
Room-temperature fabrication of defective CoOxHy nanosheets with abundant oxygen vacancies and high porosity as efficient 5-hydroxymethylfurfural oxidation electrocatalysts†
Ruyi
Zhong
 a,
Puwei
Wu
a,
Qi
Wang
b,
Xiting
Zhang
a,
Lei
Du
a,
Yunhua
Liu
a,
Huakang
Yang
a,
Meng
Gu
b,
Z. Conrad
Zhang
c,
Limin
Huang
*d and
Siyu
Ye
*a
a,
Puwei
Wu
a,
Qi
Wang
b,
Xiting
Zhang
a,
Lei
Du
a,
Yunhua
Liu
a,
Huakang
Yang
a,
Meng
Gu
b,
Z. Conrad
Zhang
c,
Limin
Huang
*d and
Siyu
Ye
*a
aHuangpu Hydrogen Energy Innovation Center/Guangzhou Key Laboratory for Clean Energy and Materials, School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, P.R. China. E-mail: siyu.ye@gzhu.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, P.R. China
cDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P.R. China
dDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, P.R. China. E-mail: huanglm@sustech.edu.cn
First published on 18th April 2023
Abstract
Recently, cobalt oxides/hydroxides have attracted increasing attention in the electrocatalytic oxidation reaction of 5-hydroxymethylfurfural (HMFOR) under ambient conditions for 2,5-furandicarboxylic acid (FDCA) production, but understanding of the interplay of defective sites (i.e., oxygen vacancies and porosity) remains lacking. Herein, a series of defective cobalt oxide hydrate (CoOxHy) nanosheets were fabricated via room-temperature reductive treatments with methylamine (MA) and/or NaBH4 (BH). These defective CoOxHy nanosheets possessed abundant oxygen vacancies in relation to high Co2+/Co3+ ratios and high porosity, with a largely maintained ultrathin lamellar framework, and thus exhibited markedly improved catalytic activity and selectivity for HMFOR. DFT calculations also verified the beneficial role of oxygen vacancies towards HMF adsorption and activation, preferentially via the aldehyde group of HMF. In the optimal CoOxHy-MA, 98% FDCA yield and a faradaic efficiency of 83% were achieved within 200 min at a constant potential of 1.52 V vs. RHE. The mesoporosity mainly induced by MA improved the mass transportation of reactants and products, leading to a higher rate of HMFOR. Meanwhile, with the additional in-sheet micropores mainly induced by BH, the selectivity towards the oxidation intermediate 5-formyl-2-furancarboxylic acid (FFCA) significantly increased, probably due to the accelerated penetration of FFCA instead of further oxidation to FDCA. This work highlights the simultaneous regulation of the oxygen vacancies and porosity of metal oxide/hydroxide catalysts by facile reductive treatments for efficient electrochemical biomass conversion.
1. Introduction
In recent decades, the production of high value-added chemicals or biofuels from widely available biomass has become an important strategy to alleviate energy and environmental crises.1,2 Thermochemical methods have been widely employed in the biomass refining process, but they often entail high temperature, high pressure, and additional redox agents.2 Meanwhile, electrochemical methods, such as electrochemical oxidation processes, provide an efficient, economical, and clean means to realize the conversion of biomass-derived platform molecules under low temperature and atmospheric pressure conditions.2–4 The reactivity of the biomass-derived platform molecules can be facilely regulated by the electrode potential and transformed by the in situ generated oxidizing agents.5,6 Therefore, the electrocatalytic method has received intensive interest as an emerging and burgeoning approach for bio-refinery.3,7,85-Hydroxymethylfurfural (HMF) is one of the representative platform furanic molecules from biomass valorization.1 The electrocatalytic oxidation reaction of HMF (HMFOR) is a promising strategy for producing high-value fine chemicals, including 2,5-furandicarboxylic acid (FDCA), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furancarboxylic acid (FFCA), and diformylfuran (DFF).9,10 Among them, FDCA is a near-market alternative monomer to petroleum-derived terephthalic acid for the polymeric industry.11,12 The electrocatalytic oxidation reaction of HMFOR under alkaline conditions to produce FDCA as the main product with a high faradaic efficiency (FE) is a challenging process.13,14 Furthermore, the thermodynamic oxidation potential of HMF is 0.30 V, which is much lower than 1.23 V of the oxygen evolution reaction (OER),3,14 making HMFOR an effective alternative anodic reaction to the OER, when constructing a hybrid water electrolysis system for the co-production of hydrogen at a low overall energy consumption.15
In recent years, Earth-abundant cobalt-based electrocatalysts, such as their sulfides (Co-CoSx16), phosphides (CoP17) and borides (CoB18), have been proposed as highly efficient HMFOR electrocatalysts to produce FDCA.4 These non-oxides are inclined to surface reconstruction into their oxide/hydroxide counterparts, which play a real role in HMF oxidation.19 Luo et al. used an electrochemistry-assisted hydrolysis approach to transform NiCo-MOF into a NiCo-layered double hydroxide (LDH) structure20 and Kang et al. similarly reported a CoOOH catalyst from an in situ electrochemical oxidation strategy, demonstrating the real active species for HMFOR with cobalt oxides/hydroxides.21 However, the cobalt oxides/hydroxides still show low catalytic efficiency, due to the limited number of catalytic sites13 and the poor conductivity.22 Aside from strategies such as interfacial electronic engineering (Co-CoSx,16 CoP-CoOOH23) and introducing multimetallic components (NiCoFe-LDH,22 NiCo2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24), constructing mesoporous Co3O412 with facile mass diffusion and oxygen vacancy-doped Co3O411,25,26 has been proposed to increase the electrochemically active surface area and electrical conductivity, thus optimizing the adsorption behavior of intermediates and the catalytic performance of HMFOR.8 It can be seen that these two types of defective sites, i.e., oxygen vacancies and porosity play a key role in HMF performance. Nevertheless, the interplay of oxygen vacancies and porosity and their co-influence on the HMFOR process have not been studied previously.
24), constructing mesoporous Co3O412 with facile mass diffusion and oxygen vacancy-doped Co3O411,25,26 has been proposed to increase the electrochemically active surface area and electrical conductivity, thus optimizing the adsorption behavior of intermediates and the catalytic performance of HMFOR.8 It can be seen that these two types of defective sites, i.e., oxygen vacancies and porosity play a key role in HMF performance. Nevertheless, the interplay of oxygen vacancies and porosity and their co-influence on the HMFOR process have not been studied previously.
Vacancy incorporation has been demonstrated to be one of the most promising strategies for modulating electronic structures.7,8 Our previous work reported a topotactic conversion of two-dimensional (2D) cobalt oxide hydrate (CoOxHy) to surfactant-free and polycrystalline Co3O4 nanosheets with enriched oxygen vacancies at grain boundaries via rapid calcination at 300 °C for 5 min to improve the HMFOR performance.10 The temperature of 300 °C was chosen to remove the surfactant to expose the catalyst surface and simultaneously maintain the ultrathin 2D structure. Nevertheless, heat treatment still caused the loss of active surfaces with increased crystallinity. Here, we report a facile room-temperature fabrication strategy towards the synthesis of defective CoOxHy nanosheets with abundant oxygen vacancies and high porosity through reductive treatments with methylamine (MA) and/or NaBH4 (BH) (Fig. 1a), and the surfactant can be substituted during this ligand-exchange process for efficient electron transfer.27 Compared to the pristine CoOxHy nanosheets, lower onset potential and faster kinetics of HMFOR can be observed with these reductively-treated nanosheets. Density functional theory (DFT) calculations demonstrate the optimized adsorption of HMF intermediates with the presence of oxygen vacancies. Mesoporosity provided accelerated mass transportation, increasing the accessibility to active sites, whereas the incorporation of in-sheet micropores increased the selectivity of 5-formyl-2-furancarboxylic acid (FFCA), probably by the tuned diffusive transport of the reaction intermediates.
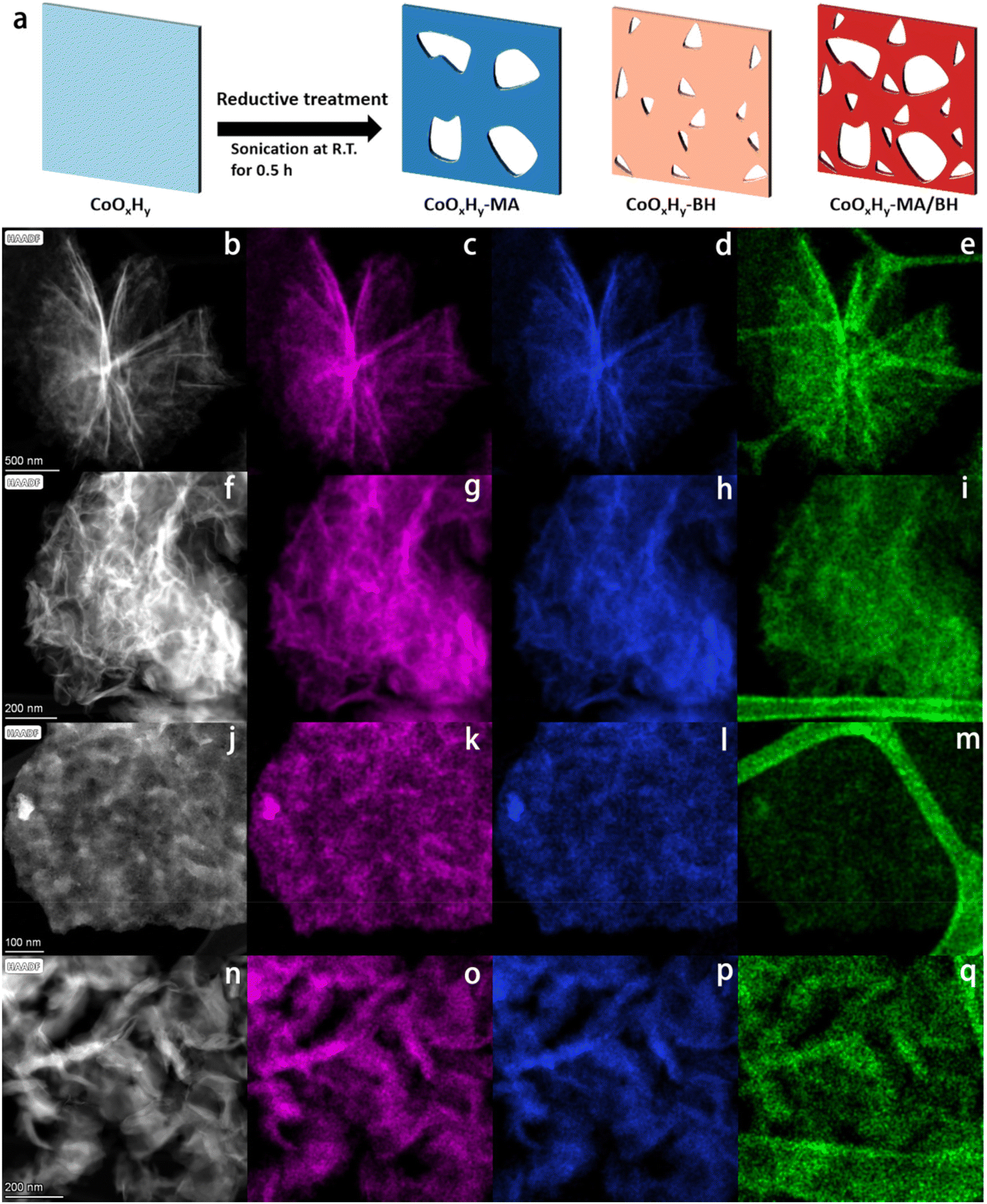 | ||
| Fig. 1 Schematic illustration and TEM characterization of the pristine and reductively-treated CoOxHy nanosheets. (a) Schematics of the reductive treatments at room temperature for the formation of CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH. (b, f, j, n) The HAADF-STEM images, and the corresponding EDS mappings of (c–e) CoOxHy, (g–i) CoOxHy-MA, (k–m) CoOxHy-BH, and (o–q) CoOxHy-MA/BH. Co, O, and C elements are represented in magenta, blue, and green, respectively. | ||
2. Experimental
2.1 Preparation of catalysts
All chemicals with analytical grade were directly used without further purification. The CoOxHy nanosheets were synthesized by a simple template-assisted solvothermal method.10 Concretely, 0.5 g P123 (EO20PO70EO20, Mw = 5800 g mol−1) was dissolved in a mixed solvent composed of 7.5 g ethanol, 2.5 g H2O, and 30 mL ethylene glycol by sonication to form the template solution. 1.25 mmol of cobalt acetate tetrahydrate (Co(OOCCH3)2·4H2O) and 1.25 mmol of hexamethylenetetramine (HMTA) were then added in the P123 solution under vigorous stirring for 30 min. The precursor solution was transferred into a 100 mL Teflon-lined stainless steel autoclave and heated at 170 °C for 2 h. After cooling to room temperature, the solid was collected by centrifugation, washed with ethanol and H2O, and finally dried at 60 °C overnight.For room-temperature treatments, 50 mg of the as-synthesized CoOxHy nanosheets were dispersed in a 10 mL ethanol solution containing 1 mL of methylamine (MA) aqueous solution (40%). The molar ratio of MA to Co was approximately 20. After sonication for 0.5 h, the catalyst was obtained by washing with ethanol and drying at 60 °C overnight. Similarly, a freshly-prepared 10 mL ethanol solution containing 7.6 mg NaBH4, or methylamine/NaBH4 (1 mL of methylamine and 7.6 mg NaBH4) was applied to the 50 mg as-prepared CoOxHy nanosheets under sonication for 0.5 h at room temperature, during which H2 was released from NaBH4 hydrolysis.28 The catalysts were named CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH, respectively, according to the added reductant. Besides, Co3O4-300-MA, Co3O4-300-BH, and Co3O4-300-MA/BH were prepared in the same way except that Co3O4-300 nanosheets from the rapid calcination of CoOxHy nanosheets10 were applied as the catalyst precursor.
2.2 Characterizations
The morphology and structure of the resulting materials were characterized by transmission electron microscopy (TEM, Titan Themis) at 200 kV, and the corresponding energy-dispersive X-ray spectroscopy (EDS) mapping was carried out using 4 in-column Super-X detectors. The specimen was prepared by ultrasonically suspending the sample in ethanol. A drop of the suspension was deposited on a carbon-coated copper grid and dried in air. The organic residue content was measured on a thermogravimetric analyzer at a heating rate of 10 °C min−1 in air. The porous structure of the samples was characterized by nitrogen sorption at −196 °C on a Micromeritics ASAP 2020M analyzer. Prior to adsorption, the samples were outgassed at 80 °C overnight. The specific surface area (SBET) was calculated using the Brunauer–Emmett–Teller (BET) method. The total pore volume (Vtot) was obtained according to the single point method at relative pressure P/P0 = 0.99. The micropore surface area (Smicro) and micropore volume (Vmicro) were determined by the t-plot method. The pore size distributions were estimated according to the non-local density functional theory (NLDFT) method. Zeta potential (ζ) was measured on a Zetasizer (Nano ZS) in an isopropanol–water solution. X-ray diffraction (XRD) analysis was performed by using an X'Pert-MPD diffractometer (PANalytical) using Cu Kα monochromatic radiation (λ = 0.15418 nm) as an X-ray source. Fourier transform infrared spectroscopy (FTIR) was performed on a Thermo Scientific Nicolet iS50 spectrometer equipped with a MCT detector in the diffuse reflectance mode in the wavelength range of 4000–400 cm−1. Raman spectra were collected using a Horiba spectrometer (LabRAM HR Evolution) and probed with a 532 nm laser. X-ray photoelectron spectra (XPS) were recorded on an ESCALab250Xi X-ray spectrometer with a monochromatic excitation source of Al Kα (1486.8 eV). All the peaks were adjusted using the hydrocarbon C 1s peak at 284.8 eV from adventitious carbon as the binding energy reference. Electron paramagnetic resonance (EPR) measurements were performed on a Bruker A300 spectrometer at −196 °C.2.3 Electrochemical measurements
All the electrochemical measurements were performed in a standard three-electrode system on a CHI-760E electrochemical workstation. The working electrode was glassy carbon (3 mm in diameter) with the catalysts while the Pt plate (1 × 1 cm2) and Hg/HgO electrode worked as the counter electrode and the reference electrode, respectively. Firstly, 3 mg of the catalyst was dispersed in 1.5 mL of isopropanol/water (1:3) solution and 10 μL of Nafion (5%). After the mixture was well dispersed by sonication for 0.5 h, 6 μL of the catalyst ink was dropped onto the clean glassy carbon electrode and dried in air at ambient temperature for a catalyst loading of 0.167 mg cm−2. Linear sweep voltammetry (LSV) measurements for HMF electrooxidation (HMFOR) and the OER were performed at a scanning rate of 5 mV s−1 in 1 M KOH electrolyte solution with and without 5 mM HMF, respectively. The potential values were converted to potential versus the reversible hydrogen electrode (RHE) using eqn (1):| ERHE = EHg/HgO + 0.059 pH + 0.098 V. | (1) |
The Tafel plots were applied to assess the kinetics during the electrooxidation reactions, which were plotted as potential (ERHE) vs. Log(J/(mA cm−2)). The electrochemically active surface area (ECSA) was estimated by the double-layer capacitance (Cdl) via cyclic voltammetry (CV) in a non-faradaic potential region at scanning rates ranging from 10 to 50 mV s−1 in 1 M KOH. The ECSA was calculated from the corresponding Cdl, based on eqn (2):
| ECSA = Cdl/Cs. | (2) |
The Cdl was calculated as the double-layer charging current divided by the scanning rate (Cdl = Δj/(2ν), where Δj = (ja − jc) and ν is the scanning rate). The Cs is the specific capacitance, typically reported to be 0.040 mF cm−2 in 1 M KOH.12 The durability of the catalyst was evaluated by the chronoamperometric (CA) method in 1 M KOH at a constant potential of 1.52 V vs. RHE. The electrochemical impedance spectroscopy (EIS) tests were performed in the frequency range from 105 to 1 Hz with an AC voltage amplitude of 5 mV. The obtained Nyquist plots were fitted with a simplified Randles circuit. For reference, the electrocatalytic performance of commercial IrO2 was also checked under the same reaction conditions. It should be noted that no iR compensation was applied in all the above measurements.
To determine the HMF conversion, FDCA yield, and FE for FDCA, an H-type electrolyzer separated by a Nafion 117 membrane was applied for long-term HMF electrolysis. 200 μL of the catalyst ink was transferred to a piece of carbon fiber cloth (1 × 1 cm2, 0.35 mm thick, HCP331N), dried in air, and employed as the working electrode. The catalyst mass loading was 0.40 mg cm−2. A Pt plate (1 × 1 cm2) and a Hg/HgO electrode were employed as the counter and reference electrodes, respectively. After passing a certain amount of charges, the sample was taken from the anode compartment for HPLC analysis. The HPLC was equipped with a Biorad 87H column (300 mm × 7.8 mm) and a refractive index detector (RID), using sulfuric acid (5 mM) as the mobile phase at a flow rate of 0.60 mL min−1 and 65 °C. The concentrations of HMF, FDCA, DFF, HMFCA, and FFCA were calculated with respect to external standards.
Definitions of HMF conversion and product yield are as follows:
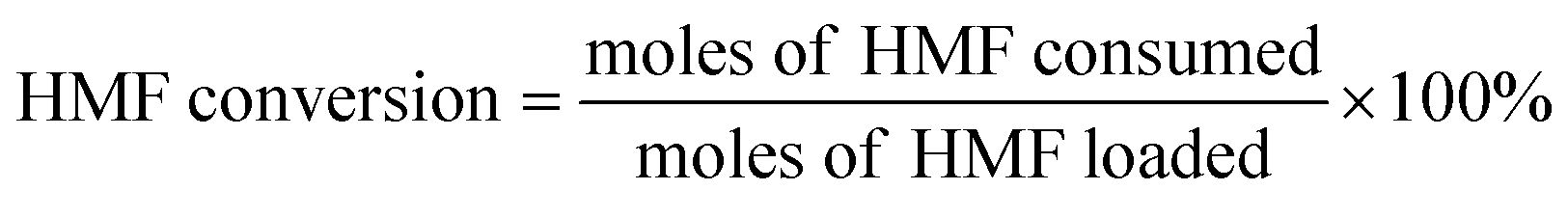 | (3) |
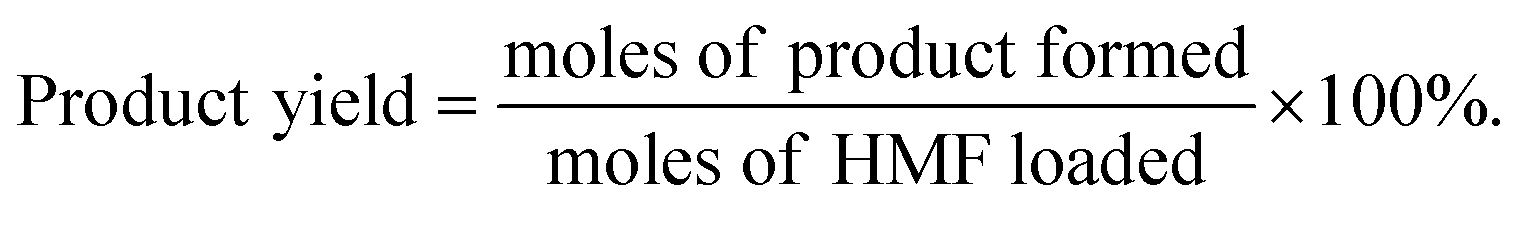 | (4) |
The definition of faradaic efficiency (FE) of product formation is as follows:
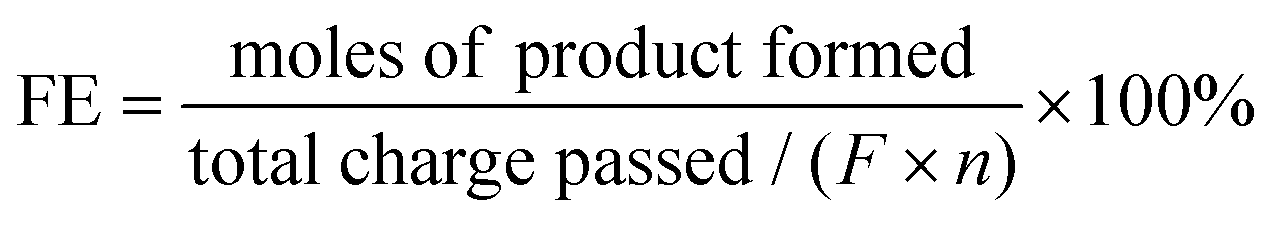 | (5) |
485 C mol−1) and n is the electron transfer per mole of product.
2.4 DFT calculations
Perdew–Burke–Ernzerhof (PBE)29 DFT calculations were performed for evaluating the adsorption behavior with the projector augmented wave (PAW)30 on the Vienna Ab initio Simulation Package (VASP)31 platform. PBE/PAW calculations were used to optimize the geometries and compute the energies of the adsorbate molecule, the surface adsorbent and the adsorption complex at a convergence energy threshold of 10−5 eV. The vacuum space was set to be 15 Å along the z direction to avoid the interaction between the two neighboring cubic periodic boxes. The Hubbard U (DFT+U) corrections were considered with a U value of 3 for the localized 3d orbitals of transition metal Co.32The adsorption energy (Eads) of the adsorbate molecule is defined as eqn (6):
| Eads = Ecomplex − Esurface − Emolecule | (6) |
3. Results and discussion
Ultrathin cobalt oxide hydrate (CoOxHy) nanosheets were synthesized by a template-assisted solvothermal method, which have a flower-like layered assembly structure (Fig. 1b–e).10 Due to the structure-directing by polymeric P123 inverse lamellar micelles,33 the as-prepared CoOxHy nanosheets were encapsulated in organic residues of 31.2 wt% according to the TGA results (Fig. S3, ESI†). Subsequently, room-temperature reductive treatments were used to fabricate defective CoOxHy-MA, CoOxHy-BH and CoOxHy-MA/BH by soaking the as-prepared CoOxHy in an ethanol solution of methylamine (MA) and/or NaBH4 (BH) and sonicating for 0.5 h. The polymeric organic residues significantly reduced after the reductive treatments by MA and/or BH, proving the simultaneous ligand exchange to expose the catalyst surface (Table 1, Fig. S3, ESI†).34| Catalyst | Organicsa (wt%) | S BET (m2 g−1) | S micro (m2 g−1) | V tot (cm3 g−1) |
V
mesod (cm3 g−1) |
V
microc (cm3 g−1) |
D
poree (nm) |
ζ (mV) | Co2+/Co3+g |
ECSAh |
|---|---|---|---|---|---|---|---|---|---|---|
| a The organic residual content was determined by TGA as shown in Fig. S3.† b The surface area (SBET) was calculated by using the Brunauer–Emmett–Teller (BET) equation. c The area of micropores (Smicro) and the micropore volume (Vmicro) were calculated by the t-plot method. d The total pore volume (Vtot) was calculated from the saturation plateau at high relative pressures. The mesopore volume (Vmeso) was equal to Vtot minus Vmicro. e The pore size (Dpore) was calculated from the adsorption branch of the isotherms using the NLDFT model. f Zeta potential ζ was measured in a dispersion of an isopropanol–water mixture. g Co2+/Co3+ ratio was calculated from the peak deconvolution results of XPS Co 2p spectra in Fig. 2d. h ECSA value was obtained from the double-layer capacitance (Cdl). | ||||||||||
| CoOxHy | 31.2 | 166 | 71 | 0.61 | 0.58 | 0.03 | 18.2 | 33 | 1.36 | 37.5 |
| CoOxHy-MA | 21.0 | 101 | 40 | 0.79 | 0.77 | 0.02 | 32.8 | 30 | 1.53 | 65.5 |
| CoOxHy-BH | 24.8 | 165 | 152 | 0.23 | 0.16 | 0.07 | 13.0 | 2 | 1.50 | 62.6 |
| CoOxHy-MA/BH | 16.0 | 147 | 130 | 0.37 | 0.31 | 0.06 | 26.8 | −7 | 1.48 | 73.5 |
Scanning transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (STEM-EDS) mappings were used to verify the morphology and elemental distribution. As shown in Fig. 1f–q, CoOxHy in the reductively-treated samples formed different assembly structures, which is in line with the different surface charges, as verified by the different zeta potentials in Table 1, but remained in the ultrathin nanosheet framework structure. In addition, the elemental mappings of Co showed an isolated point-like distribution of deep contrast depth, which indicates the etching of CoOxHy by MA and/or BH. The dissolution reactions could follow the Hard Soft Acids Bases coordination theory27 for the Co2+ and Co3+ sites in the as-prepared CoOxHy; Co2+ was more inclined to dissolute than Co3+. Due to the vulnerability of CoOxHy upon electron beam exposure, Co3O4-300 from the rapid calcination of the as-prepared CoOxHy at 300 °C for 5 min, and the catalysts after the reductive treatments of Co3O4-300 nanosheets were studied in parallel. Based on the STEM (Fig. S1†) and HRTEM images (Fig. S2†) of Co3O4-300, Co3O4-300-MA, Co3O4-300-BH, and Co3O4-300-MA/BH, MA and BH created nanodots by etching, where the lattice fringes were blurred, indicating the distortion of the lattice related to the oxygen vacancies.35 MA was inclined to dissolute nanograin edges,36 bringing in more mesopores, while BH led to more in-sheet micropores below 2 nm. These micropores should be formed during the addition of BH following hydrogen generation.28,37
The different etching effects on the porous structure by MA and BH were also evidenced by the N2 adsorption–desorption results in Fig. 2a, b, and Table 1. As shown in Fig. 2a, these catalysts all exhibited IV-type isotherms with an H1-type hysteresis loop, which confirms the accumulation of thin CoOxHy nanosheets with slit-shaped pores.10,12 After the MA treatment, the micropore volume decreased while the mesopore volume increased. It is possible that the CoOxHy nanosheets in CoOxHy-MA could reduce their size and re-assemble. The large mesopore volume of 0.77 cm3 g−1 of CoOxHy-MA can facilitate the mass transport of the reactants and products in the HMFOR reaction. In contrast, the BH treatment caused a significant increase in micropores while decreasing the mesopore volume. The MA/BH treatment superimposed the effects of MA and BH, leading to a hierarchically porous CoOxHy nanosheet assembly.
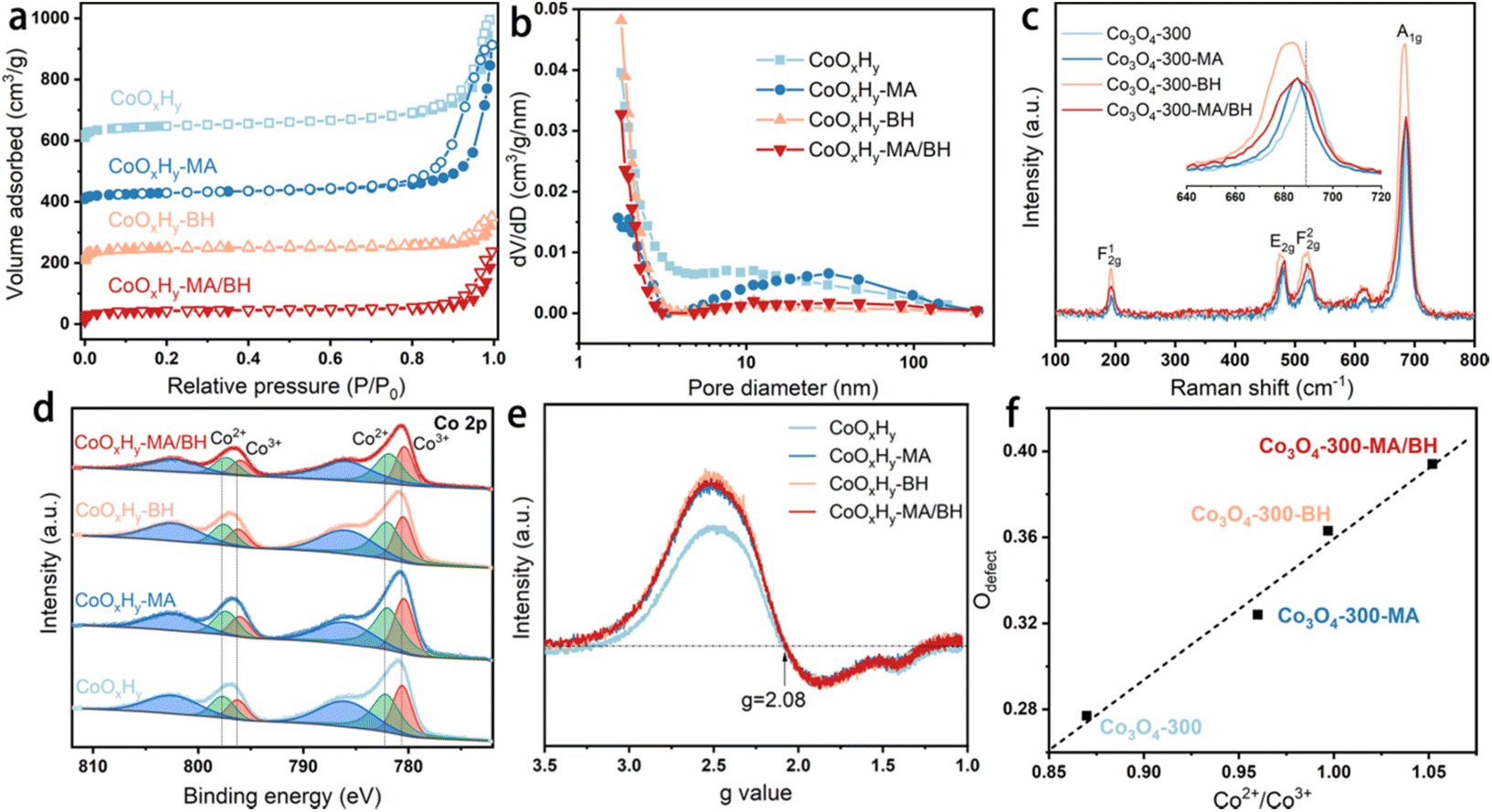 | ||
| Fig. 2 Structural characterization results of the pristine and reductively-treated CoOxHy and Co3O4-300 nanosheets. (a) N2 sorption isotherms and (b) the corresponding pore size distributions of CoOxHy, CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH; (c) Raman spectra of Co3O4-300, Co3O4-300-MA, Co3O4-300-BH, and Co3O4-300-MA/BH; (d) XPS Co 2p spectra and (e) EPR signals against the g values of CoOxHy, CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH; (f) The linear relationship between Odefect and Co2+/Co3+ ratios, calculated from the XPS Co 2p and O 1s spectra of Co3O4-300, Co3O4-300-MA, Co3O4-300-BH, and Co3O4-300-MA/BH, as shown in Fig. S6.† | ||
The crystal structure of the as-prepared CoOxHy nanosheets remained amorphous upon these reductive treatments, as confirmed by the XRD patterns (Fig. S4†). The functional groups were probed with FTIR spectroscopy (Fig. S5†). CoOxHy-MA showed the decrease of the bands at 3450 and 3345 cm−1, which were assigned to O–H, indicating the loss of surface OH after MA treatment.36 CoOxHy-BH and CoOxHy-MA/BH showed relatively significant modifications in the shapes of the C–H bending peak at 1400 cm−1 and the Co2+–O stretching peak at 580 cm−1.38 These results support the in-sheet etching of CoOxHy nanosheets by BH. The overall P123 residual-associated peaks were less intensive in these reductively-treated CoOxHy nanosheets, demonstrating the partial removal of organic residues via ligand exchange.
As for the investigation of the chemical constituents and oxygen defects, Raman spectroscopy, XPS, and EPR were performed.39 Due to the vulnerability of CoOxHy nanosheets under the 532 nm laser, Co3O4-300 nanosheets before and after the reductive treatments were measured as a reference (Fig. 2c). The blue-shifts of the A1g Raman peak induced by the reductive treatments indicate the presence of the defective structures of oxygen vacancies.39 From the high-resolution Co 2p XPS spectra in Fig. 2d, the peaks at around 782.16 and 797.73 eV can be attributed to the Co2+ 2p3/2 and 2p1/2 of CoOxHy nanosheets.9 The other two peaks at about 780.59 and 796.28 eV belong to Co3+. The binding energies of Co 2p were negatively shifted about 0.1–0.2 eV after the reductive treatments by MA and/or BH, revealing the modification in the electronic states of Co by MA and/or BH. In the O 1s XPS spectra (Fig. S6†), the O 1s peak can be deconvoluted into three characteristic peaks, which are located at 533.60, 531.21, and 529.72 eV, corresponding to adsorbed H2O, O adsorbed in the defective sites (Odefect), and lattice O,40 respectively. As shown in Fig. 2f, the percentage of Odefect can be positively related to the Co2+/Co3+ ratio, which agrees well with the previous observations.10,42,43 Part of Co2+ was induced by an oxygen vacancy defective site to balance the charge, and the oxygen vacancy facilitated the reconstruction of a higher valence state of Co species such as CoOOH to improve the HMFOR performance.42,43 The Co2+/Co3+ ratios were 1.53, 1.50, and 1.48 for CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH, respectively, higher than 1.36 for the pristine CoOxHy. The EPR signals were contributed by the unpaired electrons from both Co2+ species and oxygen vacancies,10,39 and the EPR lineshape of the transition metal is typically asymmetric arising from a mixture of absorption and dispersion effects.44,45 A g-value of 2.08 was obtained (Fig. 2e) from the extreme points of the magnetic field strength diagram in Fig. S7.† The same g-values of these CoOxHy nanosheets evidenced similar signal sources from both Co2+ species and oxygen vacancies. However, the reductively-treated CoOxHy nanosheets exhibited stronger EPR signals compared to the pristine CoOxHy, confirming the higher Co2+/Co3+ ratios and the correspondingly higher surface oxygen vacancies in these samples.10,43 The above characterization results demonstrated that the treatment by MA and/or BH can create high porosity and abundant oxygen vacancies in CoOxHy nanosheets, which provides a promising opportunity for promoting the electrocatalytic activity. Additionally, the residual surface N contents were quite low, about 2 at% according to the N 1s XPS spectra in Fig. S8,† implying that the electron-donating effect from NH2 coordination should not be a dominating factor for modifying the electronic properties of MA and MA/BH treated catalysts.
The electrocatalytic performance of the as-prepared and reductively-treated CoOxHy nanosheets for HMFOR was evaluated in 1 M KOH containing 5 mM HMF by LSV. As shown by the different HMFOR LSV plots in Fig. S10,† the catalyst performance was influenced by the catalyst loading. Therefore the catalyst loading on the glassy carbon electrode was controlled to be 0.167 mg cm−2 for the convenience of comparison. In the results presented in Fig. 3a–c, the pristine CoOxHy showed overpotentials of 338 and 350 mV to reach the current density of 10 mA cm−2 for HMFOR and OER activity, respectively. The MA and/or BH reductively-treated CoOxHy nanosheets exhibited better electrocatalytic oxidation performance, with more significant improvement in HMFOR than the OER. A similar phenomenon was also observed when comparing the electrooxidation activities of Co3O4-300 and CoOxHy; the more significantly improved HMFOR performance was ascribed to the increased accessible active sites and oxygen vacancies.10 At 10 mA cm−2, the overpotentials of CoOxHy-MA and CoOxHy-MA/BH were only 284 and 279 mV for HMFOR, respectively, much lower than those of the reported CoOOH thin film13 and the pure Co LDH catalyst.22 Moreover, the Tafel slopes of all CoOxHy nanosheets in HMFOR were about 90 mV dec−1, larger than the values in the OER, indicating the more complex electron transfer kinetics for HMFOR.10 The Tafel slopes agree well with the reported Co-based electrocatalysts with the partial formation of Co4+ as the catalytically active species.9 The exchange current densities of CoOxHy-MA (6.7 × 10−3 mA cm−2), CoOxHy-BH (4.6 × 10−3 mA cm−2), and CoOxHy-MA/BH (9.6 × 10−3 mA cm−2) were higher than that of the pristine CoOxHy (1.8 × 10−3 mA cm−2), implying the higher intrinsic electron transfer rates at equilibrium23,40 with these reductively-treated CoOxHy nanosheets. MA and/or BH created abundant defective active sites by ligand exchange and etching, which governed the superior HMFOR reactivity. As shown in Fig. 4, by increasing the MA/Co molar ratio from 0 to 20, the increased extent of ligand exchange and etching resulted in steadily improved HMFOR performance.
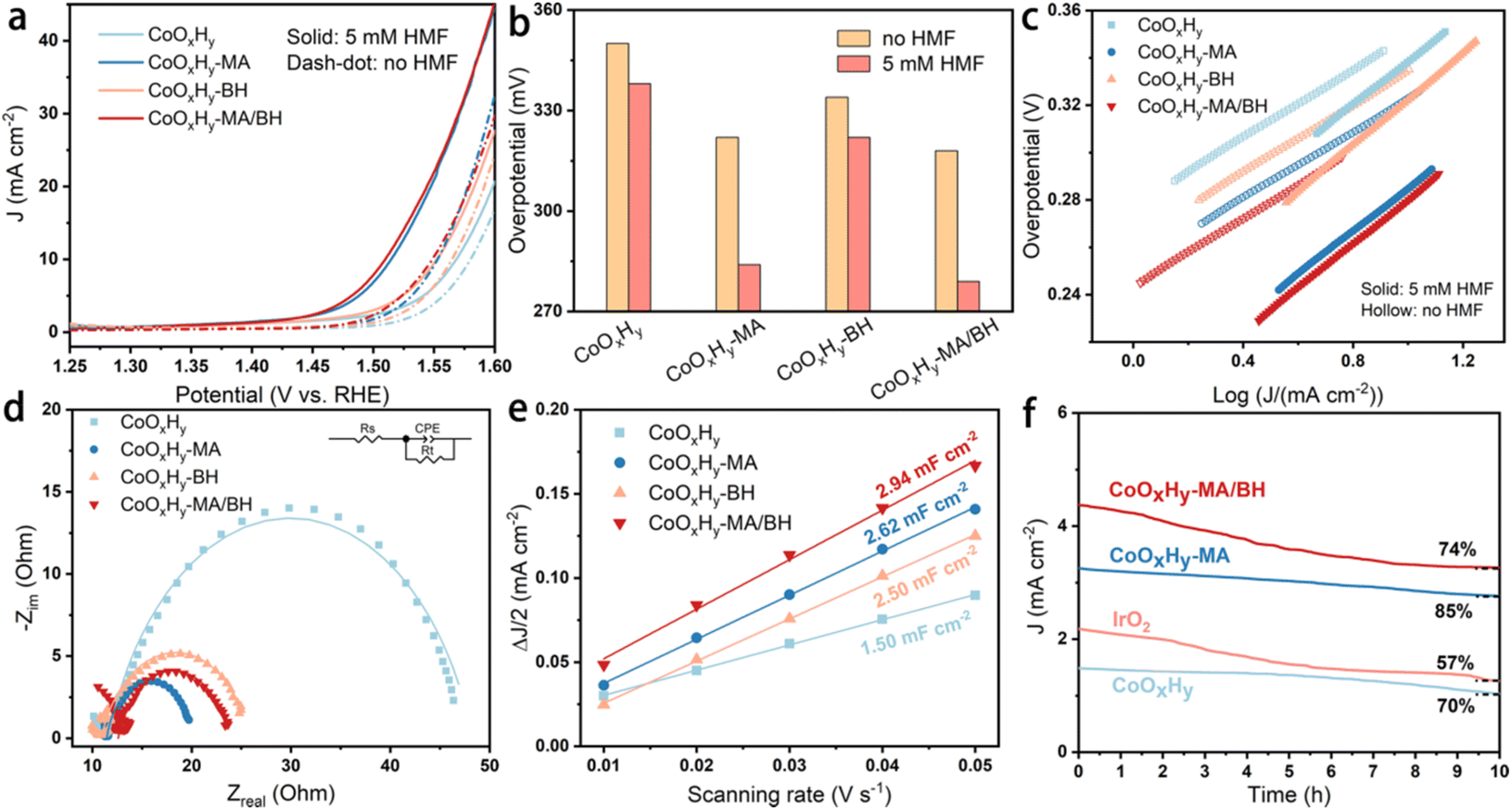 | ||
| Fig. 3 Electrocatalytic performance of CoOxHy, CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH. (a) Polarization curves at a scanning rate of 5 mV s−1 in 1 M KOH with and without 5 mM HMF, (b) overpotentials at a current density of 10 mA cm−2, and (c) the corresponding Tafel plots; (d) EIS Nyquist plots at 1.62 V vs. RHE, (e) linear fitting of half of the difference between the oxidation and reduction current densities against the scanning rate, derived from CVs in Fig. S9,† and (f) i–t tests in 1 M KOH at 1.52 V vs. RHE. | ||
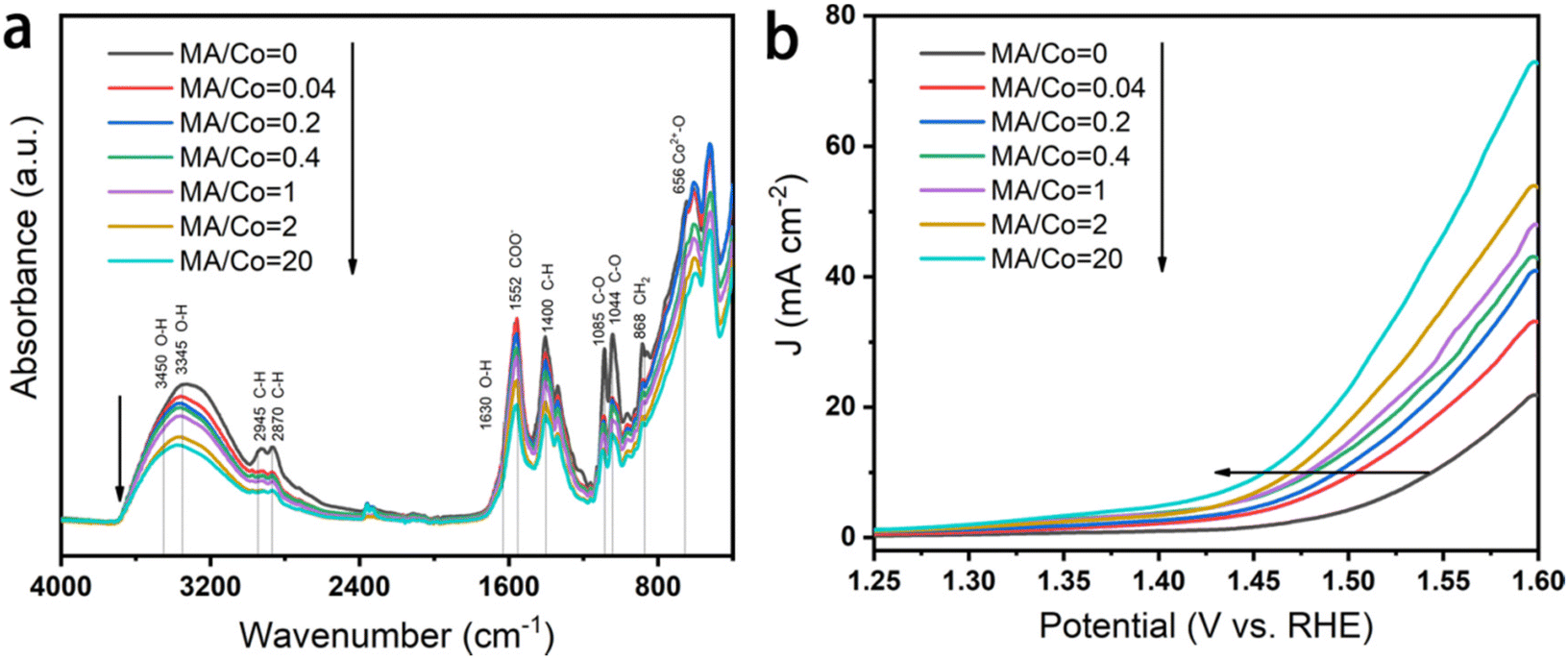 | ||
| Fig. 4 Influence of the MA/Co molar ratio on the electrocatalytic performance of CoOxHy. (a) FTIR spectra and (b) LSVs in 1 M KOH solution containing 10 mM HMF at a scanning rate of 10 mV s−1. | ||
The origin of the remarkable HMFOR performance was further investigated by electrochemical impedance spectra (EIS) and the electrochemical surface area (ECSA). As shown in Fig. 3d and Table S1,† the charge transfer resistances (Rct) of CoOxHy-MA (8.1 Ω), CoOxHy-BH (15.1 Ω), and CoOxHy-MA/BH (11.1 Ω) were smaller than that of CoOxHy (36.7 Ω), implying more rapid electronic transport in these reductively-treated CoOxHy nanosheets. The ECSA obtained by double-layer capacitances (Cdl) showed the Cdl values of CoOxHy (1.50 mF cm−2), CoOxHy-MA (2.62 mF cm−2), CoOxHy-BH (2.50 mF cm−2), and CoOxHy-MA/BH (2.94 mF cm−2), indicating higher densities of exposed catalytically active sites like oxygen vacancy-induced active sites26 after these reductive treatments. At an overpotential of 300 mV, the CoOxHy, CoOxHy-MA, CoOxHy-BH, and CoOxHy-MA/BH displayed current densities of 0.10, 0.22, 0.09, and 0.21 mA cm−2ECSA for HMFOR (Fig. S11†), respectively. These results indicate that oxygen vacancies increase the intrinsic HMFOR activities, and pore structures influence the catalytic performance by changing the mass transport. With the abundant oxygen vacancies and large mesopore volume in CoOxHy-MA and CoOxHy-MA/BH, the mass transport of the reactants and products to the oxygen vacancy-induced active sites can be increased and the normalized HMFOR activity was enhanced. However, in CoOxHy-BH, although with abundant oxygen vacancies, the normalized HMFOR activity was even slightly lower than the pristine CoOxHy, indicating that micropores mainly induced by BH can significantly influence the mass transport during the HMFOR process. The electrocatalytic stability was evaluated by chronoamperometry at a constant potential of 1.52 V vs. RHE. The deactivation commonly occurs at a high anodic potential due to structural changes or detachment of redox-active species.41 Nevertheless, after a 10 h chronoamperometric test, the current density decreased by 15% and 24% for CoOxHy-MA and CoOxHy-MA/BH, respectively, indicating the limited change and decent stability of these defective CoOxHy nanosheets (Fig. 3f).
HMF electrolysis was also conducted in 1 M KOH containing 5 mM HMF at a constant potential of 1.52 V vs. RHE. The degradation of HMF to humin-type products in 1 M KOH is shown in Fig. S12.† Excessive degradation of HMF can be inhibited to a certain extent due to the high electrochemical reactivity under alkaline conditions.13 Here, the thermodegradation of HMF was about 26% after 8 h. The catalytic activities and faradaic efficiencies (FEs) of the as-prepared CoOxHy, CoOxHy-MA, and CoOxHy-MA/BH towards electrochemical HMF oxidation were investigated and compared (Fig. 5). At 1.52 V vs. RHE, full conversion of HMF was realized in about 300 min for the pristine CoOxHy, while CoOxHy-MA and CoOxHy-MA/BH required less than 200 min. The significantly enhanced activities for HMFOR over CoOxHy-MA and CoOxHy-MA/BH again demonstrate the vital role of the oxygen vacancies and the mesopores that provide high accessibility to reactants and products. The FE and yield for FDCA formation were up to 39% and 30% for the pristine CoOxHy, respectively, still much higher than those of the CoOOH thin film13 and the pure Co LDH catalyst,22 owing to its ultrathin nanosheet structure. However, compared to the reductively-treated CoOxHy nanosheets in CoOxHy-MA and CoOxHy-MA/BH, the selectivity for FDCA was low due to the longer duration with more side degradation reactions of HMF and the completing oxygen evolution reaction. CoOxHy-MA can optimally achieve 98% FDCA yield and a FE of 83%, indicating the selective oxidation of HMF to FDCA with only minor losses due to decomposition and the OER. The room-temperature reductive treatment with MA can induce abundant oxygen vacancies and high mesoporosity in CoOxHy nanosheets through ligand exchange and etching, which offer highly accessible active sites for HMFOR. Varying the applied potential would decrease the FDCA yield and FE due to the increased side reactions of HMF degradation at 1.47 V or the OER at 1.57 V (Fig. S13†). On the other hand, with the additional in-sheet micropores in CoOxHy-MA/BH, the product distributions were distinct from those of CoOxHy-MA, affording FDCA and FFCA yields of 65% and 18%, respectively, at 99% conversion of HMF. It is thus speculated that these in-sheet micropores in CoOxHy-MA/BH may change the diffusion behavior of the intermediate product FFCA. An apparent proportion of FFCA could penetrate these micropores and desorb from the catalyst surface, avoiding being further oxidized.
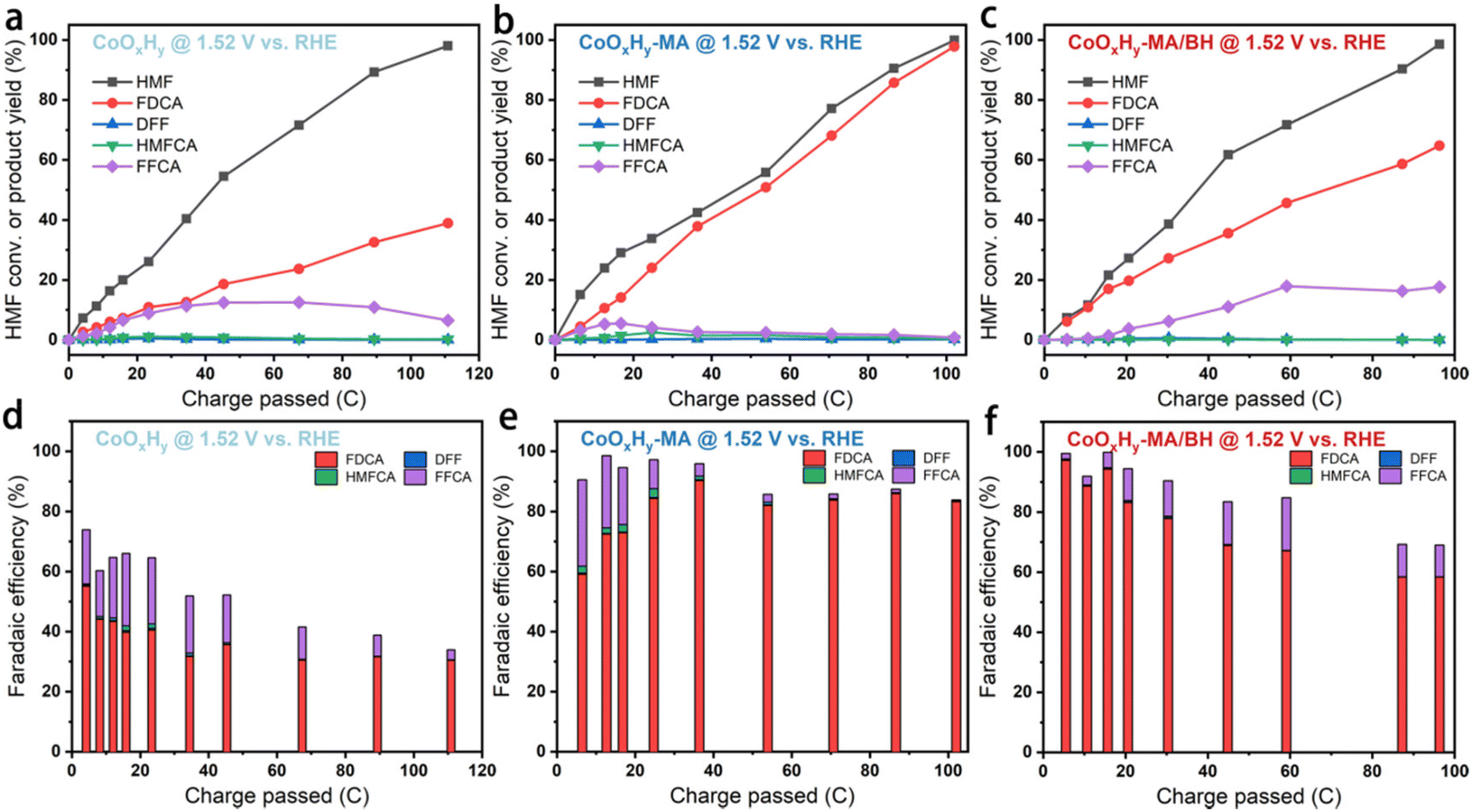 | ||
| Fig. 5 Results of HMFOR using CoOxHy, CoOxHy-MA, and CoOxHy-MA/BH at a constant potential of 1.52 V vs. RHE in 1 M KOH containing 5 mM HMF, after passing various amounts of charges. (a, b and c) The conversion of HMF and the yields of oxidation products, and (d, e and f) the corresponding faradaic efficiencies of (a and d) CoOxHy, (b and e) CoOxHy-MA, and (c and f) CoOxHy-MA/BH, respectively. | ||
Furthermore, DFT calculations were performed to establish a basic understanding of the HMFOR process over CoOxHy nanosheets and clarify the nature of the HMFOR performance enhancement by the MA and/or BH treatments. Since the adsorption of HMF on the active sites has been regarded as a rate-determining step of HMFOR,8,14,46 the adsorption free energy changes (ΔE) of HMF adsorbed on the catalyst surface with different end groups (aldehyde –CHO group and the alcohol hydroxyl –C–OH group) were calculated (Fig. 6 and S12†). As shown in Fig. 6b and e, HMF was adsorbed on the CoOxHy nanosheets in two adsorption modes via the –CHO and –C–OH groups with a ΔE of −2.03 and −1.63 eV, respectively. The introduction of an oxygen vacancy (VO) can evidently reduce the energetics to −3.21 and −2.92 eV, respectively, for the –CHO and –C–OH adsorption configurations (Fig. 6c and f). With the co-existence of MA in the neighbouring position, the energetics changed from −1.98 to −3.02 eV and from −1.66 to −2.99 eV after the introduction of VO, for the adsorption modes of –CHO and –C–OH, respectively. These results reveal that the oxygen vacancies directly promote the adsorption and activation of HMF, especially for the –CHO adsorption mode, leading to the facilitated HMFOR process. The previously proposed functions of VO like promoting OH− adsorption,11 increasing electronic conductivity,25 and forming a higher valence state of Co species,42,43 may still be present in the reductively-treated CoOxHy nanosheets and contribute to the improved intrinsic HMFOR activity. Meanwhile, the MA molecule appears to have little impact on the adsorption energies of HMF. Given the small amount of MA indicated by the XPS data, the coordination effect of residual MA in CoOxHy-MA and CoOxHy-MA/BH can therefore be neglected. The above computation results also show that the adsorption energies via –CHO were larger than via –C–OH. These energy differences indicate that the HMFOR preferentially follows the reaction pathway of forming HMFCA as an intermediate instead of DFF, as shown in Fig. 6a, in accordance with the HPLC results that much more HMFCA was formed than DFF during the electrolysis process. The main HMFCA pathway on the CoOxHy nanosheets agrees with most of the reported cobalt-based HMFOR electrocatalysts under strong alkaline conditions.11–14,25
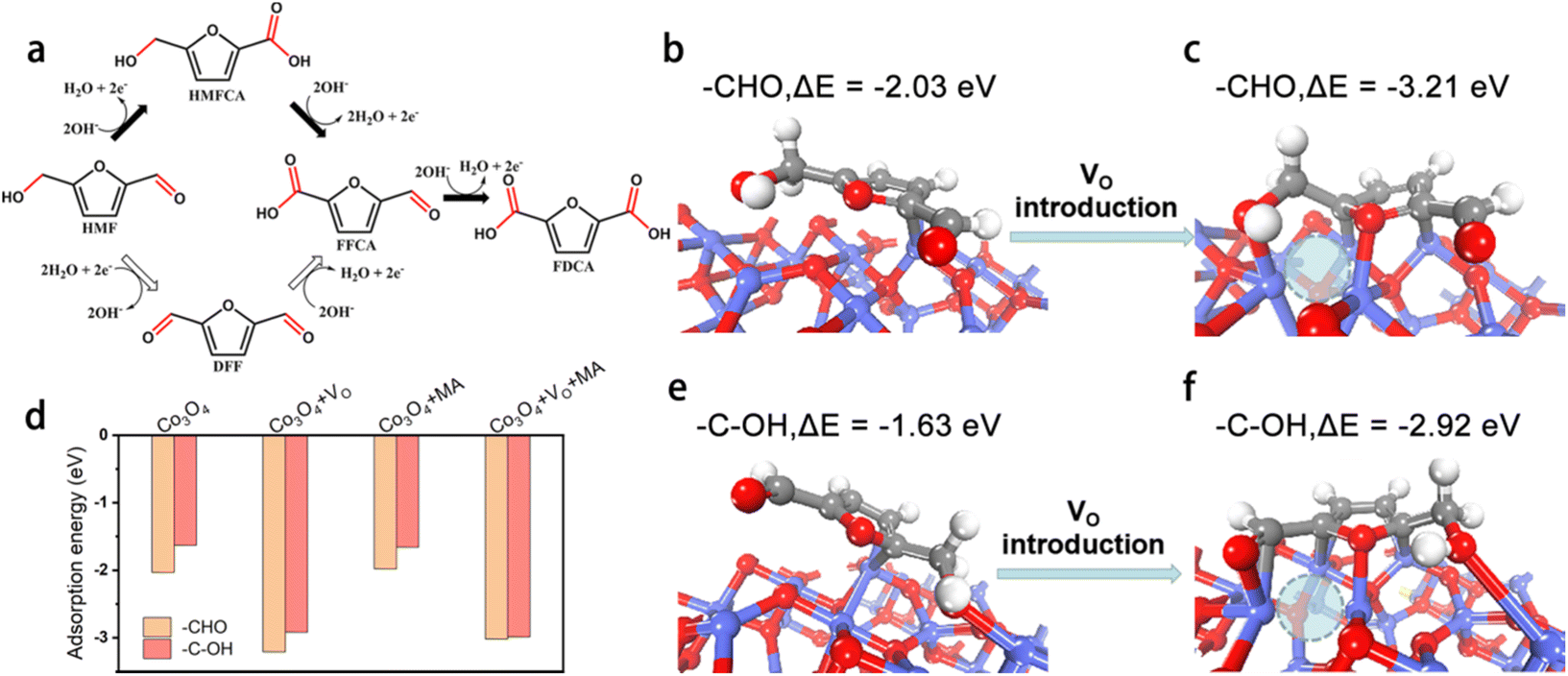 | ||
| Fig. 6 Effect of surface oxygen vacancy (VO) sites and chemisorbed methylamine (MA) on HMFOR. (a) Two reaction pathways of HMFOR; HMF adsorption modes and energies on the Co3O4 (3 1 1) surface (b, e) with no VO and (c, f) in the presence of VO. VO is denoted with dashed circles; (d) adsorption energies of HMF on various surface sites, based on the adsorption configurations in Fig. S14.† | ||
4. Conclusions
In summary, we have successfully fabricated defective CoOxHy nanosheets with abundant oxygen vacancies and high porosity as an efficient HMFOR electrocatalyst based on a room-temperature reductive treatment with methylamine (MA) and/or NaBH4 (BH). These defective CoOxHy nanosheets in CoOxHy-MA and CoOxHy-MA/BH show remarkable activity with small overpotentials of 284 and 279 mV at a current density of 10 mA cm−2 in 1 M KOH containing 5 mM HMF, respectively, nearly 60 mV lower than the that of the precursor CoOxHy. The improved catalytic performance should result from the larger electrochemically active surface areas and the faster electron transfer rates, owing to the enriched oxygen vacancies created by etching. The DFT calculations reveal the facilitated adsorption and activation with the incorporation of oxygen vacancies, preferentially through the aldehyde group. For long-term HMF electrolysis, 98% FDCA yield and a faradaic efficiency of 83% could be obtained on CoOxHy-MA with a large mesoporosity facilitating mass transport, whereas the additional in-sheet micropores in CoOxHy-MA/BH regulated the selectivity more towards the intermediate product FFCA. This facile reductive treatment with MA and/or BH can be a promising post-treatment strategy for developing cost-effective catalytic systems possessing tunable oxygen vacancies and porosity for electrochemical HMFOR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong Engineering Technology Research Center for Hydrogen Energy and Fuel Cells, the Shenzhen Science and Technology Innovation Commission (JCYJ20220818100212027), the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002), and the Pico Center at SUSTech. R. Z. acknowledges the Guangdong Provincial Department of Education Innovation Project (2022KQNCX056) and the Guangdong Basic and Applied Basic Research Foundation (2022A1515110354). Q. W. acknowledges the Shenzhen Science and Technology Innovation Committee Foundation (JCYJ20190809142019365). L. D. acknowledges the Outstanding Youth Project of the Guangdong Provincial Natural Science Foundation (2022B1515020020) and the Guangdong Basic and Applied Basic Research Foundation (2022B1515120079). S. Y. acknowledges the National Natural Science Foundation of China (22250710133).References
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- D. Yan, C. Mebrahtu, S. Wang and R. Palkovits, Angew. Chem., Int. Ed., 2023, 62, e202214333 CrossRef CAS PubMed.
- O. Simoska, Z. Rhodes, S. Weliwatte, J. R. Cabrera-Pardo, E. M. Gaffney, K. Lim and S. D. Minteer, ChemSusChem, 2021, 14, 1674–1686 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- C. Chen, L. Wang, B. Zhu, Z. Zhou, S. I. El-Hout, J. Yang and J. Zhang, J. Energy Chem., 2021, 54, 528–554 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, C. Tian and H. Fu, Small Struct., 2021, 2, 2100095 CrossRef CAS.
- C. Tang, Y. Zheng, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- R. Zhong, Q. Wang, L. Du, Y. Pu, S. Ye, M. Gu, Z. C. Zhang and L. Huang, Appl. Surf. Sci., 2022, 584, 152553 CrossRef CAS.
- Y. Lu, T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2021, 2107185 Search PubMed.
- C. Wang, H. J. Bongard, M. Yu and F. Schuth, ChemSusChem, 2021, 14, 5199–5206 CrossRef CAS PubMed.
- B. J. Taitt, D. Nam and K. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- J. Chen, Y. Wang, M. Zhou and Y. Li, Chem. Sci., 2022, 13, 4647–4653 RSC.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- J. Weidner, S. Barwe, K. Sliozberg, S. Piontek, J. Masa, U. P. Apfel and W. Schuhmann, Beilstein J. Org. Chem., 2018, 14, 1436–1445 CrossRef PubMed.
- R. Luo, Y. Li, L. Xing, N. Wang, R. Zhong, Z. Qian, C. Du, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 CrossRef CAS.
- X. Deng, M. Li, Y. Fan, L. Wang, X. Fu and J. Luo, Appl. Catal., B, 2020, 278, 119339 CrossRef CAS.
- R. Zhang, S. Jiang, Y. Rao, S. Chen, Q. Yue and Y. Kang, Green Chem., 2021, 23, 2525–2530 RSC.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- H. Wang, Y. Zhou and S. Tao, Appl. Catal., B, 2022, 315, 121588 CrossRef CAS.
- L. Gao, Y. Bao, S. Gan, Z. Sun, Z. Song, D. Han, F. Li and L. Niu, ChemSusChem, 2018, 11, 2547–2553 CrossRef CAS PubMed.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- L. Yu, P. Pellechia and M. A. Matthews, Int. J. Hydrogen Energy, 2014, 39, 442–448 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- L. Gong, D. Zhang, C. Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- Y. Dou, T. Liao, Z. Ma, D. Tian, Q. Liu, F. Xiao, Z. Sun, J. H. Kim and S. X. Dou, Nano Energy, 2016, 30, 267–275 CrossRef CAS.
- N. Naresh, F. G. S. Wasim, B. P. Ladewig and M. Neergat, J. Mater. Chem. A, 2013, 1, 8553–8559 RSC.
- R. Gao, Z. Li, X. Zhang, J. Zhang, Z. Hu and X. Liu, ACS Catal., 2015, 6, 400–406 CrossRef.
- Y. H. Lee, S. Park, K. G. Lee, M. Y. Lee, K. H. Cho, S. J. Kim and K. T. Nam, ChemCatChem, 2019, 11, 1665–1672 CrossRef CAS.
- M. Liao, G. Zeng, T. Luo, Z. Jin, Y. Wang, X. Kou and D. Xiao, Electrochim. Acta, 2016, 194, 59–66 CrossRef CAS.
- Y. Cai, J. Xu, Y. Guo and J. Liu, ACS Catal., 2019, 9, 2558–2567 CrossRef CAS.
- A. Badreldin, A. E. Abusrafa and A. Abdel-Wahab, ChemSusChem, 2021, 14, 10–32 CrossRef CAS PubMed.
- S. Bae, J. Kim, H. Randriamahazaka, S. Moon, J. Park and I. Oh, Adv. Energy Mater., 2017, 7, 1601492 CrossRef.
- A. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Perez-Ramírez, Nat. Catal., 2022, 5, 854–866 CrossRef.
- M. Sun, Y. Wang, C. Sun, Y. Qi, J. Cheng, Y. Song and L. Zhang, Chin. Chem. Lett., 2022, 33, 385–389 CrossRef CAS.
- Z. Xiao, Y. Huang, C. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- F. Dyson, Phys. Rev., 1955, 98, 349–359 CrossRef CAS.
- R. Webb, Phys. Rev., 1967, 158, 225–233 CrossRef CAS.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00588g |
| This journal is © The Royal Society of Chemistry 2023 |