
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Life cycle of single atom catalysts: a Mössbauer study on degradation and reactivation of tetrapyrrolic Fe–N–C powders†‡
Davide
Menga
a,
Friedrich E.
Wagner
b and
Tim-Patrick
Fellinger
 *c
*c
aChair of Technical Electrochemistry, Department of Chemistry and Catalysis Research Center, Technische Universität München (TUM), 85748 Garching, Germany
bDepartment of Physics, Technische Universität München (TUM), 85748 Garching, Germany
cDivision 3.6 Electrochemical Energy Materials, Bundesanstalt für Materialforschung und -prüfung (BAM), 12203, Berlin, Germany. E-mail: tim-patrick.fellinger@bam.de
First published on 4th October 2023
Abstract
The degradation of a single-site atomically dispersed, model Fe–N–C powder catalyst with high activity is investigated using cryo-Mössbauer spectroscopy. The results indicate a degradation initiated by an Fe2+ to Fe3+ oxidation due to coordination of oxygen to tetrapyrrolic Fe–N4 sites at atmospheric conditions (change between characteristic doublets) before iron(III) oxide is formed (sextet). Thermal reactivation can be used to restore substantial catalytic activity of aged Fe–N–C powders.
New conceptsThe large majority of studies on the degradation of Fe–N–C catalysts is focused on the decay of electrocatalytic activity rather than the transformation of the material. Accordingly, degradation mechanisms are usually assigned to specific potential ranges, following the idea to avoid certain potentials to avoid degradation. Catalysts, which showed activity in the relevant range, were often composed of multiple phases (atomically dispersed FeNx sites, Fe oxides and Fe carbides). Herein we investigate the degradation of a very well-defined Fe–N–C catalyst, as single-site catalysts model for Fe–N–Cs. By investigating the degradation in it's powder form, we exclude further complicating factors such as ionomers in electrodes and the role of pH. The catalyst is so active that we can investigate the electrocatalytic properties after aging, still in the relevant potential range. Interestingly, the first step of degradation (oxidation of Fe2+ to Fe3+) results in a transformation of dublets in Mössbauer spectroscopy, which were previously assigned to completely different Fe–Nx sites. The community of single-atom catalysts may become more aware of storage issues of the reactive materials and the stability of iron single sites in oxidation state +3 may become a descriptor for catalyst powder stability. |
Electrochemical devices that are able to convert chemical energy into electricity and vice versa are a key technology compatible with the aims to push the decarbonization of the energy sector further. Reactions such as oxygen reduction and evolution (ORR and OER, respectively) and CO2 reduction (CO2RR) are fundamental in order to reduce our carbon footprint and a great scientific effort is being made to improve our knowledge and understanding about these complicated processes. State-of-the-art catalysts possess good activity but rely on expensive critical raw materials such as platinum-group metals (PGMs). For economic viability, these technologies should employ more inexpensive materials with competitive activity and stability, or at least level off at increased loadings due to reduced cost. Metal and nitrogen co-doped carbons (M–N–Cs, with M being usually first-row transition metals) have been intensively studied due to the low cost and availability of their constituents combined with promising activity towards a wide range of relevant electrochemical reactions.1 The recent focus of those scientific efforts is on atomically dispersed M–N–Cs, also referred to as single-atom-catalysts (SACs) which call for a complete metal utilization and hence optimal atom efficiency.2 So far, the most studied M–N–Cs are those employing Fe as the metal center. Fe–N–Cs find applications in different reactions, from the ORR3 and CO2RR4 to, possibly, tumor therapy.5 Synthetic methods have been optimized in order to yield materials containing entirely Fe–N4 surface complexes, which are desirable active sites, being free from less active and often harmful inorganic side phases of metallic iron or iron carbide.6,7 Moreover, we reported that the formation of side phases can be avoided and a high metal loading could be achieved using a different metal (e.g. Mg or Zn) to imprint the desired N4 chelate coordination site (pyrolytic template-ion reaction), and coordinating the Fe in a later step via a transmetalation (ion-exchange) reaction.7,8 This method has been recently employed to reach record high site density and activity.9,10 Since Fe is a Mössbauer-active element, the very sensitive Mössbauer spectroscopy is widely employed to characterize Fe–N–Cs. At cryogenic conditions (T = 4.2 K), Fe–N4 sites apparently have a specific fingerprint signal that can be distinguished from side-phases.11 The sites produce quadrupole doublet signals with isomer shifts (IS) usually in the range 0.2–0.7 mm s−1 and quadrupole splitting (QS) usually between 1–4 mm s−1, depending on their exact coordination environment, which affects the spin of the Fe atoms and consequently the Mössbauer spectrum. Atomically dispersed Fe–N–Cs with the desired Fe–Nx coordination usually show one to three of those quadrupole doublet signals.6,7,12–14 In the scientific literature, there has been the long-standing question whether these quadrupole doublets arise from different active sites or from the same one with and without an additional O2 ligand.15 Employing theoretical calculations, Mineva and co-workers recently attributed the doublet with mean QS value at 0.94 mm s−1, here from now on referred to as D1, mostly to a Fe(III)N4C12 surface site with an additional O2 ligand and the doublet with mean QS value at 2.25 mm s−1, referred from now on as D2, to a different Fe(II)N4C10 moiety free from additional ligands.16 Moreover, employing in situ Mössbauer spectroscopy, Li and co-workers identified the site related to D1 as the most active but unstable one and the site related to D2 as the less active but very stable site.17 Another important difference between the two sites is the local carbon structure. The site responsible for D1 is attributed to a defect-rich site with a pyrrolic-type nitrogen coordinating the Fe, whereas the site responsible for D2 has pyridinic nitrogen to chelate the Fe and is perfectly matching the graphene structure.16–18
In the present work, we employ a phase-pure model catalyst regarding the atomically-dispersed sites, being comprised of tetrapyrrolic Fe–N4 sites that refer to a Fe(III)N4C12 structure. While the ageing of multiphase catalysts may show an overlay of different degradation mechanisms, the ageing of such single-site catalyst should reveal a distinct mechanism. We herein show by cryo-Mössbauer spectroscopy that the two doublets can convert into each other upon air exposure, indicating that identical Fe–N4 sites can result in two different doublets, depending on the atmospheric conditions. In fact, the pristine tetrapyrrolic sites show a D2 doublet, which is partially converted to D1 upon air exposure. Moreover, the life cycle of the catalyst in its powder form is investigated over time showing that the air-exposed sites degrade into nanoscopic Fe(III) oxide, with concomitant transformation of D1 into a sextet signal accompanied by loss in electrocatalytic activity. This suggests the possibility to use the lifetime of D1 as descriptor for the stability of Fe–N–Cs. Catalyst storage is therefore recommended in inert conditions, although some sites remain unchanged upon air-exposure. Moreover, the kinetic performance of the aged catalyst can be partially recovered by a thermal treatment. The proposed recovery procedure is able to increase the mass activity at 0.8 VRHE of the catalyst after about one year of shelf storage by more than 400%. Therein nanoscopic Fe(III) oxide is successfully converted back into active Fe–N4 sites, in a cycle that resembles the aquatic iron cycle in nature.
Results and discussion
NDC-Fe-HT was prepared similarly to what we previously reported.7 Briefly, a Zn–N–C material was firstly synthetized carbonizing phthalonitrile in a ZnCl2/LiCl eutectic salt melt at 800 °C in Ar atmosphere. The Lewis-acidic Zn2+ ions result in the formation of a high density of N4 moieties.19 The material obtained after acidic workup was employed as solid-state ligand in the low- and high-temperature Zn-to-Fe transmetalation.7 After high-temperature ion exchange, the material was quickly transferred into an Ar-filled glovebox, minimizing the air exposure of the catalyst. This pristine catalyst is named NDC-Fe-HT-0. After taking it out of the glovebox it starts ageing under ambient conditions. Depending on the duration of ageing, the material is called NDC-Fe-HT-X where X indicates the duration of air exposure in days. Mössbauer spectra were recorded after zero (air free), 2, 9, 30, 125 and 320 days of air exposure, on the same catalyst sample. Air-exposure was allowed by keeping the catalyst inside the absorber used in Mössbauer measurements in order to exclude effects arising from different absorber thicknesses (fitting parameters can be found in Table S1, ESI‡). Based on our previous work, the structure of the surface complexes is a tetrapyrrolic Fe–N4 motif, embedded into the graphitic backbone via compensation of pyrrolic 5-membered rings with 7-membered ring defects as in Stone–Wales defects.7 Our reported structure presents a FeN4C12 stoichiometry in the first coordination shell, however it is homogeneously embedded into carbon. Previously reported structural models were introducing pores close to the pyrrolic site and therefore had no isotropic second coordination shell.6,16 Despite the apparent single-site nature of the catalyst, the NDC-Fe-HT-0 spectrum (Fig. 1a) is composed of two quadrupole doublets, of which 58% of the total area accounts for D1 (IS = 0.27 mm s−1, QS = 1.54 mm s−1) and 42% accounts for D2 (IS = 0.63 mm s−1, QS = 2.89 mm s−1), respectively. According to literature reports,16,17 D1 is assigned to a Fe(III)-N4 site with an axial O2 ligand, while D2 is assigned to the square-planar Fe(II)–N4 moiety, free from any additional oxy ligand, due to its high QS value.7,16,17 It was previously assumed that those doublets are related to structurally different Fe–N4 sites (ref. 16–18 assign D1 to a pyrrolic and D2 to a pyridinic structure), although variations in IS and QS may also be caused by the effect of axial ligands. For square-planar tetrapyrrolic Fe–N4 surface complexes an oxidation state of +2 for iron is expected, which could theoretically turn into +3 by oxidative addition, e.g., axial coordination with O2, as observed for the respective molecular analogues. Indeed, NDC-Fe-HT-2, the first measurement after air exposure (Fig. 1b), shows that the ratio between D1 and D2 drastically changed, with D1 contributing 73% of the area and D2 accounting for the remaining 27%. The catalyst condition remains relatively stable over one week, as NDC-Fe-HT-9 (Fig. 1c) shows only minor changes (74% for D1 and 26% for D2). The fact that D2 turns into D1 upon exposure to ambient conditions, clearly shows that the different quadrupole doublets are both related to the same Fe–N4 site, since structural transformation of the FeN4-surrounding carbon backbone would require temperatures above ≈1500 °C. Therefore, the D1 and D2 signals are either a characteristic of the oxidation state of iron, or more specifically a signature of the state of axial coordination. Moreover, the fact that D2 is still present in NDC-Fe-HT-9 indicates that 26% of the Fe–N4 sites are not accessible or not sensitive to the gas phase. Even after 30 days of air exposure D2 remains stable (Fig. 1d). However, the D1 signal decreases by 8% accompanied by the formation of two sextet components, each accounting for 4% of the total area. This result indicates that after 30 days of ambient storage, a fraction of oxygen coordinated Fe(III)–N4 sites are converted into nanosized Fe(III) oxide, while the Fe(II)–N4 sites remain stable, possibly inaccessible to the environment. The conversion of D1 into the two sextet components continues over time (Fig. 1e and f), with NDC-Fe-HT-125 showing an additional ≈20% conversion of Fe–N4 sites into Fe(III) oxide. The parameters for NDC-Fe-HT-320, on the other hand, hardly changed compared to NDC-Fe-HT-125. Similarly to what is reported for the electrochemical degradation of Fe–N–C catalysts,17 also upon air exposure the sites responsible for D1 are converted into Fe(III) oxide, whereas the sites responsible for D2 are stable. This opens the possibility to use the lifetime of D1 as stability descriptor, at least for freshly prepared catalysts. The contradiction that D2 has previously been assigned to less active sites compared to D1, while we herein show that the Fe–N4 structure related to D1 and D2 hitherto is identical, can be reasonably explained by inaccessibility of remaining D2 sites to air. This is in line with previous assignments to surface-accessible sites (D1) versus buried sites (D2), however it is now evident that those can be isostructural as well.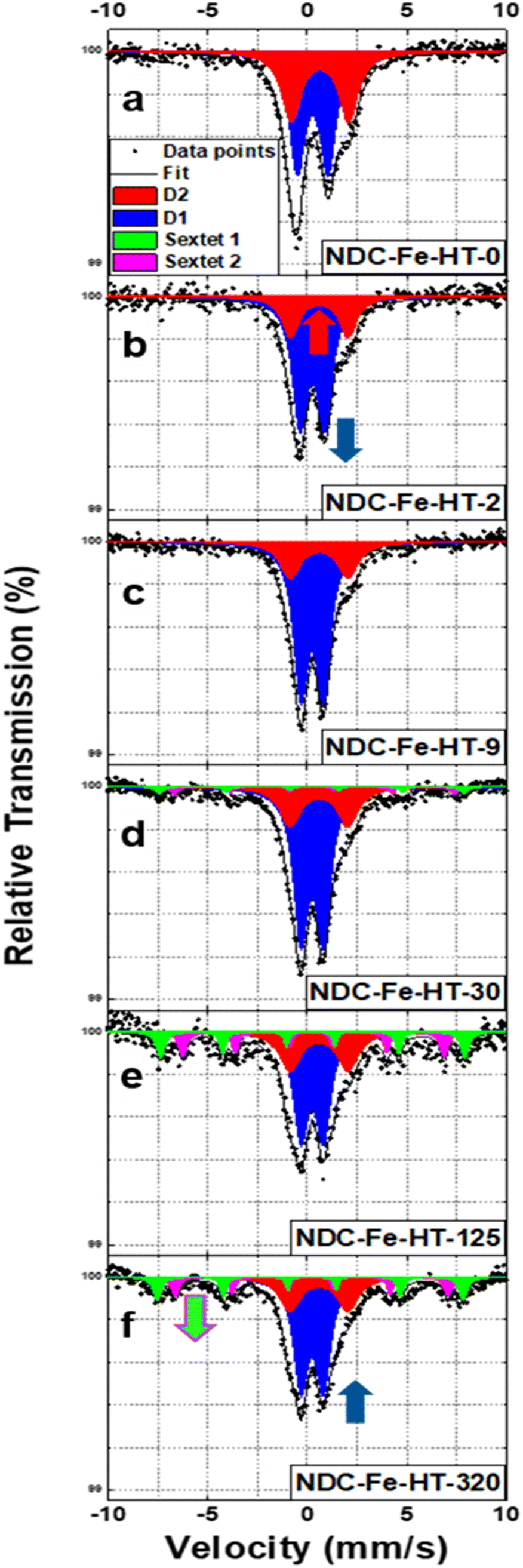 | ||
| Fig. 1 (a)–(f) Mössbauer spectra of NDC-Fe-HT-X in the 320 days of air exposure. Spectra are measured at T = 4.2 K. | ||
We conducted rotating disc electrode (RDE) experiments to assess the effect of air exposure on the ORR activity of the catalyst. Fig. 2a shows the activity of the pristine catalyst, after 200 and after 320 days of air exposure measured in O2-saturated 0.1 M HClO4 electrolyte with identical catalyst loadings of 0.29 mg cm−2. The activity clearly decreases for the air exposed catalysts with NDC-Fe-HT-200 losing ≈70 mV in half-wave potential (E1/2) and having a mass activity of ≈1.2 A g−1 at 0.8 VRHE, lower compared to the pristine mass activity of ≈3.8 A g−1 at the same potential. Even though Mössbauer spectra indicate an unchanged relative abundance of Fe phases between day 125 and day 320, NDC-Fe-HT-320 shows further decreased activity in comparison to NDC-Fe-HT-200. More specifically, an additional loss of ≈70 mV in E1/2 and ≈0.81 A g−1 at 0.8 VRHE is measured. Furthermore, reduced slopes of the polarization curve of the aged catalysts become apparent in the kinetic region (at low currents). In the Tafel plot (Fig. 2b) the corresponding increase in Tafel slope (TS) is observed only for potentials below 0.75 VRHE, indicating a regime of a mixed reaction mechanism due to the activity contribution of the carbon support. This is further supported by measuring the ORR activity of the catalyst without Fe in it, i.e. as Zn–N–C, which starts indeed around 0.75 VRHE (Fig. 2a). The pristine catalyst presents a TS of ≈75 mV dec−1, indicative of predominant contribution of Fe–N4 sites with their fast kinetics. NDC-Fe-HT-200 and NDC-Fe-HT-320 have a TS of ≈95 mV dec−1 and ≈120 mV dec−1, respectively, suggesting that the kinetics are increasingly dominated by the support material due to a reduced Fe–N4 active site density. Consequently, the measured TS represents the convoluted activity of the remaining active Fe–N4 sites and the arising electrocatalytic activity of the support in the lower potential range. The reduced contribution of Fe–N4 sites to the electrocatalytic conversion is further confirmed via rotating ring disc electrode (RRDE) experiments. A clear increase in H2O2 production is observed, pointing to the increased contribution of the support activity and its lower selectivity.
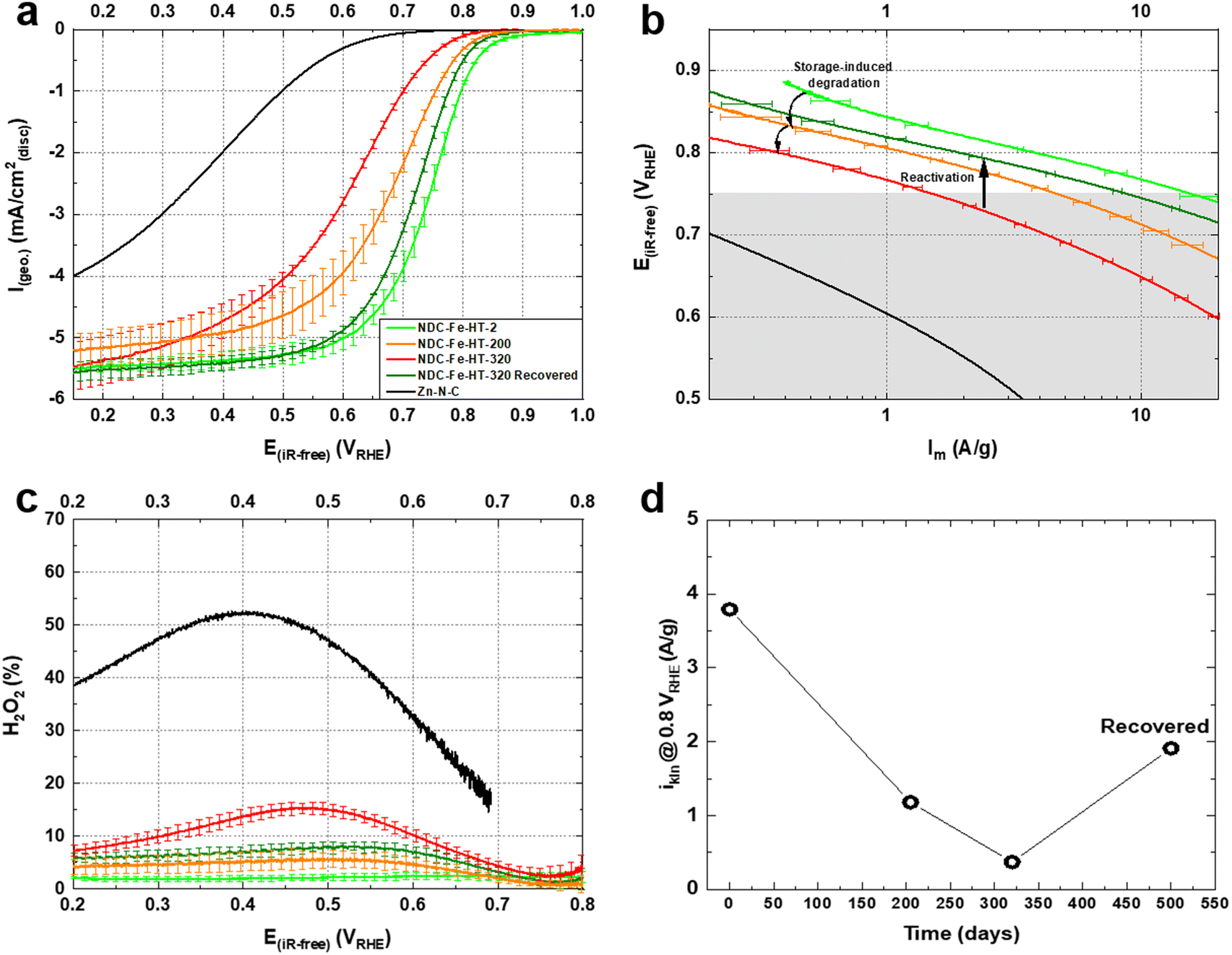 | ||
| Fig. 2 (a) Capacitance-corrected ORR curves recorded with an RDE setup at room temperature in O2-saturated 0.1 M HClO4 at 1600 rpm, 10 mV s−1 (anodic scans) for NDC-Fe-HT-X and the sample after thermal recovery. The Zn–N–C starting material is plotted to show that the activity of the carbon support starts around 0.75 VRHE. (b) Respective Tafel plots showing purely the kinetic mass activity corrected for mass-transport limitation. The shaded gray area marks the potential region where the support activity takes over. (c) Respective H2O2 yield obtained via RRDE experiments. (d) Mass activity at 0.80 VRHE of NDC-Fe-HT-X and the sample after thermal recovery. | ||
The loss of ≈3.42 A g−1 at 0.8 VRHE after 320 days corresponds to a loss of about 90% of the initial activity. This cannot be explained solely by a reduced intrinsic activity caused by the transformation of Fe–N4 sites into Fe(III) oxide, since only about 25% of D1 turn into the sextet component. Hence, other effects, possibly related to the morphology of the sample, reduce the effective activity and further compromise the catalyst efficiency after air exposure. Fig. 3a shows the N2-sorption porosimetry measurements of the pristine sample and after 320 days in air. At the beginning, the sample presents high micro- and mesoporosity and possess a Brunauer–Emmett–Teller (BET) surface area of 1247 m2 g−1. After 320 days in air a clear downshift of the isotherm is observed, and the BET surface area drops to 922 m2 g−1. Quenched solid density functional theory (QSDFT) model with slit and cylindrical pores was employed to extract the pore size distribution (PSD) from the adsorption branch of the isotherms (Fig. 3b). The results show that the PSD of the catalyst remains qualitatively unchanged, but the overall gas uptake is decreased, indicating that a fraction of the pore system is blocked, reasonably by the Fe(III) oxide particles, and hence not utilized for electrocatalysis. Even though the percentage of surface area loss is smaller than the percentage of activity loss, pore blocking may account for the additional activity loss. This may be the case, since the PSD displays no information on the pore connectivity, which is directly related to kinetics. In fact, the total pore volume loss is ≈50% (Table S2, ESI‡). This could explain why the catalyst efficiency is even lower than expected from Mössbauer spectroscopy.
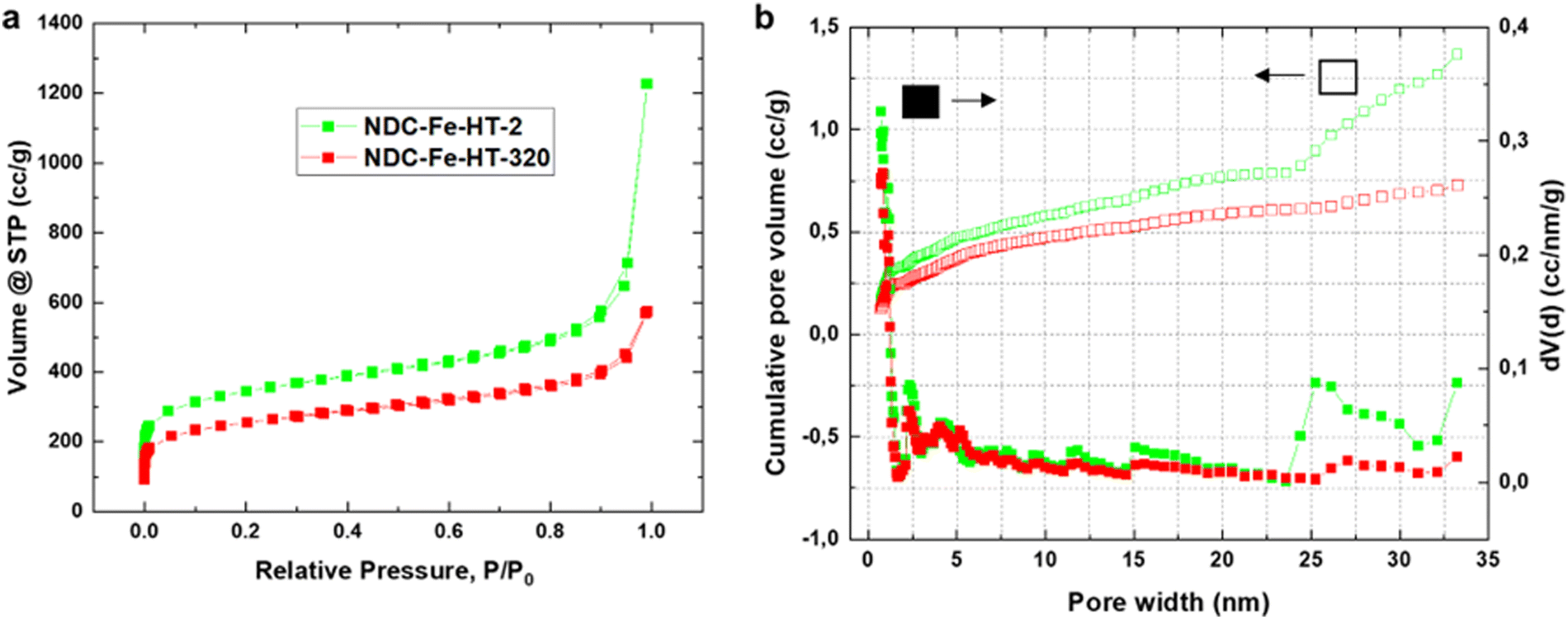 | ||
| Fig. 3 (a) Isotherms of NDC-Fe-HT-X obtained from N2 sorption measurement. (b) The respective pore-size distribution from the adsorption branch employing the QSDFT model for slit and cylindrical pores. | ||
Short shelf life of Fe–N–C catalysts may complicate reproducibility of experimental results and can explain challenges in benchmarking experiments. Therefore, our results strongly suggest the storage of atomically dispersed catalysts in inert atmosphere. Moreover, on commercial scale an inappropriate storage may result in severe loss of the material value. Heat treatments are commonly used to activate catalysts and also the conversion of metal oxides on carbon supports into atomically dispersed catalysts has been reported.18,20,21 Can the aged Fe–N–C catalyst be recycled by an additional heat treatment?
Fe(III) oxide is thermally stable, but sensitive to the carbothermal reduction in presence of carbon at high temperatures. The reaction is not conservative to the carbon support, which may be disadvantageous. However, controlled support degradation may also have advantageous effects on the catalyst efficiency.22,23 Therefore, we conducted thermogravimetric analysis coupled with mass spectrometry (TGA-MS) of the aimed thermal recovery process to search for a balance of recovery of oxidic iron and the degradation of functional Fe–N4 sites. Fig. 4a shows the thermogram in Ar atmosphere for an Fe–N–C composed of 1.34 wt% atomically dispersed Fe and 1.78 wt% of oxidic Fe (Fe2O3@Fe–N–C). Two degassing steps, one at 150 °C and the other one at 250 °C, are performed in order to remove air and adsorbed water, before the temperature is raised to 1000 °C with a heating rate of 10 K min−1. During the degassing steps signals related to CO2, H2O, NO and N2 are detected (Fig. 4b–e). At around 800 °C – the preparation temperature of the catalyst support – pyrolytic support reorganization is observed, indicated by evolution of CO2, H2O and NO (Fig. 4b–e). At ≈950 °C (with onset at ≈850 °C) N2 evolution is observed, which may go in hand with the decomposition of Fe–N4 sites. The metastability of Fe–N4 sites at the temperature of their pyrolytic formation has previously been considered the dilemma of Fe–N–C syntheses.7 Here, the preformed nature of the catalyst support (having N4 coordination sites) opens a temperature range below 850 °C in which Fe–N4 sites can be formed, as recently elegantly deployed by Myers, Jaouen and Jia et al.9 Consequently, the thermal recovery step on NDC-Fe-HT-320 was carried out at 800 °C for 15 min, in order to convert the Fe(III) oxide into active Fe–N4 sites without reaching their decomposition temperature.
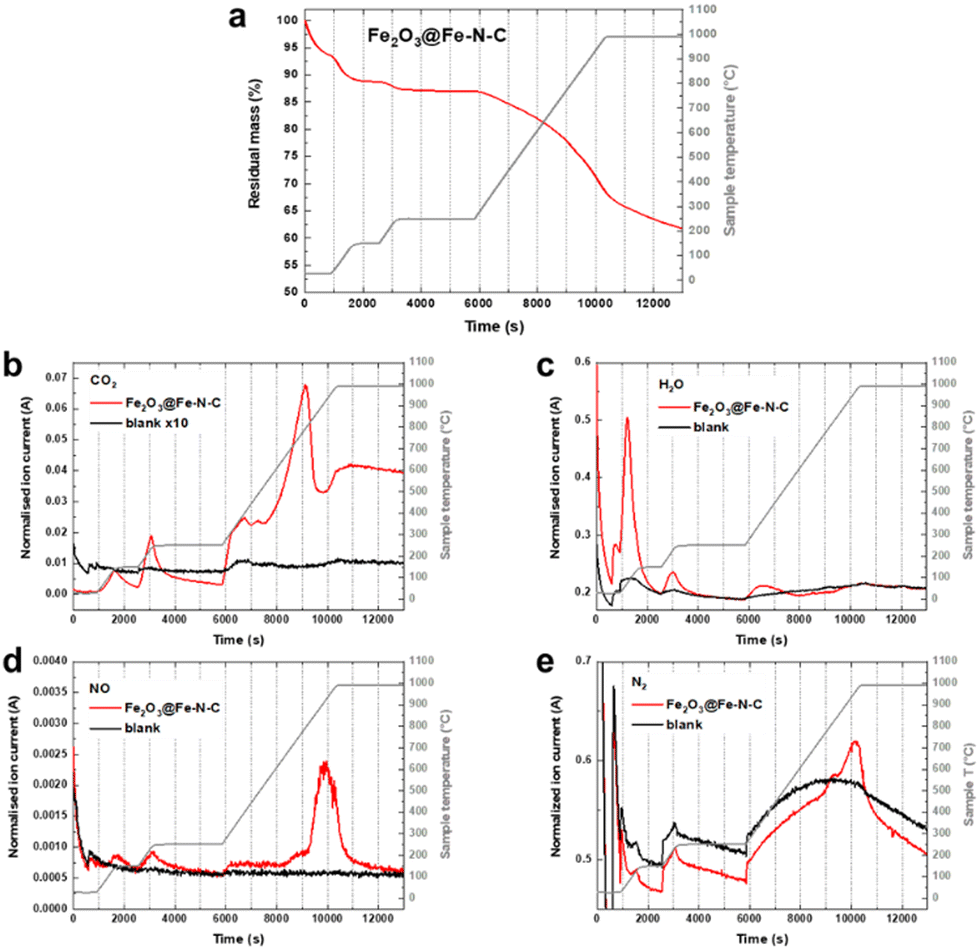 | ||
| Fig. 4 (a) Results of the TGA-MS investigation of Fe2O3@Fe–N–C upon heating to 1000 °C under Ar atmosphere. TGA curve displaying the sample mass in percentage (red) and sample temperature (grey). (b) CO2, (c) H2O, (d) NO and (e) N2 MS signals for the sample (red) in comparison with a blank measurement where only the crucible without sample is employed (black). The sample temperature is displayed in grey. | ||
Fig. 2a shows the ORR activity measured with RDE setup in O2-saturated 0.1 M HClO4 electrolyte with the same catalyst loading of 0.29 mg cm−2. After the recovery step, the catalyst shows an E1/2 only 20 mV lower compared to the pristine catalyst and a mass activity of 1.93 A g−1 at 0.8 VRHE. This reflects a high degree of recovery (only a factor of 2 less when comparing the mass activity at 0.8 VRHE) or an improvement compared to the aged NDC-Fe-HT-320 of 120 mV in E1/2 and 1.56 A g−1 at 0.8 VRHE, which correspond to an increase of more than 400% of its mass activity at 0.8 VRHE. Even though the H2O2 production remains slightly higher compared to the pristine state (Fig. 2c), it is 8% less compared to NDC-Fe-HT-320, where the H2O2 production reaches its maximum (at 0.45 VRHE). The recovery of the good catalytic kinetics of the material is further confirmed by the TS of ≈75 mV dec−1, which equals the pristine state (Fig. 2b). Mössbauer spectroscopy further confirms the conversion of Fe(III) oxide formed during almost one year of ageing under air exposure into active Fe–N4 sites (Fig. 5). After recovery the spectrum shows inferior statistical accuracy due to low amount of sample and hence low total iron content. The variation of the Mössbauer parameters compared to non-activated samples may be a consequence of low spectrum quality. Based on a fit with two quadrupole doublets and one sextet, despite the low signal-to-noise-ratio, D1 would be responsible for 69% of the spectral area (vs. 52% in NDC-Fe-HT-320), explaining the increased ORR activity, whereas D2 would decrease to 15%. As shown from TGA-MS analysis, the conversion of Fe(III) oxide into Fe–N4 sites can be accompanied by carbon oxidation to CO2. It is reasonable to assume that some previously buried D2 sites, are now accessible to air, hence contributing to D1. The sextet component, on the other hand, decreases from a total area of 24% to 16%. This indicates that some Fe(III) oxide is still present and explains why the catalyst does not recover entirely its activity. Further work should target the optimization of the recovery step in terms of time, temperature and gas atmosphere employed. The fact that the Fe–N–Cs obtained after low temperature transmetalation (NDC-Fe) are composed of atomically-dispersed Fe–N4 sites and Fe(III) oxide, makes the material a reasonable precursor which can be stored and activated just before use, ultimately saving one energy-consuming thermal step.19
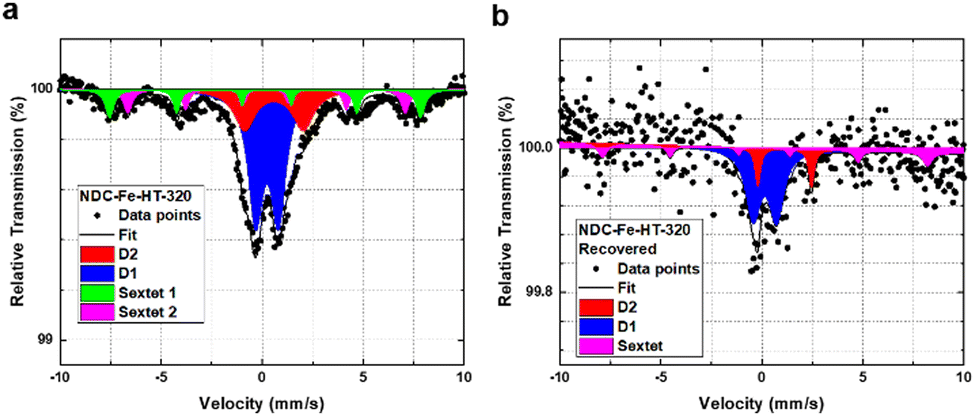 | ||
| Fig. 5 Mössbauer spectra of NDC-Fe-HT-320 measured at 4.2 K (a) before and (b) after thermal recovery. | ||
Similarly to what was recently shown by K.T. Santos et al.24 and Boldrin et al.,25 we reported the drop in ORR activity over time due to storage under atmospheric conditions of the catalyst powder and reactivation upon thermal treatment. Our study confirms their general findings specifically for tetrapyrrolic Fe–N4 sites employing the very sensitive Mössbauer spectroscopy at cryogenic conditions (T = 4.2 K). Here it is further revealed that the doublet signal D2 of the degrading sites over time indeed converts into the D1 doublet signal. Since the degradation was earlier shown to be related to oxygen, this points to a degradation mechanism starting with the adsorption of oxygen molecules to the tetrapyrrolic Fe–N4 site. The Fe(II) to Fe(III) transition is the requirement for the observed formation of iron oxide, indicated by the conversion of D1 to the sextet signal.
Conclusions
In conclusion, we showed experimentally that the D1 and D2 Mössbauer signals observed for atomically dispersed Fe–N–C catalysts are not characteristics of different Fe–N4 sites, but characteristics of the state of axial coordination of Fe–N4 sites. Different IS and QS may be related to different axial ligands in general. Specifically, for herein investigated tetrapyrrolic Fe(II)–N4 sites, the quadrupole doublet D2 with IS and QS of ≈0.60 mm s−1 and ≈2.90 mm s−1, can be turned into D1 (IS and QS ≈0.24 mm s−1 and ≈1.13 mm s−1) by air exposure and assigned to O-coordinated Fe(III)–N4 sites. Moreover, it revises literature reports that assign D1 specifically to Fe–N4 sites located on the catalyst surface, whereas D2 is assigned to a site which is not accessible to the gas phase. This statement seems to be true only for catalysts that have been exposed to atmospheric conditions, although the coordination chemistry may vary for different Fe–N4 sites. Mössbauer spectroscopy analysis of the calendar ageing of tetrapyrrolic Fe–N–Cs over almost a year reveals the chemical decomposition mechanism that also results in electrocatalytic activity loss. Analogous to the well-known aquatic iron cycle in nature, the Fe(II) species is oxidized in presence of oxygen to Fe(III), which triggers the precipitation of Fe(III) oxide. The powder degradation mirrors the reported degradation in electrochemical tests of this class of materials and strongly suggests avoidance of oxidative environment, and the storage of freshly prepared catalysts under inert atmosphere.17 The lifetime of D1 for freshly prepared Fe–N–C catalysts may be utilized as stability descriptor. Thermal treatment below 850 °C has the potential for a thermal recovery in which Fe(III) oxide is reconverted into active Fe–N4 sites, which remain stable at this temperature. We present a more than 400% increase of the mass activity at 0.8 VRHE after almost one year of ageing when performing the thermal recovery step. Considering that the discovered ageing process presents the opposite process of the commonly used thermal catalyst post-treatment, this step is recommended to be done shortly before use. Because of the equivalent structure and chemistry of their active sites, we assume a closely related ageing and recovery option for other atomically dispersed M–N–C materials or SACs, depending on the stability of the metal inside the active sites versus its oxidic form.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Federal Ministry BMWK is acknowledged for funding the project innoKA (03ET6096A). The authors D.M and T.-P. F. would like to acknowledge the analytical lab at the TUM catalyst research centre, the Chair for Technical Electrochemistry TEC (Prof. H. Gasteiger) for hosting the Fellinger group. T.-P. F. would like to acknowledge the former Head of Department 3 at BAM (Dr Thomas Goedecke) for the support to continuation of electrocatalysis research.References
- Z. Shi, W. Yang, Y. Gu, T. Liao and Z. Sun, Adv. Sci., 2020, 7, 2001069 CrossRef CAS.
- Editiorial, Nat. Commun., 2021, 12, 5884 CrossRef PubMed.
- F. Jaouen, D. Jones, N. Coutard, V. Artero, P. Strasser and A. Kucernak, Johnson Matthey Technol. Rev., 2018, 62, 231–255 CrossRef CAS.
- S. Paul, Y.-L. Kao, L. Ni, R. Ehnert, I. Herrmann-Geppert, R. van de Krol, R. W. Stark, W. Jaegermann, U. I. Kramm and P. Bogdanoff, ACS Catal., 2021, 11, 5850–5864 CrossRef CAS.
- M. Huo, L. Wang, Y. Wang, Y. Chen and J. Shi, ACS Nano, 2019, 13, 2643–2653 CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- D. Menga, J. L. Low, Y.-S. Li, I. Arčon, B. Koyutürk, F. Wagner, F. Ruiz-Zepeda, M. Gaberšček, B. Paulus and T.-P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- A. Mehmood, J. Pampel, G. Ali, H. Y. Ha, F. Ruiz-Zepeda and T.-P. Fellinger, Adv. Energy Mater., 2018, 8, 1701771 CrossRef.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623 CrossRef PubMed.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef CAS PubMed.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS.
- U. I. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69 CrossRef CAS.
- Q. Jia, E. Liu, L. Jiao, S. Pann and S. Mukerjee, Adv. Mater., 2019, 31, 1805157 CrossRef PubMed.
- T. Mineva, I. Matanovic, P. Atanassov, M.-T. Sougrati, L. Stievano, M. Clémancey, A. Kochem, J.-M. Latour and F. Jaouen, ACS Catal., 2019, 9, 9359–9371 CrossRef CAS.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 652–663 CrossRef CAS.
- D. Menga, F. Ruiz-Zepeda, L. Moriau, M. Šala, F. Wagner, B. Koyutürk, M. Bele, U. Petek, N. Hodnik, M. Gaberšček and T.-P. Fellinger, Adv. Energy Mater., 2019, 9, 1902412 CrossRef CAS.
- U. I. Kramm, I. Herrmann-Geppert, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2011, 115, 23417–23427 CrossRef CAS.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS PubMed.
- T. Lazaridis and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 114517 CrossRef CAS.
- A. Kongkanand and M. K. Carpenter, US Pat., US 9947935 B1, 2018 Search PubMed.
- K. T. Santos, K. Kumar, L. Dubau, H. Ge, S. Berthon-Fabry, C. S. A. Vasconcellos, F. H. B. Lima, T. Asset, P. Atanassov, V. A. Saveleva, P. Glatzel, X. Li, F. Jaouen and F. Maillard, J. Power Sources, 2023, 564, 232829 CrossRef CAS.
- P. Boldrin, D. Malko, A. Mehmood, U. I. Kramm, S. Wagner, S. Paul, N. Weidler and A. Kucernak, Appl. Catal., B, 2021, 292, 120169 CrossRef CAS.
Footnotes |
| † This article is dedicated to our co-author Prof. em. Fritz Wagner who unfortunately passed away before this work was published. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00308f |
| This journal is © The Royal Society of Chemistry 2023 |