. The density of Ce(III) was proportional to the oxygen amount, and was related to the catalytic hydrogenation activity.51 After introducing Au nanoparticles on the CeO2, the content of Ce(III) in Au/CeO2 (Au/CeO2-rod, 21.73%) was higher than that of CeO2 (CeO2-rod, 11.50%). Further, Au/CeO2-rod had the highest intensity of Ce(III), which indicated that the interaction between Au nanoparticles and CeO2 with the optimal crystal facets could reduce the total energy of the Au–CeO2 catalyst, and then cerium would be kept in a lower valence state, thus making the coordination of the unsaturated Ce(III) sites increase.52 In other words, at the interface of Au–O–Ce, electron transfer from the Au atom to Ce was more conducive to the formation of Ce(III), which led to an increase in the oxygen vacancy density. Considering that CeO2 with different shapes have different redox abilities due to the different cyclic alternation abilities between Ce(III) and Ce(IV), combined with the XPS characterization analysis results for Ce(III) for CeO2-based materials, we can infer that the catalytic performances of Au/CeO2 with different morphologies of CeO2 should also be related to the concentration of Ce(III). Fig. S4d (ESI†) exhibits the XPS spectra of O 1s at 532.5 and 529.3 eV, which were attributed to the adsorbed oxygen species and lattice oxygen. In general, the content of adsorbed oxygen was related to the intensity of oxygen vacancies.53,54 Au/CeO2-rod showed a relatively high concentration of adsorbed oxygen (33.86%) in comparison with Au/CeO2-flower (31.26%), Au/CeO2-cube (26.84%), and Au/CeO2-octa (4.25%), which could be ascribed to the high surface energy of the optimal exposed crystal facet (110) for boosting the formation of active sites. Importantly, the concentration of adsorbed oxygen species on the series of CeO2-based materials followed the same order as that of the Ce(III) amount, i.e., rod > flower > cube > octa, which further revealed that the exposed crystal facet of CeO2 in Au/CeO2 is crucial to the formation of active sites.
The TGA results are shown in Fig. S5a and b (ESI†), where a decrease in the mass fraction can be seen from room temperature to 200 °C, which was due to the loss of the adsorbed water. From 200 °C to 500 °C, there was another decrease in weight (%), which was due to the decomposition of a small amount of CeO2, whereby the mass losses of Au/CeO2-rod and CeO2-rod were about 6%, whereas Au/CeO2-octa and CeO2-octa showed almost no mass loss. These results show that, in general, these CeO2-based materials had relatively high thermal stability.
To further investigate the structure of the series of CeO2-based materials prepared, Raman analysis was conducted, and the results are shown in Fig. 2(c). A tiny peak for Ag was observed located at 250 cm−1, which was related to the second-order transverse acoustic mode of the fluorite phase of CeO2.55,56 The intense peak located at approximately 465 cm−1 was assigned to the first-order F2g vibration mode of the lattice expanded O–Ce–O bond with an oxygen vacancy,57 and the peak located at around 595 cm−1 was assigned to the defect-induced D mode, which was the oxygen defect site. The ratio of ID/IF2g was used to express the concentration or amount of oxygen vacancy in the various CeO2-based materials. Au/CeO2-rod (0.110) exhibited the highest density of oxygen vacancies among the Au/CeO2 catalysts, including Au/CeO2-flower (0.096), Au/CeO2-cube (0.068), and Au/CeO2-octa (0.031), which was possibly related to different oxygen vacancy formation energies in CeO2 with the different morphologies. As depicted in Fig. S5c (ESI†), the concentration of oxygen vacancies in CeO2 followed the same order as for the Au/CeO2 catalyst: CeO2-rod (0.054) > CeO2-flower (0.039) > CeO2-cube (0.029) > CeO2-octa (0.014). Moreover, the amount of oxygen vacancies in CeO2 was lower than that of the Au/CeO2 catalysts, which suggests that the interaction between Au and CeO2 may promote the growth of oxygen vacancies. To further prove the existence of oxygen vacancies, ESR measurements were used and the results are shown in Fig. 2(e) and Fig. S5e (ESI†). All the CeO2 and Au/CeO2 catalysts showed a Lorentzian curve at the Lande factor g = 1.97, which is a signal of unpaired electrons. The stronger the signal, the more oxygen vacancies exist on the catalyst surface.58 Nano Au/CeO2-rod showed the strongest signal strength, indicating that it contained the most oxygen vacancies, while Au/CeO2-octa showed the weakest signal strength. The ESR results were consistent with the results from the Raman analysis.
In order to study the redox property of the catalyst, H2-TPR experiments were performed and the results are shown in Fig. S5d (ESI†) and Fig. 2(d). Fig. S5d (ESI†) depicts the H2-TPR results of CeO2 with different shapes, whereby the α peak ranging from 280 °C to 550 °C could be assigned to the reduction of the outer layer Ce4+ to Ce3+, and the β peak ranging from 600 °C to 900 °C corresponded to the bulk reduction, which was related to the inner Ce4+ layer or lattice oxygen, and the shoulder peak at around 388 °C was attributed to the reduction of adsorption oxygen.59 CeO2-rod showed the strongest reduction at a relatively low temperature (425 °C) in comparison with CeO2-flower (460 °C), CeO2-cube (480 °C), and CeO2-octa (540 °C). This indicated that CeO2-rod with the preferentially exposed (110) facet was more active than that with the (100) and (111) crystal facets.
As depicted in Fig. 2(d), after introducing Au nanoparticles on CeO2, the α reduction peak shifted to lower temperatures, ranging from 50 °C to 260 °C, attributed to the reduction peak γ of the chemisorbed active oxygen species. The peak intensity of Au/CeO2-rod was stronger than that of the other Au/CeO2 catalysts, indicating that the interaction between Au and CeO2 could improve the reduction of Au/CeO2 catalyst in the following order: Au/CeO2-rod > Au/CeO2-flower > Au/CeO2-cube > Au/CeO2-octa. The new peak δ at 450 °C was related to the active oxygen species at the interface of Au–O–Ce,60 wherein the synergy effect between Au and CeO2 is beneficial for creating more active oxygen species, which is crucial to improving the catalytic reduction activity of Au/CeO2 catalysts.
Catalytic hydrogenation of 4-nitrophenol
Based on our previous work,61 the catalytic hydrogenation of 4-nitrophenol (4-NP) was studied as a model reaction to screen the Au/CeO2 catalysts because of its simple operation steps.62 Fig. S6 (ESI†) exhibits the UV-Vis spectra obtained from CeO2 with different shapes, where it can be seen that the catalytic 4-nitrophenol hydrogenation hardly occurred, thus eliminating the physical adsorption on the CeO2 supports. As depicted in Fig. 3(a), Au/CeO2-rod achieved 100% 4-nitrophenol conversion in 7 min, which was the highest catalytic activity compared with Au/CeO2-flower (10 min, Fig. 3(b)), Au/CeO2-cube (18 min, Fig. 3(c)), Au/CeO2-octa (>60 min, Fig. 3(d)), respectively. The normalized absorbance plots of the various Au/CeO2 catalysts are depicted in Fig. 3(e), and Au/CeO2-rod had the highest the degradation rate k, which was 0.3082 min−1, compared with Au/CeO2-flower (0.2346 min−1), Au/CeO2-cube (0.1775 min−1), Au/CeO2-octa (0.0047 min−1), respectively.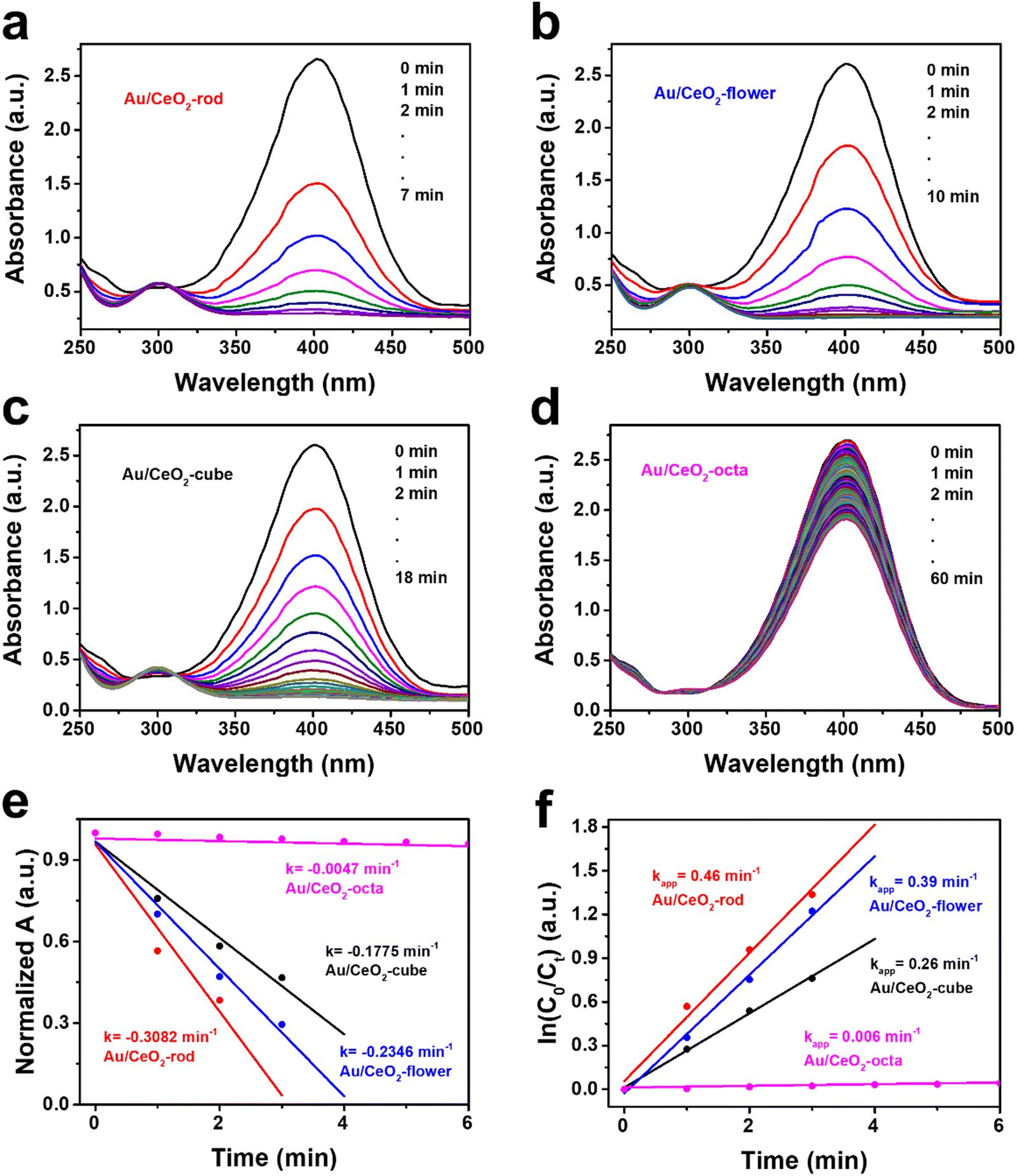 | ||
| Fig. 3 Time-dependent UV-Vis spectra results acquired from 4-NP reduction by NaBH4 with Au/CeO2-rod (a), Au/CeO2-flower (b), Au/CeO2-cube (c), Au/CeO2-octa (d). Normalized absorbance versus time (e), and plots of ln(C0/Ct) versus time (f) for the various Au/CeO2 catalysts. | ||
Moreover, Fig. 3(f) presents the plot of ln(C0/Ct) versus reaction time, showing a linear relationship, which further confirmed the first-order reaction. The apparent rate constant kapp decreased in the order: Au/CeO2-rod (0.46 min−1) > Au/CeO2-flower (0.39 min−1) > Au/CeO2-cube (0.26 min−1) > Au/CeO2-octa (0.006 min−1). The above experimental data demonstrate that Au/CeO2-rod catalyst with the optimal exposed crystal facet (110) showed superior catalytic performance than that of the other Au/CeO2 catalysts. In combination with the results in Table 1, it could be found that the catalytic hydrogenation activity of Au/CeO2 was not directly related to the BET surface area; in other words, it indicated that the BET surface area and total pore volume of the various CeO2-based materials were not the most important factors for the catalytic hydrogenation reaction. However, this was correlated with the content of Ce(III) and the amount of adsorbed oxygen. The results further illustrate that morphology engineering is an effective strategy for the design of high-efficiency hydrogenation catalysts.
Catalytic transfer hydrogenation of nitrobenzene
To further evaluate the effect of various CeO2 shapes on the activities of the corresponding Au/CeO2 catalysts, nitrobenzene (NB) hydrogenation was used as a probe reaction. Fig. S7 (ESI†) displays a photo of the experimental setup for NB hydrogenation. To investigate the effect of the reaction temperature, detailed reaction and kinetics tests were performed. Fig. S8a–c (ESI†) show the catalytic activity of NB at different reaction temperatures for Au/CeO2-rod, Au/CeO2-flower, and Au/CeO2-cube, respectively. With the increase in reaction temperature, the conversion of NB and the selectivity of AN increased, which suggested the reaction temperature is a major factor affecting NB hydrogenation, and the increase in temperature can accelerate the hydrogenation reaction. Temperature is a parameter of thermodynamics and plays a key role in chemical reactions. The NB hydrogenation reaction has a certain reaction energy barrier, and the reaction can occur at a certain temperature. Fig. S8d (ESI†) compares the catalytic performance of the various Au/CeO2 catalysts. Au/CeO2-rod gave the highest NB conversion of 60.3% after 6 h at 120 °C, while 35.7% was achieved over Au/CeO2-flower, whereas Au/CeO2-cube showed a NB conversion of only 25.2%. When the temperature increased to 180 °C, the NB conversion could reach 100% for both Au/CeO2-rod and Au/CeO2-flower, whereas that for Au/CeO2-cube was less at 93.5%. Meanwhile, it can be seen from these results that the different kinds of Au/CeO2 forms had different catalytic activities with the increase in temperature, and Au/CeO2-rod exhibited the best catalytic performance. As the conversion deviation can be neglected at high NB conversion, in the subsequent experiments we chose 180 °C as the reaction temperature for the catalytic NB hydrogenation reaction, and the detailed experimental data for the temperature exploration are summarized in Table S1 (ESI†).To investigate the influence of the catalyst amount on the catalytic transfer hydrogenation of NB, as depicted in Fig. 4, the conversion of NB and the selectivity of AN were assessed and were found to increase with the increase in the amount of Au/CeO2 catalyst, which was ascribed to the increase in catalytic active sites. The selectivity of the intermediate products, such as NSB, PHA, AOB, and AB, showed a trend of increasing, and then decreasing until all were converted to AN for the Au/CeO2-rod (Fig. 4(a)) and Au/CeO2-flower (Fig. 4(b)) catalysts. When the amount of catalyst was 30 mg, the conversions of NB could reach 100% over both Au/CeO2-rod and Au/CeO2-flower, while it was 72% for Au/CeO2-cube (Fig. 4(c)). When 10 mg catalyst was used, the Au nanoparticles supported on CeO2-rod exhibited comprehensive advantages over those supported on CeO2-flower, and CeO2-cube (Fig. 4(d)), as evidenced by the highest NB conversion and AN selectivity with Au/CeO2-rod. Aniline is the final product for the catalytic NB transfer hydrogenation, while NSB, PHA, AOB, and AB are all intermediate products. From the selectivity trend in Fig. 4, we can see that within the range of the experimental use of the catalysts, with the increase in the catalyst dosage, the amount of NSB increased, and a small amount of PHA could also be detected. The further hydrogenation of PHA could produce aniline, while at the same time, PHA could also react with NSB through condensation to generate AOB, so the amount of AOB also increased. The further hydrogenation of AOB could form AB. When there was no accumulation of AOB, AB gradually decreased to produce the final product aniline, which thus increased.
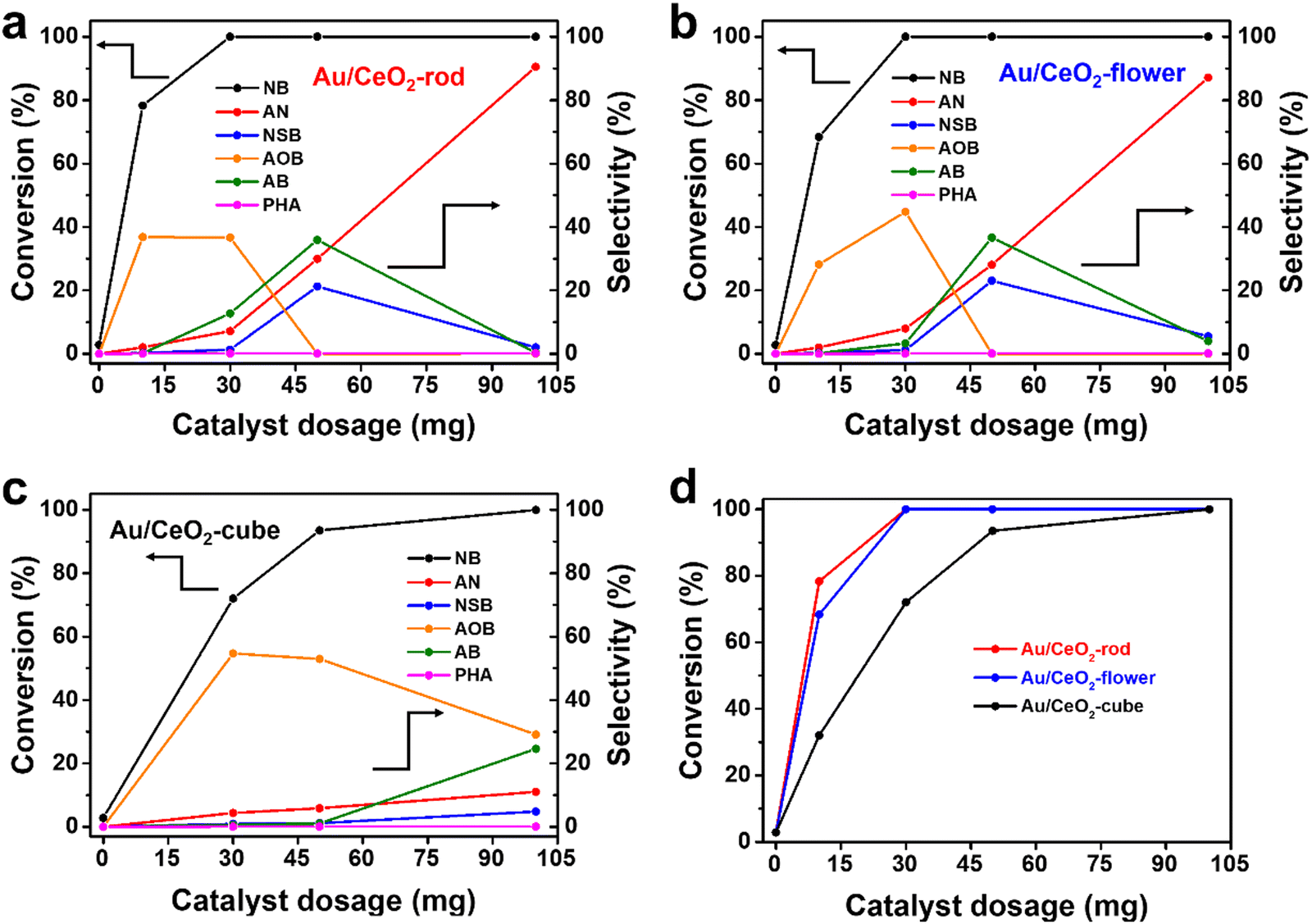 | ||
| Fig. 4 Effect of the catalyst dosage on the catalytic transfer hydrogenation of nitrobenzene over Au/CeO2-rod (a), Au/CeO2-flower (b), Au/CeO2-cube (c), and comparison of the conversions of nitrobenzene over the different Au/CeO2 catalysts (d). Reaction conditions: 0.06 mL of NB, 5 mL of isopropanol, Au/CeO2 catalyst, 180 °C, 6 h. | ||
For the nano Au/CeO2-rod and Au/CeO2-flower catalysts with excellent catalytic performance, within the range of 0–30 mg of catalyst usage, with the increase in catalyst usage, the catalytic active sites increased, and the selectivity of the reaction intermediates showed a trend of gradually increasing. When the catalyst dosage was 50 mg, the selectivity of AOB decreased to 0, and the selectivity of NSB and AB reached the highest degree. Further, when the catalyst usage was 100 mg, the reaction took place very quickly due to the large number of catalytic active sites, thereby causing the intermediate products to be rapidly converted into the final product aniline. Therefore, the intermediate products could hardly be detected, whereas the selectivity of aniline was the highest. Additionally, compared to the Au/CeO2-rod and Au/CeO2-flower catalysts, Au/CeO2-cube had a slightly lower catalytic performance, whereby when the Au/CeO2-cube dosage was 100 mg, the selectivity for the intermediate AOB gradually decreased, and the selectivity for AB and NSB gradually increased. This trend was similar to that of nano Au/CeO2-rod and Au/CeO2-flower but with catalyst dosages of 0–30 mg, since the catalytic activity of Au/CeO2-cube was lower than that of Au/CeO2-rod and Au/CeO2-flower. The detailed experimental data are listed in Table S2 (ESI†).
To further study the effect of the reaction time, Au/CeO2-rod and Au/CeO2-flower were chosen for the detailed reaction. As depicted in Fig. S9 (ESI†), with the reaction time proceeding, the NB conversion, and the selectivity of the products, including NSB, PHA, AOB, AB, and AN, increased. NB conversion reached 100% for Au/CeO2-rod, while it was 96% for Au/CeO2-flower in 4 h reaction time. As the reaction continued to 6 h, the NB conversion for both reached 100% with selectivities to AN of around 7%, while the selectivity toward AB was 12.8% for Au/CeO2-rod but 3.3% for Au/CeO2-flower, respectively. The above results suggest that Au/CeO2-rod had better catalytic performance than that of Au/CeO2-flower. The detailed experimental data are summarized in Table S3 (ESI†). It was also found that both a high temperature and long reaction time could boost the NB conversion and AN formation.
To investigate the morphological characteristics, the catalytic transfer hydrogenation of NB over CeO2 with different shapes was performed. Experiments were performed with the corresponding Au/CeO2 catalysts, and the results are displayed in Fig. 5(a). After 6 h, CeO2-rod exhibited the highest conversion of NB (100%), compared with CeO2-flower (98.05%), CeO2-cube (40.13%), and CeO2-octa (15.65%). Also, the experimental results for the corresponding Au/CeO2 catalysts are presented in Fig. 5(b), with Au/CeO2-rod and Au/CeO2-flower showing identical 100% conversion of NB, while their selectivities toward AOB and AB differed, with Au/CeO2-rod showing 36.67% selectivity for AOB and 12.75% for AB, and Au/CeO2-flower showing 44.8% selectivity for AOB and 3.28% for AB. The intermediate AOB was further reduced to AB, which also indicated that Au/CeO2-rod had better catalytic activity than Au/CeO2-flower. In addition, the NB conversions for Au/CeO2-cube and Au/CeO2-octa were 72%, and 34.1%, respectively. Compared with CeO2, the Au/CeO2 catalysts exhibited better catalytic performance, and the activity orders of the Au/CeO2 catalysts were as follows: Au/CeO2-rod > Au/CeO2-flower > Au/CeO2-cube > Au/CeO2-octa. The selectivities to the products AOB, AB, and AN obtained from the Au/CeO2 catalysts were higher than those obtained by CeO2. To get further insight into the morphology difference between Au/CeO2-rod and Au/CeO2-flower, the reaction time was decrease to 4 h, and the results are listed in Table S4 (ESI†) and depicted in Fig. 5(c). It can be seen that the series of CeO2 catalysts with different morphologies and the corresponding Au/CeO2 catalysts showed the same sequence of catalytic NB transfer reduction activities in the following order: rod > flower > cube > octa. Fig. 5(d) shows a photo of the mixture after 4 h reaction, where AB itself is brownish yellow, but the color of the mixed solution was related to the NB conversion and the amount of AB, whereby the darker the color, the higher the content of AB, and the higher the conversion. This illustrates that the color was related to the depth of NB hydrogenation, and the higher the catalytic performance of the Au/CeO2 catalyst, the more complete the reaction. In many reports, the oxygen vacancies in catalysts can affect the catalytic activity.63–66 The higher the density of defects, the higher the Ce3+ proportion, and the higher the content of adsorbed oxygen species, which were all related to the much higher conversion of NB. Here, the Au/CeO2 materials had different morphologies of CeO2 supports with different crystal planes exposed, namely Au/CeO2 rod {(110), (100), (111)}, Au/CeO2 flower {(100), (111)}, Au/CeO2 cube (100), and Au/CeO2 octa (111). The stronger interaction between nano Au and CeO2 with a high O vacancy content was conducive to the anchoring and dispersion of Au nanoparticles, and therefore, the corresponding catalytic activity of the Au/CeO2 catalyst was higher. Besides, with a higher concentration of Ce(III) in CeO2, the concentration of Ce(IV) is relatively lower, and the content of lattice O is lower, and the content of adsorbed O species is higher, which is more conducive to the adsorption of reactant molecules on the surface of the Au/CeO2 catalyst, thus obtaining higher catalytic efficiency. We also compared the nitrobenzene hydrogenation activities with that of other research groups, and the detailed results are listed in Table 2. For the nitrobenzene hydrogenation reaction, the hydrogen source used in this work was isopropanol, which is safer than H2 from a high-pressure steel cylinder. Comparing Au/CeO2-rod with Au@CeO2, the reaction time for the nitrobenzene hydrogenation was shorter. Hence, the reaction system in this work is simpler and milder than the other reaction systems on the list.
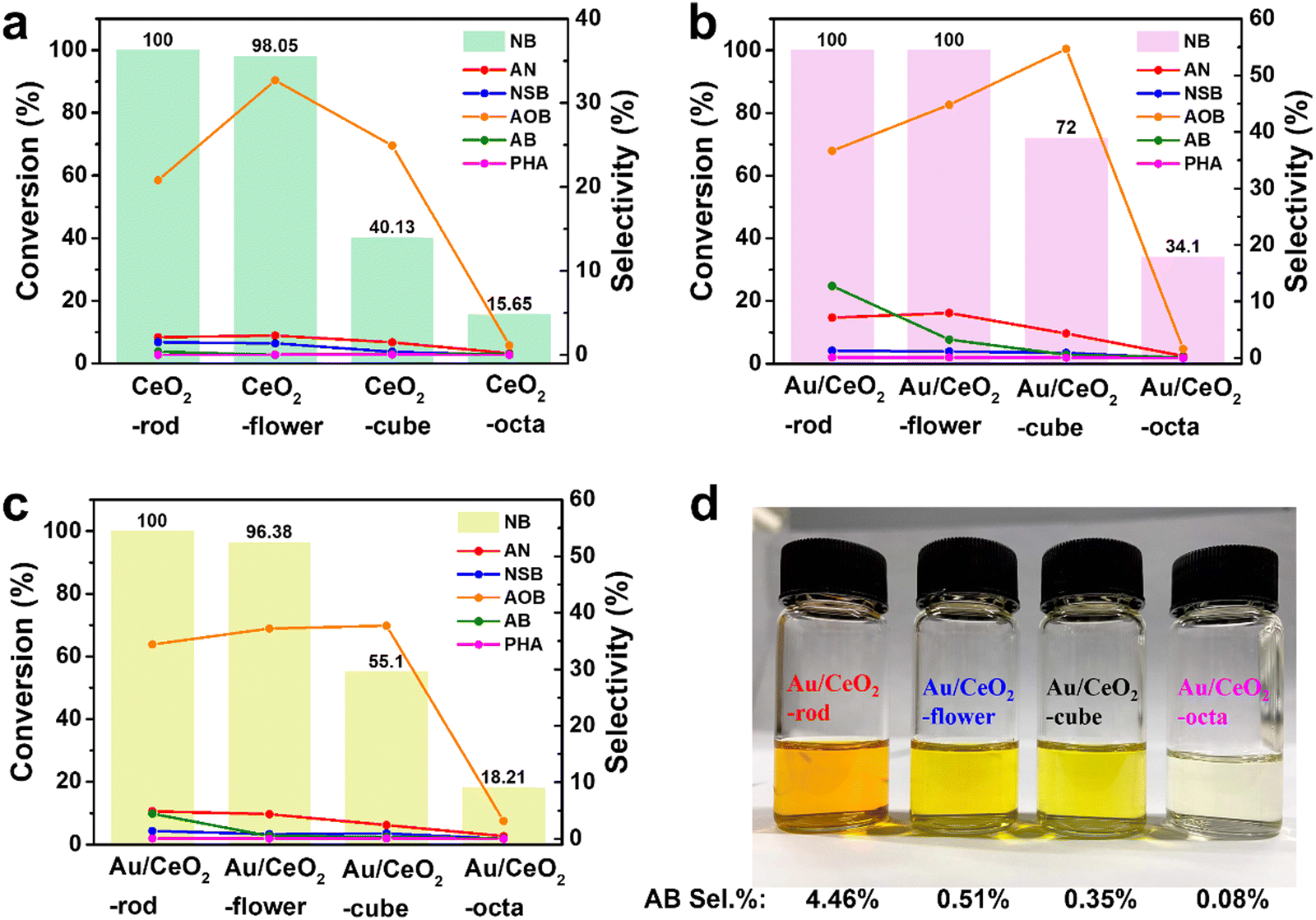 | ||
| Fig. 5 Comparison of various CeO2 (a) and Au/CeO2 (b and c) catalysts for the catalytic transfer hydrogenation of NB. (d) Photos of the catalyst mixtures after NB hydrogenation reaction under the conditions described below for c. Reaction conditions: 0.06 mL of NB, 30 mg of Au/CeO2, 5 mL of isopropanol, 180 °C, 6 h (a and b); reaction conditions: 0.06 mL of NB, 30 mg of Au/CeO2, 5 mL of isopropanol, 180 °C, 4 h (c). | ||
| Catalyst | Reductant | Temperature (°C) | Time (h) | NB Conv.% | AB Sel.% | AOB Sel.% | AN Sel.% | Ref. |
|---|---|---|---|---|---|---|---|---|
| Au/CeO2-rod | Isopropanol | 180 | 6 | 100 | 0.31 | 0.08 | 90.5 | This work |
| Pd/C | 2 MPa H2 | 40 | 16 | 81 | — | — | — | 67 |
| CeO2 | 1 MPa CO | 150 | 7 | 100 | — | 89.8 | — | 68 |
| Pt/CeO2 | 1 MPa H2 | 25 | 2 | 100 | 0.1 | 2.3 | 87.2 | 69 |
| Ru-SAs/CeO2 | 3 MPa H2 | 100 | 4 | 65 | — | 78 | — | 70 |
| Pt1/PO4–CeO2-500 | 0.1 MPa H2 | 60 | 0.75 | 100 | — | — | 100 | 71 |
| Au@CeO2 | Isopropanol | 50 | 13 | 100 | — | — | — | 72 |
Au nanoparticles have high catalytic activity as an active metal component. However, Au nanoparticles tend to become unstable due to agglomeration during heat treatment or reaction. The recyclability of the four kinds of Au/CeO2 catalysts was measured by reusing the catalysts at 180 °C for 6 h, and the results are demonstrated in Fig. S10 (ESI†). Fresh Au/CeO2-rod catalyst achieved an NB conversion of 100% in the first use, and the NB conversion still remained high at 94.2% in later cycles, indicating the good activity and stability of the Au/CeO2-rod. After 6 cycles, the NB conversion decreased from 34.1% to 5.1% for Au/CeO2-octa, indicating it had the worst stability. The spent Au/CeO2 catalysts were characterized by TEM (Fig. S11, ESI†), where it could be seen that the morphology of the spent Au/CeO2-rod and Au/CeO2-flower remained unchanged, while the size of the Au nanoparticles almost was unchanged. Nevertheless, for Au/CeO2-cube and Au/CeO2-octa, the catalysts become clustered after the reaction; in particular, the Au nanoparticles in Au/CeO2-octa showed serious aggregation and clustering. Taken together, these results confirmed that Au/CeO2-rod exhibited the highest stability, recyclability, and catalytic hydrogenation activity.
DFT calculation for the mechanism
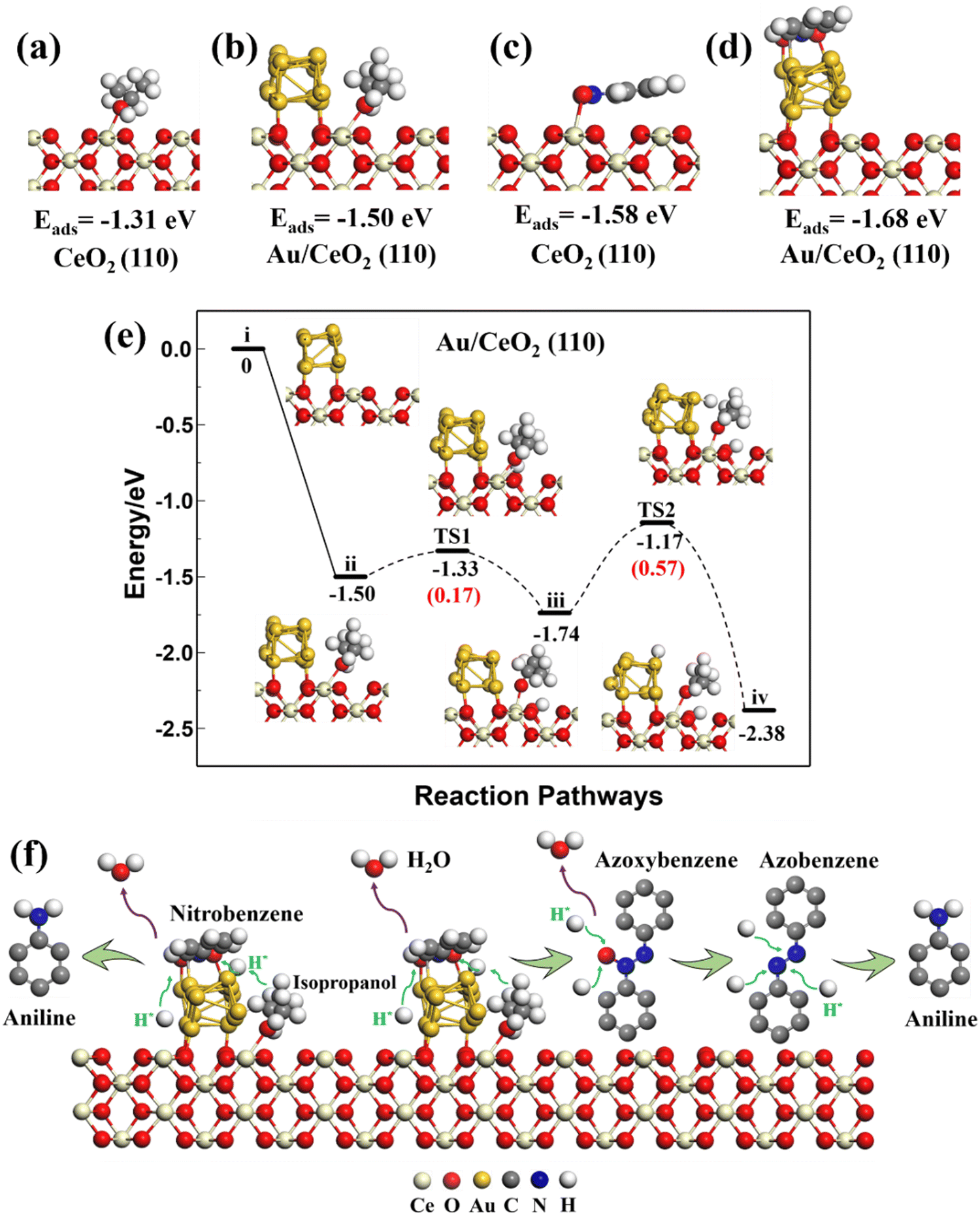 | ||
| Fig. 6 Optimized adsorption structures and calculated adsorption energies (Eads) for isopropanol on CeO2 (110) (a) and Au/CeO2 (110) (b) surfaces, and for nitrobenzene on CeO2 (110) (c) and Au/CeO2 (110) (d) surfaces. Reaction pathways for the dehydrogenation of isopropanol on the Au/CeO2 (110) (e) surface, and the possible pathways and mechanisms for the catalytic nitrobenzene transfer hydrogenation over Au/CeO2 (110) (f). | ||
The optimized structure of nitrobenzene lay parallel to the Au (111) and CeO2 (111), (110), and (100) surfaces with adsorption energies of −1.22, −1.11, −1.58, and −1.12 eV, respectively. Nitrobenzene preferred to adsorb parallel to the top of the Au strip on the Au/CeO2 interfacial structures, rather than on Ce. Isopropanol and nitrobenzene had different adsorption sites on the Au/CeO2 interfacial structures. The adsorption energies of the nitrobenzene on the Au/CeO2 (111), (110), and (100) surfaces were −1.63, −1.68, and −1.52 eV, stronger than that of nitrobenzene on related CeO2 surfaces and the Au surface. So the interfacial structures promoted the adsorption of nitrobenzene. Such large adsorption energies on the Au/CeO2 interfacial structures and separate reaction sites could benefit isopropanol and nitrobenzene adsorption and subsequent reactions.
Therefore, the theoretical results demonstrated an obvious synergistic effect on the Au/CeO2 interfacial structures for the dehydrogenation of isopropanol and the reactant adsorption. Among them, the (110) surfaces had the best isopropanol dehydrogenation performance, which contributes to the subsequent hydrogenation of nitrobenzene.
Conclusions
In this work, we prepared four kinds of eco-friendly CeO2 with different shapes exposing different crystal planes, namely CeO2-rod (110), (100), (111); CeO2-flower (100), (111); CeO2-cube (100); and CeO2-octa (111). The corresponding environmentally benign Au/CeO2 catalysts were synthesized successfully using polyphenol tannic acid as a reductant, chelating agent, and stabilizer instead of the traditional agents, which curbs the use and production of hazardous chemicals, thus making the process more environmentally clean. The oxygen vacancy concentration and Ce(III) proportion were closely related to the catalytic activities of the Au/CeO2 catalysts. However, the BET specific surface area and total pore volume were not directly correlated with the morphology of CeO2. Notably, the Au/CeO2 interface could effectively promote the catalytic performance.Using isopropanol as a hydrogen source instead of the traditional use of high-pressure H2 from a cylinder, the experimental safety was greatly improved. Moreover, isopropanol decomposed into hydrogen directly in the reactor, which increases the contact between H2 and nitrobenzene, thereby enhancing the efficiency of the catalytic nitrobenzene transfer hydrogenation. Compared with CeO2, the Au/CeO2 catalysts exhibited stronger catalytic nitrobenzene transfer hydrogenation activity, and the order of activity and stability both followed the order: Au/CeO2-rod > Au/CeO2-flower > Au/CeO2-cube > Au/CeO2-octa. Furthermore, high-value-added chemicals (aniline, azoxybenzene, and azobenzene) could be produced and their proportions could be regulated by tuning the reaction conditions. Combining the experimental and DFT calculation results revealed the mechanism of the catalytic nitrobenzene transfer hydrogenation. Isopropanol preferred to adsorb on CeO2 and nitrobenzene preferentially adsorbed on Au. Moreover, compared with Au/CeO2 (100) and Au/CeO2 (111), Au/CeO2 (110) had the best adsorption capacity of reactants and dehydrogenation capacity toward isopropanol. As a consequence, the interface of the Au/CeO2 catalysts played a synergistic role in nitrobenzene hydrogenation, whereby the different catalytic active sites at the interface of Au/CeO2 synergistically catalyzed nitrobenzene transfer hydrogenation by the Au/CeO2 catalyst, thus enhancing the catalytic performance. This work can provide a new idea for the structural engineering of catalysts for use in the chemical industry.
Materials and methods
Materials
Ce(NO3)3·6H2O (ceric nitrate hexahydrate) (AR, 99.9%), glucose (99%), and nitrosobenzene (98%) were purchased from Shanghai Macklin Biochemical Co., Ltd. KOH (potassium hydroxide) (95%) was bought from Shanghai Meryer Chemical Technology Co., Ltd. Na3PO4·12H2O (99.99%) was obtained from Shanghai Yien Chemical Technology Co., Ltd, NH3·H2O (AR, 25–28 wt%) was purchased from Beijing Mreda Technology Co., Ltd. Azoxybenzene (≥98%, GC) and azobenzene (97%) were received from Shanghai Aladdin Biochemical Technology Co., Ltd. HAuCl4 (chloroauric acid) (48–50% Au), analytical grade TA (tannic acid), and acrylamide (AR) were provided by Fuchen Chemical Reagent Co., Ltd (Tianjin, China). Isopropanol (≥99.9%, ACS grade) was provided by Anhui Zesheng Technology Co., Ltd. NaBH4 (sodium borohydride) (AR) was obtained from Xilong Chemical Co., Ltd. 4-Nitrophenol (99%, HPLC grade), aniline (99.5%), and phenylhydroxylamine (97%) were purchased from Saen Chemical Technology Co., Ltd (Shanghai, China). Nitrobenzene (99%) was obtained from damas-beta Reagent Co., Ltd. Acetone (≥99.5%, AR) was bought from Beijing Tongguang Fine Chemical Co., Ltd. Ethanol (AR, ≥99.7%) was purchased from Tianjin Damao Chemical Reagent Factory. All the chemical reagents were used without further purification. Water used in all the experiments was deionized water.Methods
Characterization
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained on a JEOL JEM-2100F microscope with a maximum operating voltage of 200 kV. The preparation steps for the samples for TEM characterization were as follows: a certain amount of sample was weighed and dispersed in ethanol solution, with sonicating for 30 min at room temperature, until the mixture presented a uniform state. Then, a rubber dropper was used to suck the sample and drop it onto the copper mesh of an ultrathin carbon film, and the ethanol was allowed to volatilize completely; after this, the TEM sample measurements were conducted. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping was performed on an OXFORD 51-XMX, UK instrument. The crystalline phase was recorded by X-ray diffraction (XRD) analysis on a Rigaku RU-200b powder X-ray diffractometer using Cu Kα radiation operated at 40 kV and 40 mA in the 2θ range of 10°–85°. The steps for preparing the samples for XRD analysis were: a certain amount of catalyst sample was ground with a mortar and pestle into a fine powder until there were no obvious particles left, and then a tablet of the sample was made for testing. X-Ray photoelectron spectroscopy (XPS) measurements of the chemical state of the different elements in the samples were obtained on a Thermo Fisher Escalab 250Xi instrument equipped with Al Kα (1486.6 eV) as the monochromatic X-ray source. The steps for preparing the samples for XPS analysis were: a double-sided adhesive was stuck on the front of the foil, and cut into a suitable size, and then the powdered catalyst sample was placed on the double-sided adhesive to fix it, and then to form a tablet, blowing off the unfixed powder. The foil was then cut to a suitable size and stuck on the sample table for testing. N2 adsorption–desorption isotherms, the specific surface area (Brunauer–Emmett–Teller, BET method), pore volume, and pore-size distribution (Barrett–Joyner–Halenda, BJH method) characterizations of the samples were performed on a Micromeritics ASAP2460 instrument with nitrogen adsorption at 77 K. Before analysis, the samples were pretreated for 12 h at 150 °C in a vacuum. Raman analysis was conducted on a Thermo Dxi confocal laser with 532 nm excitation (Ar+ laser). The amount of Au in the samples was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) (PerkinElmer Optima 7300 DV). Before the measurements, the samples were dissolved in aqua regia and diluted with deionized water. Thermogravimetric analysis (TGA) experiments were performed on a SETARAM Labsys evo (France) instrument. Temperature-programmed reduction by hydrogen (H2-TPR) analysis was performed on a Micromeritics Autochem II 2920 instrument with a thermal conductive detector (TCD). Electron spin resonance (ESR) spectroscopy was performed on a Bruker EMX-PLUS spectrometer, operating in the 9.84 GHz microwave frequency with a field modulator frequency of 100 kHz.Catalytic hydrogenation of 4-nitrophenol
The prepared catalysts were used for the reduction of 4-NP to 4-AP in a 3.5 mL quartz cuvette. In a typical reaction, 2 mg of NaBH4 was added into 2 mL aqueous solution of 0.14 mM 4-NP (the initial n4-NP/nNaBH4 = 1/188.8). Next, 0.8 mL of deionized water was added to the above mixture to keep the total volume to 2.8 mL. Finally, 1 mg of sample was added to the mixture to trigger the reaction at room temperature. To record the reaction process, UV-Vis spectra were recorded at every minute interval in the range of 250–500 nm using a HITACHI U-3900 spectrometer. In the recycling tests, the used catalyst was collected by centrifugation, washed with deionized water and ethanol several times, and then dried at 70 °C in air overnight for the recycling trials.Catalytic transfer hydrogenation of nitrobenzene
The liquid-phase hydrogenation of nitrobenzene was performed in a 25 mL stainless steel autoclave equipped with a magnetic stirrer. In a typical experiment, the autoclave was loaded with 0.58 mmol nitrobenzene, 5 mL of isopropanol, and 30 mg of catalyst. The reactor was purged with N2 at 0.5 MPa three times to displace the air in the reactor system, and then the reactor was sealed after releasing all the N2 left, and heated to the specified temperature (120–180 °C) while stirring at 1000 rpm. Once the reaction reached the set time, the autoclave was quickly removed from the heating device and rapidly cooled to stop the reaction. The obtained liquid reaction mixture was analyzed using a Shimadzu HPLC system with a UV/Vis detector and C18 column. In the analysis, the column temperature was 40 °C, the mobile phase was methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (volume to volume) = 75:25, flow rate was 1 mL min−1, and the detection wavelength was 232 nm. In addition, there are also some literature reports of studies that employed GC-MS or HPLC to evaluate the products and intermediates of nitrobenzene hydrogenation, and notably, the results from the GC-MS and HPLC analyses were consistent.74,75 Moreover, the route for nitrobenzene hydrogenation has been reported in many papers, which also confirmed our conjecture in this work.
:water (volume to volume) = 75:25, flow rate was 1 mL min−1, and the detection wavelength was 232 nm. In addition, there are also some literature reports of studies that employed GC-MS or HPLC to evaluate the products and intermediates of nitrobenzene hydrogenation, and notably, the results from the GC-MS and HPLC analyses were consistent.74,75 Moreover, the route for nitrobenzene hydrogenation has been reported in many papers, which also confirmed our conjecture in this work.
The nitrobenzene conversion and product selectivity were calculated as follows:
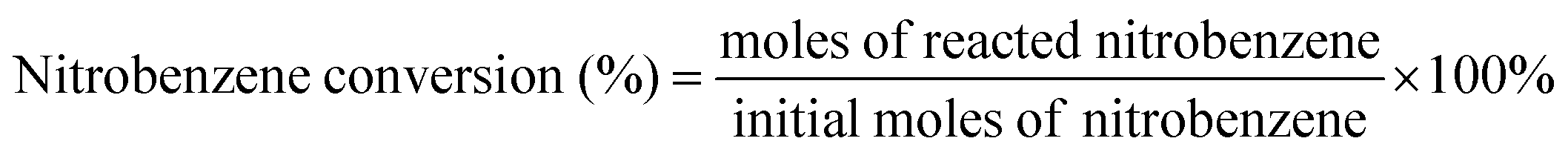 | (1) |
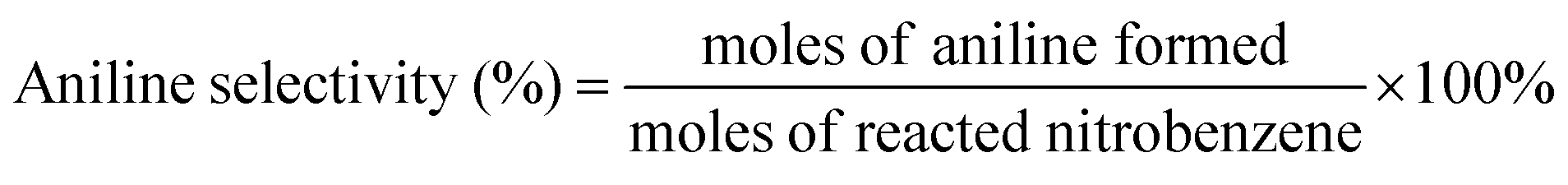 | (2) |
Density functional theory calculations
To further study the reaction mechanism of catalytic NB transfer hydrogenation, DFT calculations were performed with the Vienna ab initio simulation package code.76,77 The exchange–correlation energy was approximated by using the PBE functional within the generalized gradient approximation (GGA) with a basis set of plane waves up to an energy cutoff of 400 eV.78 The PAW method was used to simulate the core electrons.79,80 Dispersion forces were considered according to the Grimme approach (DFT+D3).81,82 The convergence criteria for the energy calculation and structure optimization were set to 1.0 × 10−4 eV and a force tolerance of 0.05 eV Å−1, respectively. The Brillouin-zone integration was sampled at the gamma point.83 We also employed DFT + U corrections to describe the CeO2 with the Ueff value of 4.5 eV for Ce 4f orbitals.84The Au (111) surface was modeled using a three-layer periodic slab model with a (4 × 4) super cell including a 15 Å vacuum slab, within which the adsorption and dehydrogenation occurred. According to the XRD results, three common CeO2 surface terminations of (111), (110), and (100) were chosen for the simulations and the effect of the crystal plane for the reactions was investigated.85,86 The CeO2 (111) surface consisted of a p(4 × 3) six-atomic-layer supercell with the bottom three layers fixed, while the CeO2 (110) surface was modeled by a p(3 × 3) four-atomic-layer supercell with the bottom two layers fixed, and the CeO2 (100) surface was modeled by an O-terminated p(4 × 3) seven-atomic-layer supercell with the bottom two layers fixed. The CeO2-supported Au interfacial system was modeled by adding a two-layer Au strip on the top of the CeO2 surfaces with different terminations. To eliminate the interactions between slabs, the vacuum region was set to 15 Å in the z-direction.
The adsorption energy was calculated by the equation: Eads = E(adsorbate/surface) − E(adsorbate) − E(surface), where E(adsorbate/surface), E(adsorbate), and E(surface) are the energies of the surface interacting with the adsorbate, the isolated adsorbate, and bare surface, respectively. With this definition, a negative value of Eads means a release of energy or a stable adsorption on the surface. The reaction transition states (TSs) were found with the climbing image nudged elastic band (CI-NEB) method.87,88 Frequency analysis was used to confirm that the transition states had only one imaginary frequency. The energy barrier (ETS) was defined as the total energy difference between the transition state and the initial structure.
Author contributions
Junqing Ye conducted most of the experimental work, writing the original draft and editing. Meizan Jing made the DFT calculation. Yu Liang, Wenjin Li, and Wanting Zhao contributed to collecting the experimental data. Jianying Huang helped with the material characterization and data analysis, Yuekun Lai and Jian Sun supervised Junqing Ye, and were responsible for writing, reviewing, and editing. Weiyu Song and Jian Liu provided the software of theoretical calculation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (51972063 and 22075046), National Key Research and Development Program of China (2022YFB3804905, 2022YFB3804900 and 2019YFE0111200), Natural Science Fund for Distinguished Young Scholar of Fujian Province (2020J06038), Higher Education Discipline Innovation 111 Project (No. D17005), and the Startup Foundation of Beijing Institute of Technology (3160013532102, and 3160011182007). The authors thank Biological and Medical Engineering Core Facilities, and Analysis & Testing Center, Beijing Institute of Technology, for supporting experimental tests.References
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21–33 CrossRef.
- S. Han, Y. Cheng, S. Liu, C. Tao, A. Wang, W. Wei, H. Yu and Y. Wei, Angew. Chem., Int. Ed., 2021, 60, 6382–6385 CrossRef CAS PubMed.
- J. Qin, Y. Long, F. Sun, P. P. Zhou, W. D. Wang, N. Luo and J. Ma, Angew. Chem., Int. Ed., 2022, 61, e202112907 CAS.
- S. Doherty, J. G. Knight, T. Backhouse, R. J. Summers, E. Abood, W. Simpson, W. Paget, R. A. Bourne, T. W. Chamberlain, R. Stones, K. R. J. Lovelock, J. M. Seymour, M. A. Isaacs, C. Hardacre, H. Daly and N. H. Rees, ACS Catal., 2019, 9, 4777–4791 CrossRef CAS.
- H. K. Kadam and S. G. Tilve, RSC Adv., 2015, 5, 83391–83407 RSC.
- P. Zhou, L. Jiang, F. Wang, K. Deng, K. Lv and Z. Zhang, Sci. Adv., 2017, 3, e1601945 CrossRef PubMed.
- T. Hou, Y. Wang, J. Zhang, M. Li, J. Lu, M. Heggen, C. Sievers and F. Wang, J. Catal., 2017, 353, 107–115 CrossRef CAS.
- H. Chong, P. Li, J. Xiang, F. Fu, D. Zhang, X. Ran and M. Zhu, Nanoscale, 2013, 5, 7622–7628 RSC.
- M. Turáková, T. Salmi, K. Eränen, J. Wärnå, D. Y. Murzin and M. Králik, Appl. Catal., A, 2015, 499, 66–76 CrossRef.
- E. A. McLoughlin, K. M. Waldie, S. Ramakrishnan and R. M. Waymouth, J. Am. Chem. Soc., 2018, 140, 13233–13241 CrossRef CAS PubMed.
- S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS PubMed.
- F. Alonso, P. Riente and M. Yus, Nickel nanoparticles in hydrogen transfer reactions, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS PubMed.
- V. Escande, C. Poullain, G. Clavé, E. Petit, N. Masquelez, P. Hesemann and C. Grison, Appl. Catal., B, 2017, 210, 495–503 CrossRef CAS.
- J. Hu, S. Zhang, R. Xiao, X. Jiang, Y. Wang, Y. Sun and P. Lu, Bioresour. Technol., 2019, 279, 228–233 CrossRef CAS PubMed.
- H. C. Yao and Y. Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- B. Liu, C. Li, Y. Zhang, Y. Liu, W. Hu, Q. Wang, L. Han and J. Zhang, Appl. Catal., B, 2012, 111–112, 467–475 CrossRef CAS.
- Y. Zhang, W. Feng, F. Yang and X. Bao, Chin. J. Catal., 2019, 40, 204–213 CrossRef CAS.
- F. Cao, M. Zhang, K. Yang, Z. Tian, J. Li and Y. Qu, Nano Res., 2020, 14, 715–719 CrossRef.
- Z. Zhao, G. Gao, Y. Xi, J. Wang, P. Sun, Q. Liu, W. Yan, Y. Cui, Z. Jiang and F. Li, Chem, 2022, 8, 1–16 Search PubMed.
- T. R. Reina, M. Gonzalez-Castano, V. Lopez-Flores, T. L. Martinez, A. Zitolo, S. Ivanova, W. Xu, M. A. Centeno, J. A. Rodriguez and J. A. Odriozola, J. Am. Chem. Soc., 2022, 144, 446–453 CrossRef CAS PubMed.
- M. Cargnello, V. V. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- D. Combita, P. Concepción and A. Corma, J. Catal., 2014, 311, 339–349 CrossRef CAS.
- X. Shi, X. Wang, X. Shang, X. Zou, W. Ding and X. Lu, ChemCatChem, 2017, 9, 3743–3751 CrossRef CAS.
- W. She, T. Qi, M. Cui, P. Yan, S. W. Ng, W. Li and G. Li, ACS Appl. Mater. Interfaces, 2018, 10, 14698–14707 CrossRef CAS PubMed.
- G. Spezzati, A. D. Benavidez, A. T. DeLaRiva, Y. Su, J. P. Hofmann, S. Asahina, E. J. Olivier, J. H. Neethling, J. T. Miller, A. K. Datye and E. J. M. Hensen, Appl. Catal., B, 2019, 243, 36–46 CrossRef CAS.
- P. Zhou, G. Hai, G. Zhao, R. Li, X. Huang, Y. Lu and G. Wang, Appl. Catal., B, 2023, 325, 122364 CrossRef CAS.
- Y. J. Hao, Y. G. Ma, X. Zhang, J. Li, S. Wang, X. Chen and F. Li, Chem. Eng. J., 2022, 433, 134619 CrossRef CAS.
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS.
- Y. Guo, Y. Qin, H. Liu, H. Wang, J. Han, X. Zhu and Q. Ge, ACS Catal., 2022, 12, 2998–3012 CrossRef CAS.
- B. Hu, K. Sun, Z. Zhuang, Z. Chen, S. Liu, W. C. Cheong, C. Chen, M. Hu, X. Cao, J. Ma, R. Tu, X. Zheng, H. Xiao, X. Chen, Y. Cui, Q. Peng, C. Chen and Y. Li, Adv. Mater., 2022, 34, 2107721 CrossRef CAS PubMed.
- J. Li, Z. Liu, D. A. Cullen, W. Hu, J. Huang, L. Yao, Z. Peng, P. Liao and R. Wang, ACS Catal., 2019, 9, 11088–11103 CrossRef CAS.
- Y. A. May, W. W. Wang, H. Yan, S. Wei and C. J. Jia, Chin. J. Catal., 2020, 41, 1017–1027 CrossRef CAS.
- Z. Wu, M. Li and S. H. Overbury, J. Catal., 2012, 285, 61–73 CrossRef CAS.
- V. Muravev, G. Spezzati, Y. Q. Su, A. Parastaev, F. K. Chiang, A. Longo, C. Escudero, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2021, 4, 469–478 CrossRef CAS.
- W. Wan, J. Geiger, N. Berdunov, M. Lopez Luna, S. W. Chee, N. Daelman, N. Lopez, S. Shaikhutdinov and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2022, 61, e202112640 CAS.
- N. Ta, J. J. Liu, S. Chenna, P. A. Crozier, Y. Li, A. Chen and W. Shen, J. Am. Chem. Soc., 2012, 134, 20585–20588 CrossRef CAS PubMed.
- Y. L. Lee, A. Mnoyan, H. S. Na, S. Y. Ahn, K. J. Kim, J. O. Shim, K. Lee and H. S. Roh, Catal. Sci. Technol., 2020, 10, 6299–6308 RSC.
- J. Lee, D. Shin, E. Lee, C. Li, J. M. Kim, J. W. Han and D. H. Kim, Appl. Catal., B, 2022, 305, 121038 CrossRef CAS.
- Y. G. Wang, D. Mei, V. A. Glezakou, J. Li and R. Rousseau, Nat. Commun., 2015, 6, 6511 CrossRef CAS PubMed.
- H. S. Jeong, S. H. Ryu, Y. Myung, S. M. Lee, H. J. Kim and S. U. Son, ACS Appl. Nano Mater., 2022, 5, 4603–4608 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- H. Li, F. Qin, Z. P. Yang, X. M. Cui, J. F. Wang and L. Z. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- Q. Fu, A. Weber and M. Flytzani-Stephanopoulos, Catal. Lett., 2001, 77, 87–95 CrossRef CAS.
- Y. Bu, Y. Chen, G. Jiang, X. Hou, S. Li and Z. Zhang, Appl. Catal., B, 2020, 260, 118138 CrossRef CAS.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., Int. Ed., 2008, 47, 2884–2887 CrossRef CAS PubMed.
- Q. Wang, Y. Li, B. Liu, Q. Dong, G. Xu, L. Zhang and J. Zhang, J. Mater. Chem. A, 2015, 3, 139–147 RSC.
- A. Kundu, B. Park, C. Ray, J. Oh and S. C. Jun, New J. Chem., 2019, 43, 900–906 RSC.
- J. Zhang, G. Chen, M. Chaker, F. Rosei and D. Ma, Appl. Catal., B, 2013, 132–133, 107–115 CrossRef CAS.
- Y. Wei, Y. Zhang, P. Zhang, J. Xiong, X. Mei, Q. Yu, Z. Zhao and J. Liu, Environ. Sci. Technol., 2020, 54, 2002–2011 CrossRef CAS PubMed.
- J. Park, J. Graciani, J. Evans, D. Stacchiola, S. Ma, P. liu, A. Nambu, J. Sanz, J. Hrbek and J. Rodriguez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4975–4980 CrossRef CAS PubMed.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- Y. Zhang, J. Lu, L. Zhang, T. Fu, J. Zhang, X. Zhu, X. Gao, D. He, Y. Luo, D. D. Dionysiou and W. Zhu, Appl. Catal., B, 2022, 309, 121249 CrossRef CAS.
- Y. Wei, Z. Zhao, J. Liu, C. Xu, G. Jiang and A. Duan, Small, 2013, 9, 3957–3963 CrossRef CAS PubMed.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- J. Lin, L. Li, Y. Huang, W. Zhang, X. Wang, A. Wang and T. Zhang, J. Phys. Chem. C, 2011, 115, 16509–16517 CrossRef CAS.
- S. Jiang, C. Li, Y. Muhammad, Y. Tang, R. Wang, J. J. Li, J. Li, Z. Zhao and Z. X. Zhao, Appl. Catal., B, 2023, 328, 122509 CrossRef CAS.
- P. Sudarsanam, B. Hillary, M. H. Amin, N. Rockstroh, U. Bentrup, A. Bruckner and S. K. Bhargava, Langmuir, 2018, 34, 2663–2673 CrossRef CAS PubMed.
- Y. Wei, J. Liu, Z. Zhao, A. Duan and G. Jiang, J. Catal., 2012, 287, 13–29 CrossRef CAS.
- J. Ye, S. Wang, G. Li, B. He, X. Chen, Y. Cui, W. Zhao and J. Sun, Chem. – Asian J., 2021, 16, 3371–3384 CrossRef CAS PubMed.
- T. Yu, J. Zeng, B. Lim and Y. Xia, Adv. Mater., 2010, 22, 5188–5192 CrossRef CAS PubMed.
- X. Zhou, H. Zhao, S. Liu, Y. Yang, R. Qu, C. Zhen and X. Gao, Chin. Chem. Lett., 2021, 32, 761–764 CrossRef CAS.
- H. Zhao, C. I. Ezeh, S. Yin, Z. Xie, C. H. Pang, C. Zheng, X. Gao and T. Wu, Appl. Catal., B, 2020, 263, 117829 CrossRef CAS.
- H. Zhao, G. Yang, X. Gao, C. H. Pang, S. W. Kingman and T. Wu, Environ. Sci. Technol., 2016, 50, 1056–1064 CrossRef CAS PubMed.
- Y. Hao, Y. Ma, X. Zhang, J. Li, S. Wang, X. Chen and F. Li, Chem. Eng. J., 2022, 433, 134619 CrossRef CAS.
- W. Li, J. Artz, C. Broicher, K. Junge, H. Hartmann, A. Besmehn, R. Palkovits and M. Beller, Catal. Sci. Technol., 2019, 9, 157–162 RSC.
- X. Zhou, H. Zhao, S. Liu, Y. Yang, R. Qu, C. Zhen and X. Gao, Chin. Chem. Lett., 2021, 32, 761–764 CrossRef CAS.
- Q. Zhang, J. Bu, J. Wang, C. Sun, D. Zhao, G. Sheng, X. Xie, M. Sun and L. Yu, ACS Catal., 2020, 10, 10350–10363 CrossRef CAS.
- B. Wu, T. Lin, R. Yang, M. Huang, H. Zhang, J. Li, F. Sun, F. Song, Z. Jiang, L. Zhong and Y. Sun, Green Chem., 2021, 23, 4753 RSC.
- Y. Ma, B. Chi, W. Liu, L. Cao, Y. Lin, X. Zhang, X. Ye, S. Wei and J. Lu, ACS Catal., 2019, 9, 8404–8412 CrossRef CAS.
- J. Li, S. Song, Y. Long, L. Wu, X. Wang, Y. Xing, R. Jin, X. Liu and H. Zhang, Adv. Mater., 2018, 30, 1704416 CrossRef PubMed.
- H. X. Mai, L. D. Sun, Y. W. Zhang, R. Si, W. Feng, H. P. Zhang, H. C. Liu and C. H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- D. Combita, P. Concepción and A. Corma, J. Catal., 2014, 311, 339–349 CrossRef CAS.
- Z. Zhang, H. Gai, Q. Li, B. Feng, M. Xiao, T. Huang and H. Song, Chem. Eng. J., 2022, 429, 132224 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- L. Zhang, X. M. Cao and P. Hu, Appl. Surf. Sci., 2017, 392, 456–471 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- Y. Tang, Y. G. Wang and J. Li, J. Phys. Chem. C, 2017, 121, 11281–11289 CrossRef CAS.
- M. Jing, W. Song, Y. Li, Z. Zhao, J. Liu and G. Henkelman, Chem. Eng. J., 2022, 433, 133599 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00103b |
| This journal is © The Royal Society of Chemistry 2023 |