
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDebonding-on-demand adhesives based on photo-reversible cycloaddition reactions†
Moeka
Inada
a,
Tatsuhiro
Horii
b,
Toshinori
Fujie
 b,
Takuya
Nakanishi
c,
Toru
Asahi
*ac and
Kei
Saito
*d
b,
Takuya
Nakanishi
c,
Toru
Asahi
*ac and
Kei
Saito
*d
aGraduate School of Advanced Science and Engineering, Department of Advanced Science and Engineering, Waseda University, Tokyo, 162-8480, Japan
bSchool of Life Science and Technology, Tokyo Institute of Technology B-50, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8501, Japan
cComprehensive Research Organization, Waseda University, Shinjuku, Tokyo 169-8555, Japan
dGraduate School of Advanced Integrated Studies in Human Survivability, Kyoto University, Higashi-Ichijo-Kan, Yoshida-nakaadachicho 1, Sakyo-ku, Kyoto, 6068306, Japan. E-mail: saito.kei.1y@kyoto-u.ac.jp
First published on 4th January 2023
Abstract
Debonding-on-demand (DoD) adhesives, which have the ability to repeatedly adhere and release in response to external stimuli, are attracting attention as sustainable functional materials. DoD adhesives can be designed by fabricating dynamic covalent bonds and using polymer cleavage and flow generation by dynamic mechanisms that respond autonomously to external stimuli, such as heat and light. However, the typical DoD adhesives using dynamic covalent bonds mainly utilize heat-induced systems, and the practical application of these adhesives at room temperature and ambient pressure is challenging. In this study, we report a DoD adhesive system based on a polymer that is fabricated by the reversible cycloaddition reaction of coumarin-terminated four-arm siloxane monomers. Thermophysical property analysis of the obtained polymers confirmed that photoinduced fluidization was based on the reversible crosslinking and decrosslinking reactions of the monomers. These thermophysical properties can directly control the stiffness of the polymers, providing mechanistic evidence of DoD adhesion. This simple siloxane-based reversible cycloaddition system exhibits significant potential as a DoD adhesive that functions at room temperature.
1. Introduction
Adhesives are widely used in mechanical bonding methods and have been implemented in various daily applications. Over the past decade, debonding-on-demand (DoD) systems with a variety of bond strengths according to requirements have been developed in the medical, automotive, micro, and soft electronics fields.1–5 These DoD adhesives are required to possess switchable adhesiveness, that is, they should be sufficiently strong to hold and bond adherends when in use, while decreasing in strength sufficiently to be peeled off from the adherends after use. For example, acrylic- and silicone-based adhesives are mainly used as peelable wound dressings in the biomedical field.6–8 In the automotive field, a temporary adhesive tape is typically used as an in-process masking tape; recently, a DoD mechanism has been considered for easy material retrieval from end-of-life electronic devices.9–13 The most attractive mechanism for developing DoD adhesives that are reversibly detachable on demand is incorporating a system of reversible bond breaking and forming into the material design.3–5 Dynamic covalent bonds, which are distinguished by their reversibility and responsiveness to external stimuli such as heat, light, electricity, and pH, can control the degree of polymerization, crosslink density, and viscoelastic properties of polymeric materials.14,15 Recently, stimulus-responsive dynamic covalent bonding has been shown to be applicable to adhesives in terms of controlling adhesiveness.16,17 Notably, current DoD adhesives are mainly based on thermo-reversible reactions such as Diels–Alder18–21 and transesterification reactions,22,23 which limit their application at room temperature. In contrast, photoinduced processes that enable “direct” remote manipulation of the macroscopic properties of materials by irradiating light of specific wavelengths and intensities are useful for DoD adhesives under ambient conditions.1Typically, photo-reversible covalent bonding reactions include [2+2]- and [4+4]-type photochemical cycloadditions of thymine,24 coumarin,25–27 and anthracene,28–33trans–cis photoisomerization of azobenzene,34 and photolysis of trithiocarbonates.35–38 Dynamic covalent polymer networks incorporating these light-induced reversible reactions have a wide range of applications, including self-healing polymers39–41,46–50 and shape memory materials.42–45 Herein, the cycloaddition reaction of coumarin and its derivatives is one of the most promising reactions applicable to DoD adhesives. It is expected to provide a light-responsive reversible change in adhesiveness and exhibit stability under sunlight owing to its properties, such as irradiation with ultraviolet (UV) light with a wavelength of 365 nm or longer, inducing the cyclodimerization of coumarin, leading to the formation of a dimer with a cyclobutane ring, whereas the reverse photocleavage reaction occurs by irradiation with light at wavelengths below 254 nm, recovering the original monomers. Although polymers with coumarins have been used in self-healing and shape-memory materials, only a few have been used to create reversible adhesives. In addition, most of the adhesives developed with coumarins are based on block polymers, and their synthetic pathways are exceedingly complex.25,26
In this study, we report a DoD system based on a coumarin-terminated four-arm monomer with a siloxane (–Si–O–Si–) backbone. Four-arm monomers are known to induce macromolecular architectural transformations (MAT) by increasing the number of dynamic covalent bonding sites, forming denser and more reproducible polymer networks.51–55 To achieve MAT, two features are important: (i) flowability (ability to be fluidized); and (ii) intermolecular bonding (ability to form networks). In the molecule we synthesized (SS1, Fig. 1a), a central moiety consisting of cyclic siloxane appears suitable as a liquid monomer and has a high glass transition temperature (Tg) in its polymerized state, and each terminal coumarin motif includes the photo-reversible intermolecular bonding; this structure is designed to enable light-induced change in the rigidity and fluidity of the materials. The synthesis of the four-arm monomer SS1 was achieved by selecting a simple epoxy ring opening reaction of 2,4,6,8-tetramethyl-2,4,6,8-tetrakis(propylglycidylether)cyclotetrasiloxane with 7-hydoxycoumarin. A dynamic covalent polymer network derived from SS1 was demonstrated to possess reversible adhesive properties, exhibiting a strong potential as a DoD adhesive.
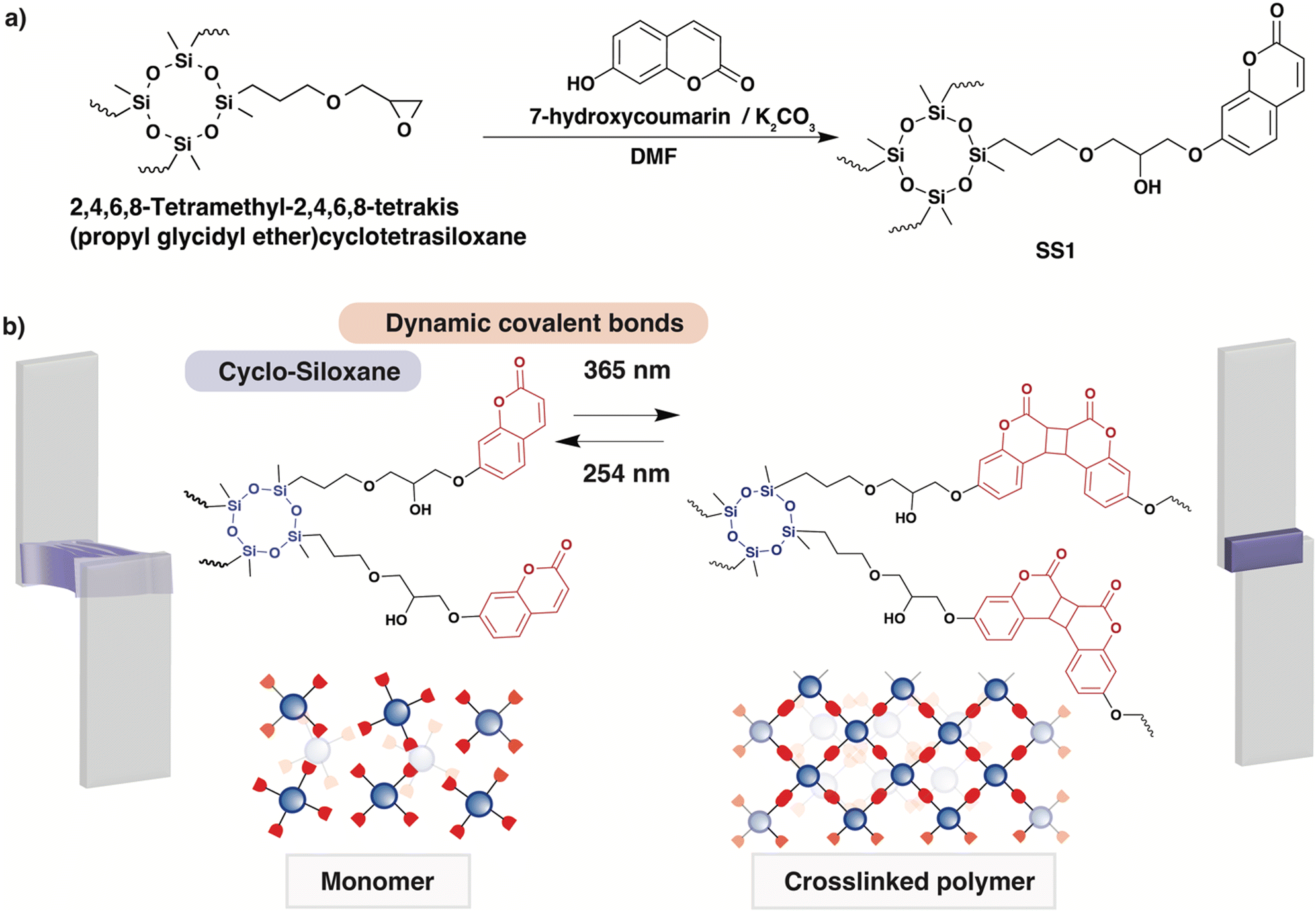 | ||
| Fig. 1 Four-arm SS1 adhesives. (a) Synthetic scheme for SS1, and (b) schematic diagram of a photoinduced debonding-on-demand system of SS1. | ||
2. Results and discussion
2.1 Spectroscopic characterization of light-induced reversible reactions of SS1
As mentioned in the Introduction, a tetrafunctional monomer (SS1) was designed with a structure consisting of a cyclic siloxane core and a photoreactive coumarin attached to the terminus. SS1 was synthesized by a one-step epoxy ring opening at the end of the side chain with the addition of coumarins. The product was obtained as a liquid and characterized using 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S1, ESI†)To obtain crosslinked cured polymers, the SS1 monomer was cast onto glass slides and irradiated with 365 nm UV light. The monomer was placed directly on a glass slide and irradiated in a UV-crosslinking device under ambient temperature and pressure. After irradiation at 365 nm, the resultant polymer film was self-standing (Fig. 2a). The relevant photoreaction processes were tracked using infrared (IR) and UV-visible (UV-vis) spectroscopy separately, and the reversibility of the polymers crosslinked by dynamic bonding was demonstrated. The IR spectra of the three types of SS1 samples, prior to light irradiation, and irradiated at 365 and 254 nm, are shown in Fig. 2b. Compared to the spectrum prior to UV irradiation, the peak assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the coumarin moiety, observed at 1749 cm−1 in pre-UV, shifted to 1723 cm−1 with an increase in peak intensity owing to the influence of dimer formation (inset of Fig. 2b). The peak assigned to the cyclobutane ring, not observed in the pre-UV spectrum, appeared at 1555 cm−1 owing to the formation of a cyclobutane-based dimer. The peak at 1580 cm−1, assigned to CC stretching of the pyrone moiety decreased, whereas the peaks at 1620 and 1510 cm−1, assigned to the CC stretching of the aromatic ring, were observed both before and after irradiation. After subsequent irradiation at 254 nm, causing photocleavage of the cyclobutane ring, the CO peak shifted back to 1749 cm−1 with a decreased intensity, the peak assigned to the cyclobutene ring at 1555 cm−1 disappeared, and the peak originating from the pyrone moiety at 1580 cm−1 became clear again. These FT-IR results accompanied by the irradiation at 365 and 254 nm confirm the crosslinking and decrosslinking of SS1.46–48,55
O stretching of the coumarin moiety, observed at 1749 cm−1 in pre-UV, shifted to 1723 cm−1 with an increase in peak intensity owing to the influence of dimer formation (inset of Fig. 2b). The peak assigned to the cyclobutane ring, not observed in the pre-UV spectrum, appeared at 1555 cm−1 owing to the formation of a cyclobutane-based dimer. The peak at 1580 cm−1, assigned to CC stretching of the pyrone moiety decreased, whereas the peaks at 1620 and 1510 cm−1, assigned to the CC stretching of the aromatic ring, were observed both before and after irradiation. After subsequent irradiation at 254 nm, causing photocleavage of the cyclobutane ring, the CO peak shifted back to 1749 cm−1 with a decreased intensity, the peak assigned to the cyclobutene ring at 1555 cm−1 disappeared, and the peak originating from the pyrone moiety at 1580 cm−1 became clear again. These FT-IR results accompanied by the irradiation at 365 and 254 nm confirm the crosslinking and decrosslinking of SS1.46–48,55
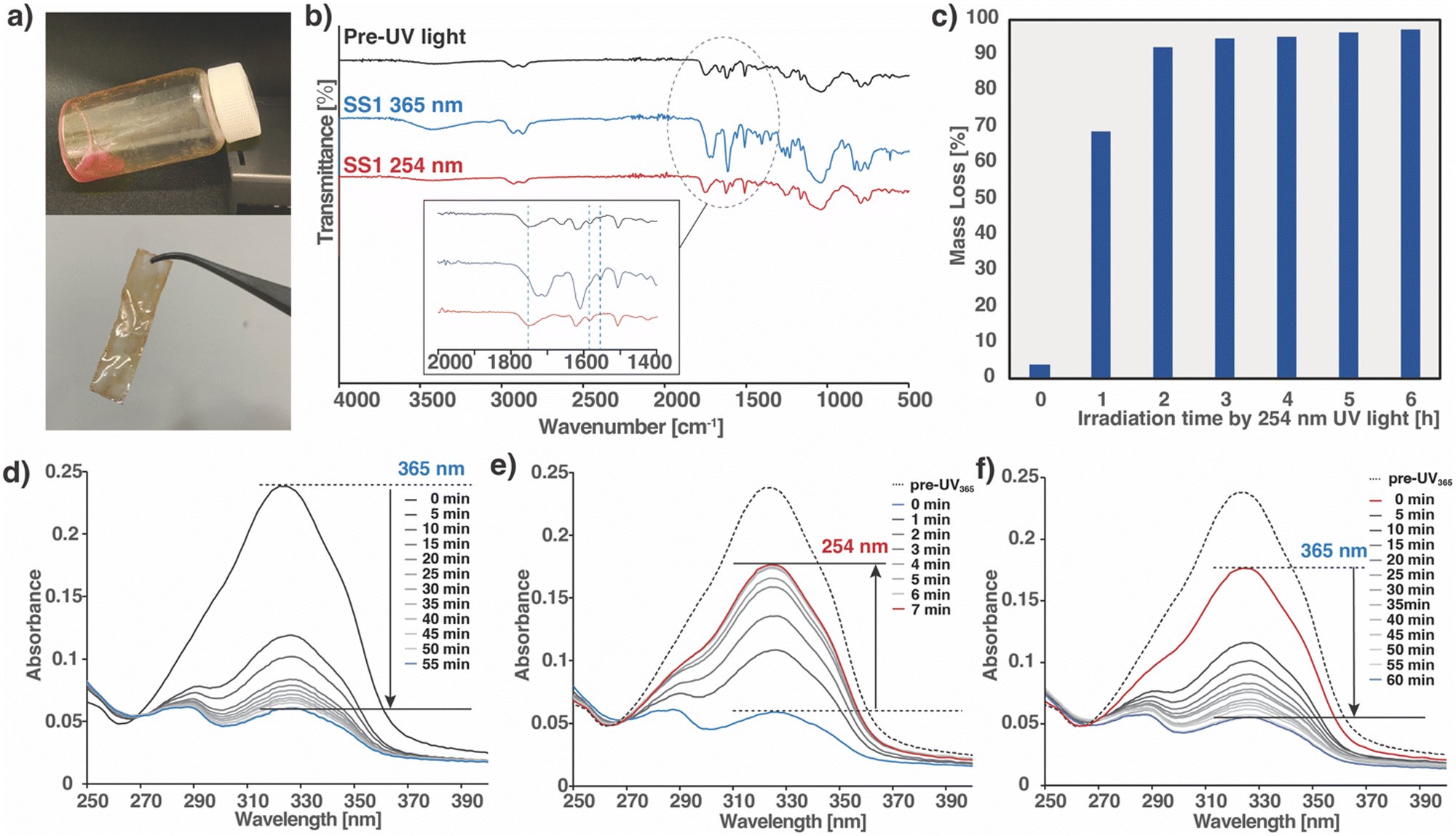 | ||
| Fig. 2 Characterization of photoinduced reactions of SS1 films. (a) Photo of four-arm SS1 before and after UV Irradiation, (b) Fourier-transform infrared spectrum, and (c) solubility test of SS1. UV irradiation at 254 nm increased the mass of the crosslinked polymer dissolved in THF and decreased the mass left over. UV-Vis spectra of the samples after UV irradiation with (d) 365 nm for 55 min, (e) 254 nm for 7 min, and (f) 365 nm (again) for 60 min. The blue and red lines indicate the minimum intensity after 365 nm UV irradiation and the maximum intensity after irradiation with 254 nm, respectively. The dotted line shows the absorbance before 365 nm UV irradiation. | ||
The progress of the polymerization reaction of SS1 was determined from the UV-vis spectra by comparing the intensity of the absorption peak before and after light irradiation (Fig. 2d–f), for which samples were prepared by spin-coating the SS1 monomer solution on quartz glass. A series of UV-vis spectra were obtained after each exposure to 365 nm UV light (irradiation intensity: 0.28 J cm−2 min−1) every 5 min in a UV crosslinker. As shown in Fig. 2d, a gradual decrease in the absorbance at 321 nm, corresponding to the π–π* transition of the pyrone structure with the double bond in coumarin, was observed upon light irradiation at 365 nm. The absorption decrease was attributed to the cleavage of the double bond in the pyrone structure and the collapse of the conjugated π-system, indicating that the [2+2] cyclic addition reaction proceeded and a cyclobutene dimer was formed.56,57 The photochemical conversion of these coumarin moieties within SS1 was estimated using eqn (1):48
| Coumarin conversion (%) = 1 − (At321/A0321) × 100 | (1) |
2.2 Analysis of thermal properties
The thermal behavior induced by the [2+2] cycloaddition of the terminal coumarin moiety upon photoirradiation of SS1 confirmed differential scanning calorimetry (DSC) (Fig. S3, ESI†). The Tg of the four-arm monomers and prepared polymers from those monomers can be controlled by changing the wavelength.1,47,52–54 The Tg of SS1 before and after UV irradiation at 365 nm (polymerized) and 254 nm (depolymerized) were determined by DSC. The Tg of the monomer was −14.2 °C and that of the crosslinked polymer was as high as 128.3 °C, confirming the achievement of a significant difference in Tg before and after polymerization as designed. After being irradiated with 254 nm UV light to enable the reverse photocleavage of the cyclobutene ring, the Tg of the decrosslinked polymer was determined to be 16.5 °C, which is higher than that of the original monomer but lower than room temperature. This is attributed to the monomer not completely returning to the decrosslinked state and the oligomers being mixed in the polymer. Gel permeation chromatography (GPC) revealed the presence of oligomers (Table S1, ESI†). It was demonstrated that the irradiation with UV light at ambient conditions could reversibly convert between two states of SS1: the hard crosslinked state with a Tg of ∼130 °C, and the soft decrosslinked state with a Tg below room temperature.2.3 Solubility test
The dependence of the degree of polymer decrosslinking on the duration of UV irradiation was monitored by the solubility testing of SS1 in THF.42 From the UV-vis and DSC results discussed in the preceding section, UV irradiation at 254 nm for opening the cyclobutane ring of the coumarin motif in the crosslinked part is considered to result in the decrosslinking of the monomers and oligomers. Because these low molecular-weight oligomers are soluble, the mass loss of the samples by dissolution in the solvent was monitored every 1 h during irradiation. After every 1 h, the samples were irradiated with 254 nm UV, completely dissolved in THF solvent, dried, and weighed. The mass loss of the samples was plotted vs. the irradiation exposure time (Fig. 2c). The mass loss was ∼70% after irradiation for 1 h, was as high as ∼90% after 2 h, and gradually increased to 96% after 6 h. As suggested from the GPC results (Table S1, ESI†), it is considered that decrosslinking progressed and monomers and oligomers were formed. Furthermore, the moisture sensitivity of the material was analyzed by measuring the moisture tolerance of SS1 polymer and the contact angle of its surface (Fig. S4, ESI†). Nor like polyethylene glycol-based hydrogel, the weight of the SS1 polymer under the moisture tolerance analysis did not increase. In addition, the contact angle of the polymer film was 59.8°.2.4 Light-controlled reversible adhesion
The results of the thermal measurement described in Section 2.2 confirm that the photoresponsivity of the four-arm polymer adjusted its Tg below room temperature by cleaving the monomer end-junctions into oligomers and monomers after 254 nm UV irradiation. Therefore, considering the potential of SS1 films as DoD adhesives, the effects of this reversible photoresponsive property at room temperature on adhesiveness were investigated. The quartz glass substrates were bonded together by irradiating them with 365 nm UV light, and applying a weight of 1 kg (two 500 mL plastic bottles filled with water was suspended at the end of the quartz glass) (Fig. 3b); the weight remained suspended for more than 30 min. The glass was then irradiated with 254 nm light and again suspended with a weight; the bonded quartz glass peeled off owing to the weight. In addition, lap-shear tests were performed to quantitatively evaluate adhesive strength behavior. A 10 wt% SS1 solution dissolved in THF was prepared for spin-coating, and the reversible light-controlled adhesive strength of the resultant films was evaluated by uniaxial shear tests and stress–strain measurements by a five-step procedure (Fig. 3a). The monomer solution (10 μL) was cast onto a single glass slide (quartz) with an area of 250 mm2 divided with masking tape (Step 1) and spin-coated at 5000 rpm (Step 2). Another glass plate was then placed on the cast area of the SS1 monomer and pressure was applied to the contact area (Step 3). Subsequently, 365 nm UV light was irradiated for 5, 10, 15, and 30 min (Step 4), and the shear strength was measured (Step 5).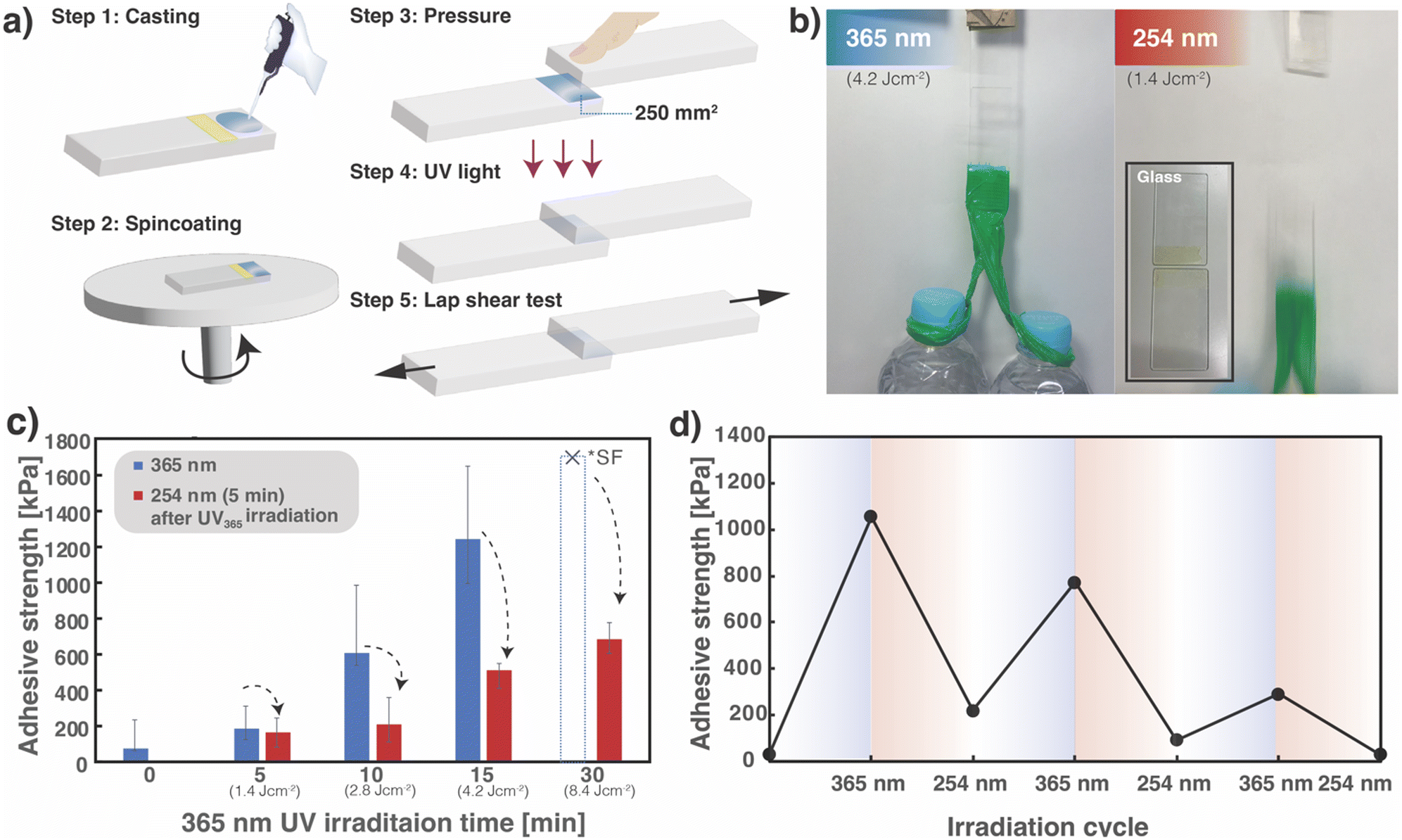 | ||
| Fig. 3 Characterization of DoD adhesion properties of SS1 films. (a) Schematic diagram of shear tensile test preparation, (b) photo of photoinduced DoD adhesives and the surface of the glass after a shear tensile test. (left) 365 nm irradiation for 15 min (4.2 J cm−2), (right) 254 nm irradiation for 5 min (1.4 J cm−2), (c) average adhesion strengths of shear stress test upon prolonged UV exposure (>360 nm); the cross mark in the graph denotes the average of the glass breaking (substrate failure strength (see text). (d) Shear adhesion of SS1 reversibly crosslinked by repeated photo crosslinking. The SS1 adhesives were irradiated by 365 nm UV light for 15 min and 254 nm for 5 min. | ||
As shown in Fig. 3c, the shear adhesion strength increased with increasing 365 nm light irradiation and a strength of 1.0–1.7 MPa (similar to that reported for other light-responsive adhesives, such as 0.6–5.0 MPa for photoisomerization-based systems5), was achieved after the 15 min irradiation. Notably, the adhesion forces of the samples after irradiation for more than 30 min appeared too strong to be measured without substrate failure; glass breaking occurred at an average force of ∼1.7 MPa, which is lower than the shear adhesion force of the samples. Moreover, surface observations of the samples after the lap-shear test revealed that the adhesives remained on both substrates (adherends) in all cases, confirming that the mode of failure was cohesive (Fig. 3b).
For each sample irradiated at 365 nm for a specific period, subsequent 254 nm UV irradiation for 5 min decreased the bond strength by ∼33% (Fig. 3c). Notably, the samples after 30 min irradiation at 365 nm, of which the adhesive strength was unmeasurable owing to substrate failure, exhibited an adhesiveness as high as ∼0.7 MPa, even after subsequent irradiation for 5 min at 254 nm. The decrease in adhesiveness is related to the change in the crosslinked density, and to the change in the stiffness of the film. In addition, Fig. S5 (ESI†) shows the frequency dependence of the dynamic modulus (G′: storage modulus, G′′: loss modulus) in the nonirradiated and UV-irradiated samples at 365 and 254 nm. In the nonirradiated state, G′ was significantly lower than G′′, suggesting that the monomer was in a liquid state. Subsequently, following crosslinking by photoirradiation, the corresponding values of G′ and G′′ were reversed, implying that the stiffness increased. After further irradiation at 254 nm, G′ was higher than G′′ in the low-frequency region; however, after 30 Pa, G′ was lower than G′′ in the high-frequency region. This rheological behavior indicates that the adhesive strength is not only owing to microscopic intermolecular bonding, but also to light-induced changes in the macroscopic stiffness of the overall material.
Furthermore, a series of lap-shear tests were performed by returning the two separated glass slides to an originally bonded state and alternately switching the irradiation wavelength between 365 and 254 nm; this is done in particular by repeating Steps 3–5 in Fig. 3a by alternately switching the two wavelengths for UV irradiation. Representative results are shown in Fig. 3d. The adhesive strength of the SS1 film, determined to be ∼1.1 and 0.2 MPa for the initial crosslinked and subsequent decrosslinked states, respectively, recovered to ∼0.8 MPa by recrosslinking (after the second 365 nm irradiation). Thus, the reworkability of the SS1 film as a detachable adhesive was successfully demonstrated. The recovery of adhesiveness from ∼0.1 MPa at the second decrosslinked state to ∼0.3 MPa by the third crosslinking indicates that the bond strength can be cyclically controlled, even though the bond strength gradually decreased. As suggested by the UV-vis spectroscopy and solubility results, photodimerization and photocleavage transitions occurred in only 70% of cases, leading to an accumulated decrease in strength with repeated test cycles. This repeated reduction and recovery of adhesiveness is suggestive of the promising potential of photo-reversible cycloaddition systems, such as SS1 in this study, for DoD adhesives.
3. Conclusions
We propose the utilization of photo-reversible cycloaddition reactions to DoD adhesives and demonstrate its potential using a newly synthesized coumarin-functionalized four-arm cyclosiloxane monomer (SS1). This simple SS1 system exhibited photoinduced reversible crosslinking and decrosslinking with irradiation at 365 and 254 nm under ambient conditions, and the resultant two states exhibited Tg values higher and lower than room temperature, respectively. In addition, the SS1 films exhibited alternating increases and decreases in adhesiveness with changes in crosslink density upon light irradiation at two different wavelengths. The results of this study enable the feasible application of simple siloxane-based reversible cycloaddition systems, represented by SS1, as sustainable DoD adhesives in industrial processes.4. Experimental section
4.1 Materials
7-Hydroxycoumarin was purchased from Tokyo Chemical Industry Co., Ltd; anhydrous potassium carbonate and N,N-dimethylformamide (DMF) were purchased from FUJIFILM Wako Pure Chemical Corporation; and 2,4,6,8-tetramethyl-2,4,6,8-tetrakis(propylglycidylether)cyclotetrasiloxane was purchased from Sigma-Aldrich. The other solvents were purchased from FUJIFILM Wako Pure Chemical Corporation.4.2 Equipment and characterization
Photochemical reactions were performed using a UV crosslinker (CL1000 365 nm, UVP and CL1000 254 nm, UVP) with a lamp that produced UV light at wavelengths centered at 365 nm and 254 nm. A UV optical filter (P/N 50230FBB, eSource optics) was used. 1H-NMR spectroscopy was performed on a Varian INOVA 400 spectrometer. 29Si-NMR spectroscopy was performed on a Bruker AVANCE III 600. The molecular weight was analyzed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (AVANCE 600 NEO, BRUKER). Fourier-transform infrared (FT-IR) spectra were acquired using a Nicolet 6700 FT-IR spectrometer. The spectra were measured directly on the sample in the frequency range 4000–500 cm−1. Differential scanning calorimetry (DSC) analyses were conducted using a PerkinElmer DSC8000 at a rate of 50 °C min−1 for heating and cooling under an N2 atmosphere. The UV spectra were measured using a JASCO V-550 UV-vis spectrometer. The molecular loss was determined by gel permeation chromatography (GPC at 40 °C using a Showa Denko Shodex KF-804L column and a JASCO RI-4030 refractive index detector calibrated with a polystyrene standard. Tetrahydrofuran (THF) with a stabilizer was used as the eluent at a flow rate of 1.0 mL min−1 and a calibration curve was prepared using Shodex PMMA standards. The lap-shear test was performed using a universal testing machine equipped with a 5 kN load cell (Autograph AG 25 TB, Shimadzu Co.) with a stretching rate of 10 mm min−1. Dynamic rheology was performed using a rotary rheometer (MCR 101, Anton Paar) with a 50 mm parallel plate at a gap of 0.5 mm. During the measurements, a frequency sweep (0.1–100 rad s−1) was performed at a strain of 50 Pa. The water contact angle of SS1 polymer was analyzed by using a contact angle meter (DMs-301, Kyowa Interface Science Co. Ltd).4.3 Synthesis of SS1 monomer
7-Hydroxycoumarin was dissolved in DMF, and anhydrous potassium carbonate was added. The mixture was stirred for 5 min and heated to 105 °C under N2. 2,4,6,8-Tetramethyl-2,4,6,8-tetrakis(propylglycidylether)cyclotetrasiloxane was dissolved in DMF (15 mL), added to the flask over 3 h, and stirred for 48 h under constant heating. The reaction mixture was cooled to room temperature and diluted with DMF. The solids were removed by filtration, and the sediments of the product were dropped in ether at set intervals. The ether was poured out and the residues (sticky) were redissolved in DCM and stirred overnight at room temperature. The solution was washed thrice with water (pH 10) and then dried over MgSO4. The solvent was evaporated, and the product was purified by column chromatography with methanol and dichloromethane (20/1). Finally, the sticky brown products were collected by evaporation. Final mass = 0.9 g, 24% yield. 1H-NMR (CDCl3): 0.07 ppm (12H, br, –Si(CH3)–); 0.53 ppm (8H, m, –SiCH2CH2CH2–); 1.63 ppm (8H, m, –SiCH2CH2CH2–); 3.46 ppm (8H, m, –SiCH2CH2CH2–); 3.58–4.1 ppm (20H, O–CH2–CH(OH)–CH2–O–); 6.23 ppm (4H, d, ArH); 6.81–6.91 ppm (8H, m, ArH); 7.34 ppm (4H, d, ArH); and 7.60 ppm (4H, d, ArH). 13C-NMR (CDCl3): 1.65, 13.65, 23.68, 69.49, 70.22, 72.11, 74.68, 102.18, 113.44, 113.88, 114.28, 129.45, 143.92, 156.33, 161.76, 162.42 ppm. 29Si-NMR (CDCl3): −19.89 pm. FT-IR: 3411, 2931, 2870, 1724, 1706, 1666, 1608, 1556, 1508, 1479, 1456, 1429, 1402, 1350, 1292, 1281, 1279, 1230, 1198, 1159, 1119, 1045, 989, 891, 831, 798, 750, 685, 664, 6323, 615, 544–565, and 521 cm−1 (Fig. 2b). MALDI-TOF/mass: m/z 1368.51 [M + Na]+.4.4 Preparing photocrosslinked polymer
The synthesized SS1 monomer was used to produce a crosslinked polymer. The monomer was cast directly onto glass slides and irradiated with UV light at 365 nm in a UV crosslinker at room temperature. For the UV-vis spectra and adhesion tests, the monomer solution (THF, 10 wt%) was spin-coated onto the quartz glass. After irradiation at 365 nm for 1 h, the crosslinked polymers were obtained.4.5 Tensile test
To prepare for the adhesion test, the SS1 monomer (THF, 0.05 M) was spin-coated onto a 25 × 10 mm2 masked glass (quartz, 1 mm thick). Another substrate was then placed over the previously spin-coated substrate and the joint was hand-pressed. The crosslinked polymers were then irradiated with 365 nm light. To measure the adhesion strength of the polymers, a lap-shear test was performed using a universal testing machine (Autograph AG 25 TB, Shimadzu) equipped with a 5 kN load cell. At a stretching rate of 10 mm min−1, each substrate was stretched until it peeled off, and the lap-shear strength was measured. After the test, adhesive strength (kPa) was calculated by dividing the maximum load (N) by the lap area (A). Further attempts were made to produce an adhesive after curing. After the shear test, a THF solution (1 μL) was dropped to one of the separated interfaces to add fluidity to the sample on the surface. The glass was placed on top of each other, and hand-pressed pressure was applied. Subsequently, repeated shear tensile tests were performed under the same conditions.Author contributions
Data collection and assembly were conducted by M. I. The draft of the paper was written by M. I. and K. S. T. H. performed the adhesive measurements. Data analysis and interpretation were conducted by M. I., T. F., T. N., K. S., and T. A. All the authors approved the final version of the paper. K. S. was involved in the writing and editing, supervision, and conceptualization of the study. T. A. funded the acquisition and supervised the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by JST SPRING (grant number, JPMJSP2128) and Nano-Energy Projects, Waseda University. M. I. would like to thank the Graduate Program for Power Energy Professionals, Waseda University, from the MEXT WISE Program. T. F. acknowledges support from the Fusion Oriented Research for disruptive Science and Technology (FOREST) from the Japan Science and Technology Agency (JST) (grant number, JPMJFR203Q) and JSPS KAKENHI Grant-in-Aid for Scientific Research(B) JP21H03815. K. S. acknowledges the support from the JSPS KAKENHI Grant-in-Aid for Transformative Research Areas (B) JP22H05046.Notes and references
- D. K. Hohl and C. Weder, Adv. Opt. Mater., 2019, 7, 1900230 CrossRef.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CrossRef CAS PubMed.
- H. Zhu, A. Demirci, Y. Liu, J. Gong and M. Mitsuishi, ACS Appl. Polym. Mater., 2022, 4, 1586–1594 CrossRef CAS.
- Z. Liu and F. Yan, Adv. Sci., 2022, 9, 2200264 CrossRef PubMed.
- N. D. Blelloch, H. J. Yarbrough and K. A. Mirica, Chem. Sci., 2021, 12, 15183–15205 RSC.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Chem. Rev., 2022, 122, 5604–5640 CrossRef CAS PubMed.
- K. R. Mulcahy, A. F. R. Kilpatrick, G. D. J. Harper, A. Walton and A. P. Abbott, Green Chem., 2021, 24, 36–61 RSC.
- Y. Choi, K. Kang, D. Son and M. Shin, ACS Nano, 2022, 16, 1368–1380 CrossRef CAS PubMed.
- Y. Lu, J. Broughton and P. Winfield, Int. J. Adhes. Adhes., 2014, 50, 119–127 CrossRef CAS.
- G. Gerhard and W. Karmann, ed. Adhesives and Adhesive Tapes, John Wiley & Sons, 2008 Search PubMed.
- M. D. Banea, L. F. M. da Silva and R. J. C. Carbas, Int. J. Adhes. Adhes., 2015, 59, 14–20 CrossRef CAS.
- C. Zhang, M. Wang, C. Jiang, P. Zhu, B. Sun, Q. Gao, C. Gao and R. Liu, Nano Energy, 2022, 95, 106991 CrossRef CAS.
- C. Shao, L. Meng, C. Cui and J. Yang, J. Mater. Chem. C, 2019, 7, 15208–15218 RSC.
- H. Memon, Y. Wei and C. Zhu, Polym. Test., 2022, 105, 107420 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- M. A. Rahman, C. Bowland, S. Ge, S. R. Acharya, S. Kim, V. R. Cooper, X. C. Chen, S. Irle, A. P. Sokolov, A. Savara and T. Saito, Sci. Adv., 2021, 7, eabk2451 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- S. Das, S. Samitsu, Y. Nakamura, Y. Yamauchi, D. Payra, K. Kato and M. Naito, Polym. Chem., 2018, 9, 5559–5565 RSC.
- J. H. Aubert, J. Adhes., 2003, 79, 609–616 CrossRef CAS.
- K. K. Oehlenschlaeger, N. K. Guimard, J. Brandt, J. O. Mueller, C. Y. Lin, S. Hilf, A. Lederer, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4348–4355 RSC.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- H. Y. Tsai, Y. Nakamura, T. Fujita and M. Naito, Mater. Adv., 2020, 1, 3182–3188 RSC.
- H. Zhang, C. Cai, W. Liu, D. Li, J. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 11833 CrossRef PubMed.
- N. Ishikawa, M. Furutani and K. Arimitsu, ACS Macro Lett., 2015, 4, 741–744 CrossRef CAS PubMed.
- L. Wang, X. Ma, L. Wu, Y. Sha, B. Yu, X. Lan, Y. Luo, Y. Shi, Y. Wang and Z. Luo, Eur. Polym. J., 2021, 144, 110213 CrossRef CAS.
- S. R. Trenor, T. E. Long and B. J. Love, J. Adhes., 2005, 81, 213–229 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- L. Shen, J. Cheng and J. Zhang, Eur. Polym. J., 2020, 137, 109927 CrossRef CAS.
- H. Akiyama, Y. Okuyama, T. Fukata and H. Kihara, J. Adhes., 2018, 94, 1–15 CrossRef.
- S. Kaiser, S. V. Radl, J. Manhart, S. Ayalur-Karunakaran, T. Griesser, A. Moser, C. Ganser, C. Teichert, W. Kern and S. Schlögl, Soft Matter, 2018, 14, 2547–2559 RSC.
- Z. Wang, L. Guo, H. Xiao, H. Cong and S. Wang, Mater. Horiz., 2019, 7, 282–288 RSC.
- Z. Liu, G. Wang, J. Cheng and J. Zhang, Eur. Polym. J., 2021, 152, 110472 CrossRef CAS.
- Z. Liu, J. Cheng and J. Zhang, Macromol. Chem. Phys., 2021, 222, 2000298 CrossRef CAS.
- G. Xu, S. Li, C. Liu and S. Wu, Chem. – Asian J., 2020, 15, 547–554 CrossRef CAS PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- K. J. Arrington, S. C. Radzinski, K. J. Drummey, T. E. Long and J. B. Matson, ACS Appl. Mater. Interfaces, 2018, 10, 26662–26668 CrossRef CAS PubMed.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2016, 4, 3379–3387 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- A. V. Menon, G. Madras and S. Bose, Polym. Chem., 2019, 10, 4370–4388 RSC.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- S. Peng, Y. Sun, C. Ma, G. Duan, Z. Liu and C. Ma, e-Polym., 2022, 22, 285–300 CrossRef CAS.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- T. Defize, J.-M. Thomassin, H. Ottevaere, C. Malherbe, G. Eppe, R. Jellali, M. Alexandre, C. Jérôme and R. Riva, Macromolecules, 2019, 52, 444–456 CrossRef CAS.
- T. Hughes, G. P. Simon and K. Saito, Polym. Chem., 2019, 10, 2134–2142 RSC.
- T. Hughes, G. P. Simon and K. Saito, Mater. Horiz., 2019, 6, 1762–1773 RSC.
- M. Abdallh, C. Yoshikawa, M. T. W. Hearn, G. P. Simon and K. Saito, Macromolecules, 2019, 52, 2446–2455 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373–18380 RSC.
- S. Banerjee, R. Tripathy, D. Cozzens, T. Nagy, S. Keki, M. Zsuga and R. Faust, ACS Appl. Mater. Interfaces, 2015, 7, 2064–2072 CrossRef CAS PubMed.
- H. Zhu, A. Demirci, Y. Liu, J. Gong and M. Mitsuishi, ACS Appl. Polym. Mater., 2022, 4, 1586–1594 CrossRef CAS.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- S. Honda and T. Toyota, Nat. Commun., 2017, 8, 502 CrossRef PubMed.
- D. Aoki, S. Uchida and T. Takata, Angew. Chem., Int. Ed., 2015, 54, 6770–6774 CrossRef PubMed.
- A. U. Alrayyes, Y. Hu, R. F. Tabor, H. Wang and K. Saito, J. Mater. Chem. A, 2021, 9, 21167–21174 RSC.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- S. Inacker, J. Fanelli, S. I. Ivlev and N. A. Hampp, Macromolecules, 2022, 55, 8461–8471 CrossRef CAS.
- D.-T. Van-Pham, M. T. Nguyen, K. Ohdomari, H. Nakanishi, T. Norisuye and Q. Tran-Cong-Miyata, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2017, 8, 025003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma01048h |
| This journal is © The Royal Society of Chemistry 2023 |