
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of alkaline 5-HMF electrolytes via Cannizzaro reaction for the electrochemical oxidation to FDCA†
Moritz Lukas
Krebs
 a,
Alexander
Bodach
a,
Changlong
Wang
*b and
Ferdi
Schüth
*a
a,
Alexander
Bodach
a,
Changlong
Wang
*b and
Ferdi
Schüth
*a
aDepartment of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: schueth@kofo.mpg.de
bInstitute of Circular Economy, Faculty of Materials and Manufacturing, Beijing University of Technology, Beijing 100124, PR China. E-mail: changlongwang1987@gmail.com
First published on 8th February 2023
Abstract
The electrochemical oxidation of 5-(hydroxymethyl)furfural (HMF) to 2,5-furandicarboxylic acid (FDCA), a monomer for biopolymer production, caught attention as a route to renewable materials. However, this process is mostly performed in alkaline media, which causes HMF to degrade into humins. In this study, we demonstrate that alkaline degradation of HMF yielded 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) and dihydroxymethylfuran (DHMF), which are both stable in alkaline media, and both of which can be electrooxidized to FDCA. Furthermore, the stability of the Cannizzaro products allowed the “indirect” electrooxidation of HMF to FDCA at unprecedentedly high concentrations of substrate and base, leading to current densities on the order of technical processes (∼1 A cm−2) and increased space–time-yields.
Introduction
Substituting oil-derived products by renewable feedstocks is one of the main challenges associated with reducing CO2 emissions and environmental pollution.1–3 5-Hydroxymethylfurfural (HMF) could develop into one of the most important biomass-derived building blocks for the synthesis of plastics, chemicals, and fuels.4–7 Among its derivatives, 2,5-furandicarboxylic acid (FDCA) is of special interest since it can be used as a monomer to synthesize biopolymers, including poly(ethylene 2,5-furandicarboxylate) (PEF).8,9 PEF is widely considered a suitable replacement for polyethylene terephthalate (PET), which is produced on a 70 Mt a−1 scale with a yearly increasing demand.10–12 Currently, HMF is oxidized to FDCA in alkaline solutions (pH ≥ 13) at elevated oxygen pressures and temperatures, often using noble metal catalysts.13,14 Electrochemical oxidation of HMF is a promising alternative approach as it can be done continuously, at ambient pressure and temperature, while also avoiding the use of hazardous additives or oxidants.1 Further, electrochemical HMF oxidation is typically coupled with electrocatalytic hydrogen evolution reaction (HER) on the cathode side.14–17 Thus, green hydrogen can be produced efficiently while producing a value-added product in the anode compartment, replacing the kinetically demanding and non-value-adding oxygen evolution reaction (OER).18–23 The electrochemical oxidation of HMF to FDCA is thought of as a three-step reaction involving two different functional groups, an alcohol and an aldehyde (Fig. 1a).24,25 To date, mainly transition metal electrodes, especially Ni-based materials, have been reported to be active and selective for the oxidation of HMF to FDCA in alkaline media.1,15,24–27 Based on a Web of Science search, using the keywords “electrochemical HMF oxidation”, more than 120 papers were published in the last 3-years alone.28 To the best of our knowledge, in alkaline electrooxidation of HMF, the well-known fact that HMF rapidly degrades in alkaline solution on time scales much shorter than those of the electrooxidation itself, is completely ignored.24,27,29,30 This is probably due to the fact that high yields of FDCA are typically obtained in bench set-ups, and thus HMF stability is not considered problematic. Only Huber et al. mentioned as a side remark that decomposition products of HMF can be oxidized to FDCA, without giving any details.31 This is practically, in fact, very important, since HMF readily decomposes into humins, polymeric species of HMF, at pH ≥ 12, which cannot be oxidized to FDCA anymore and are thus lost.26,32–38 The formation of humins, reduces the yields of FDCA, and significantly hampers industrial application due to additional challenges in separation and purification.1 Lowering the pH increases the stability of HMF, however, environments with higher OH− concentrations accelerate the electrochemical oxidation due to improved ad- and desorption of HMF and FDCA, respectively.35,36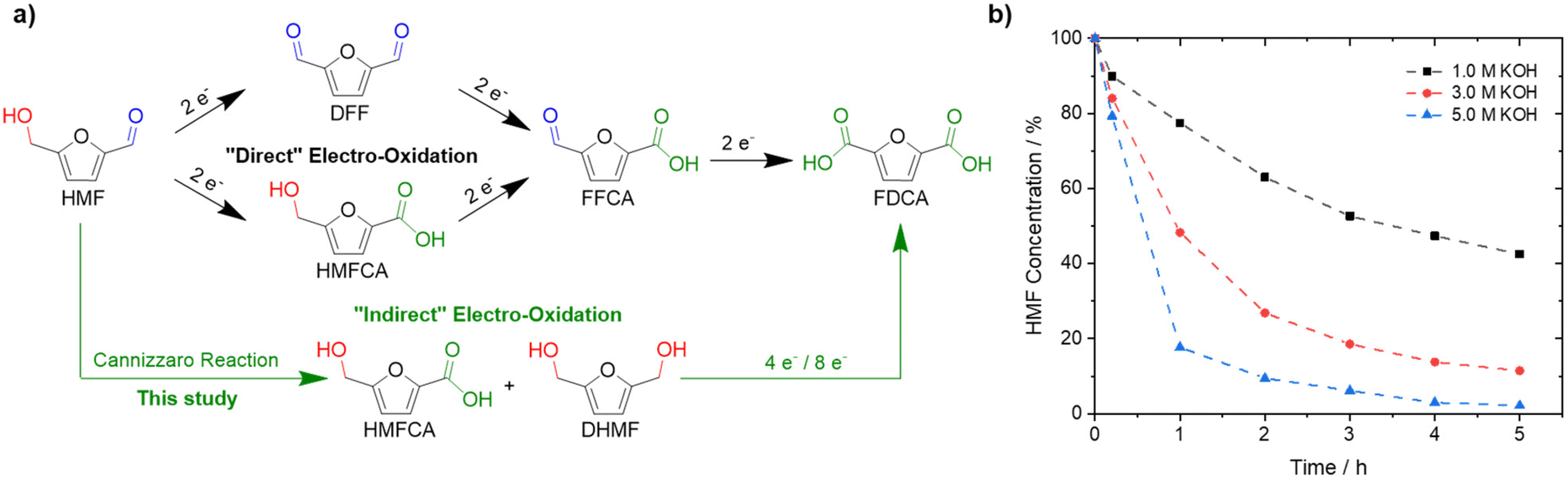 | ||
| Fig. 1 Reaction scheme of electrochemical HMF oxidation in alkaline media. (b) Degradation of a 0.05 M HMF solution in the presence of different KOH concentrations at ambient conditions. 100% is based on the initial mass of HMF and assumed to be the starting point at t = 0 h. Dashed lines are used as a guide to the eyes. | ||
If the high yields under alkaline conditions can be rationalized, further improvement based on this knowledge appears possible. Recently, it was demonstrated that electrochemical furfural oxidation competes with the solution phase Cannizzaro reaction, which is the base-mediated disproportionation of aldehydes to their carboxylic acids and alcohols.39,40 Similarly, we suggest the Cannizzaro reaction as the decisive degradation step for HMF in alkaline media,41 with further implications for process optimization. The products of this reaction, dihydroxymethylfuran (DHMF) and 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), can be electrooxidized to FDCA as well and allow almost quantitative conversion of the starting HMF. In highly alkaline media, HMF is selectively transformed to the respective Cannizzaro products (Fig. 1a), instead of humin formation, to yield a stable electrolyte under strongly alkaline conditions (pH ∼ 14). Both substrates can then be oxidized to FDCA. This led us to investigate the effect of KOH and HMF concentration on the formation of the Cannizzaro products and humin formation, followed by the electrochemical oxidation of the most promising electrolyte obtained. “Indirect” electrooxidation of HMF at different initial concentrations of HMF and KOH is thus possible at high substrate and base concentrations in spite of the instability of the HMF.
Results and discussion
To demonstrate the instability of HMF, a 50 mM HMF solution was stirred in the presence of different KOH concentrations at ambient conditions. After just 1 h, a loss of about 20% of the initial HMF concentration was observed (Fig. 1b). This result is in stark contrast to the yields and FE close to 100%, which are often reported for the alkaline electrochemical oxidation of HMF to FDCA at time scales of ∼1 h.24,27,29–31 To identify formed products during alkaline HMF decomposition, we employed quantitative NMR spectroscopy (qNMR), measured in H2O using a water suppression sequence and DMSO as an internal standard (for further information, see the ESI†). Here, it is seen that HMF is partly converted to the Cannizzaro products DHMF and HMFCA (Fig. S1†). The formed Cannizzaro products never closed the carbon balance, which indicates the formation of other products during the decomposition not detectable via NMR measurements (likely humins). The Cannizzaro reaction is a base catalyzed reaction.40 As a result, the yield of the Cannizzaro products, as shown in Fig. 2a, does significantly increase with the base concentration. Consequently, lower base concentration promotes humin formation, indicated by a lower carbon balance. After stirring a 50 mM HMF solution in 1 M KOH for 20 h, less than 30% of the original carbon amount could be detected in qNMR, compared to the ∼55% obtained in 5 M KOH (Fig. 2a). In addition, increasing the HMF concentration also has a positive impact on the yield of Cannizzaro products. This effect is saturated for HMF concentrations higher than 0.3–0.4 M in 3 M and 5 M KOH, with a carbon balance of ∼60% and ∼70%, respectively. A further increase in carbon balance can be achieved by performing the reaction at 0 °C. Yields of 80–90%, with an average of about ∼85% conversion were detected for concentrations of 1 M HMF and 5 M KOH. Time resolved measurements reveal that ∼90% of the initial HMF is already transformed during the first 3 h (Fig. S2†). At elevated temperatures, the carbon balance decreased considerably, again suggesting increased humin formation. Overall, these results show that lower KOH concentrations increase the stability of HMF in basic solution, but favor decomposition into humins instead of Cannizzaro products. Higher KOH concentrations do result in rapid decomposition of HMF but a more selective transformation into the Cannizzaro products DHMF and HMFCA. The obtained mixture of the Cannizzaro products is stable in strongly alkaline media (5 M KOH) for several days and therefore allows for a much better storage lifetime (Fig. 2b). The resulting solution is also stable at elevated temperatures (8 h at 80 °C), which is industrially relevant.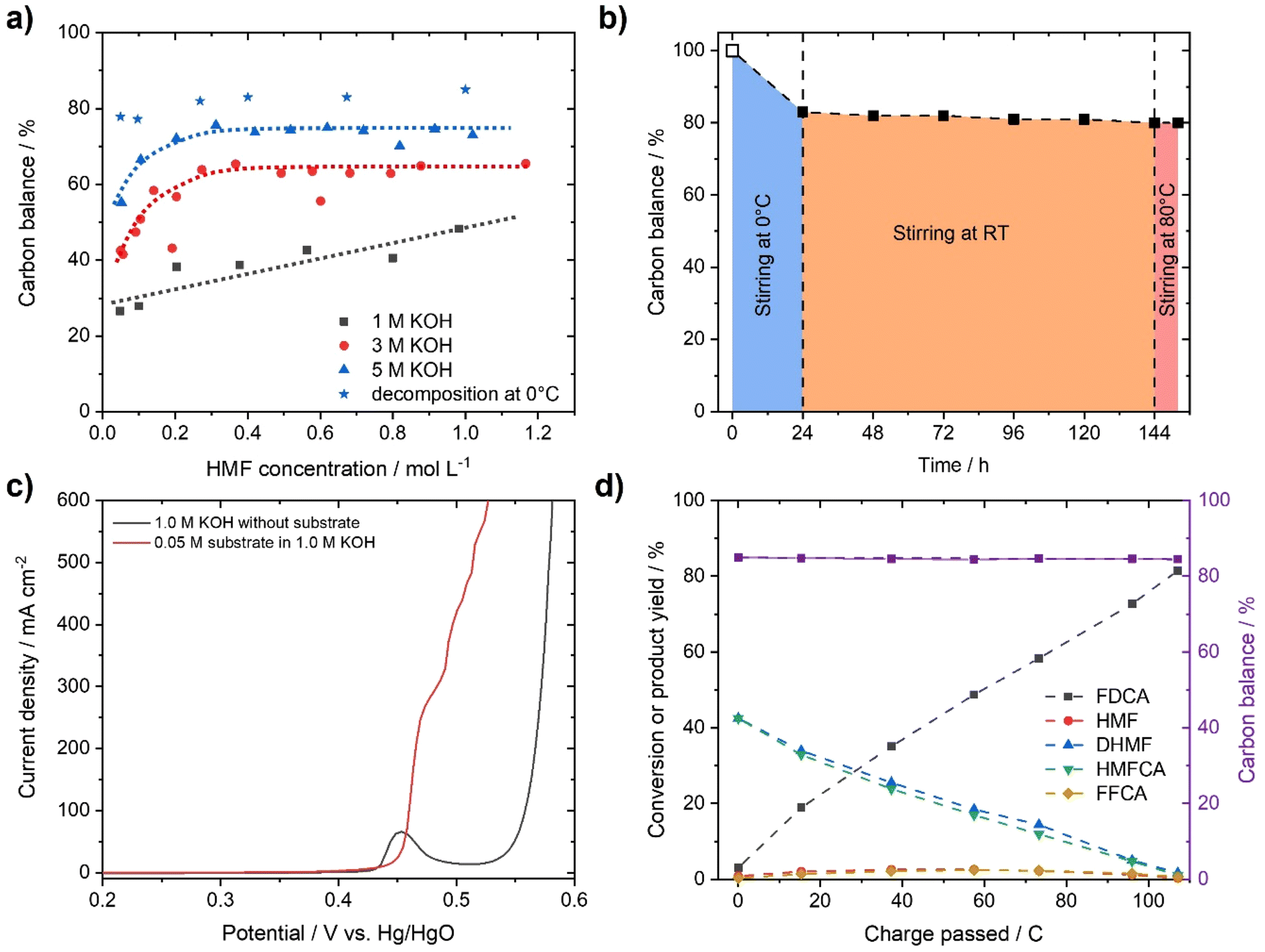 | ||
| Fig. 2 HMF decomposition at different concentrations of HMF and KOH, and temperatures. Results are measured after 20 h. The determined carbon balance is based on the sum of HMF, DHMF and HMFCA present in the solution. Dashed lines are used as a guide to the eyes. (b) Stability measurement of a 1 M HMF solution in 5 M KOH. Decomposition of HMF to the Cannizzaro products, DHMF and HMFCA, was performed at 0 °C during the first 24 h. After 120 h of stirring at RT, temperature stability was evaluated by stirring the solution for 8 h at 80 °C. Quantification was performed using qNMR with DMSO as an internal standard. 100% is based on the initial mass of HMF and assumed to be the starting point at t = 0 h. (c) LSV curves measured with and without 0.05 M of the resulting Cannizzaro products in 1 M KOH after decomposition (scan rate: 5 mV s−1). Potentials are corrected for the uncompensated solution resistance. (d) Reaction intermediates monitored for the oxidation of a 0.05 M solution of the Cannizzaro products (based on initial HMF input) in 1 M KOH during constant potential electrolysis (0.50 V vs. Hg/HgO) for 15 min. | ||
The stability of the Cannizzaro products in alkaline media is of importance since it would allow the oxidation of HMF equivalents at elevated substrate and KOH concentrations, and therefore reducing liquid waste while it might also rationalize the high yields and FE in alkaline HMF oxidation.15,24,27,31 Partial conversion of HMF via the Cannizzaro reaction stabilizes the electrolyte during electrochemical oxidation in alkaline media. To prove that the Cannizzaro products are indeed oxidized to FDCA via the “indirect” oxidation pathway (Fig. 1a), including the initial conversion of HMF via the Cannizzaro reaction and subsequent electrochemical oxidation, we investigated the electrochemical oxidation of HMF and its reaction intermediates. Fe-modified Ni-foams were prepared as reported in our previous study.42 In short, Ni-foams were treated in a mixture of FeCl3 and H2O2 to yield the Fe modified Ni foams. This results in nanosheet-like structures on the surface of the Ni-foam, which were identified as NiFe-layer double hydroxide/FeOOH heterostructures (a detailed study is described in ref. 42). The electrochemical oxidation of the respective organic substrates was studied using high performance liquid chromatography (HPLC) and qNMR.
In order to test whether a mixture of HMFCA and DHMF, resulting from the Cannizzaro reaction of HMF in KOH, can also be electrooxidized, linear sweep voltammetry (LSV) measurements (Fig. 2c) were performed. For these experiments, we used an electrolyte with an initial conversion of HMF (1 M) to HMFCA and DHMF of about ∼80% in 5 M KOH. Further dilution with a KOH solution yielded the desired KOH and substrate concentrations in the tested electrolytes. As observed for the oxidation of HMF (Fig. S10–S13), ∼400 mA cm−2 at 0.50 V vs. Hg/HgO were reached when an electrolyte containing 50 mM concentration of the organic substrates (based on the HMF input before degradation in alkaline media) is oxidized in 1 M KOH (Fig. 2c). As expected for Ni-catalysts, a strong increase in current in the presence of the organic substrates is observed at potential ranges where Ni2+ is typically oxidized to Ni3+, visible by the pre-OER peak in the absence of the organic substrates.33,43,44 The formation of the higher oxidation state of the transition metal is necessary for the electrochemical oxidation of alcohols and aldehydes. Thus, the absence of the pre-OER signal is described to be a result of the altered electrochemical response originating from oxidation of the organic molecules overlapping with the oxidation of the transition metal.14,15,32 To confirm that the organic substrates are oxidized to FDCA, constant potential measurements at different potentials (Fig. S14†) were used to determine the faradaic efficiency (FE) as well as to quantify formed products by HPLC and qNMR. Both measurements confirmed that DHMF and HMFCA are oxidized to FDCA. FE towards FDCA of 98%, assuming an average 6 e− oxidation for each molecule FDCA, and conversions of 96% were reached after 15 min of electrolysis at 0.50 V vs. Hg/HgO (Fig. S15†). While higher potentials lead to lower FE, likely originating from OER activity, lower potentials reduced the space–time yields without changing the selectivity (Fig. S14†). It is important to note that the electrolyte is also stable during electrolysis, revealed by the constant carbon balance. Investigation of the reaction intermediates confirms simultaneous oxidation of both DHMF and HMFCA to FDCA (Fig. 2d). The presence of HMF during the oxidation, only accessible via oxidation of DHMF, further validates this finding. The electrochemical oxidation of the Cannizzaro products was also studied using mesoporous cobalt oxide (m-Co3O4) on carbon paper or unmodified Ni-foams as the working electrode (section 7). These results demonstrate that the oxidation of the Cannizzaro products is a more general phenomenon, which also works for other catalytic systems. Since the Cannizzaro products are stable in highly alkaline media, electrochemical measurements can be performed at elevated KOH and substrate concentrations without being affected by rapid degradation.37,38 Conducting the electrochemical oxidation at fivefold KOH and substrate concentration does result in a shift of the currents to lower potentials (Fig. 3a). Current densities of 100 mA cm−2 are now reached at 0.43 V vs. Hg/HgO compared to 0.46 V vs. Hg/HgO, respectively. The shift of the anodic currents is likely to result from the shift of the Ni2+/Ni3+ pre-OER peak to lower anodic potentials at higher alkalinity.25 Further, the fivefold increase in the concentration of the Cannizzaro products and KOH led to current densities of >1 A cm−2, reaching the level of technical processes. Constant potential electrolysis (0.47 V vs. Hg/HgO; Fig. S16†) resulted in 94% conversion and a FE towards FDCA of 98% after 50 min of electrolysis. Investigation of the reaction intermediates (Fig. 3b) again confirms simultaneous oxidation of both substrates to FDCA as observed for the oxidation in 1 M KOH. Overall, yields of ∼80% based on the initial HMF concentration were achieved, mainly limited by the conversion of HMF to the Cannizzaro products. Obvious extrapolation allows even higher current densities and space–time yields during upscaling when using suitable electric equipment, which is, however, beyond the scope of this study. The carbon balance remained at ∼80%, indicating selective oxidation to FDCA.
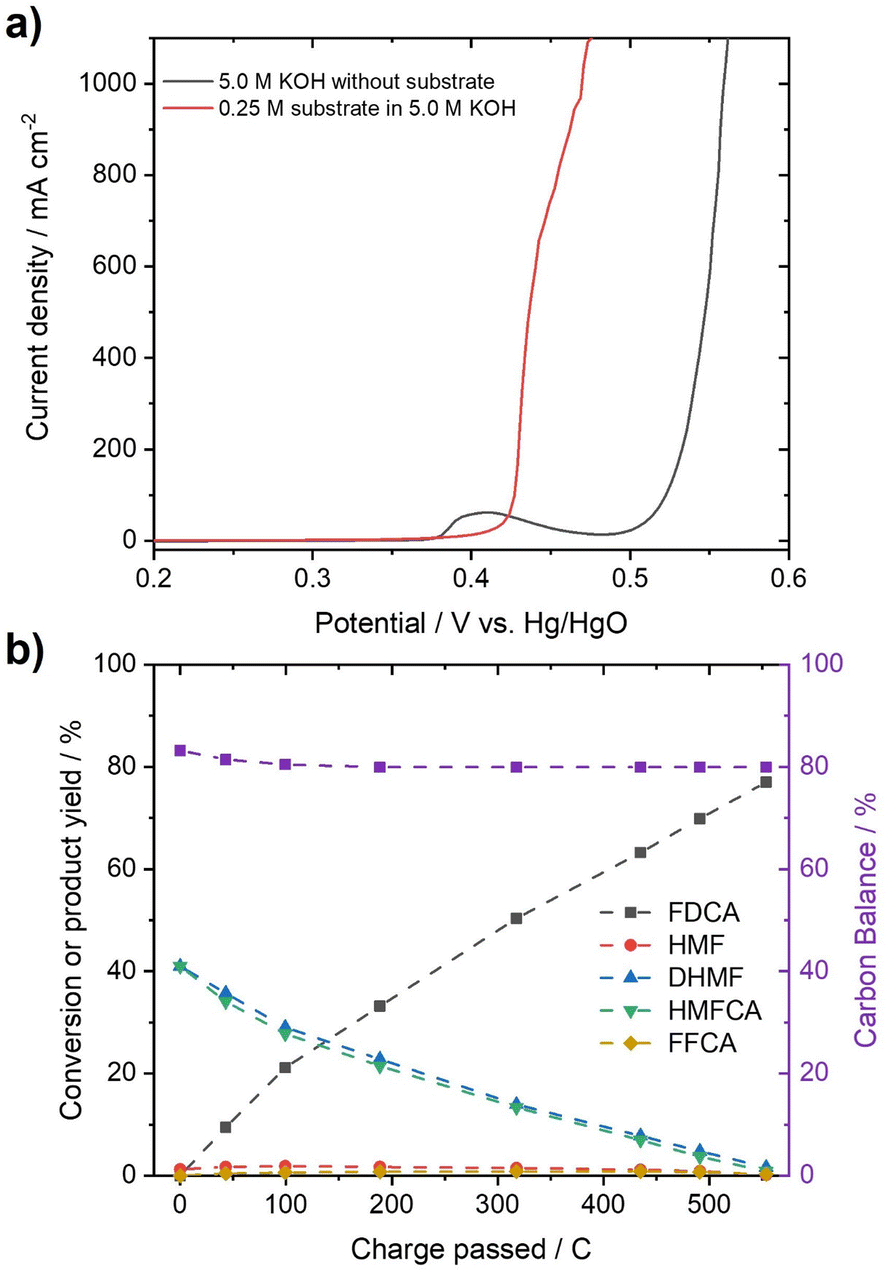 | ||
| Fig. 3 (a) LSV curves measured with and without 0.25 M of the resulting Cannizzaro products in 5 M KOH after decomposition (scan rate: 5 mV s−1). Potentials are corrected for the uncompensated solution resistance. (b) Reaction intermediates monitored for the oxidation of a 0.25 M solution of the Cannizzaro products (based on initial HMF input) in 5 M KOH during constant potential electrolysis (0.43 V vs. Hg/HgO). | ||
Conclusions
In this study, the surprisingly high FDCA yields from HMF electrooxidation under alkaline conditions were investigated. In highly alkaline media, HMF is selectively converted through the Cannizzaro reaction to DHMF and HMFCA. Both products can be electrooxidized to FDCA, rationalizing the high FDCA yields reported in the literature in spite of the known instability of HMF in alkaline solution. Based on these findings, we present an alternative, “indirect” route for the electrooxidation of HMF to FDCA. Both DHMF and HMFCA are oxidized simultaneously to FDCA while achieving high FE of ∼98% at conversions >90%. Due to the stability of the Cannizzaro products, “indirect” oxidation of HMF to FDCA is possible at both elevated concentrations of base and HMF while avoiding rapid degradation into humins, improving the atom economy of alkaline HMF electrooxidation to FDCA. Utilizing this, we report the “indirect” HMF oxidation to FDCA at current densities >1 A cm−2. This study thus reveals a new approach in the highly important electrochemical oxidation of HMF to FDCA by introducing DHMF and HMFCA as stable HMF-equivalents in an industrial environment at a performance comparable to the initial electrolyte.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank H. Hinrichs for conducting HPLC analysis and Dr M. Leutzsch for assistance with qNMR measurements and valuable discussions on product quantification. The authors also thank S. Palm for scanning electron microscopy and Dr E. Budiyanto for the discussions and support in the laboratory. Financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy-Cluster of Excellence 2186 “The Fuel Science Center”-ID: 390919832 and FOR 2982-UNODE, Project number 413163866 is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- D. A. Giannakoudakis, J. C. Colmenares, D. Tsiplakides and K. S. Triantafyllidis, ACS Sustainable Chem. Eng., 2021, 9, 1970–1993 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S. Department of Energy, 2004.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, Green Chem., 2021, 23, 1716–1733 RSC.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- S. Rajendran, R. Raghunathan, I. Hevus, R. Krishnan, A. Ugrinov, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., Int. Ed., 2015, 54, 1159–1163 CrossRef CAS PubMed.
- J. Tomaszewska, D. Bieliński, M. Binczarski, J. Berlowska, P. Dziugan, J. Piotrowski, A. Stanishevsky and I. A. Witońska, RSC Adv., 2018, 8, 3161–3177 RSC.
- X. Liu, D. C. Y. Leong and Y. Sun, Green Chem., 2020, 22, 6531–6539 RSC.
- C. Wang, H. Han, Y. Wu and D. Astruc, Coord. Chem. Rev., 2022, 458, 214422 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- Z.-M. Xu, J.-Y. Luo and Y.-B. Huang, Green Chem., 2022, 24, 3895–3921 RSC.
- M. T. Bender, X. Yuan, M. K. Goetz and K.-S. Choi, ACS Catal., 2022, 12, 12349–12368 CrossRef CAS.
- Z. Yan, J. L. Hitt, J. A. Turner and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12558–12563 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- B. Garlyyev, S. Xue, J. Fichtner, A. S. Bandarenka and C. Andronescu, ChemSusChem, 2020, 13, 2513–2521 CrossRef CAS PubMed.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- G. Chen, X. Li and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209014 CAS.
- Z. Li, X. Li, H. Zhou, Y. Xu, S.-M. Xu, Y. Ren, Y. Yan, J. Yang, K. Ji, L. Li, M. Xu, M. Shao, X. Kong, X. Sun and H. Duan, Nat. Commun., 2022, 13, 5009 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem. A, 2021, 9, 20164–20183 RSC.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- Web of Science, available at: https://www.webofscience.com/wos/woscc/summary/8fc51df6-70e9-464d-ac48-74e75a4268b8-61cc9c74/relevance/1, accessed 28 November 2022.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- C. Wang, H.-J. Bongard, M. Yu and F. Schüth, ChemSusChem, 2021, 14, 5199–5206 CrossRef CAS PubMed.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- S. R. Kubota and K.-S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- X. Deng, G.-Y. Xu, Y.-J. Zhang, L. Wang, J. Zhang, J.-F. Li, X.-Z. Fu and J.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- N. Shi, Q. Liu, R. Ju, X. He, Y. Zhang, S. Tang and L. Ma, ACS Omega, 2019, 4, 7330–7343 CrossRef CAS PubMed.
- D.-H. Nam, B. J. Taitt and K.-S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- H. Liu, N. Agrawal, A. Ganguly, Y. Chen, J. Lee, J. Yu, W. Huang, M. Mba Wright, M. J. Janik and W. Li, Energy Environ. Sci., 2022, 15, 4175–4189 RSC.
- S. Cannizzaro, Ann. Chem. Pharm., 1853, 88, 129–130 CrossRef CAS.
- S. Subbiah, S. P. Simeonov, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Green Chem., 2013, 15, 2849 RSC.
- C. Wang, Y. Wu, A. Bodach, M. L. Krebs, W. Schuhmann and F. Schüth, Angew. Chem., Int. Ed., 2023, 62, e202215804 CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef CAS.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, liquid phase analysis, and supplementary experiments. See DOI: https://doi.org/10.1039/d2gc04732b |
| This journal is © The Royal Society of Chemistry 2023 |