
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmarking the performance of lithiated metal oxide interlayers at the LiCoO2|LLZO interface†
André
Müller
 *a,
Faruk
Okur
ab,
Abdessalem
Aribia
a,
Nicolas
Osenciat
a,
Carlos A. F.
Vaz
c,
Valerie
Siller
d,
Mario
El Kazzi
d,
Evgeniia
Gilshtein
a,
Moritz H.
Futscher
a,
Kostiantyn V.
Kravchyk
ab,
Maksym V.
Kovalenko
ab and
Yaroslav E.
Romanyuk
*a
*a,
Faruk
Okur
ab,
Abdessalem
Aribia
a,
Nicolas
Osenciat
a,
Carlos A. F.
Vaz
c,
Valerie
Siller
d,
Mario
El Kazzi
d,
Evgeniia
Gilshtein
a,
Moritz H.
Futscher
a,
Kostiantyn V.
Kravchyk
ab,
Maksym V.
Kovalenko
ab and
Yaroslav E.
Romanyuk
*a
aLaboratory for Thin Films and Photovoltaics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Uberlandstrasse 129, CH-8600 Dübendorf, Switzerland. E-mail: andre.mueller@empa.ch; yaroslav.romanyuk@empa.ch
bLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, CH-8093 Zurich, Switzerland
cSwiss Light Source, Paul Scherrer Institute, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland
dElectrochemistry Laboratory, Paul Scherrer Institute, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland
First published on 17th April 2023
Abstract
Integrating Li7La3Zr2O12 (LLZO) solid-state electrolytes in combination with a high-energy cathode remains a major challenge in developing all-solid-state batteries. In particular, diffusion processes and solid-state reactions at the cathode–electrolyte interface during the co-sintering of the oxide materials at elevated temperatures result in high interfacial impedances. In this work, we study the performance of lithiated Nb, Al, and Ti metal oxide interlayers as diffusion barriers to prevent the formation of deleterious interphases at the cathode–electrolyte interface during fabrication, thus enabling easy Li-ion transfer between LiCoO2 and LLZO. Specifically, we characterize the impact of the different interlayers on the morphology and elemental distribution at the interface and evaluate their influence on the electrochemical behavior of the battery stacks after the high-temperature process. We find that the mixing of Co/La cations at the interface is reduced by using the metal oxide diffusion barriers. It is shown that the interfacial impedance can be reduced from 8 kΩ cm2 to 1 kΩ cm2 and that the electrochemical performance of all cells with interlayers exceeds that of the battery without interlayer. In particular, the Li–Nb–O modification outperforms the other metal oxide interlayers in terms of the discharge capacities achieved.
1 Introduction
Today's commercially available lithium-ion battery (LIB) systems are reaching their limits in terms of energy density and no longer meet the increasing demands of grid energy systems or electric vehicles. For this reason, the development of batteries with high energy density combined with acceptable safety standards and affordable cost is urgently needed. One of the most promising ways to increase the safety and energy density of LIBs is to replace the organic liquid electrolyte with a solid conductive Li-ion electrolyte, resulting in an all-solid-state battery (ASSB) configuration.1,2 To date, several different electrolyte materials have been developed with a wide range of chemical properties. Among the different classes, garnet and NASICON electrolytes are promising candidates.3,4The garnet Li7La3Zr2O12 (LLZO) and its doped compositions are promising as they have comparable conductivities as liquid electrolytes (σ > 1 × 10−4 S cm−1),5 have high chemical stability,6,7 are easy to handle in the environment, and therefore offer great opportunities for use in ASSBs. While the application of Li metal as an anode has been extensively studied and a sound progress has been achieved for its implementation,8,9 the integration of high-energy cathodes remains a challenge in ASSB development.1,10 The main obstacles include electrolyte oxidation and the formation of Li-deficient interphases, mechanical instability such microgaps due to volume changes of the active materials, and cation interdiffusion due to co-sintering for intimate bonding.11
The most studied active cathode materials are layered cathodes such as LiCoO2 (LCO) or the LiNixMnyCozO2 (NMC) classes. Co-sintering of the various transition metal oxides and ceramic electrolytes at temperatures >500 °C is required to ensure uniform physical contact.11 On the one hand, without such a high-temperature process, the interface would be severely limited to point contacts, complicating the transfer of Li+ charge carriers involved in the redox reaction.12 On the other hand, such high-temperature processes also lead to the interdiffusion of elements and the formation of phases from decomposition products13–15 with high interfacial impedances.16 In their recent work, Yildiz and her team have shown that the formation of secondary phases of decomposition products such as LaCoO3 and La2Zr2O7 significantly impacts the dynamics of the cathode–electrolyte interface.17,18 Furthermore, it is shown that such phases can form not only at sintering conditions (>700 °C), which are common for ceramics, but also at much lower temperatures (<500 °C).19,20
To mitigate diffusion, so-called artificial solid electrolyte interlayers can be deposited to act as a protective barrier at the cathode–electrolyte interface.21 Metal oxide barriers can improve the structural integrity of the interphase and prevent the dissolution of transition metals and side reactions, as well as limit cathode degradation and electrolyte decomposition.22,23 Understanding the oxidation kinetics of SEs and the suppression of interfacial degradation during high-temperature fabrication and cycling is one of the keys to determine the lifetime of an interlayer.
In their first-principles computational study, Nolan et al.24 systematically investigated the thermodynamic stability of LLZO and high-energy cathodes to identify the most promising materials that can stabilize the interface. The most stable interlayer materials were predicted to have compositions along the interface between Li2O and a metal oxide. In particular, ternary Li–Me–O compositions were found to be stable. However, experimental benchmarking and verification of these interlayers is needed to stabilize the cathode–electrolyte interfaces and make ASSBs with high energy cathode materials feasible.
In the present study, we investigate the effects of interlayers predicted by Nolan et al.24 at the cathode–electrolyte interface. We benchmark three different material Li–Me–O interlayers in terms of their compatibility with the high-temperature fabrication and resulting electrochemical performance. By combining a thin film LCO cathode with a bulk LLZO electrolyte as a solid-state battery model, we can disentangle the effects of each interlayer on battery performance. In addition, the effects of interfacial modification become more prominent, as the effects on a thin cathode layer are more pronounced than on a micrometer-thick bulk cathode. Reducing the thickness of the cathodic layer also improves access to the interfacial area. This allows us to use a wide range of characterization techniques to understand the properties of the interface.
We selected the lithiated Nb, Al and Ti metal oxides for their high chemical stability and high performance in liquid Li-ion electrolytes. Intermediate Li–Me–O layers of 10 nm were deposited between the LCO and LLZO pellets by RF magnetron sputtering to serve as an interlayer. The thickness of the interlayers was chosen based on literature values showing that too thin layers do not provide sufficient protection, while too thick layers lead to an increase in resistance due to low conductivity.9,25–27
2 Results and discussion
2.1 Cathode and interlayer characterization
To understand the chemical composition of all Li–Me–O interlayers, we performed XPS on films deposited on a Si reference substrate (Fig. 1). For the Li–Nb–O film, the Nb 3d spectra (Fig. 1a) shows three distinct peaks at 208.9 eV, 206.1 eV, and 203.5 eV. The main contributions can be assigned to the +4 and +5 oxidation states of Nb and a slight contribution from metallic Nb in the lower energy range.28,29 In contrast, the Al 2p spectrum for the Li–Al–O composition (Fig. 1b) shows a single peak at 73.2 eV with a slight shoulder in the lower binding energy region. The higher intensity peak was assigned to the +3 oxidation state and the formation of Al–O bond while the low intensity peak is attributed to metallic Al.30,31 The binding energy of the latter intensity is about 0.4 eV higher than that of metallic Al. We attribute this to the fact that the binding energy of a small metal cluster, such as in thin films, shifts to a higher value compared to that of the bulk metal.32–34Fig. 1c shows the spectra of the Ti 2p signal for the Li–Ti–O thin film. The two peaks at 462.9 eV and 457.1 eV correspond to the characteristic peaks for Ti3+.35 While the contribution of the main peaks is assigned to Ti3+, the small shoulder in the lower binding energy regions at 454.5 eV and 461 eV and comes from Ti2+ and metallic Ti.36 Although the thin interlayers at the cathode–electrolyte interface do not have an exact stoichiometric composition, they still exhibit a pattern within the ternary Li–Me–O composition space. Subsequent co-sintering is apt to change the composition in either direction. However, the influence of the respective ternary Li–Me–O interlayers on cation mixing, especially of Co/La, and cell performance remains to be investigated.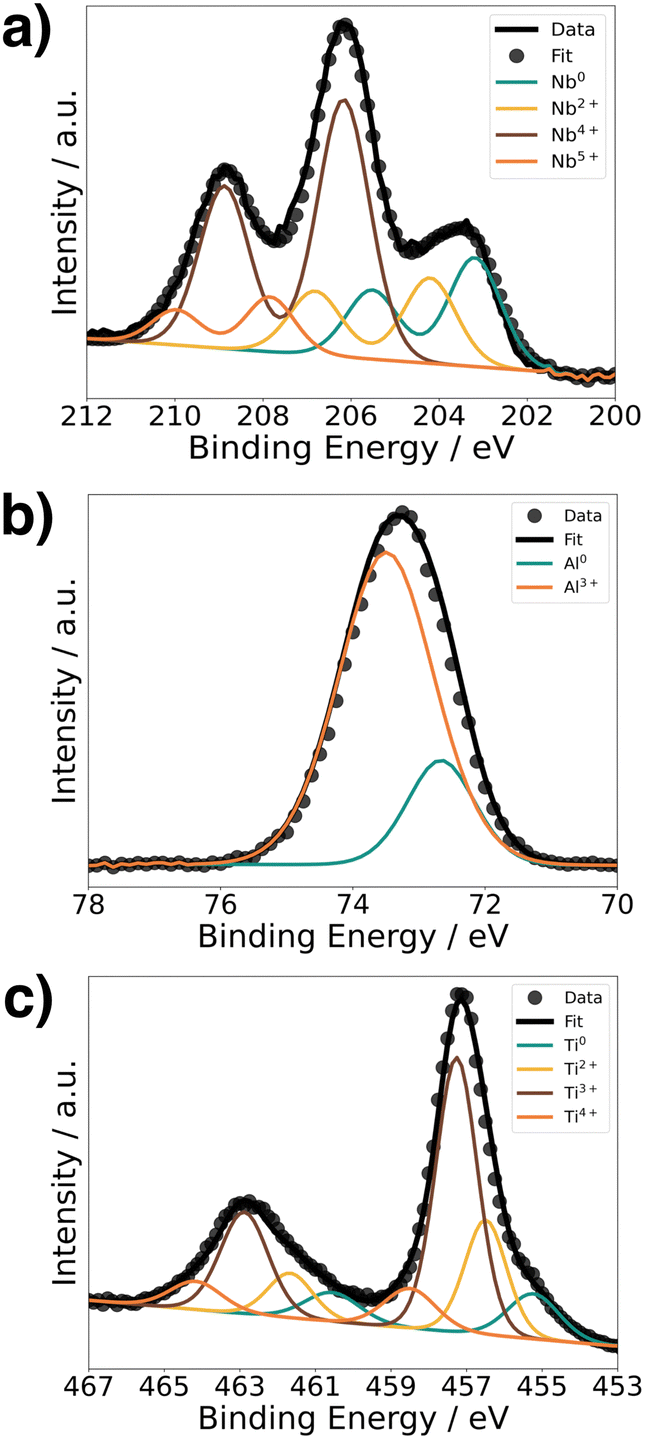 | ||
| Fig. 1 XPS spectra of the core-level metal elements for (a) Li–Nb–O, (b) Li–Al–O, and (c) Li–Ti–O. | ||
To not obscure the morphology of the LCO cathode layers, the SEM studies were carried out on stacks without the Au contact layer (Fig. 2a). An apparent color change at the edge of individual cathode electrolyte layers after co-sintering can already be seen before the SEM examinations (Fig. 2b). The color change is evident in samples without an interlayer and those with a Li–Ti–O interlayer, while it is only subtly visible in the Li–Nb–O and Li–Al–O samples. This observation suggests that a potential solid-state reaction may be occurring at the interface. To examine whether the morphology of the cathode layer also undergoes changes during co-sintering, the stacks were examined in the as-deposited and sintered states. Fig. 2c shows an SEM image of a stack as it was deposited. The image shows that the LCO cathode film covers the entire surface and has an amorphous, homogeneous structure, which is crucial for the electrochemical functionality of the battery. In contrast, Fig. 2d shows the top view of a co-sintered stack with micrometer-sized LCO crystallites. Thus, not only an apparent color change at the edges of the cathode electrolyte layers but also a change in the morphological structure of the uppermost cathode layer can be seen.
 | ||
| Fig. 2 (a) Schematic of an LCO|LLZO stack, (b) image after co-sintering the pellets with LCO, (c) top view of an as-deposited LCO layer, and (d) top view of the LCO layer after co-sintering at 500 °C. | ||
XAS measured in fluorescence mode (TFY) was used to detect changes in the electronic structure occurring with the co-sintering process (Fig. 3a). To reach the interface, the thickness of the LCO layers was reduced to 50 nm. The XAS spectrum for the Co L2,3-edge is shown in Fig. 3b for an LCO|LLZO sample after co-sintering at 500 °C in oxygen compared to the deposited state. Looking at the XAS Co L2,3-edge, one can see two absorption lines at 795.8 eV and 781.7 eV corresponding to the L2 and L3 edges, respectively.37 In the spectra of cobalt oxides, a shoulder on the low energy side at 779.8 eV of the Co L2,3-edge indicates reduced cobalt (Co2+), while a shoulder at higher energies indicates more oxidized states of cobalt.37 As can be seen from Fig. 3b, the contribution of the low-energy shoulder decreases with annealing temperature in the co-sintered LCO|LLZO sample. This change in the peak shape of the L3-edge indicates an increase in the Co oxidation state during co-sintering. In addition, there is a significant change in peak full width half maximum (FWHM) after co-sintering at 500 °C compared to the as-deposited state. Both edges – Co L2 and L3 – became significantly sharper when heated to 500 °C. This change is consistent with an increase in local order in the cobalt environment due to co-sintering, and supports the earlier hypothesis that micrometer-sized crystallites form.17
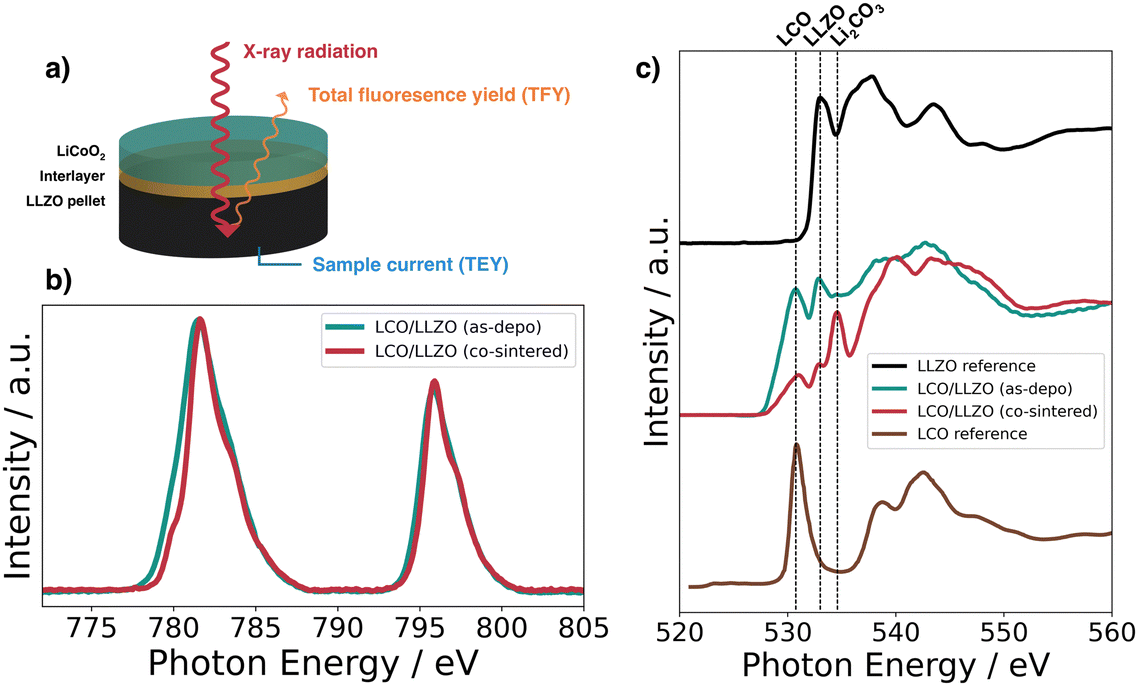 | ||
| Fig. 3 (a) Schematic of the XAS measurement geometry, (b) Co L2,3-edge, and (c) O K-edge in fluorescence mode (TFY) for LLZO, LCO, and 50 nm LCO|LLZO as-deposited and annealed at 500 °C in oxygen. | ||
Fig. 3c shows the O K-edge spectra for the LCO|LLZO sample without interlayer, before and after thermal treatment, as well as reference measurements on LCO and LLZO. The feature typical of LiCoO2 at about 530.8 eV can be clearly identified for the as-deposited LCO|LLZO sample.38 However, annealing at 500 °C leads to a significant decrease in this feature, which is most likely related to the sudden appearance of the large Li2CO3 feature (534.6 eV).39 Moreover, the peak associated with LLZO at 532.9 eV lost intensity compared to LCO, suggesting that LLZO may decompose to La2Zr2O7 or LaCoO3/La2CoO4.36,38 No significant differences were observed between the various inter-layered samples, hence the remaining XAS graphs are shown in the ESI.†
2.2 Elemental distribution at the interface
Fig. 4 compares the elemental distribution at the interface between cathode and electrolyte in co-sintered stacks. By studying the interface with a combined FIB-SEM/EDX approach, it is possible to gain insight into the cation intermixing at the interface. To compare the different samples, we show the cross-sectional SEM images in conjunction with the EDX elemental distribution maps for Co and La. Fig. 4a shows the interface with no interlayer. As shown previously by Vardar et al.17 and Park et al.,40 direct crystallization of LCO on garnet electrolytes such as LLZO is detrimental to a chemically stable interface. | ||
| Fig. 4 FIB-SEM image and elemental mapping of Co K series and La L series of a stack (a) without interlayer, and with (b) Li–Nb–O, (c) Li–Al–O, and (d) Li–Ti–O interlayer. | ||
The high-temperature co-sintering at 500 °C leads to uniform physical contact between the electrolyte and the cathode but results in cross-diffusion of the elements. As can be seen, the boundaries in the Co and La mappings at the cathode–electrolyte interface were diluted by co-sintering when no interlayer is present. This is indicative of partial diffusion of Co from the LCO into the electrolyte and likewise of La from the electrolyte into the cathode film.40
In contrast, the interface maps for stacks with Li–Me–O interlayers show a clearer, sharper boundary (Fig. 4b–d). In particular, the signal intensity of Co at the interface is found to be much more intense in the stacks with the interlayers. This confirms that the interlayers act as an effective diffusion barrier against Co diffusion. These observations are supported by the intensity lines of all samples. We observe a much steeper intensity drop in the Co signal for the stacks with interlayer than for the stack without. In contrast, the intensity for the stack without interlayer drops only gradually, consistent with Co diffusion into the LLZO electrolyte. We attribute the high La signals in the LCO layers (for all stacks) to the fact that the interaction volume for EDX measurements is much larger than the 300 nm thick cathode. La is thus also detected through the thin LCO cathode film. However, besides the EDX measurements, all cross-sections show a structurally reliable interface. This is further evidence of good conformal physical bonding between the different layers after co-sintering. The question arises about what influence the deposited Li–Me–O interlayers have on Li+ diffusion and whether this is also influenced.
2.3 Interfacial impedance at the cathode|electrolyte interface
In this study, we used electrochemical impedance spectroscopy41,42 (EIS) to quantify the charge transfer resistance at the cathode–electrolyte interface. For this purpose, we studied half-cells with and without interlayers (Fig. 5a) to evaluate the effects of the different Li–Me–O interlayers on charge transport. To analyze the impedance spectra of the samples, we used an equivalent circuit model shown in (Fig. 5b) previously used by Zhang et al.43 as a model for the LCO|LGPS solid-electrolyte interface (Fig. 5b). In the following, we refer to the interface1 as the interface between the cathode and the electrolyte, and the interface2 as the interface between the current collector (CC) and the electrolyte. The Nyquist plots of the samples (Fig. 5c–f) compare the impedance spectra of half-cells with and without interlayers in magnified view to show the mid-frequency impedance region corresponding to the impedance contribution of the cathode–electrolyte interface.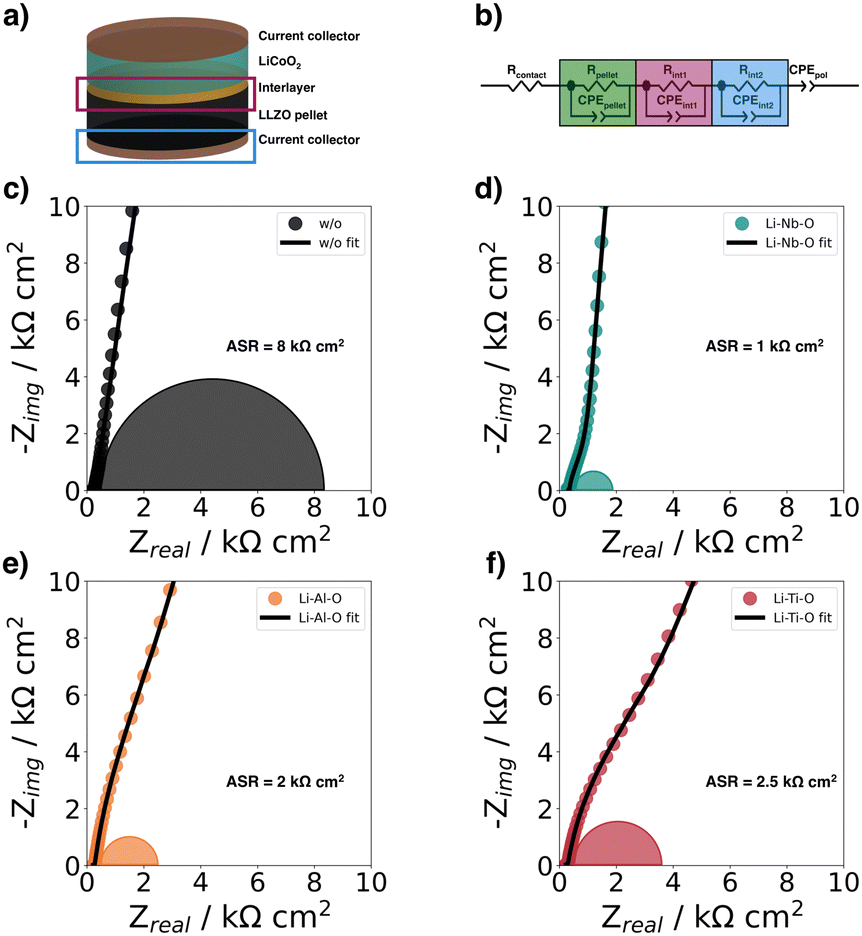 | ||
| Fig. 5 (a) Schematic of half-cells structure for electrochemical impedance spectroscopy at room-temperature, (b) equivalent circuit model used to fit the impedance spectra by Zhang et al.,43 EIS spectra of (c) Au|LCO|LLZO|Au (w/o), (d) Au|LCO|Li–Nb–O|LLZO|Au (Li–Nb–O), (e) Au|LCO|Li–Al–O|LLZO|Au (Li–Al–O), and f) Au|LCO|Li–Ti–O|LLZO|Au (Li–Ti–O). The measured data are shown together with the fits from the equivalent circuit. | ||
The typical semicircle contribution of bulk LLZO electrolytes appears at frequencies above 1 MHz, and the intrinsic impedance and capacitance components for the LLZO pellets can be extracted from the equivalent circuit. The resulting refined capacitance values for the total resistance of LLZO average 90 nF cm−2 and range between 10−12 and 10−9 F cm−2, thus are consistent with the expected capacitance values of the respective charge transport phenomena.44–47 The ionic conductivity was extracted from the EIS data and is within the expected range at 5 mS cm−1.48
The semicircle for the electrolyte is followed by a second semicircle in the mid-frequency range (from 500 kHz to 1 kHz), which is characteristic for the cathode–electrolyte interface (interface1).43 Due to the large influence of interface2 and the resulting large semicircle in the low-frequency range (from 1 kHz to 1 Hz), the semicircle for interface1 becomes less distinguishable in the mid-frequency range. The semicircles plotted in the figures correspond to the calculated semicircles from the values of the fits. A detailed deconvolution of the different semicircles is explained in more detail using the Li–Nb–O sample as an example in the ESI.†
The charge transfer resistance at the interface between cathode and electrolyte is about 8 kΩ cm2 for the unmodified stack and results from the degraded solid cathode–electrolyte interface.49 This value is in the same order of magnitude as that reported by Sastre et al.50 in their thin-film system and Vardar et al.17 The slightly higher value compared to Sastre et al.50 can be explained by the extended co-sintering time at 700 °C leading to a higher degree of interface degradation. In addition, the value is consistent with the observation by Vardar et al.17 that the area resistivity (ASR) increases upon annealing.
We find that the Li–Nb–O interlayer reduces the charge transfer resistance at the cathode–electrolyte interface by an order of magnitude. With a value of 1 kΩ cm2, it is significantly lower than the stack with no interlayer, again demonstrating the positive effect of a Li–Nb–O interface.26,50–53 The impedance spectra show no clear difference in electrolyte resistance between the stacks without and with the Nb-oxide interlayer, while the charge transfer resistances differ significantly.26 The other lithium-containing metal oxide layers (Li–Al–O or Li–Ti–O) have a similar effect on the interface. The Nyquist diagrams of these interface-modified half-cells (Fig. 5e and f) show three semicircles as before. It can also be seen that each modification leads to a decrease in ASR compared to samples with no interlayer, namely, 2 kΩ cm2 and 2.5 kΩ cm2 for Li–Al–O and Li–Ti–O, respectively.
2.4 Electrochemical characteristics of full cells
Using cyclic voltammetry (CV) and galvanostatic charge/discharge experiments on a full cell configuration with Li metal foil as anode, we investigate the charge transport dynamics at the cathode–electrolyte interface. Fig. 6a shows the CVs of the full cells with and without interlayers. The scans for Li–Al–O, Li–Ti–O, and with no interlayer are characterized by two corresponding reversible redox peaks with similar shapes, matching the main lithiation/delithiation processes in LCO at approximately 3.9 V vs. Li/Li+.54,55 However, one can notice an increased split between the peak positions of the modified full cells compared with the stack with no interlayer. We attribute the higher peak split to the increase in electrode thickness as we add an additional interphase between the electrolyte and the cathode.56,57 Additionally, it is observed that the cells with interlayer have higher peak cathodic and anodic currents. This indicates a higher electrochemical activity of the full cells – an indication of faster and unhindered ion transport across the interface. Furthermore, no additional redox peaks are observed in any of the cyclic voltammograms, indicating that no secondary phases are involved in the redox process.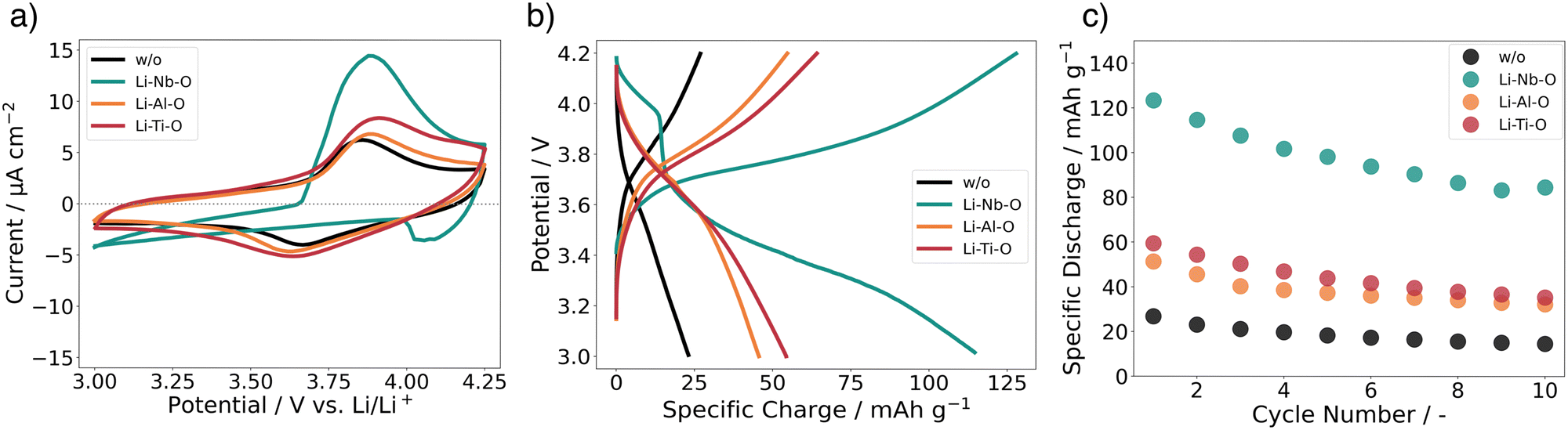 | ||
| Fig. 6 (a) Cyclic voltammetry scans measured with a sampling rate of 0.1 mV s−1 between 3 and 4.25 V vs. Li/Li+, (b) charge–discharge curves with 1.75 μA cm−2 between 3 V and 4.2 V at 80 °C, and (c) specific discharge capacities for full cells with and without interface modification. | ||
Surprisingly, the CV scan of the Li–Nb–O interlayer shows a different pattern. While the cathodic peak is still at about 3.9 V vs. Li/Li+, the anodic peak shifts to about 4 V vs. Li/Li+. Both peaks have a much steeper slope in the scan compared to other samples. Nevertheless, it can be seen that the electrochemical activity is still present and outperforms that of the reference cell with no interlayer in the cathodic peak.
To better understand and account for the anomalous behavior of the Li–Nb–O interlayer, as well as to evaluate the performance of the Li–Me–O modifications compared to the reference with no interlayer, galvanostatic cycling was performed in the next step. Fig. 6b summarizes the charge–discharge characteristics of the full battery stacks. The stacks have been cycled with a current of 1.75 μA cm−2 (C/10) for ten cycles. The lower and upper cutoff potentials were chosen to be 3 V and 4.2 V, respectively, as they cause the smallest degree of damage to the positive electrode.58 For all cells, we can observe a voltage “plateau” at 3.9 V vs. Li/Li+ in the charging step. As with CVs, the plateau is characteristic of the lithiation/delithiation processes in LCO. However, the discharge plateaus vary greatly among cells. While it remains at 3.9 V vs. Li/Li+ for Li–Al–O and Li–Ti–O, it drops to 3.6 V Li/Li+ for cells with no interlayer and to 3.4 V vs. Li/Li+ for Li–Nb–O. This is consistent with previous findings of high impedance at the cathode–electrolyte interface.59
In general, all cells with lithiated metal oxide interlayers exhibit higher specific discharge capacities (Fig. 6c). The specific discharge capacity for the first cycle of the unmodified cell only exhibits 26 mAh g−1, steadily decreasing to approximately 15 mAh g−1 over ten cycles. As expected, the presence of the interlayers significantly improves the performance of the cells. While the Li–Al–O interlayer has an initial specific discharge capacity of 50 mAh g−1, the capacity more than doubles to 60 mAh g−1 with the Li–Ti–O interlayer. In contrast, the cell with Li–Nb–O interface modification achieves a specific discharge of 125 mAh g−1 in the first cycle. However, there is a distinct voltage drop at about 4 V vs. Li/Li+ in the discharge curve, consistent with the kink in the CV scan. Similar kinks were also seen in several other samples, e.g. without interlayer, and are included in the ESI.† Liu et al.60 and Luo et al.61 report similar voltage drops in the discharge process between 3.9 and 3.8 V. However, the feature remains elusive and is not further explained. Yet, it is surprising that the electrochemical activity is only observed during the discharge cycle and not during the charge cycle. Notwithstanding, our reported values are in line with earlier reports of the achieved capacities for similar material structures.59,62 The gradual decrease in capacities is attributed to both degradations of the Li|LLZO and of the LCO|LLZO interface.
We speculate that the effect just described may be due to electrochemical and/or mechanical instability at the LCO|LLZO or LLZO|Li interface. For instance, the loss of contact between LCO and LLZO caused by cracking and delamination at the LCO|LLZO interface may reduce the capacity. Wang et al.63 suggested that the main reason is microcracks caused by the volumetric changes of the cathode during the electrochemical process. The mechanical degradation at the interfaces accumulates over several cycles and occurs despite the coatings. In addition, it is possible that electrochemically induced cation diffusion during cycling can affect the battery performance. It is worth noting that the capacities achieved still fall short of those reported in liquid LIB systems and other solid-state studies.58 However, the very thin cathode films used in this study compared to normal bulk cathodes (single cathode particle >3 μm),64 imply that small changes in the cathode–electrolyte interphase are markedly noticeable.
Nolan et al.24 describe large stability phases for Li–Nb–O and Li–Ti–O compositions compared to the Li–Al–O system, which are chemically and electrochemically stable against the garnet LLZO, and are expected to be well suited to stabilize the interface. In the case of the Li–Al–O system, Al-rich compositions near the Al corner have poor stability with LLZO due to the high decomposition energy of the Al-LLZO pseudo-binary. The systems tend to form deleterious products such as LaAlO3, La2Zr2O7, etc., due to the tendency of LLZO to lose Li and the corresponding lithiation of the TM oxides. Therefore, the unoxidized Al metal contribution, evident in the XPS studies, can react with the garnet. The reasons behind the lower discharge capacities of Li–Ti–O compared to Li–Nb–O remain unclear. We can only speculate that our interlayers are in the stable region of the Li–Nb–O system and our Li–Ti–O system is in or near the reactive zone.
3 Conclusions
In summary, we find that thin interlayers of Li–Me–O at the LLZO|LCO interface improve battery performance after co-sintering. We have shown that the interlayers prevent undesirable diffusion of Co/La cations intermixing at the interface, confirming previous theoretical predictions. As an outcome, all interlayers exhibit higher specific discharge capacities – especially in the first cycles. Despite the initial improvement, the achieved first discharge capacities are still below the expected discharge capacity of 140 mAh g−1 for LCO. It can be concluded that there is still a resistive interphase between LCO and LLZO, which restricts the movement of Li+ charge carriers at the interface.We find that the presence of an interlayer does not prevent degradation during cycling, as the capacity still decreases significantly. We ascribe this effect to the volume expansion and contraction of the cathode active materials. Due to the compositional change during charge/discharge, which continuously induces stress at the interface between LLZO and LCO, microgaps are formed at the interface, leading to contact loss. Electrochemically induced cation diffusion and the resulting electrolytic oxidation can also lead to cell degradation. These effects are particularly evident in thin cathode films, where the influence of deleterious intermediate phases is more pronounced than in bulk cathodes. This work shows that an interlayer is essential for cathode co-sintering on LLZO electrolytes and highlights that the choice of interlayer significantly affects cell performance. Li–Nb–O is the most suitable and can substantially impact the fabrication of future ASSBs. To further isolate the underlying reaction mechanisms, additional studies are needed. These may include operando or post-mortem analysis.
4 Experimental details
4.1 Materials and fabrication
4.2 Characterization
The structure of the battery stacks was studied with a multifunctional field-emission scanning electron microscope (FEI Quanta 650) combined with a Thermo Fischer EDX system. A Gaussian filter was applied for the EDX mappings (standard deviation for Gaussian kernel: ΣCo = 2, ΣLa = 3) using python.The surface composition was analyzed by X-ray photoelectron spectroscopy (XPS, PHI Quantera SXM). The Al Kα X-ray source is monochromated at 1486.6 eV and equipped with an Ar+ sputter source. The spectra were analyzed and processed using CasaXPS software. The experimental data were analyzed using curve fitting. All XPS spectra were corrected for charge effects by setting the C 1s binding energy to 285.0 eV and we used a Shirley background subtraction.
The X-ray absorption spectroscopy (XAS) spectra for the O K-edge and the Co L2,3-edge were recorded at the SIM beamline (surface/interface microscopy) at the Swiss Light Source (SLS) in Switzerland. The total electron yield (TEY) signal originates from photoelectrons from the top surface layer. In addition, to compare surface species with bulk components, the total fluorescence yield (TFY) signal was acquired with a depth analysis of several hundred nanometers. To reach the interface, the thickness of the top LCO layer was reduced to 50 nm. Data were analyzed using Athena software and processed in Python.
Electrochemical measurements were performed in a split coin cell on a hot plate in an Ar-filled glovebox at 80 °C using a Squidstat potentiostat (Admiral Instruments) with a prior OCV step for 3 h. Electrochemical impedance spectroscopic measurements were performed between 5 MHz and 1 Hz with an AC amplitude of 50 mV using a Paios instrument (Fluxim AG) at room temperature. Impedance spectra were fitted using Zview4 software (Scribner Associates Inc.).
Author contributions
A. M.: conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, visualization; F. O.: formal analysis, investigation, resources; A. A., N. O., M. H. F.: formal analysis; C. A. F. V, V. S., M. E. K.: investigation; E. G.: investigation, formal analysis; K. V. K, M. V. K.: resources, supervision, funding acquisition; Y. R.: conceptualization, resources, supervision, funding acquisition; all authors contributed to the manuscript writing and revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic Focus Area (SFA) Advanced Manufacturing of the ETH Domain (project “SOL4BAT”). Part of this work was performed at the Surface and Interface Microscopy (SIM beamline of the Swiss Light Source, Paul Scherrer Institut (PSI), Villigen, Switzerland). We thank Joel Casella for performing the XRD measurements.References
- M. Balaish, J. C. Gonzalez-Rosillo, K. J. Kim, Y. Zhu, Z. D. Hood and J. L. M. Rupp, Nat. Energy, 2021, 6, 227–239 CrossRef CAS.
- C. Li, Z.-Y. Wang, Z.-J. He, Y.-J. Li, J. Mao, K.-H. Dai, C. Yan and J.-C. Zheng, Sustainable Mater. Technol., 2021, 29, e00297 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- X. Han, Y. Gong, K. K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- Q. Liu, Z. Geng, C. Han, Y. Fu, S. Li, Y.-B. He, F. Kang and B. Li, J. Power Sources, 2018, 389, 120–134 CrossRef CAS.
- T. Thompson, S. Yu, L. Williams, R. D. Schmidt, R. Garcia-Mendez, J. Wolfenstine, J. L. Allen, E. Kioupakis, D. J. Siegel and J. Sakamoto, ACS Energy Lett., 2017, 2, 462–468 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- Y. Lu, X. Huang, Y. Ruan, Q. Wang, R. Kun, J. Yang and Z. Wen, J. Mater. Chem. A, 2018, 6, 18853–18858 RSC.
- R. Dubey, J. Sastre, C. Cancellieri, F. Okur, A. Förster, L. Pompizii, A. Priebe, Y. E. Romanyuk, L. P. H. Jeurgens, M. V. Kovalenko and K. V. Kravchyk, Adv. Energy Mater., 2021, 11, 2102086 CrossRef CAS.
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599–4604 CrossRef CAS PubMed.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- P. Barai, T. Rojas, B. Narayanan, A. T. Ngo, L. A. Curtiss and V. Srinivasan, Chem. Mater., 2021, 33, 5527–5541 CrossRef CAS.
- K. Nie, Y. Hong, J. Qiu, Q. Li, X. Yu, H. Li and L. Chen, Front. Chem., 2018, 6, 616 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama and Y. Tateyama, ACS Appl. Mater. Interfaces, 2017, 9, 286–292 CrossRef CAS PubMed.
- J. Xu, E. Hu, D. Nordlund, A. Mehta, S. N. Ehrlich, X.-Q. Yang and W. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 31677–31683 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC.
- G. Vardar, W. J. Bowman, Q. Lu, J. Wang, R. J. Chater, A. Aguadero, R. Seibert, J. Terry, A. Hunt, I. Waluyo, D. D. Fong, A. Jarry, E. J. Crumlin, S. L. Hellstrom, Y.-M. Chiang and B. Yildiz, Chem. Mater., 2018, 30, 6259–6276 CrossRef CAS.
- Y. Kim, D. Kim, R. Bliem, G. Vardar, I. Waluyo, A. Hunt, J. T. Wright, J. P. Katsoudas and B. Yildiz, Chem. Mater., 2020, 32, 9531–9541 CrossRef CAS.
- H. Liu, Y. Yang and J. Zhang, J. Power Sources, 2006, 162, 644–650 CrossRef CAS.
- A. Sharafi, S. Yu, M. Naguib, M. Lee, C. Ma, H. M. Meyer, J. Nanda, M. Chi, D. J. Siegel and J. Sakamoto, J. Mater. Chem. A, 2017, 5, 13475–13487 RSC.
- H. M. K. Sari and X. Li, Adv. Energy Mater., 2019, 9, 1901597 CrossRef.
- S.-T. Myung, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7074–7095 RSC.
- Z. Chen, Y. Qin, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7606–7612 RSC.
- A. M. Nolan, E. D. Wachsman and Y. Mo, Energy Storage Mater., 2021, 41, 571–580 CrossRef.
- A. Aribia, J. Sastre, X. Chen, E. Gilshtein, M. H. Futscher, A. N. Tiwari and Y. E. Romanyuk, J. Electrochem. Soc., 2021, 168, 040513 CrossRef CAS.
- T. Kato, T. Hamanaka, K. Yamamoto, T. Hirayama, F. Sagane, M. Motoyama and Y. Iriyama, J. Power Sources, 2014, 260, 292–298 CrossRef CAS.
- M. Müller, J. Schmieg, S. Dierickx, J. Joos, A. Weber, D. Gerthsen and E. Ivers-Tiffee, ACS Appl. Mater. Interfaces, 2022, 14, 14739–14752 CrossRef PubMed.
- N. Kaufherr, D. J. Eichorst and D. A. Payne, J. Vac. Sci. Technol., A, 1996, 14, 299–305 CrossRef CAS.
- J. C. Shank, M. B. Tellekamp and W. A. Doolittle, Thin Solid Films, 2016, 609, 6–11 CrossRef CAS.
- T. Tago, N. Kataoka, H. Tanaka, K. Kinoshita and S. Kishida, Procedia Eng., 2017, 216, 175–181 CrossRef.
- T. Wei, F. Yan and J. Tian, J. Alloys Compd., 2005, 389, 169–176 CrossRef CAS.
- S. J. Heo, B. Hu, V. Manthina, A. Hilmi, C.-Y. Yuh, A. Surendranath and P. Singh, Int. J. Hydrogen Energy, 2016, 41, 18884–18892 CrossRef CAS.
- F. Parmigiani, E. Kay, P. S. Bagus and C. J. Nelin, J. Electron Spectrosc. Relat. Phenom., 1985, 36, 257–267 CrossRef CAS.
- M. G. Mason, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 748–762 CrossRef CAS.
- H. Xu, J. Chen, Y. Li, X. Guo, Y. Shen, D. Wang, Y. Zhang and Z. Wang, Sci. Rep., 2017, 7, 2960 CrossRef PubMed.
- Y. Cai, Y. Huang, W. Jia, X. Wang, Y. Guo, D. Jia, Z. Sun, W. Pang and Z. Guo, J. Mater. Chem. A, 2016, 4, 9949–9957 RSC.
- H. C. Choi, S. Y. Lee, S. B. Kim, M. G. Kim, M. K. Lee, H. J. Shin and J. S. Lee, J. Phys. Chem. B, 2002, 106, 9252–9260 CrossRef CAS.
- J. Wang, Y. Ji, N. Appathurai, J. Zhou and Y. Yang, Chem. Commun., 2017, 53, 8581–8584 RSC.
- L. Cheng, E. J. Crumlin, W. Chen, R. Qiao, H. Hou, S. F. Lux, V. Zorba, R. Russo, R. Kostecki, Z. Liu, K. Persson, W. Yang, J. Cabana, T. Richardson, G. Chen and M. Doeff, Phys. Chem. Chem. Phys., 2014, 16, 18294–18300 RSC.
- K. Park, B.-C. Yu, J.-W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS.
- U. Tröltzsch, O. Kanoun and H.-R. Tränkler, Electrochim. Acta, 2006, 51, 1664–1672 CrossRef.
- P. Vadhva, J. Hu, M. J. Johnson, R. Stocker, M. Braglia, D. J. L. Brett and A. J. E. Rettie, ChemElectroChem, 2021, 8, 1930–1947 CrossRef CAS.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schroeder, R. Koerver, T. Leichtweiss, P. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS PubMed.
- Y. Kim, A. Yoo, R. Schmidt, A. Sharafi, H. Lee, J. Wolfenstine and J. Sakamoto, Front. Energy Res., 2016, 4, 20 Search PubMed.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- T. Thompson, J. Wolfenstine, J. L. Allen, M. Johannes, A. Huq, I. N. David and J. Sakamoto, J. Mater. Chem. A, 2014, 2, 13431–13436 RSC.
- A. Sharafi, H. M. Meyer, J. Nanda, J. Wolfenstine and J. Sakamoto, J. Power Sources, 2016, 302, 135–139 CrossRef CAS.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- S. Ohta, T. Kobayashi, J. Seki and T. Asaoka, J. Power Sources, 2012, 202, 332–335 CrossRef CAS.
- J. Sastre, X. Chen, A. Aribia, A. N. Tiwari and Y. E. Romanyuk, ACS Appl. Mater. Interfaces, 2020, 12, 36196–36207 CrossRef CAS PubMed.
- X. Li, L. Jin, D. Song, H. Zhang, X. Shi, Z. Wang, L. Zhang and L. Zhu, J. Energy Chem., 2020, 40, 39–45 CrossRef.
- N. Ohta, K. Takada, I. Sakaguchi, L. Zhang, R. Ma, K. Fukuda, M. Osada and T. Sasaki, Electrochem. Commun., 2007, 9, 1486–1490 CrossRef CAS.
- H. Guo, F. Shen, W. Guo, D. Zeng, Y. Yin and X. Han, Mater. Lett., 2021, 301, 130302 CrossRef CAS.
- D. Aurbach, B. Markovsky, A. Rodkin, E. Levi, Y. Cohen, H.-J. Kim and M. Schmidt, Electrochim. Acta, 2002, 47, 4291–4306 CrossRef CAS.
- C.-L. Liao and K.-Z. Fung, J. Power Sources, 2004, 128, 263–269 CrossRef CAS.
- M. Reddy, B. Pecquenard, P. Vinatier and A. Levasseur, Electrochem. Commun., 2007, 9, 409–415 CrossRef CAS.
- T. Kim, W. Choi, H.-C. Shin, J.-Y. Choi, J. M. Kim, M.-S. Park and W.-S. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 14–25 CrossRef CAS.
- K. J. Nelson, J. E. Harlow and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A456–A462 CrossRef CAS.
- Y. Ren and E. D. Wachsman, J. Electrochem. Soc., 2022, 169(4), 040529 CrossRef.
- T. Liu, Y. Ren, Y. Shen, S.-X. Zhao, Y. Lin and C.-W. Nan, J. Power Sources, 2016, 324, 349–357 CrossRef CAS.
- W. Luo, Y. Gong, Y. Zhu, Y. Li, Y. Yao, Y. Zhang, K. K. Fu, G. Pastel, C. Lin, Y. Mo, E. D. Wachsman and L. Hu, Adv. Mater., 2017, 29, 1606042 CrossRef PubMed.
- M. Kato, T. Hayashi, G. Hasegawa, X. Lu, T. Miyazaki, Y. Matsuda, N. Kuwata, K. Kurihara and J. Kawamura, Solid State Ionics, 2017, 308, 54–60 CrossRef CAS.
- D. Wang, Q. Sun, J. Luo, J. Liang, Y. Sun, R. Li, K. Adair, L. Zhang, R. Yang, S. Lu, H. Huang and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 4954–4961 CrossRef CAS PubMed.
- M. Müller, L. Schneider, N. Bohn, J. R. Binder and W. Bauer, ACS Appl. Energy Mater., 2021, 4, 1993–2003 CrossRef.
- A. N. Filippin, T.-Y. Lin, M. Rawlence, T. Zünd, K. Kravchyk, J. Sastre-Pellicer, S. G. Haass, A. Wäckerlin, M. V. Kovalenko and S. Buecheler, RSC Adv., 2018, 8, 20304–20313 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00155e |
| This journal is © The Royal Society of Chemistry 2023 |