
Cu and Si co-doping on TiO2 nanosheets to modulate reactive oxygen species for efficient photocatalytic methane conversion†
Jun
Ma
ab,
Jingxiang
Low
 a,
Di
Wu
a,
Wanbing
Gong
a,
Hengjie
Liu
a,
Dong
Liu
*ab,
Ran
Long
*a and
Yujie
Xiong
*a
a,
Di
Wu
a,
Wanbing
Gong
a,
Hengjie
Liu
a,
Dong
Liu
*ab,
Ran
Long
*a and
Yujie
Xiong
*a
aHefei National Research Center for Physical Sciences at the Microscale, School of Chemistry and Materials Science, National Synchrotron Radiation Laboratory, School of Nuclear Science and Technology, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: dongliu@ustc.edu.cn; longran@ustc.edu.cn; yjxiong@ustc.edu.cn
bSuzhou Institute for Advanced Research, University of Science and Technology of China, Suzhou, Jiangsu 215123, China
First published on 11th November 2022
Abstract
In this study, we successfully construct Cu and Si co-doped ultrathin TiO2 nanosheets. As confirmed by comprehensive characterizations, Cu and Si co-doping can rationally tailor the electronic structure of TiO2 to maneuver reactive oxygen species for effective photocatalytic methane conversion. In addition, this co-doping greatly enhances the utilization efficiency of photogenerated charges. Furthermore, it is revealed that Cu and Si co-doping can significantly boost the adsorption and activation of methane on TiO2 nanosheets. As a result, the optimized catalyst achieves a C2H6 production rate of 33.8 μmol g−1 h−1 with a selectivity of 88.4%. This work provides insights into nanocatalyst design toward efficient photocatalytic methane conversion into value-added compounds.
New conceptsMethane (CH4), the predominant constituent of natural gas, shale gas and combustible ice, has provoked increasing attention as an essential feedstock for energy supply and chemical production. However, current industrial routes for methane transformation are highly energy-intensive. Less than 10% of the global methane production is used for chemical manufacture, which mainly ascribes to the inert C–H bond and highly symmetrical structure of methane molecule. Herein, we provide an efficient and selective candidate for photocatalytic methane conversion into ethane through Cu and Si co-doping into ultrathin TiO2 nanosheets. As determined by comprehensive spectroscopic characterizations, the presence of Cu and Si could rationally regulate the electronic structure to modulate reactive oxygen species for C–H bond activation of methane molecule. Impressively, the as-synthesized catalyst possesses a remarkable catalytic activity and selectivity with a 33.8 μmol g−1 h−1 C2H6 generation rate and 88.4% C2H6 selectivity at room temperature under light irradiation. This co-doping approach provides a feasible route for designing low-cost and highly active photocatalysts for practical methane conversion applications under mild conditions. |
1. Introduction
With the rapid development of exploration and exploitation technology, natural gas reserves and production have been continuously growing over decades.1–4 Methane has received tremendous attention from both scientific and industrial communities due to its abundance and potential as a feedstock for manufacturing high-value fuels and chemicals.5,6 However, the inert C–H bond and highly symmetrical structure of methane molecule make it substantially challenging to transform methane into target products. Conventional methane activation is a severe energy-intensive process with carbon emissions. It requires critical conditions (i.e., high temperature and pressure) to activate the C–H bond combined with sophisticated post-processes.7–9 Thus, it is urgent to develop efficient and selective approaches for the transformation of methane into value-added compounds under moderate conditions.Recently, photocatalysis emerges as a prospective approach for methane conversion under mild conditions because it can utilize reactive oxygen species (ROS) to dissociate the C–H bond of methane upon photoexcitation, and greatly decrease the energy barrier of C–H bond activation.1,2,10–12 In 1998, direct photocatalytic methane coupling was first achieved over SiO2–Al2O3–TiO2 ternary catalyst.13 Since then, various semiconductors-based photocatalysts have been employed for catalytic coupling of methane into ethane at room temperature.1,14,15 Among these photocatalysts, TiO2-based catalysts are one of the most widely applied materials for photocatalytic methane conversion due to their suitable energy band structure and malleable qualities.16–18 For example, Tang and co-workers constructed Pt and CuOx co-decorated TiO2 for the photocatalytic oxidative coupling of methane (OCM), which achieved a 6.8 μmol h−1 yield of C2+ products (C2H6 and C2H4) with approximately 60% selectivity.19 Despite these advances, the efficiency and selectivity of photocatalytic methane conversion are still unsatisfactory due to scarce surface active sites. Therefore, efficient and selective photocatalysts should be further developed to satisfy the requirements for practical applications.
Copper (Cu) is generally regarded as the active center of particulate methane monooxygenases enzyme in the nature while silica matrix can improve the photocatalytic stability.20–22 Here, we successfully fabricated Cu and Si co-doped ultrathin TiO2 nanosheets for efficient photocatalytic methane conversion at room temperature and ambient pressure. As demonstrated by comprehensive characterizations, the presence of Cu and Si could rationally regulate the electronic structure of TiO2 nanosheets and modulate the reactive oxygen species for the activation of C–H bonds, accompanied with the presence of oxygen vacancies and Ti3+ species in the TiO2 matrix. Furthermore, this co-doping could substantially improve the separation and migration of the photogenerated charges. As a result, the optimized catalyst achieves a C2H6 production rate of 33.8 μmol g−1 h−1 with a selectivity of 88.4% at room temperature.
2. Results and discussion
The Cu and Si co-doped ultrathin TiO2 nanosheets were synthesized through a simple solvothermal method in ethanol solution (shown in Fig. 1) using Cu(NO3)2 and tetraethyl orthosilicate (TEOS) as doping precursors, respectively. The as-synthesized samples are denoted as TiO2(Si)–X%Cu, where X is the molar percentage of Cu atoms in the precursors. The crystal structures of TiO2 based catalysts were identified using powder X-ray diffraction (XRD) characterization. As shown in Fig. S1 (ESI†), the XRD patterns of all TiO2(Si)–X%Cu nanosheets are attributed to anatase TiO2 (JCPDS-21-1272), without any secondary patterns related to Cu/Si or its oxide phases, suggesting that co-doping does not alter the crystal structure of TiO2 nanosheets. As evidenced by the transmission electron microscopy (TEM) characterization (Fig. 1b and Fig. S2, ESI†), the ultrathin TiO2 nanosheets display a lateral sheet size of ca. 10 nm and a thickness of ca. 3 nm, exposing abundant sites for photocatalytic reactions. The high-resolution TEM (HRTEM) image (Fig. 1c) shows lattice fringes of 0.19 nm and 0.20 nm, indexing to the (200) and (020) facets of anatase TiO2, respectively. These results reveal that the as-synthesized ultrathin TiO2 nanosheets exhibit a high ratio of reactive (001) faces.23,24 The element distribution of the co-doped TiO2 nanosheets is then analysed by energy-dispersive X-ray spectroscopy (EDS). As shown in Fig. 1d, the homogeneous distribution of Ti, O, Cu, and Si elements is confirmed throughout the nanosheets. Combined with the XRD results, this indicates that Cu and Si were successfully and uniformly doped into TiO2.25 Furthermore, UV-vis diffuse reflectance spectroscopy (Fig. 1e) revealed that the absorption edges of the TiO2 nanosheets redshift with the incorporation of Cu due to the presence of oxygen vacancies and 2Eg → 2T2g transitions from O to Cu atoms,23 while the absorption edges of TiO2 show negligible relevance to Si doping. Interestingly, Cu doping could increase the near-infrared absorption up to 800 nm, assigned to the d–d transitions of doped Cu,23,25,26 which would obviously enhance the light absorption for the photocatalytic reaction. In addition, the absorption in the near-infrared region can increase the surface temperature of the photocatalyst due to photothermal effect (Fig. S3, ESI†), facilitating methane activation on the TiO2 surface.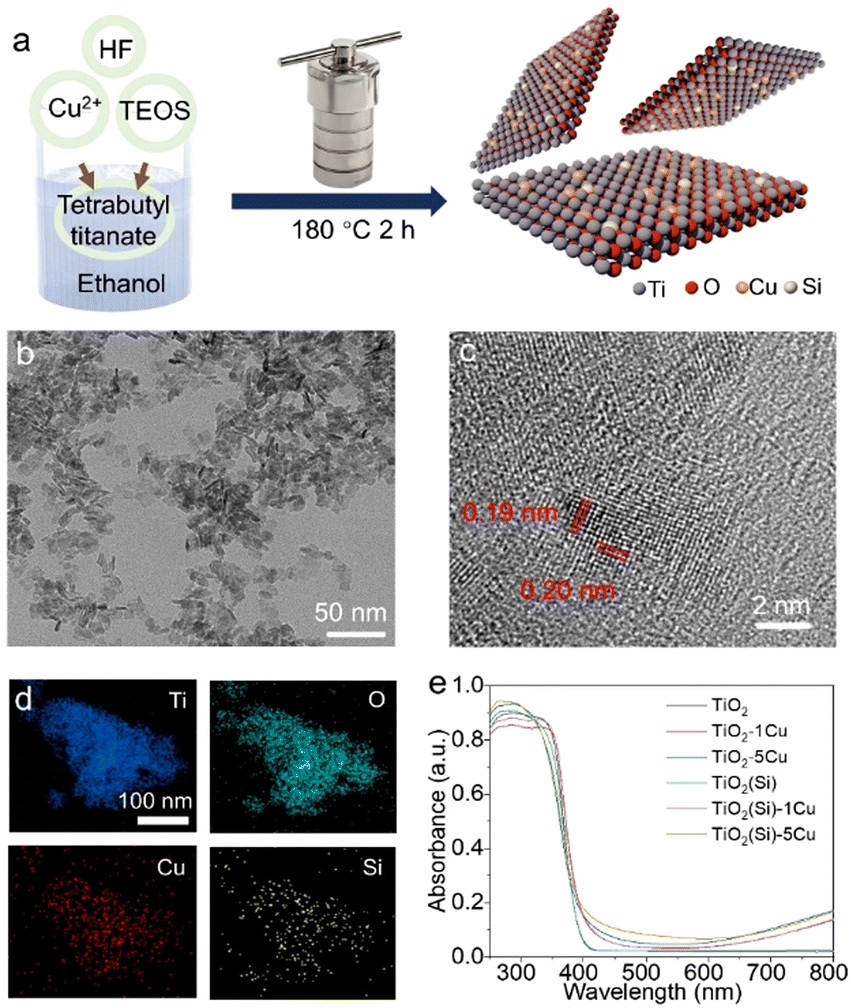 | ||
| Fig. 1 (a) Schematic illustration of the preparation of Cu & Si co-doped ultrathin TiO2 nanosheets. (b) TEM, (c) HRTEM images and (d) EDS mapping of TiO2(Si)–1%Cu nanosheets. (e) UV-vis diffuse reflectance spectra of the prepared Cu & Si co-doped TiO2 nanosheets. | ||
X-ray photoelectron spectroscopy (XPS) was employed to further investigate the electronic structure of the co-doped TiO2 nanosheets. The refined Cu 2p XPS spectra of TiO2–1%Cu and TiO2(Si)–1%Cu (Fig. S4, ESI†) both exhibit a +2 oxidation state, confirmed by bimodal peaks at 933.2 eV (Cu 2p3/2) and 953.1 eV (Cu 2p1/2).23 The Si 2p spectra of TiO2(Si) and TiO2(Si)–1%Cu (Fig. S5, ESI†) are mainly attributed to +4 oxidation state.27,28 As shown in Fig. 2a, the majority of Ti is in the Ti4+ state for all samples, while Ti3+ can be distinctly observed in the Ti 2p spectra of TiO2–1%Cu and TiO2(Si)–1%Cu due to the introduction of oxygen vacancies by Cu doping. Interestingly, the intensity of Ti3+ in the Ti 2p XPS spectrum decreased after the incorporation of Si into the TiO2 nanosheets, which resulted from the modulation of the electronic structure between Si and Ti atoms.28,29 To further elucidate the oxidation states of the doped Cu, electron paramagnetic resonance (EPR) was performed to recognize the electronic structure and coordination environment of the dopant.30 As shown in Fig. S6 (ESI†), the EPR spectra of TiO2–1%Cu and TiO2(Si)–1%Cu exhibit a characteristic Cu2+ hyperfine signal with resonance parameters at g‖ = 2.33 and g⊥ = 2.07, which indicates that Cu2+ ions substitute Ti sites in the TiO2 lattice.30,31
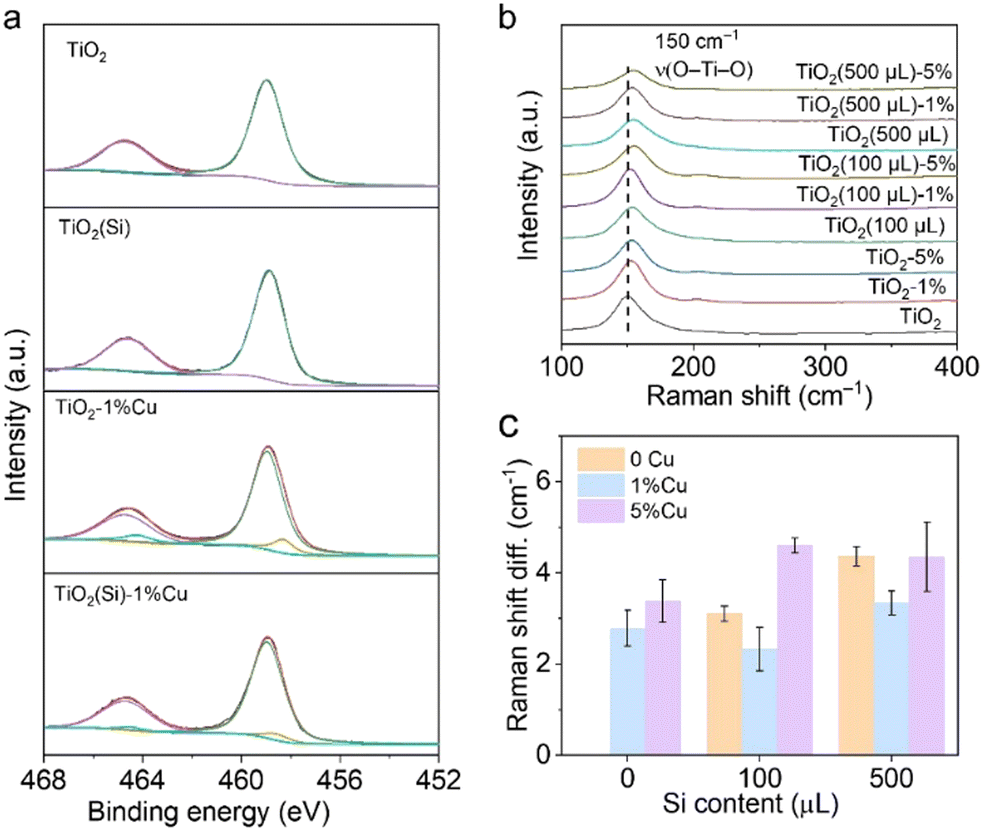 | ||
| Fig. 2 (a) Ti 2p XPS spectra and (b) Raman spectra of TiO2(Si)–X%Cu nanosheets. (c) Raman shift difference of TiO2(Si)–X%Cu nanosheets compared with TiO2 nanosheets. | ||
Moreover, Raman spectroscopy was carried out to investigate the local structure alteration upon the incorporation of Cu and Si. As shown in Fig. 2b, the Raman spectra exhibit intensive peaks at 149 cm−1 attributed to the Eg mode of anatase TiO2, consistent with the XRD results.25,32 The Eg mode, assigned to the symmetric stretching vibration of O–Ti–O in TiO2,32 shifts towards a higher wavenumber upon the incorporation of Cu or Si. This phenomenon could occur due to the introduction of Cu and Si induces lattice distortion in TiO2 and the formation of Cu–O–Ti or Si–O–Ti bonds.25,32 The lattice distortion could maneuver the adsorption configuration of reactant molecules on the surface.25,33 In particular, the peak shift achieves its smallest when 1% molar Cu and 100 μL Si are doped into TiO2 nanosheets (Fig. 2c), originating from the distortion balance between Cu and Si,34 which may boost the photocatalytic activity and stability over the co-doped TiO2.
To clarify the co-doping effect on photocatalytic reactivity of TiO2, in situ EPR was carried out to detect the ROS generated on the prepared TiO2 nanosheets under light irradiation using 5, 5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapping agent. As revealed in Fig. 3a, the EPR spectra for TiO2 and TiO2(Si) nanosheets show quartet signals with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1, which could be assigned to the adduct of hydroxyl radicals with spin-trapping agent (DMPO–˙OH), while the EPR spectrum for TiO2–1%Cu exhibits negligible DMPO–˙OH signals. In contrast, a sextet signal is generated in the EPR spectra for TiO2(Si)–1%Cu, which may result from the N–C bond cleavage and ring opening of DMPO over TiO2(Si)–1%Cu under light irradiation,35,36 indicating the significant oxidation ability of the co-doped TiO2. The EPR results demonstrate that the incorporation of Cu and Si can rationally modulate the oxidation capability of TiO2 and the ROS for photocatalytic reactions, benefiting from the alteration of the electronic structure of TiO2 and the presence of Ti3+ and oxygen vacancies.
:2:2:1, which could be assigned to the adduct of hydroxyl radicals with spin-trapping agent (DMPO–˙OH), while the EPR spectrum for TiO2–1%Cu exhibits negligible DMPO–˙OH signals. In contrast, a sextet signal is generated in the EPR spectra for TiO2(Si)–1%Cu, which may result from the N–C bond cleavage and ring opening of DMPO over TiO2(Si)–1%Cu under light irradiation,35,36 indicating the significant oxidation ability of the co-doped TiO2. The EPR results demonstrate that the incorporation of Cu and Si can rationally modulate the oxidation capability of TiO2 and the ROS for photocatalytic reactions, benefiting from the alteration of the electronic structure of TiO2 and the presence of Ti3+ and oxygen vacancies.
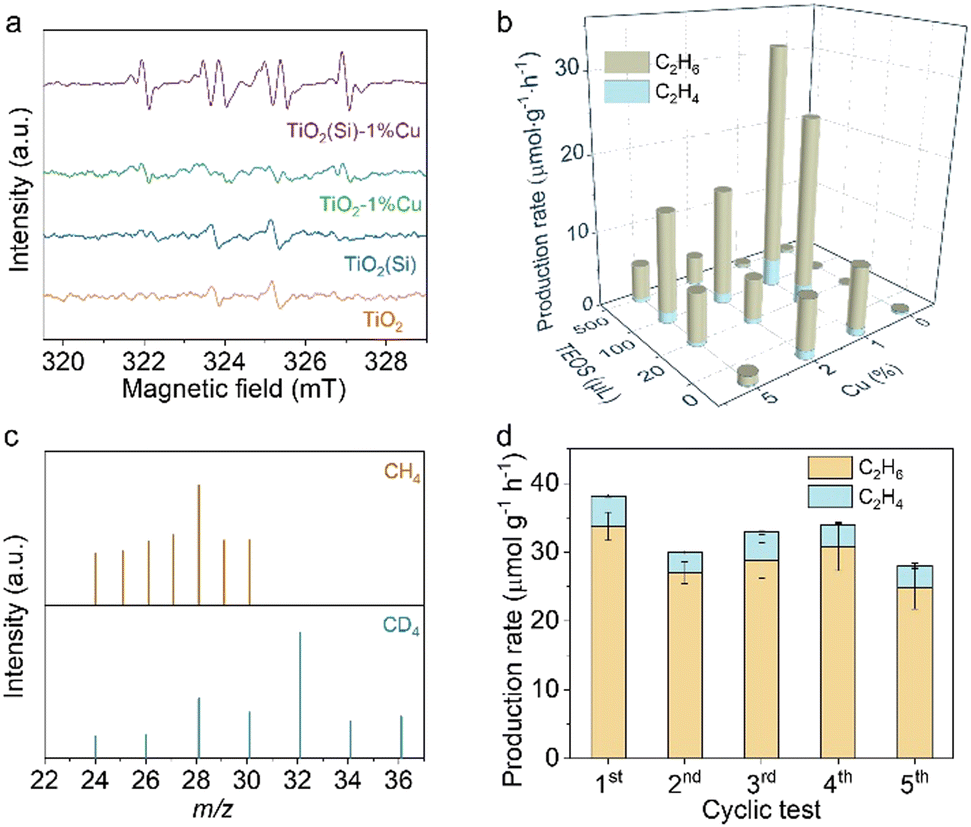 | ||
| Fig. 3 (a) In situ EPR spectra for the prepared TiO2 nanosheets under irradiation using DMPO as a spinning trapping agent. (b) Photocatalytic performance of the prepared TiO2 nanosheets after 4 h of light irradiation. (c) Mass spectra of ethane produced over TiO2(Si)–1%Cu using CH4 and CD4 as the feed gas. (d) Photocatalytic production rate in the cyclic test of TiO2(Si)–1%Cu. | ||
Upon recognizing the status of doped Cu and Si in TiO2 nanosheets, we are now in a position to evaluate the performance of the prepared TiO2 nanosheets for photocatalytic methane conversion. The photocatalytic experiments were carried out in CH4 saturated water under 300 W xenon lamp (PLS-SXE300, Perfect Light) irradiation under ambient conditions, where the solvation effect of H2O could facilitate the desorption of products from the surface of the catalyst to avoid overoxidation.37,38 As detected by gas chromatography (GC, Agilent 7890B), the pure TiO2 nanosheets are inactive for methane conversion (shown in Fig. 3b and Fig. S7, ESI†). Interestingly, the introduction of Cu or Si into the TiO2 nanosheets endow the distinctly enhanced activity for methane conversion. Moreover, the synergetic effect of Cu and Si would further facilitate photocatalytic methane conversion over TiO2 nanosheets. In particular, TiO2(100μL Si)–1%Cu achieves a C2H6 production rate of 33.8 μmol g−1 h−1 with a selectivity of 88.4% upon 4 h light irradiation, consistent with the Raman and in situ EPR characterizations. These results clearly demonstrate the importance of the ROS and the regulation of electronic structure in photocatalytic methane conversion. Nevertheless, the photocatalytic performance of the prepared catalyst declines with further increase in Cu or Si content, due to the shielding effect and photogenerated charge recombination.
To further elucidate the accuracy of the photocatalytic results, isotope labelling experiments are performed using CH4 or CD4 as the feed gas to conduct photocatalytic methane conversion over TiO2(Si)–1%Cu. As shown in Fig. 3c, the m/z values of ethane vary upon changing the reactant gas. In detail, the location of the strongest peak shifts from m/z = 28 (using CH4 feed gas) to m/z = 32 (using CD4 as feed gas), and a new peak with constant relative intensity at m/z = 36 appears, demonstrating the produced ethane originating from the fed CH4. Moreover, control experiments showed that no products could be detected in the absence of methane, photocatalyst, or light irradiation (Fig. S8, ESI†), suggesting that the products originate from photocatalytic methane conversion over co-doped TiO2 nanosheets. Moreover, the co-doped TiO2 nanosheets exhibit significant photocatalytic stability, and the photocatalytic performance of TiO2(Si)–1%Cu is well maintained for five cycles with each run of 4 h (Fig. 3d and Fig. S9, ESI†).
To comprehend the utilization efficiency of photogenerated charge carriers by co-doping, we performed photoelectrochemical measurements to observe the interfacial charge kinetics of TiO2 nanosheets through the transient photocurrent response. As shown in Fig. 4a, the photocurrents of TiO2(Si), TiO2–1%Cu and TiO2(Si)–1%Cu are obviously larger than that of pristine TiO2. More importantly, TiO2(Si)–1%Cu achieves the highest transient photocurrent, indicating that Cu and Si doping can greatly facilitate photogenerated charge separation and migration for photocatalytic methane conversion. Moreover, electrochemical impedance measurements further evaluate the charge transport. All TiO2 nanosheets exhibit a positive slope in the Mott–Schottky plot (Fig. S10, ESI†). Specifically, the slopes for doped TiO2 nanosheets are more obviously gradual than that of the pristine TiO2 nanosheets, which qualitatively indicates a higher charge carrier density according to the Mott-Schottky equation.39 Overall, the improved photocatalytic performance for doped TiO2 could be mainly attributed to the enhanced charge separation and migration.
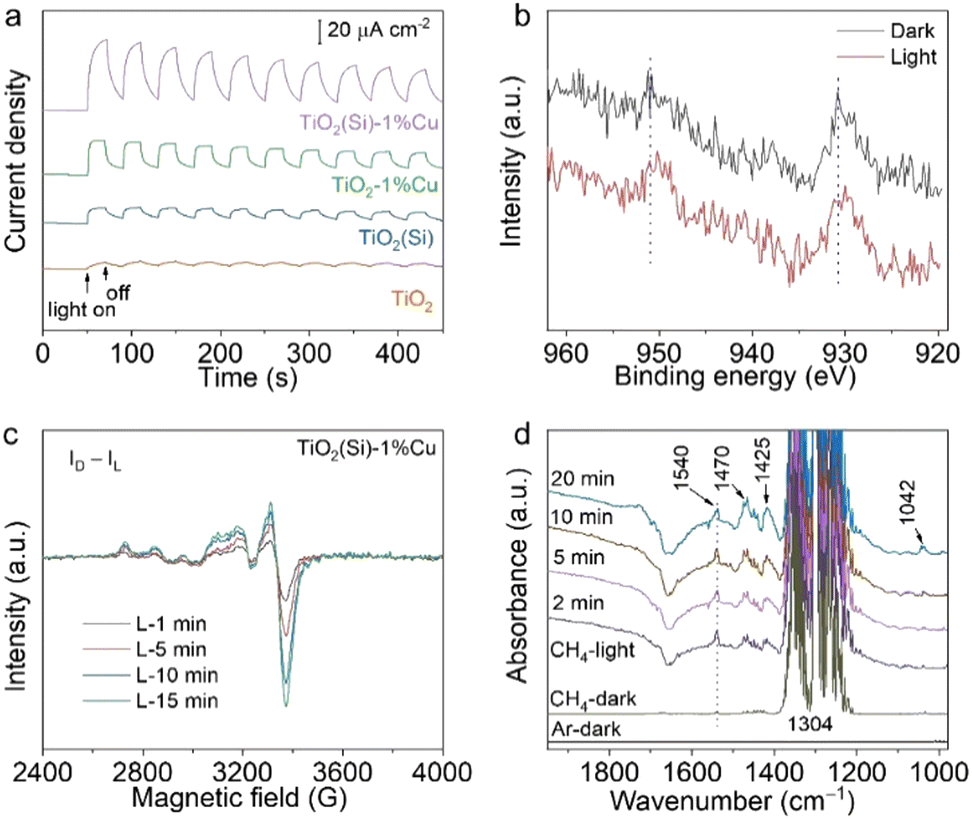 | ||
| Fig. 4 (a) Transient photocurrent response of the prepared TiO2 nanosheets. (b) The light-irradiated Cu 2p XPS spectra for TiO2(Si)–1%Cu. (c) The EPR difference spectra between light irradiated spectra and pristine spectrum for TiO2(Si)–1%Cu. (d) In situ DRIFTS spectra for photocatalytic methane conversion over TiO2(Si)–1%Cu under light irradiation. | ||
To gain insight into the electron transfer of TiO2 nanosheets upon Cu and Si doping, we monitored the oxidation state changes of doped Cu and Si in TiO2 nanosheets during the photocatalytic reaction using irradiated XPS. Specifically, the Cu 2p XPS spectrum shifts towards a lower binding energy upon light irradiation (Fig. 4b), while no obvious shift can be observed in the Si 2p XPS spectrum (Fig. S11, ESI†). In addition, light-irradiated EPR measurements were carried out to further elucidate the change in the oxidation state of Cu under light irradiation. The differences between the EPR spectra under light irradiation (Fig. S12, ESI†) and the spectrum in the dark (Fig. S6, ESI†) are shown in Fig. 4c. The positive and increasing values of the EPR difference reveal that Cu2+ is reduced to EPR-silent Cu+ by photogenerated electrons under light irradiation. Interestingly, the EPR spectrum for the irradiated TiO2(Si)–1%Cu would recover to its initial state after exposure to air for 20 min (Fig. S13, ESI†), suggesting the high photocatalytic stability of the co-doped TiO2 nanosheets. Taken together, these results illustrate that doping could significantly enhance the separation of photogenerated carriers and the activation of methane molecules on the surface.
We then performed in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to observe the intermediates in the photocatalytic methane conversion process to analyse the reaction mechanism. The in situ DRIFTS in the presence of CH4 and water vapour for TiO2(Si)–1%Cu (Fig. 4d) exhibit obvious peaks at 1304 and 1540 cm−1, assigned to the C–H deformation vibration and symmetric deformation vibration of CH4, respectively.40,41 It is worth mentioning that the C–H symmetric vibration of CH4 at 1540 cm−1 is normally infrared-forbidden for free methane molecules, which manifests the efficient adsorption of CH4 molecules on the TiO2(Si)–1%Cu surface.42 The C–H symmetric stretching vibration of CH4 intensified immediately upon illumination, indicating photoenhanced methane adsorption. Moreover, the vibration modes of CH2/CH3 deformation at new peaks at 1470/1425 cm−1, and the C–O stretching vibrational mode at 1042 cm−1 appear and grow gradually in the DRIFTS of TiO2(Si)–1%Cu, demonstrating the dissociation of CH4 molecules over the TiO2(Si)–1%Cu catalyst surface under light irradiation.43 In sharp contrast, the CH2/CH3 and C–O species can hardly be resolved in the DRIFTS for pristine TiO2 nanosheets (Fig. S14, ESI†). These results firmly reveal that the Cu and Si co-doping can rationally modulate adsorption configuration of reactant molecules on the surface and significantly boost the cleavage of methane on the TiO2 nanosheets.
3. Conclusions
In conclusion, we have provided an efficient candidate for photocatalytic methane conversion into ethane through Cu and Si co-doping into ultrathin TiO2 nanosheets. It is demonstrated that the co-doping strategy can rationally alter the reactive oxygen species for efficient activation of the C–H bond according to the in situ EPR. Furthermore, Cu and Si doping greatly improves the efficiency of photogenerated charge separation and migration. As a result, the optimized TiO2(Si)–1%Cu achieves a C2H6 production rate of 33.8 μmol g−1 h−1 with a selectivity of 88.4% upon 4 h light irradiation. This work presents a fresh perspective on the photocatalytic activation of C–H bonds under mild conditions and paves the way for efficient methane conversion into value-added chemicals through a highly sustainable approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (2020YFA0406103), NSFC (22232003, 21725102, 22122506, 22279128, 91961106, 22075267, 21950410514, 22109148), Strategic Priority Research Program of the CAS (XDPB14), Anhui Provincial Natural Science Foundation (2008085J05), Youth Innovation Promotion Association of CAS (2019444), China Postdoctoral Science Foundation (2021M703122), Jiangsu Funding Program for Excellent Postdoctoral Talent, Gusu Innovation and Entrepreneurship Leading Talents Program (ZXL2022386) and Users with Excellence Program of Hefei Science Center CAS (2020HSC-UE003). In situ DRIFTS measurements were performed at the Infrared Spectroscopy and Microspectroscopy Endstation (BL01B) of NSRL. We thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication.References
- H. Song, X. Meng, Z.-J. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- J. Low, J. Ma, J. Wan, W. Jiang and Y. Xiong, Acc. Mater. Res., 2022, 3, 331–342 CrossRef CAS.
- J. Ma, R. Long, D. Liu, J. Low and Y. Xiong, Small Struct., 2022, 3, 2100147 CrossRef CAS.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- X. Shen, D. Wu, X.-Z. Fu and J.-L. Luo, Chin. Chem. Lett., 2022, 33, 390–393 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- W.-J. Jang, J.-O. Shim, H.-M. Kim, S.-Y. Yoo and H.-S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- J. Ma, K. Mao, J. Low, Z. Wang, D. Xi, W. Zhang, H. Ju, Z. Qi, R. Long, X. Wu, L. Song and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 9357–9361 CrossRef CAS.
- S. Song, H. Song, L. Li, S. Wang, W. Chu, K. Peng, X. Meng, Q. Wang, B. Deng, Q. Liu, Z. Wang, Y. Weng, H. Hu, H. Lin, T. Kako and J. Ye, Nat. Catal., 2021, 4, 1032–1042 CrossRef CAS.
- Y. Li, L. Wang, J. Low, D. Wu, C. Hu, W. Jiang, J. Ma, C. Wang, R. Long, L. Song, H. Xu and Y. Xiong, Chin. Chem. Lett., 2020, 31, 231–234 CrossRef CAS.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355–8362 CrossRef CAS.
- L. Li, Y.-Y. Cai, G.-D. Li, X.-Y. Mu, K.-X. Wang and J.-S. Chen, Angew. Chem., Int. Ed., 2012, 51, 4702–4706 CrossRef CAS.
- Z. Chen, S. Wu, J. Ma, S. Mine, T. Toyao, M. Matsuoka, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11901–11909 CrossRef CAS PubMed.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- X. Li, J. Xie, H. Rao, C. Wang and J. Tang, Angew. Chem., Int. Ed., 2020, 59, 19702–19707 CrossRef CAS PubMed.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355–8362 CrossRef CAS.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed.
- H. Yoshida, C. Murata and T. Hattori, Chem. Commun., 1999, 1551–1552, 10.1039/A904886C.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef.
- Z. Zheng, B. Huang, X. Qin, X. Zhang, Y. Dai, M. Jiang, P. Wang and M.-H. Whangbo, Chem. – Eur. J., 2009, 15, 12576–12579 CrossRef CAS.
- B. Choudhury, M. Dey and A. Choudhury, Int. Nano Lett., 2013, 3, 25 CrossRef.
- G. Colón, M. Maicu, M. C. Hidalgo and J. A. Navío, Appl. Catal., B, 2006, 67, 41–51 CrossRef.
- J. Ma, C. Gao, J. Low, D. Liu, X. Lian, H. Zhang, H. Jin, X. Zheng, C. Wang, R. Long, H. Ji, J. Zhu and Y. Xiong, J. Phys. Chem. C, 2021, 125, 5542–5548 CrossRef CAS.
- Y. Su, S. Chen, X. Quan, H. Zhao and Y. Zhang, Appl. Surf. Sci., 2008, 255, 2167–2172 CrossRef CAS.
- C. Chen, Y. Wei, G. Yuan, Q. Liu, R. Lu, X. Huang, Y. Cao and P. Zhu, Adv. Funct. Mater., 2017, 27, 1701575 CrossRef.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. A. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CrossRef CAS.
- S. Neubert, D. Mitoraj, S. A. Shevlin, P. Pulisova, M. Heimann, Y. Du, G. K. L. Goh, M. Pacia, K. Kruczała, S. Turner, W. Macyk, Z. X. Guo, R. K. Hocking and R. Beranek, J. Mater. Chem. A, 2016, 4, 3127–3138 RSC.
- F. Tian, Y. Zhang, J. Zhang and C. Pan, J. Phys. Chem. C, 2012, 116, 7515–7519 CrossRef CAS.
- N. Ulagappan and H. Frei, J. Phys. Chem. A, 2000, 104, 7834–7839 CrossRef CAS.
- S. Duhalde, M. F. Vignolo, F. Golmar, C. Chiliotte, C. E. R. Torres, L. A. Errico, A. F. Cabrera, M. Rentería, F. H. Sánchez and M. Weissmann, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 161313 CrossRef.
- P. Kuppusamy and J. L. Zweier, J. Biol. Chem., 1989, 264, 9880–9884 CrossRef CAS.
- A. F. Carley, H. A. Edwards, B. Mile, M. W. Roberts, C. C. Rowlands, F. E. Hancock and S. D. Jackson, J. Chem. Soc., Faraday Trans., 1994, 90, 3341–3346 RSC.
- P. G. Lustemberg, R. M. Palomino, R. A. Gutiérrez, D. C. Grinter, M. Vorokhta, Z. Liu, P. J. Ramírez, V. Matolín, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, J. Am. Chem. Soc., 2018, 140, 7681–7687 CrossRef CAS PubMed.
- H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507–20515 CrossRef CAS.
- G. Yuan, K. Aruda, S. Zhou, A. Levine, J. Xie and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2334–2338 CrossRef CAS.
- X. Yu, V. De Waele, A. Löfberg, V. Ordomsky and A. Y. Khodakov, Nat. Commun., 2019, 10, 700 CrossRef CAS PubMed.
- W. Jiang, J. Low, K. Mao, D. Duan, S. Chen, W. Liu, C.-W. Pao, J. Ma, S. Sang, C. Shu, X. Zhan, Z. Qi, H. Zhang, Z. Liu, X. Wu, R. Long, L. Song and Y. Xiong, J. Am. Chem. Soc., 2021, 143, 269–278 CrossRef CAS.
- D. Scarano, S. Bertarione, G. Spoto, A. Zecchina and C. Otero Areán, Thin Solid Films, 2001, 400, 50–55 CrossRef CAS.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280–9286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00457g |
| This journal is © The Royal Society of Chemistry 2023 |