
Decarboxylative arylation with diaryliodonium(III) salts: alternative approach for catalyst-free difluoroenolate coupling to aryldifluoromethyl ketones†
Kotaro
Kikushima
 *a,
Kohei
Yamada
a,
Narumi
Umekawa
a,
Natsumi
Yoshio
a,
Yasuyuki
Kita
*b and
Toshifumi
Dohi
*ab
*a,
Kohei
Yamada
a,
Narumi
Umekawa
a,
Natsumi
Yoshio
a,
Yasuyuki
Kita
*b and
Toshifumi
Dohi
*ab
aCollege of Pharmaceutical Sciences, Ritsumeikan University, 1-1-1, Nojihigashi, Kusatsu Shiga, 525-8577, Japan. E-mail: kixy@fc.ritsumei.ac.jp; td1203@ph.ritsumei.ac.jp
bResearch Organization of Science and Technology, Ritsumeikan University, 1-1-1, Nojihigashi, Kusatsu Shiga, 525-8577, Japan. E-mail: kita@ph.ritsumei.ac.jp
First published on 24th January 2023
Abstract
Catalyst-free access to aryldifluoromethyl ketones has been achieved through decarboxylative arylation of α,α-difluoro-β-ketoacid salts using diaryliodonium(III) salts. The products were successfully transformed into the corresponding esters, amides, and difluoromethyl compounds. This strategy provides access to fluorine-containing drugs, thus contributing to drug design.
Introduction of fluorine atoms into small molecules is a widely accepted strategy in medicinal chemistry for enhancing the lipophilicity, metabolic stability, and bioavailability relative to those of non-fluorinated parent compounds.1 Considerable efforts have been devoted to constructing fluorine-containing molecules. Aryldifluoromethyl carbonyls are attractive synthetic intermediates for the corresponding esters, amides, alcohols, and difluoromethyl compounds. These units are present in biologically active compounds, such as the AMPAR allosteric modulator,2a FKB12 inhibitor,2b fungal CYP51 inhibitor,2c and BACE 1 inhibitor2d (Fig. 1). Furthermore, the difluoromethylene (-CF2-) groups serve as bioisosteres for carbonyl compounds and oxygen and sulfur atoms.3,4
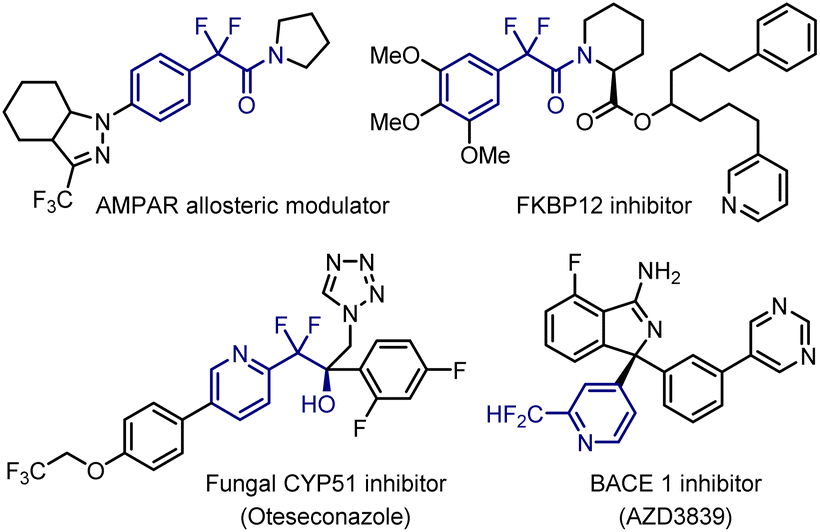 | ||
| Fig. 1 Biologically active compounds containing difluoromethylene units. | ||
Difluoromethylene units have been introduced by α-fluorination or deoxofluorination of the corresponding carbonyl compounds using electrophilic fluorination reagents5 or aminosulfur trifluorides.6 However, these reagents are expensive and unstable in water, generating toxic compounds such as HF. In contrast, transformation of fluorine-containing building blocks is an efficient approach as it does not require hazardous fluorination reagents.7,8 Aryldifluoroacetates have been synthesized via transition metal-catalyzed coupling reactions of commercially available bromodifluoroacetate and aryl metal species containing zinc,9 boron,10 or silicon (Fig. 2a).11 The combination of aryl halides with metal enolates of α,α-difluoroacetates or α,α-difluoroamides is also an effective approach for accessing α,α-difluorinated carbonyl compounds (Fig. 2b).12,13 These methods require the use of organometallic species and transition metal catalysts, which increases costs and generates stoichiometric amounts of metallic byproducts containing zinc, boron, or silicon salts. Direct C–H bond functionalization of arenes (Fig. 2c) or difluoroacetophenone (Fig. 2d) using Fenton reagents,14 photocatalysts,15 or palladium catalysts can generate aryldifluoromethyl carbonyl compounds.16 Nevertheless, these methods require metal catalysts and/or organometallic species.
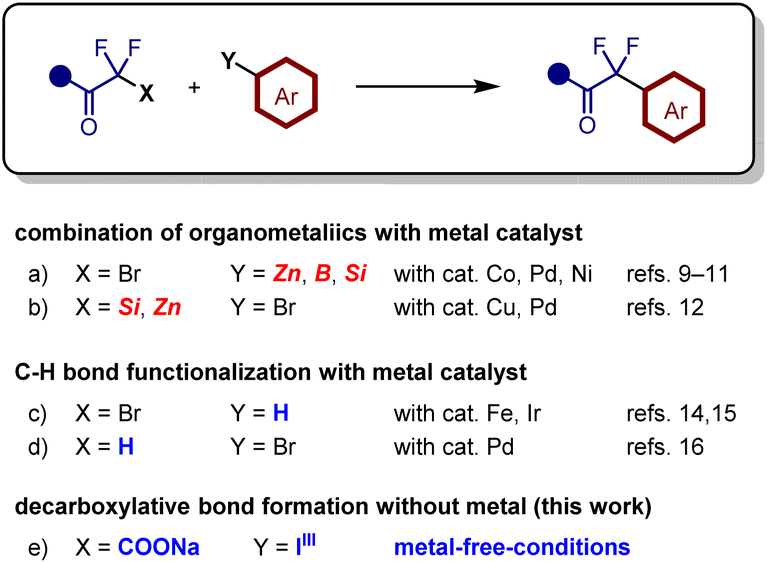 | ||
| Fig. 2 Various approaches to α-aryl-α,α-difluorinated carbonyl compounds. | ||
As an alternative approach to avoid chemical wastes, decarboxylative coupling reactions are an attractive strategy because they generate only carbon dioxide as the byproduct during bond formation.17 Decarboxylative fluoroalkylation using α-fluorinated carboxylate can produce the corresponding organofluorines, although these reactions require transition metals, heat, or photoirradiation in most cases.17j,18 Fluorine-containing activated methylene derivatives undergo decarboxylative C–C bond formation under relatively mild conditions,19 although the generation of aryldifluoromethyl ketones via decarboxylative arylation is unprecedented so far.20 Given these circumstances, the development of catalyst-free arylation of fluorinated carboxylates, without chemical waste, would be an economical and environmentally friendly method for the synthesis of fluorine-containing biologically active compounds.
To achieve catalyst-free decarboxylative fluoroalkylation, we focused on the use of diaryliodonium(III) salts (Ar1Ar2I+X−) as arylation reagents. This will offer an alternative approach to transition metal-free arylations of various nucleophiles forming aryl–heteroatom and aryl–carbon bonds.21 Our group has developed an efficient method for the arylation of carboxylic acids,22a phenols,22b and hydroxyamines22c using easily accessible trimethoxyphenyl (TMP) iodonium salts (Ar(TMP)I+X−).23,24 Herein, we describe the first example of the decarboxylative arylation of difluoroacetate derivatives using TMP-iodonium tosylates to afford aryldifluoromethyl carbonyl compounds (Fig. 2e). The selective C–H bond functionalization of uracil through the present strategy to incorporate fluorine-containing functional groups was also demonstrated.
Initially, we examined the reaction of α,α-difluoro-β-ketoacid sodium salt 1a-Na with (4-nitrophenyl)(TMP)iodonium tosylate 2a-OTs-TMP in toluene at 100 °C to afford the desired decarboxylative coupling product 3aa in 86% yield (Table 1, entry 1). In this reaction, formation of the corresponding aryl ester via a direct coupling reaction was not observed. When the reaction was carried out at 70 °C, the yield dropped to 34% (entry 2). Combining acid 1a-H, instead of 1a-Na, with Na2CO3 decreased the yield to 48%, and a large quantity of the decarboxylative protonation product was generated as a side product presumably reacting with the in situ generated H2O as the proton source (entry 3). In fact, the reaction of 1a-Na with 2a-OTs-TMP in the presence of H2O increased the yield of the protonation product (Fig. S1†). Thus, sodium salt 1a-Na was employed as the starting material for subsequent examinations. The use of TMP-iodonium trifluoroacetate afforded a comparable yield of 3aa (entry 4). In contrast, the yield decreased when TMP-iodonium salts bearing triflate, acetate, and tetrafluoroborate were used (entries 5–7). Next, we screened the solvents using 2a-OTs-TMP. The use of benzotrifluoride was comparable to that of toluene (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | L | Ar | Solvent | NMR Yield |
| a Reaction conditions: 1a-Na (0.30 mmol) and 2a-L-Ar (0.20 mmol) in solvent (1.0 mL) at 100 °C for 20 h: 19F NMR yield. b Conducted at 70 °C. c The corresponding acid 1a-H (0.30 mmol) and Na2CO3 (0.30 mmol) were used instead of 1a-Na. d Isolated yield. | ||||
| 1 | OTs | TMP | Toluene | 86% |
| 2b | OTs | TMP | Toluene | 34% |
| 3c | OTs | TMP | Toluene | 48% |
| 4 | OCOCF3 | TMP | Toluene | 83% |
| 5 | OTf | TMP | Toluene | 26% |
| 6 | OAc | TMP | Toluene | 15% |
| 7 | BF4 | TMP | Toluene | 60% |
| 8 | OTs | TMP | PhCF3 | 85% |
| 9 | OTs | TMP | DMF | 23% |
| 10 | OTs | TMP | MeCN | 67% |
| 11 | OTs | TMP | i-BuOAc | 76% |
| 12 | OTs | TMP | i-BuOH | 11% |
| 13 | OTs | TMP | 1,4-Dioxane | 65% |
| 14 | OTs | TMP | 4-MeTHP | 89% (85%)d |
| 15 | OTs | DMP | 4-MeTHP | 87% |
| 16 | OTs | Mes | 4-MeTHP | 45% |
| 17 | OTs | DCP | 4-MeTHP | 35% |

|
||||
The reactivity in N,N-dimethylformamide (DMF), in which both starting materials were highly soluble, was low, affording the product in 23% yield (entry 9). Using acetonitrile and isobutyl acetate, which are recommended solvents in the CHEM21 selection guide,25 acceptable yields of 3aa (entries 10 and 11) were obtained, whereas the yield was low in isobutyl alcohol (entry 12). The present reaction proceeded in THF at 100 °C to afford 3aa in high yield (not shown in the Table), albeit with low reproducibility, presumably because of the incompatibility of the reaction with the boiling point of THF. Thus, we tested cyclic ethers with high boiling points. Consequently, the reaction in 1,4-dioxane afforded a moderate yield of 65% (entry 13), whereas the reaction in 4-methyltetrahydropyrane (4-MeTHP)26 afforded 3aa in good yields (89%; entry 14). The dimethoxyphenyl (DMP) group was also employed as a dummy aryl ligand instead of the TMP group to afford the desired product 3aa in a comparable yield (entry 15). In addition, the mesityl (Mes) and dichlorophenyl (DCP) groups allowed selective bond formation, albeit with decreased yields (entries 16 and 17).
We speculated that the present reaction proceeded via the initial ligand exchange of fluorinated carboxylate 1a-Na with the tosylate anion of 2a-OTs, followed by decarboxylative C–C bond formation. The expected intermediate 1a2a-TMP was prepared via the general synthetic method for TMP-iodonium salts from 4-nitoroiodobenzene with α,α-difluoro-β-keto acid 1a-H using mCPBA as an oxidant (Fig. S2†). Treatment of isolated iodonium salt 1a2a-TMP in toluene at 100 °C afforded the decarboxylative arylation product 3aa in 70% yield (Fig. 3a). The reaction of 1a-Na with 2a-OTs-TMP in CD3CN at room temperature was monitored by NMR spectroscopy to confirm the generation of 1a2a-TMP (Fig. 3b and Fig. S3†). Decarboxylative ligand coupling of 1a2a-TMP proceeded continuously at 100 °C to afford the coupling product 3aa in 74% yield (Fig. S4†). The reaction of 1a-Na and 2a-OTs-TMP in the presence of TEMPO produced 3aa in 84% yield (Fig. 3c), indicating that the present reaction does not involve radical species generated by Hunsdiecker-type decarboxylation17i,27 and/or single-electron-transfer (SET) process.12f,28 When the reaction of 1a-Na and 2a-OTs-TMP was carried out with 4-(trifluoromethyl)benzaldehyde, the decarboxylative arylation reaction preferentially proceeded to afford 3aa in 66% yield, along with quantitative recovery of the added aldehyde (Fig. 3d); formation of a β-hydroxy ketone via decarboxylative aldol reaction was not observed. In contrast, 1a-Na reacted with the aldehyde in the absence of 2a-OTs-TMP to produce the corresponding β-hydroxy ketone in 76% yield.19c,d,e We assume that sodium difluoroenolate is not a major intermediate for the present decarboxylative bond formation. Based on these results, a plausible reaction mechanism involving decarboxylative arylation is outlined in Fig. 3e. Initially, the ligand exchange of 1a-Na with tosylate anion of 2a-OTs-TMP preferentially proceeds before the decarboxylation, generating intermediate 1a2a-TMP along with the release of TsONa. The precipitation of TsONa can promote this step if nonpolar solvents are used. We envisioned that the decarboxylation of 1a2a-TMP proceeded to form the corresponding iodonium difluoroenolate salt, containing O-bound and/or C-bound difluoroenolates.29 Subsequent ligand coupling generates the arylation product 3aa. The heating is presumably required for the decarboxylation step; silyl difluoroenolates react with diaryliodonium salts at room temperature to afford the corresponding arylation products.13 Alternatively, concerted decarboxylative ligand coupling could also be a possible reaction mechanism for the present system.
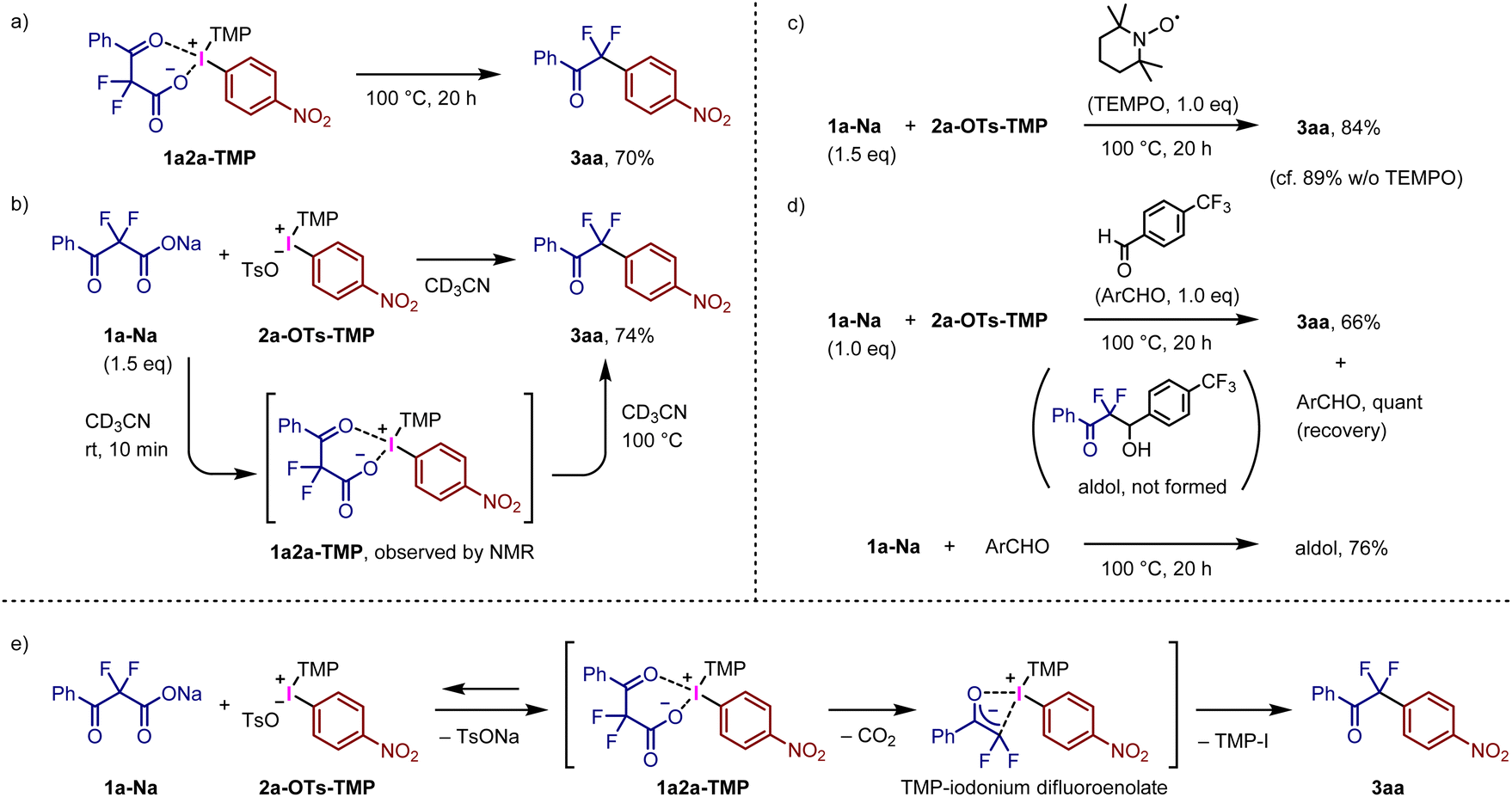 | ||
| Fig. 3 Mechanistic studies for decarboxylative ligand coupling. | ||
The decarboxylative arylation of benzoylacetate derivatives is limited to a few examples, which require copper catalysts.20a,c Therefore, the present reaction is a complementary method for accessing difluoromethyl aryl ketones. The limited examples prompted us to examine various combinations of α,α-difluoro-β-ketoacid sodium salts 1-Na and TMP-iodonium tosylate salts 2-OTs-TMP (Fig. 4). Functionalized aryl rings bearing nitro, cyano, ester, acetyl, and trifluoromethyl groups were introduced via decarboxylative arylation reactions to afford 3aa–3ae. The ester group was not hydrolysed and survived during the reaction, indicating that the present reaction proceeds under mild conditions at least below pH 10. The introduction of an aryl group bearing an ortho-substituent showed a low reactivity (3af, 36%) due to steric hindrance. The use of TMP-iodonium tosylates bearing electron-rich aromatic rings proved challenging under standard conditions. Reaction monitoring by NMR spectroscopy suggested that the decarboxylative coupling step was influenced by the low electrophilicity of aryl groups (Fig. S5†). Additional optimization of the reaction conditions (Fig. S6†) revealed that aryl rings bearing methyl and methoxy groups could be successfully introduced at a higher temperature (130 °C) in toluene to afford 3ag–3ai in moderate yields. α,α-Difluoro-β-ketoacid sodium salts bearing electron-donating groups, such as methoxy, tert-butyl, and methyl groups, were employed to afford the corresponding products 3ba, 3bb, 3bj, 3ca, 3da, 3ej and 3bk. The reactivity was unaffected by ortho-methyl group to afford 3da in 72% yield. In contrast, ortho-methoxy group decreased the yield (3ej: 42%), presumably because the corresponding difluoroenolate intermediate is hardly generated due to the steric hindrance. The metal-free conditions enable the preparation of reactive aryl halide-bearing products 3af, 3fa, 3ga, 3bk, 3al and 3am. Heteroarenes, such as pyridine and thiophene groups, were incorporated as aryl groups to obtain 3an and 3ha. Notably, the nucleophilic aromatic substitution of fluorine and chlorine atoms did not proceed under our conditions (see 3am and 3an). Aryl halides 3af, 3fa, 3ga, 3bk, 3al, 3am, and 3an can be used as starting materials for transition metal-catalyzed coupling or nucleophilic aromatic substitutions. In addition to the aryl ketones, alkyl ketones 3ia and 3ja were synthesized from the corresponding α,α-difluoro-β-ketoacid sodium salts, albeit in moderate yields. In all the cases depicted in Fig. 4, arylation products bearing the TMP group were not observed.
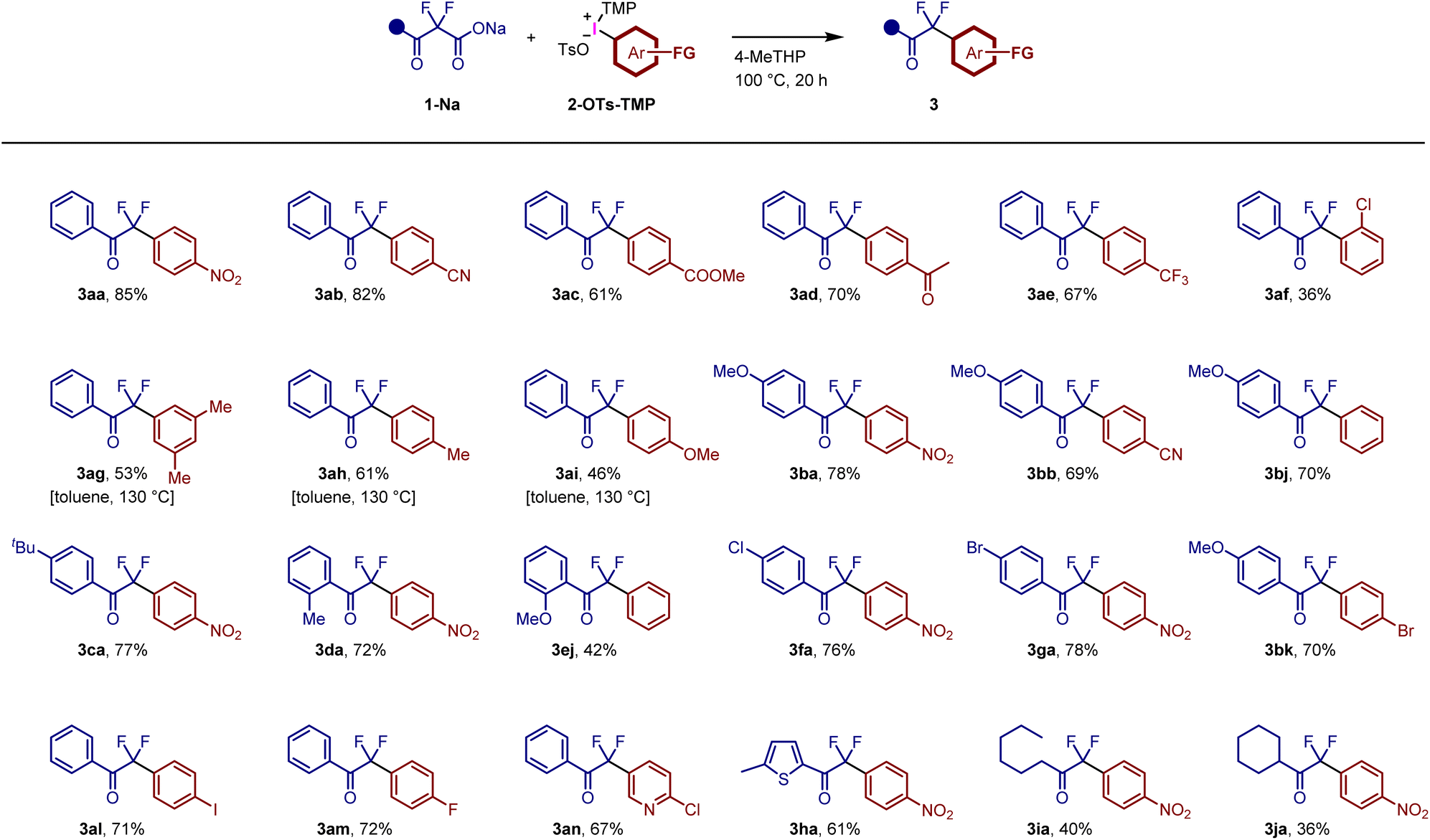 | ||
| Fig. 4 Synthesis of various aryldifluoromethyl ketones using α,α-difluoro-β-ketoacid sodium salts and TMP-iodonium tosylates. | ||
The reaction of N,N-dimethyluracil(DCP)iodonium tosylate 2o-OTs-DCP with 1a-Na resulted in uracil-selective C–C bond formation, affording the corresponding difluoromethyl ketone 3ao in 65% yield (Fig. 5a). Notably, a C–C bond was formed at the 6-position of N,N-dimethyluracil instead of the 5-position, wherein 1,4-addition presumably occurred in the conjugated enone system of the uracil derivative. N,N-Dimethyluracil iodonium salts can be synthesized via the direct introduction of iodonium moieties, starting from N,N-dimethyluracil.30,31 In fact, 2o-OTs-DCP was prepared by the reaction of N,N-dimethyluracil and 2,6-dichloroiodobenzene (DCP-I) in the presence of mCPBA and p-TsOH (Fig. 5b). After the decarboxylative coupling with 1a-Na, DCP-I was successfully recovered in its pure form, which could be recycled as the starting material for 2o-OTs-DCP. This strategy enables the selective C–H bond functionalization of uracil using renewable iodonium salt.
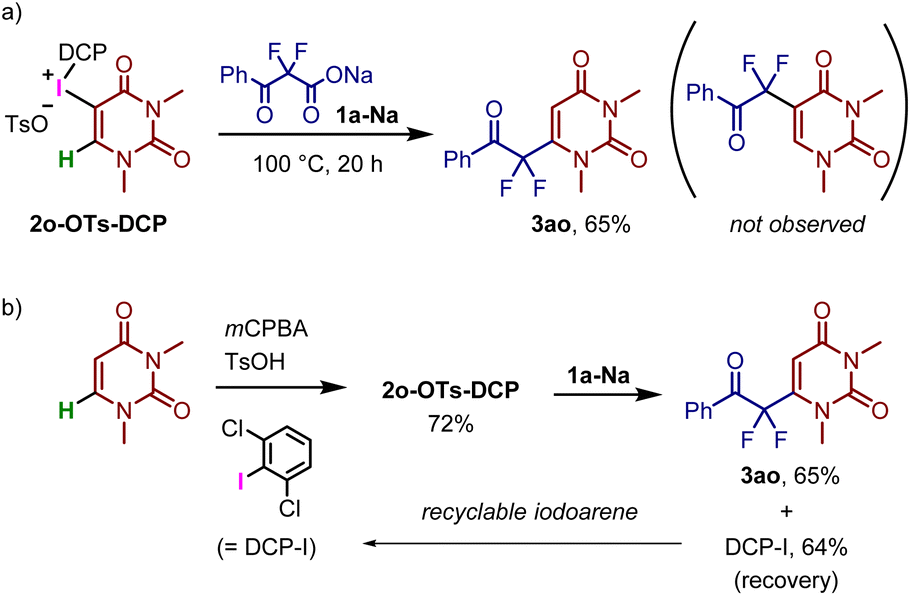 | ||
| Fig. 5 C–H functionalization of uracil using recyclable iodoarene. | ||
Next, we examined the further transformation of the decarboxylative coupling products (Fig. 6). The aryl carbonyl unit of coupling product 3ao was removed via treatment with KOH to generate difluoromethyluracil 4o in 64% yield. Aryl difluoromethyl ketone 3ae was reacted with organomagnesium reagents to afford the corresponding tertiary alcohols 5aea (85%)16b and 5aeb (61%). Treatment of 3bk with mCPBA in a mixed solvent of dichloromethane and 1,1,1,3,3,3-hexafluoro-2-propyl alcohol (HFIP) afforded quantitative yield of aryl ester 6bk, which reacted with pyrrolidine to generate the corresponding difluoroacetamide 7k in 92% yield. This sequential transformation was successfully applied to a gram-scale reaction to obtain 7k in 80% yield (1.2 g) without the isolation of the ester intermediate. Functionalized difluoroacetamide 7k was converted to the AMPAR allosteric inhibitor 8 in 89% yield via a copper-catalyzed coupling reaction.
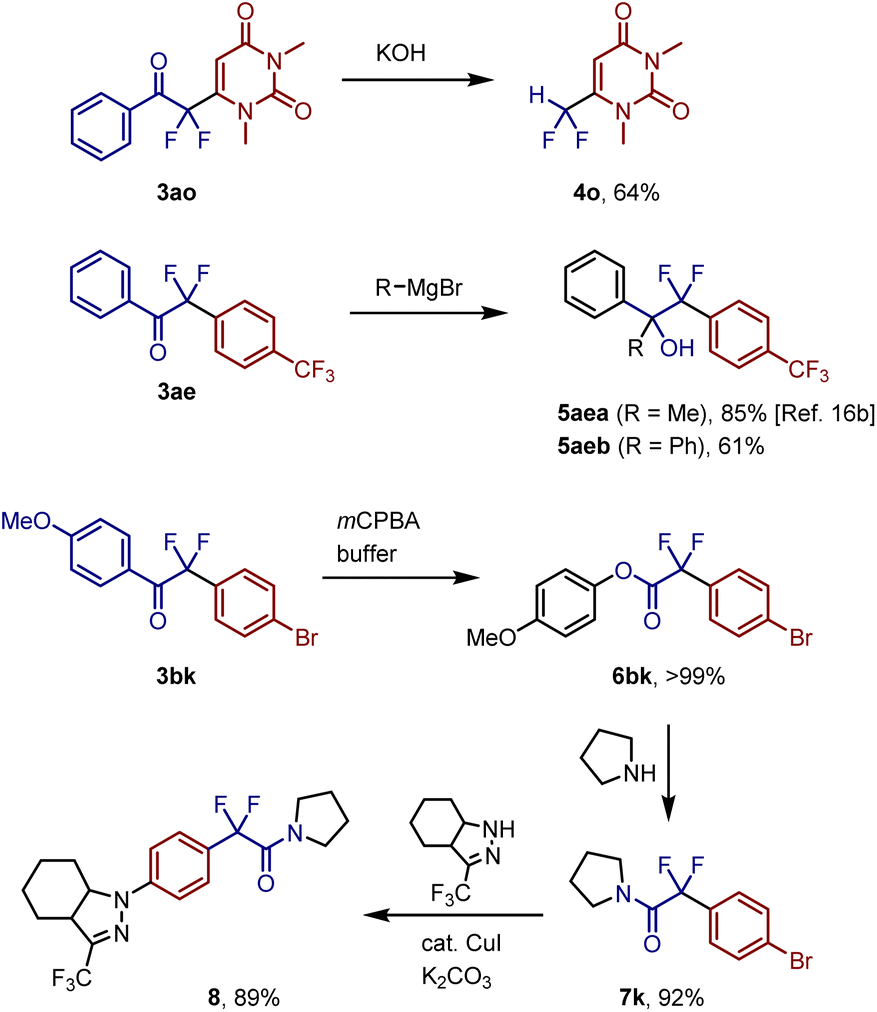 | ||
| Fig. 6 Transformation of aryldifluoromethyl ketones. | ||
Conclusions
We have developed the decarboxylative arylation of an α,α-difluoro-β-keto acid ester with diaryliodonium salts to afford the corresponding α,α-difluoroketones. The reaction involves sequential ligand exchange, followed by decarboxylative ligand coupling. Two fluorine atoms were successfully incorporated at the benzyl position in the absence of hazardous fluorination reagents, organometallic compounds, and transition metal catalysts. The resulting α,α-difluoromethyl ketone group could be converted into the corresponding ester, amide, and difluoromethyl groups, which are found in biologically active compounds. The present reaction offers an environmentally friendly synthetic approach to fluorine-containing drugs and is expected to contribute to drug design and discovery.Author contributions
K. K. and T. D. conceived and designed the experiments and directed the project. K. Y., N. U. and N. Y. performed the experiments. K. K., K. Y. and N. U. analyzed the data and checked the experimental details. K. K., Y. K. and T. D. wrote the paper. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. K. and T. D. acknowledge support from JSPS KAKENHI grant number 18H02014 (K. K.) and 19K05466 (T. D.), JST CREST grant number JPMJCR20R1, and the Ritsumeikan Global Innovation Research Organization (R-GIRO) project.References
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (b) K. Mgller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. AceÇa, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- (a) S. E. Ward, M. Harries, L. Aldegheri, N. E. Austin, S. Ballantine, E. Ballini, D. M. Bradley, B. D. Bax, B. P. Clarke, A. J. Harris, S. A. Harrison, R. A. Melarange, C. Mookherjee, J. Mosley, G. Dal Negro, B. Oliosi, K. J. Smith, K. M. Thewlis, P. M. Woollard and S. P. Yusaf, J. Med. Chem., 2011, 54, 78–94 CrossRef CAS PubMed; (b) G. M. Dubowchik, V. M. Vrudhula, B. Dasgupta, J. Ditta, T. Chen, S. Sheriff, K. Sipman, M. Witmer, J. Tredup, D. M. Vyas, T. A. Verdoorn, S. Bollini and A. Vinitsky, Org. Lett., 2001, 3, 3987–3990 CrossRef CAS PubMed; (c) A. G. S. Warrilow, C. M. Hull, J. E. Parker, E. P. Garvey, W. J. Hoekstra, W. R. Moore, R. J. Schotzinger, D. E. Kelly and S. L. Kelly, Antimicrob. Agents Chemother., 2014, 58, 7121–7127 CrossRef CAS PubMed; (d) F. Jeppsson, S. Eketjäll, J. Janson, S. Karlström, S. Gustavsson, L.-L. Olsson, A.-C. Radesäter, B. Ploeger, G. Cebers, K. Kolmodin, B.-M. Swahn, S. von Berg, T. Bueters and J. Fälting, J. Biol. Chem., 2012, 287, 41245–41257 CrossRef CAS PubMed.
- (a) G. M. Dubowchik, V. M. Vrudhula, B. Dasgupta, J. Ditta, T. Chen, S. Sheriff, K. Sipman, M. Witmer, J. Tredup, D. M. Vyas, T. A. Verdoorn, S. Bollini and A. Vinitsky, Org. Lett., 2001, 3, 3987–3990 CrossRef CAS PubMed; (b) M. O. Anderson, J. Zhang, Y. Liu, C. Yao, P.-W. Phuan and A. S. Verkman, J. Med. Chem., 2012, 55, 5942–5950 CrossRef CAS PubMed; (c) Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949–3952 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626–1631 CrossRef CAS; (b) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325–9332 CrossRef CAS PubMed; (c) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- (a) T. Umemoto, K. Kawada and K. Tomita, Tetrahedron Lett., 1986, 27, 4465–4468 CrossRef CAS; (b) S. Singh, D. D. DesMarteau, S. S. Zuberi, M. Witz and H. N. Huang, J. Am. Chem. Soc., 1987, 109, 7194–7196 CrossRef CAS.
- (a) W. J. Middleton and E. M. Bingham, J. Org. Chem., 1980, 45, 2883–2887 CrossRef CAS; (b) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 CrossRef CAS; (c) R. P. Singh, U. Majumder and J. M. Shreeve, J. Org. Chem., 2001, 66, 6263–6267 CrossRef CAS PubMed.
- For recent reviews, see: (a) J. Feng, X. Jia, S. Zhang, K. Lu and D. Cahard, Org. Chem. Front., 2022, 9, 3598–3623 RSC; (b) L. Shi, D. An and G.-J. Mei, Org. Chem. Front., 2022, 9, 4192–4208 RSC; (c) C. M. Kisukuri, V. A. Fernandes, J. A. C. Delgado, A. P. Haring, M. W. Paixao and S. R. Waldvogel, Chem. Rec., 2021, 21, 2502–2525 CrossRef CAS PubMed; (d) R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS; (e) Z. Zou, W. Zhang, Y. Wang and Y. Pan, Org. Chem. Front., 2021, 8, 2786–2798 RSC; (f) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC; (g) D.-Q. Dong, H. Yang, J.-L. Shi, W.-J. Si, Z.-L. Wang and X.-M. Xu, Org. Chem. Front., 2020, 7, 2538–2575 RSC; (h) X. Ma and Q. Song, Chem. Soc. Rev., 2020, 49, 9197–9219 RSC; (i) A. D. Dilman and V. V. Levin, Acc. Chem. Res., 2018, 51, 1272–1280 CrossRef CAS PubMed; (j) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed; (k) D. Chen, J. Jiang and J.-P. Wan, Chin. J. Chem., 2022, 40, 2582–2594 CrossRef CAS.
- Recent examples for difluoromethylenation and difluoromethylation: (a) J. Ying, T. Liu, Y. Liu and J.-P. Wan, Org. Lett., 2022, 24, 2404–2408 CrossRef CAS PubMed; (b) H. Li, Q. Sun, T.-T. Zhang, Y. Chen, J. Zhang, H. Deng and H. Jiang, Chem. – Asian J., 2022, 17, e202200448 CAS; (c) V. A. Brotsman, N. S. Lukonina, N. A. Malkin, A. V. Rybalchenko, N. M. Belov and A. A. Goryunkov, Phys. Chem. Chem. Phys., 2022, 24, 16816–16826 RSC; (d) L.-L. Mao, A.-X. Zhou, X.-H. Zhu, H. Peng, L.-X. Quan, J.-P. Wan and S.-D. Yang, J. Org. Chem., 2022, 87, 12414–12423 CrossRef CAS PubMed.
- K. Araki and M. Inoue, Tetrahedron, 2013, 69, 3913–3918 CrossRef CAS.
- (a) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS PubMed; (b) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS PubMed.
- Y. Wu, H.-R. Zhang, Y.-X. Cao, Q. Lan and X.-S. Wang, Org. Lett., 2016, 18, 5564–5567 CrossRef CAS PubMed.
- (a) Y. Guo and J. M. Shreeve, Chem. Commun., 2007, 3583–3585 RSC; (b) K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560–5563 CrossRef CAS PubMed; (c) S. Ge, S. I. Arlow, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 14401–14404 CrossRef CAS PubMed; (d) S. I. Arlow and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 4567–4572 CrossRef CAS PubMed; (e) T. Xia, L. He, Y. A. Liu, J. F. Hartwig and X. Liao, Org. Lett., 2017, 19, 2610–2613 CrossRef CAS PubMed; (f) Y.-B. Wu, G.-P. Lu, B.-J. Zhou, M.-J. Bu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 5965–5968 RSC.
- Transition-metal-free arylation of difluorinated silyl enolate with diaryliodonium salt was reported: X. Jiang, D. Meyer, D. Baran, M. A. C. González and K. J. Szabó, J. Org. Chem., 2020, 85(13), 8311–8319 CrossRef CAS PubMed.
- Y. Ohtsuka and T. Yamakawa, Tetrahedron, 2011, 67, 2323–2331 CrossRef CAS.
- (a) Y. Yuan, W. Dong, X. Gao, X. Xie and Z. Zhang, Org. Lett., 2019, 21, 469 CrossRef CAS PubMed; (b) C.-H. Qu, G.-T. Song, J. Xu, W. Yan, C.-H. Zhou, H.-Y. Li, Z.-Z. Chen and Z.-G. Xu, Org. Lett., 2019, 21, 8169–8173 CrossRef CAS PubMed.
- (a) C. Guo, R.-W. Wang and F.-L. Qing, J. Fluorine Chem., 2012, 143, 135–142 CrossRef CAS; (b) S. Ge, W. Chaładaj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149–4152 CrossRef CAS PubMed.
- (a) N. Rodríguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (b) J. Cornella and I. Larrosa, Synthesis, 2012, 653–676 CAS; (c) K. Park and S. Lee, RSC Adv., 2013, 3, 14165–14182 RSC; (d) Z.-L. Wang, Adv. Synth. Catal., 2013, 355, 2745–2755 CrossRef CAS; (e) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (f) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC; (g) T. Patra and D. Maiti, Chem. – Eur. J., 2016, 23, 7382–7401 CrossRef PubMed; (h) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (i) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (j) P. Xiao, X. Pannecoucke, J.-P. Bouillon and S. Couve-Bonnaire, Chem. Soc. Rev., 2021, 50, 6094–6151 RSC.
- Selected examples for decarboxylative coupling using trifluoroacetate generating aryl trifluoride: (a) K. Matsui, E. Tobita, M. Ando and K. Kondo, Chem. Lett., 1981, 1719–1720 CrossRef CAS; (b) G. E. Carr, R. D. Chambers, T. F. Holmes and D. G. Parker, J. Chem. Soc., Perkin Trans. 1, 1988, 921–926 RSC; (c) Y. Li, T. Chen, H. Wang, R. Zhang, K. Jin, X. Wang and C. Duan, Synlett, 2011, 1713–1716 CrossRef CAS; (d) M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628–11631 CrossRef CAS PubMed; (e) G. Shi, C. Shao, S. Pan, J. Yu and Y. Zhang, Org. Lett., 2015, 17, 38–41 CrossRef CAS PubMed; (f) J. Lin, Z. Li, J. Kan, S. Huang, W. Su and Y. Li, Nat. Commun., 2017, 8, 14353 CrossRef PubMed.
- (a) J. Saadi and H. Wennemers, Nat. Chem., 2016, 8, 276–280 CrossRef CAS PubMed; (b) E. Gupta, R. Kant and K. Mohanan, Org. Lett., 2017, 19, 6016–6019 CrossRef CAS PubMed; (c) H.-F. Mao and Z.-J. Liu, Tetrahedron Lett., 2017, 58, 3394–3397 CrossRef; (d) J.-W. Yuan, S.-N. Liua and W.-P. Mai, Org. Biomol. Chem., 2017, 15, 7654–7659 RSC; (e) A. Tarui, M. Oduti, S. Shinya, K. Sato and M. Omote, RSC Adv., 2018, 8, 20568–20575 RSC; (f) F. de Azambuja, M. H. Yang, T. Feoktistova, M. Selvaraju, A. C. Brueckner, M. A. Grove, S. Koley and P. H. Cheong, Nat. Chem., 2020, 12, 489–496 CrossRef CAS PubMed.
- Metal-catalyzed decarboxylative arylation of non-fluorinated activated methylene derivatives, see: (a) A. Bruggink and A. McKillop, Tetrahedron, 1975, 31, 2607–2619 CrossRef CAS; (b) B. Song, F. Rudolphi, T. Himmler and L. J. Gooßen, Adv. Synth. Catal., 2011, 353, 1565–1574 CrossRef CAS; (c) D. Zhao, Y. Jiang and D. Ma, Tetrahedron, 2014, 70, 3327–3332 CrossRef CAS; (d) P. J. Moon, S. Yin and R. J. Lundgren, J. Am. Chem. Soc., 2016, 138, 13826–13829 CrossRef CAS PubMed.
- For selected reviews, see: (a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123–1178 CrossRef CAS PubMed; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (c) M. S. Yusubov, A. V. Maskaev and V. V. Zhdankin, Arkivoc, 2011, 370–409 Search PubMed; (d) B. Olofsson, Top. Curr. Chem., 2016, 373, 135–166 CrossRef CAS PubMed; (e) K. Aradi, B. L. Toth, G. L. Tolnai and Z. Novák, Synlett, 2016, 1456–1485 CAS; (f) P. Villo and B. Olofsson, Arylations Promoted by Hypervalent Iodine Reagents in Patai's Chemistry of Functional Groups (Hypervalent Halogen Compounds), John Wiley & Sons, Chichester, 2018 Search PubMed; (g) N. Takenaga, R. Kumar and T. Dohi, Front. Chem., 2020, 8, 599026–599033 CrossRef CAS PubMed; (h) K. Kikushima, E. E. Elboray, J. O. C. Jiménez-Halla, C. R. Solorio-Alvarado and T. Dohi, Org. Biomol. Chem., 2022, 22, 3231–3248 RSC.
- (a) T. Dohi, D. Koseki, K. Sumida, K. Okada, S. Mizuno, A. Kato, K. Morimoto and Y. Kita, Adv. Synth. Catal., 2017, 359, 3503–3508 CrossRef CAS; (b) K. Kikushima, N. Miyamoto, K. Watanabe, D. Koseki, Y. Kita and T. Dohi, Org. Lett., 2022, 24, 1924–1928 CrossRef CAS PubMed; (c) K. Kikushima, A. Morita, E. E. Elboray, T. Bae, N. Miyamoto, Y. Kita and T. Dohi, Synthesis, 2022, 5192–5202 CrossRef CAS.
- Preparation of TMP-iodonium(III) salts; see: (a) T. L. Seidl, S. K. Sundalam, B. McCullough and D. R. Stuart, J. Org. Chem., 2016, 81, 1998–2009 CrossRef CAS PubMed; (b) V. Carreras, A. H. Sandtorv and D. R. Stuart, J. Org. Chem., 2017, 82, 1279–1284 CrossRef CAS PubMed; (c) E. Lindstedt, M. Reitti and B. Olofsson, J. Org. Chem., 2017, 82, 11909–11914 CrossRef CAS PubMed.
- For selected examples reported by other groups, see: (a) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed; (b) D. R. Stuart, Chem. – Eur. J., 2017, 23, 15852–15863 CrossRef CAS PubMed; (c) S. Basu, A. H. Sandtorv and D. R. Stuart, Beilstein J. Org. Chem., 2018, 14, 1034–1038 CrossRef CAS PubMed; (d) T. L. Seidl and D. R. Stuart, J. Org. Chem., 2017, 82, 11765–1177113 CrossRef CAS PubMed; (e) A. H. Sandtorv and D. R. Stuart, Angew. Chem., Int. Ed., 2016, 55, 15812–15815 CrossRef CAS PubMed; (f) R. T. Gallagher, S. Basu and D. R. Stuart, Adv. Synth. Catal., 2020, 362, 320–325 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- S. Kobayashi, T. Tamura, S. Yoshimoto, T. Kawakami and A. Masuyama, Chem. - Asian J., 2019, 14, 3921–3937 CrossRef CAS PubMed.
- D. Crich and K. Sasaki, the Hunsdiecker and Related Reactions, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. Molander, Elsevier, Amsterdam, 2014, 818–836 Search PubMed.
- For selected examples, see: (a) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Angew. Chem., Int. Ed., 2010, 49, 3334–3337 CrossRef CAS PubMed; (b) N. Yamaoka, K. Sumida, I. Itani, H. Kubo, Y. Ohnishi, S. Sekiguchi, T. Dohi and Y. Kita, Chem. – Eur. J., 2013, 19, 15004–15011 CrossRef CAS PubMed.
- R. Doi, K. Kikushima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 3276–3282 CrossRef CAS PubMed.
- (a) K. R. Roh, J. Y. Kim and Y. H. Kim, Chem. Lett., 1998, 27, 1095–1096 CrossRef; (b) K. R. Roh, J. Y. Kim and Y. H. Kim, Tetrahedron Lett., 1999, 40, 1903–1906 CrossRef CAS; (c) Q. Y. Toh, A. McNally, S. Vera, N. Erdmann and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 3772–3775 CrossRef CAS PubMed.
- Our group has reported the synthesis of stable uracil-iodonium salts and their transformations; see: (a) N. Takenaga, S. Ueda, T. Hayashi, T. Dohi and S. Kitagaki, Heterocycles, 2018, 97, 1248–1256 CrossRef CAS PubMed; (b) N. Takenaga, T. Hayashi, S. Ueda, H. Satake, Y. Yamada, T. Kodama and T. Dohi, Molecules, 2019, 24, 3034 CrossRef CAS PubMed; (c) N. Takenaga, S. Ueda, T. Hayashi, T. Dohi and S. Kitagaki, Heterocycles, 2019, 99, 865–874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04445e |
| This journal is © The Royal Society of Chemistry 2023 |