
Cu and Si co-doping on TiO2 nanosheets to modulate reactive oxygen species for efficient photocatalytic methane conversion†

Abstract
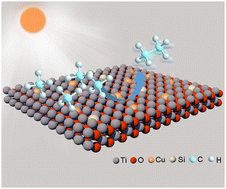
In this study, we successfully construct Cu and Si co-doped ultrathin TiO2 nanosheets. As confirmed by comprehensive characterizations, Cu and Si co-doping can rationally tailor the electronic structure of TiO2 to maneuver reactive oxygen species for effective photocatalytic methane conversion. In addition, this co-doping greatly enhances the utilization efficiency of photogenerated charges. Furthermore, it is revealed that Cu and Si co-doping can significantly boost the adsorption and activation of methane on TiO2 nanosheets. As a result, the optimized catalyst achieves a C2H6 production rate of 33.8 μmol g−1 h−1 with a selectivity of 88.4%. This work provides insights into nanocatalyst design toward efficient photocatalytic methane conversion into value-added compounds.
- This article is part of the themed collections: Nanoscale Horizons Emerging Investigator Series, Nanoscale Horizons 10th anniversary regional spotlight collection: China, 2024 Lunar New Year Collection, New horizons in materials for energy conversion, optics and electronics, Nanoscale Horizons Most Popular 2023 Articles and Nanoscale Horizons, Nanoscale, and ChemComm: Nanocatalysis
 Please wait while we load your content...
Please wait while we load your content...

