
Efficient two-step production of biobased plasticizers: dehydration-hydrogenation of citric acid followed by Fischer esterification†
Anthony
De Bruyne
 a,
Wouter
Stuyck
a,
Willem
Deleu
b,
Jarne
Leinders
a,
Carlos
Marquez
a,
Kwinten
Janssens
a,
Dimitrios
Sakellariou
a,
Ruben
Ghillebert
b and
Dirk E.
De Vos
*a
a,
Wouter
Stuyck
a,
Willem
Deleu
b,
Jarne
Leinders
a,
Carlos
Marquez
a,
Kwinten
Janssens
a,
Dimitrios
Sakellariou
a,
Ruben
Ghillebert
b and
Dirk E.
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), K.U. Leuven, Leuven, Flanders, Belgium. E-mail: dirk.devos@kuleuven.be
bCitribel, Tienen, Flanders, Belgium
First published on 21st March 2023
Abstract
We report the production of biobased plasticizers starting from citric acid (CA) by a two-step process comprising dehydration-hydrogenation of CA followed by a Fischer esterification. The use of citric acid based plasticizers is well-known in PVC. However, citrate esters tend to leach out of the PVC material over time. This problem is currently tackled by acetylating the tertiary hydroxyl group of CA via complex and environment polluting processes. Our alternative strategy consists in the removal of the tertiary hydroxyl group, resulting in propane-1,2,3-tricarboxylic acid (PTA). First, dehydration reactions of a large amount of CA (20 mmol) were performed at relatively mild reaction conditions (150 °C and 20 bar H2) using Al2(SO4)3 as a homogeneous catalyst and water as a green solvent. The catalytic system was proven to be robust in time and in the presence of other organic functionalities (e.g. amino acids with different functional groups, diacids). In a second step, the reaction mixture was transferred to a Dean–Stark setup in order to perform a Fischer esterification with n-butanol, during which the previously used Al2(SO4)3 could be recuperated. This resulted in an overall yield of 90% tributyl propane-1,2,3-tricarboxylate (TBPTC), which can be used as a plasticizer in PVC.
Introduction
In 2020, a total of 367 million tons plastic was produced, with the industry employing 1.6 million people distributed over 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 companies.1 These numbers indicate that in addition to the versatile use of plastics, the plastic industry is crucial for today's economy.2 However, plastics consist of more than just the linked monomers; they also contain numerous additives. These can be classified, according to their function, as plasticizers, stabilizers, antioxidants, lubricants or flame retardants.3,4 In general, plasticizers are the most commonly used additives. Their main function is to lower the glass transition temperature (Tg), which translates to higher flexibility and improved processability of the resulting plastic (i.e. shorter mixing time, lower pressure of extrusion, etc.).5 A well-known class of these plasticizers are low molecular weight (LMW) phthalate plasticizers, which until recently were added to polyvinyl chloride (PVC) in large amounts (up to 50 wt%).5–9 However, despite their excellent plasticizing properties, LMW phthalates have notorious adverse effects on the health and development of children due to their endocrine disrupting effects and reprotoxicity, when they leach out of the PVC matrix.10–14 As a result, the use of LMW phthalates has been strongly regulated, with the need for less toxic alternatives rising.15–17 Initially, alternatives with a similar molecular structure such as benzoates,18,19 terephthalates,18,20 trimellitates18,21 and high molecular weight phthalates5,18,22 were developed. These have a lower tendency to leach out of the PVC material. However, these components may still possess a certain toxicity. Aliphatic alternatives based on adipic acid,18,23 azelaic acid18,24 and sebacic acid18,25 are considered safe; however their field of application is limited.18,23
000 companies.1 These numbers indicate that in addition to the versatile use of plastics, the plastic industry is crucial for today's economy.2 However, plastics consist of more than just the linked monomers; they also contain numerous additives. These can be classified, according to their function, as plasticizers, stabilizers, antioxidants, lubricants or flame retardants.3,4 In general, plasticizers are the most commonly used additives. Their main function is to lower the glass transition temperature (Tg), which translates to higher flexibility and improved processability of the resulting plastic (i.e. shorter mixing time, lower pressure of extrusion, etc.).5 A well-known class of these plasticizers are low molecular weight (LMW) phthalate plasticizers, which until recently were added to polyvinyl chloride (PVC) in large amounts (up to 50 wt%).5–9 However, despite their excellent plasticizing properties, LMW phthalates have notorious adverse effects on the health and development of children due to their endocrine disrupting effects and reprotoxicity, when they leach out of the PVC matrix.10–14 As a result, the use of LMW phthalates has been strongly regulated, with the need for less toxic alternatives rising.15–17 Initially, alternatives with a similar molecular structure such as benzoates,18,19 terephthalates,18,20 trimellitates18,21 and high molecular weight phthalates5,18,22 were developed. These have a lower tendency to leach out of the PVC material. However, these components may still possess a certain toxicity. Aliphatic alternatives based on adipic acid,18,23 azelaic acid18,24 and sebacic acid18,25 are considered safe; however their field of application is limited.18,23
Much research has focused on various biobased alternatives to phthalates, aiming at low toxicity and a low migration.18 Citric acid, from fermentation, is a particularly interesting starting point to synthesize high quality biobased plasticizers. However, esters of citric acid, like tributyl citrate, have been found to leach significantly from PVC.26–29 Initial attempts to mitigate this focused on the acetylation/butyrylation of the hydroxyl group (–OH) of citric acid, aiming at decreasing the polarity of the plasticizers, hence increase their compatibility with the PVC matrix.22,30–35 Nevertheless, these reactions are carried out through environment polluting processes.30–35
An alternative strategy is to selectively deoxygenate the polar hydroxyl group of citric acid prior to esterifying the remaining carboxylic acid groups. An initial study of Verduyckt et al. reported a green synthesis route of methylsuccinic acid (MSA) directly from citric acid by using heterogeneous Pd0 or Ni0 catalysts in water as a solvent.36,37 Although high yields of methylsuccinic acid were obtained, a major disadvantage remained the formation (and thus loss) of a stoichiometric amount of CO2 and concurrently a branch of the initial citric acid skeleton. The latter might negatively affect the plasticizing properties of the corresponding methylsuccinates, although succinate plasticizers have proven to be performant in PVC.36–40 Recent studies have shifted from the production of MSA to the production of propane-1,2,3-tricarboxylic acid (PTA), hence solely removing the hydroxyl group through a dehydration-hydrogenation pathway. PTA can subsequently be esterified to tricarballylate plasticizers, which can effectively plasticize vinyl chloride polymers.41,42 Initial research showed that the combination of Pd0/C with a 12-membered ring H-Beta zeolite resulted in high propane-1,2,3-tricarboxylic acid (PTA) yields (85%) from citric acid.43 However, the strong complexation between citric acid and framework Al3+ resulted in dealumination of the zeolite material, which negatively influenced the recyclability of the catalyst. In a later study by Li et al., m-ZrO2 was combined with a Pt0/TiO2 hydrogenation catalyst. However, only moderate PTA yields (65%) were obtained and a low concentration (0.05 M) of citric acid was applied.44 More recently, a stable Pd0/Nb2O5·nH2O catalyst was synthesized which yields >90% PTA over multiple runs. Although the system proved resistant towards the corrosive citric acid, again low concentrations were applied (0.1 M) while the synthesis of niobium-based catalysts is relatively costly.45 In short, all of the previously developed systems would fail at least one of the following criteria for industrial development: high yields of PTA, stable and recyclable catalysts and a low catalyst cost.
The aim of this research is to convert citric acid into high quality biobased plasticizers via a two-step process. In a first step, citric acid will be converted into deoxygenated products, mainly PTA and small amounts of MSA, using affordable homogeneous Lewis acids as dehydration catalysts and Pd0/C as hydrogenation catalyst. In a second step, the formed PTA and MSA are converted to their corresponding esters via Fischer esterification using a Dean–Stark setup. Remarkably, the same catalyst (i.e. Al2(SO4)3) facilitates the dehydration and the esterification and later precipitates, allowing its recovery and possible recycling.
Experimental section
Catalytic reactions
For a standard dehydration-hydrogenation experiment, a glass liner (5.5 mL) was filled with citric acid (0.2 or 2 mmol), Pd0/C (0.5 mol% Pd0), a homogeneous or heterogeneous Lewis acid catalyst (50 mol% or 0.5 eq.) and deionized H2O (2 mL). A magnetic stirring bar was then added; the glass liner was partially closed with a Teflon stopper and placed in a stainless steel reactor (12 mL). The reactor was sealed and purged three times with both N2 and H2, after which a pressure of 20 bar H2 was applied. After this, the reactor was heated to 150 °C (reaction temperature) at a stirring speed of 750 rpm for 20 h. After the reaction, the reactor was cooled to room temperature before being depressurized. The liquid products were recovered by centrifugation (4000 rpm, 10 minutes) and analyzed by 1H-NMR spectroscopy and/or liquid chromatography (HPLC).Reactions were upscaled in a Hastelloy Premex batch reactor (60 mL), following an analogous procedure. Here, the glass liner (50 mL) was filled with citric acid (20 mmol), Pd0/C (0.5 mol% Pd0), Lewis acid (25 mol%) and deionized H2O (20 mL).
Fischer esterification reactions
After removal of the Pd0/C catalyst by centrifugation, the crude reaction mixtures were transferred to a glass round bottom flask (250 mL) containing 30 mL of n-butanol, which was later attached to a Dean–Stark apparatus. The Fischer esterification proceeded at 135 °C for 20 h. Over this period, n-butanol was added repeatedly (3 times 10 mL) and water was removed. After the Fischer esterification, the excess n-butanol was evaporated using a rotary evaporator (60 °C at 25 mbar) and the products with unreacted carboxylic acid groups were removed by deprotonation using a small amount of sodium carbonate (Na2CO3) and extraction in diethyl ether–water. Finally, the diethyl ether was evaporated using the rotary evaporator and the residual oil was dried with a Schlenk line overnight at room temperature under vacuum. The remaining mass was confirmed to be 93% tributyl propane-1,2,3-tricarboxylate (TBPTC) and 7% dibutyl methyl succinate (DBMS) via1H-NMR.Recycling procedure
For the catalyst recycling experiment, the scaled-up protocol was followed. The Pd0/C was separated from the reaction mixture via centrifugation (4000 rpm, 10 min) prior to the Fischer esterification (4000 rpm, 10 min). The recovered mass was washed thrice with methanol and H2O, after which it was dried in an oven at 60 °C for 96 h. Next, Al2(SO4)3 was dissolved in H2O and washed thrice with diethyl ether to remove any organics, after which Al2(SO4)3 was dried in an oven at 60 °C during 24 h. The resulting mass was dried with a Schlenk line overnight at room temperature under vacuum. The obtained catalyst was reused. After 4 runs, the resulting dry mass (75%) was complemented with an amount of fresh Al2(SO4)3 to achieve the same initial amount of 0.125 equivalents.Product analysis and identification
ICP-OES was used to determine the amount of Al2(SO4)3 in the salt precipitate, recovered during the Dean–Stark operation. The salt precipitate was dissolved in H2O and washed three times with diethyl ether. The H2O phase was separated and dried first in an oven (60 °C) and later at a Schlenk line overnight. The dried mass was once again dissolved in H2O and analyzed in a Varian 720 ICP-OES device, equipped with a glass cyclonic spray chamber, a Sea Spray glass concentric nebulizer and a high sensitivity torch.The pH measurements were performed using a SympHony VWR pH meter. A calibration was performed between pH 4.01 and 7.
To determine the amounts of CA, PTA, MSA, IA (itaconic acid) and fragmentation products, the crude reaction mixture was analyzed by 1H-NMR spectroscopy. An NMR tube was filled with 200 μL of reaction sample (liquid phase, each time diluted to 0.1 M) and 300 μL of a 0.067 M maleic acid solution in D2O (external standard). Measurements were made at room temperature on a Bruker Avance 400 MHz NMR spectrometer equipped with a BBO 5 mm probe. A modified ZGPR pulse program was used (1TD = 65536, DS = 4; NS = 32; D1 = 12 s; AQ = 3.28 s; SW = 25 ppm; O1P 4.700 ppm) to suppress the large interference of the solvent (H2O).
To determine the composition of the TBPTC and DBMS mixture, the purified product was analyzed by 1H-NMR spectroscopy. An NMR tube was filled with 250 μL of solution containing 34.4 mg of product in CD3OD (liquid phase) and 250 μL of a 0.1 M p-xylene solution in CD3OD (external standard). Measurements were made at room temperature on a Bruker Avance 400 MHz NMR spectrometer equipped with a BBO 5 mm probe. A modified ZGPR pulse program was used (1TD = 65536, DS = 4; NS = 32; D1 = 5 s; AQ = 3.28 s; SW = 25 ppm; O1P 4.700 ppm).
To determine the aluminum species 27Al liquid-state NMR experiments were performed with a Bruker 600 magnet and BBO5 probe at 25 °C and 80 °C. The FID was recorded with 15000 points and 1s of acquisition time, as well as repetition delay between each of the 64 scans. The spectral window was 400 ppm and the transmitter frequency was set to 156.38 MHz. The NMR signal was apodised with a linear broadening of 1 Hz and Fourier transformed to obtain the spectra. The total experimental time per experiment was about 2 minutes. The time required for the temperature equilibration was at least 15 min after reaching the desired temperature.
Additionally, HPLC analysis was used to determine the amount of CA and PTA after reaction. The samples were analyzed with an Agilent Technologies 1200 series SL quaternary liquid chromatograph, equipped with a G1322A degasser, a G1311A quaternary pump, a G1367A automated injector, a G1316A column compartment and a DAD detector. The separation of the solvent (H2O), CA and PTA was performed with an Acclaim Organic Acid column (250 mm × 4.0 mm i.d., 5.0 μm particles) at 40 °C. The mobile phase consisted of a 0.1 M Na2SO4 solution in Milli-Q water at a pH of 2.65 (reached after addition of methanesulfonic acid (CH3SO3H)) and was passed over the column at a flow rate of 0.6 mL min−1. The components were detected at a wavelength of 212 nm.
The free specific metal surface of the Pd0/C (i.e. hydrogenation catalyst) was examined via CO chemisorption. Both fresh Pd0/C and Pd0/C after reaction with pure CA and CA with cysteine (20 mol%) were examined. The used Pd0/C was first washed with H2O (3 times 10 mL) and dried overnight (60 °C). The measurements were performed with a ChemBET Pulsar TPR/TPD. The samples (100 mg) were prepurged with a H2 stream overnight (12 h). Next, the samples were subjected to pulses of CO (75 μL each), assuming a stoichiometry of two CO molecules per Pd0 atom.46 All measurements were performed at room temperature and atmospheric pressure. The obtained adsorption peaks were converted to free specific metal surface and Pd0 dispersion using the software Quantachrome TPRWin v4.10.
Results and discussion
Sequential dehydration-hydrogenation of citric acid to PTA
The deoxygenation of citric acid to PTA comprises a dehydration to aconitic acid followed by a hydrogenation (Scheme 1).36,43 Optimal reaction conditions were selected to ensure high PTA yields.43–45 Reactions were performed at 150 °C in water with 10 bar H2, and a reaction time of 20 h. Since the dehydration of citric acid may be catalyzed by solids possessing both Brønsted and Lewis acid sites on their surface,43–45 different homogeneous acid catalysts were tested (Table 1) in combination with a hydrogenation catalyst, Pd0/C. Previous studies already reported the use of Al3+ ions (i.e. Al(OH)3 and Al2(SO4)3) for the initial dehydration reactions of citric acid to PTA.43,44 While homogeneous catalysts generally tend to be more active, their recovery from the product stream is in many instances problematic. For this reason, we selected a series of homogeneous catalysts that may still be recovered after the next reaction step (Fischer esterification).47,48 | ||
| Scheme 1 The deoxygenation products obtained from citric acid. | ||
| Carbon yield [%] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Lewis acid | Amount Cat.b [equiv.] | X [%] | PTAd [%] | MSAe [%] | IAf [%] | Fragm.g [%] | Mass balanceh [%] | |
| a Reaction conditions: water (2 mL), 0.1 M citric acid, 0.5 mol% Pd0, 1 equivalent of H2SO4, 10 bar H2, 150 °C and a reaction time of 20 h. b Amount of Lewis acid catalyst in equivalents with respect to citric acid. c Conversion i.e. the amount of citric acid that has reacted. d Propane-1,2,3-tricarboxylic acid. e Methylsuccinic acid. f Itaconic acid. g Fragmentation products, which are represented by acetone and acetic acid. h Overall mass balance expressed in carbon yield (ESI†). i Without H2SO4. | ||||||||
| 1i | — | — | 17 | 10 | 4 | 0 | 1 | 98 |
| 2 | — | — | 24 | 15 | 4 | 0 | 1 | 96 |
| 3 | AlCl3·H2O | 0.5 | 95 | 90 | 3 | 0 | 1 | 99 |
| 4 | Al2(SO4)3 | 0.25 | 96 | 90 | 4 | 0 | 2 | >99 |
| 5 | Al(OH)3 | 0.5 | 88 | 85 | 2 | 0 | 0 | 99 |
| 6 | FeCl3·6H2O | 0.5 | 16 | 13 | 0 | 0 | 1 | 99 |
| 7 | FeO(OH) | 0.5 | 45 | 36 | 3 | 0 | 3 | 95 |
| 8 | ZrCl4 | 0.5 | 27 | 26 | 0 | 0 | 1 | 99 |
| 9 | Zr(OH)4 | 0.5 | 31 | 23 | 1 | 0 | 6 | 99 |
| 10 | LaCl3 | 0.5 | 19 | 18 | 1 | 0 | 0 | >99 |
| 11 | La(OH)3 | 0.5 | 14 | 12 | 1 | 0 | 1 | 99 |
| 12 | DyCl3·6H2O | 0.5 | 22 | 21 | 1 | 0 | 1 | >99 |
| 13 | YbCl3·6H2O | 0.5 | 26 | 24 | 0 | 0 | 1 | >99 |
| 14 | Ga(NO3)3 | 0.5 | 71 | 43 | 24 | 0 | 1 | 96 |
| 15 | MgCl2 | 0.5 | 13 | 12 | 0 | 0 | 1 | 96 |
| 16 | CaCl2 | 0.5 | 18 | 17 | 1 | 0 | 0 | 94 |
| 17 | CaCO3 | 0.5 | 23 | 18 | 1 | 0 | 4 | 96 |
| 18 | ZnCl2 | 0.5 | 16 | 15 | 1 | 0 | 0 | >99 |
| 19 | InCl3 | 0.5 | 7 | 0 | 0 | 6 | 0 | 99 |
| 20 | SnCl2·2H2O | 0.5 | 19 | 0 | 1 | 13 | 2 | 83 |
| 21 | BiCl3 | 0.5 | 27 | 0 | 2 | 20 | 1 | 78 |
| 22 | BiOCl | 0.5 | 19 | 13 | 5 | 0 | 0 | 98 |
As a starting point, several homogeneous Lewis acids as well as a Brønsted acid (H2SO4) were investigated (Table 1). In absence of both types of acids a low conversion of 17% and a PTA carbon yield of 10% were obtained (entry 1), which were only slightly increased by adding H2SO4 (entry 2). Particularly the addition of aluminum salts resulted in high conversions, with PTA carbon yields reaching 90% for Al2(SO4)3 and AlCl3, and 85% for Al(OH3) (entries 3–5). The slightly higher carbon yield of PTA, obtained with AlCl3 and Al2(SO4)3 can be explained by the higher solubility of these salts (when compared to Al(OH)3), which results in a larger number of Al3+ ions present in the solution (Table S3 and Fig. S3, ESI†).48 These results suggest that the high conversions of citric acid are a result of the interaction between dissolved Al3+ ions and citric acid, hence activating the tertiary hydroxy group and catalyzing the dehydration reaction.49–51 Since it has been reported that iron citrate (Fe-citrate) speciation in acidic solutions are similar regarding structure and stoichiometry to aluminum citrate (Al-citrate) complexes,49 FeCl3 and FeO(OH) were also evaluated for the dehydration of citric acid (entries 6 and 7). However, these iron salts resulted in low citric acid conversions. This may be attributed to the larger ionic radius of Fe3+ (compared to Al3+), giving rise to the formation of more stable complexes.49,51 This increased stability could lead to a lower dehydration activity. Next, lanthanide ions were considered (entries 10–13) in view of their high ligand exchange rates.52 However, the use of La3+, Yb3+ and Dy3+ resulted in low conversions of citric acid (26%–33%). Additionally, since Al3+ is known to be a hard ion, other hard ions, like Ga3+, Mg2+ and Ca2+, were tested (entries 14–17).53 Ga(NO3)3 and to a lesser extent CaCl2 and MgCl2 showed some dehydration activity but were inferior to Al2(SO4)3. To provide a complete overview, softer elements were also tested. Whereas ZnCl2 and BiOCl (entries 18 and 21) turned out to be mostly inactive, SnCl2·H2O and BiCl3 (entries 20 and 21) tended to deactivate the hydrogenation catalyst, since a notable amount of unsaturated itaconic acid but almost no hydrogenated products were detected. In the end Al2(SO4)3 was selected for further experiments.
Interaction between the Al3+ ion and citric acid
Results of the catalyst screening showed that aluminum salts are excellent homogeneous catalysts for the dehydration of citric acid during the formation of PTA. It could be argued that Lewis acidic metal salts (e.g. AlCl3) would be poorly active in water, due to the formation of mono- and polynuclear hydroxy species.50,51 However, several studies show that the speciation of Al3+ ions in water is highly dependent on their concentration and on the pH of the solution.49–56 Additionally, Al3+ ions and citric acid are known to strongly interact with each other resulting in the formation of various Al-citrate complexes.50,51 To investigate more closely the influence of the pH on the PTA carbon yield, reactions with Al2(SO4)3 were performed at various pH values by adding different amounts of H2SO4 or NaOH. Without the addition of H2SO4 or NaOH, the solution of citric acid and Al2(SO4)3 (0.1 M citric acid; ratio citric: Al = 2) has an initial pH of 1.76 at room temperature, resulting after reaction in a PTA carbon yield of 82% (Fig. 1). Both CA conversion (96%) and PTA carbon yield (90%) reached a maximum at a pH value of 1.3 (1 eq. of H2SO4) with very low amounts of decarboxylation products (2% MSA). A lower pH value of 1.13 (2 eq. H2SO4) led to a slightly lower CA conversion (86%) and PTA carbon yield (82%). In contrast, at higher pH values, a marked drop in CA conversion and PTA carbon yield is noticed, with a pH of 4.15 (2 eq. NaOH) resulting in a carbon yield of only 8% PTA and 16% of MSA. This indicates that there is an optimal pH range (between 1.1 and 2), at which the presence of catalytically active aluminum species is most favored, with the maximal product formation observed for a solution with a starting pH of 1.3.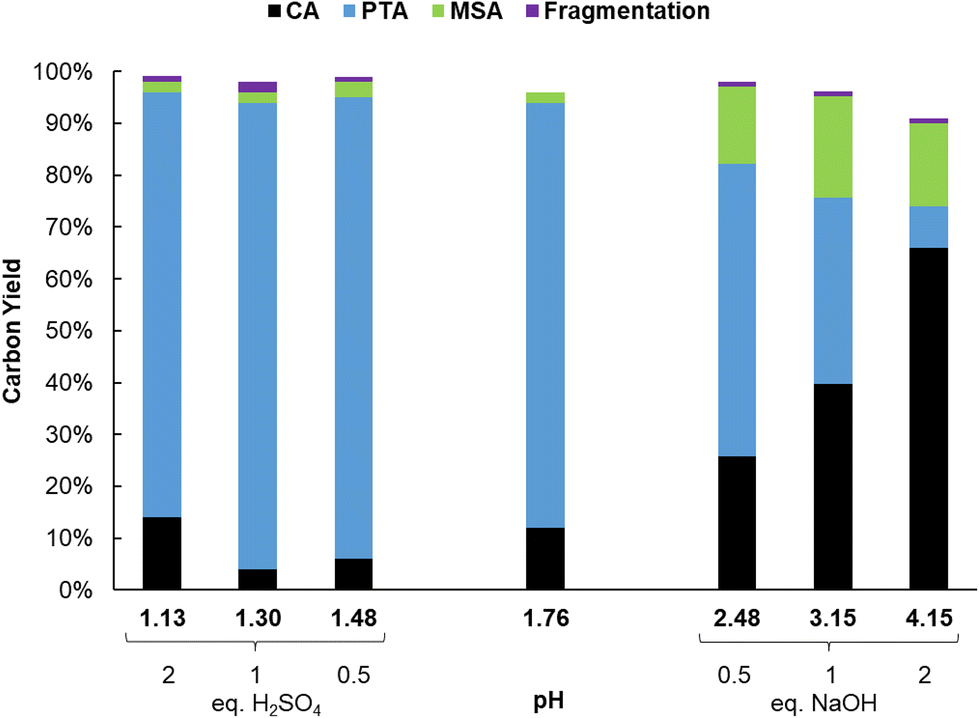 | ||
| Fig. 1 Dehydration-hydrogenation of citric acid. Reaction in water (2 mL) with citric acid (0.2 mmol), Al2(SO4)3 (0.05 mmol), Pd0/C (0.5 mol% Pd0), 20 h with 10 bar H2 at 150 °C. Different amounts (0.5; 1 and 2 equivalents) of H2SO4 and NaOH. | ||
The complexation of aluminum and citric acid has been extensively studied,49–51 resulting in different complexation models, but without a clear consensus. Fortunately, regarding the species under the most acidic conditions (pH ≤ 3), it seems that only three complexes are prevalent: [Al-Hcit]+, [Al-cit]0 and [Al-(cit)2]3− (Fig. 2; ‘cit’ represents a citrate anion with three negative charges). Given the studied conditions (pH range, concentration of Al3+ ions and citric acid), these complexes are expected to be present in the reaction mixture.51,57 In addition, free aluminum species can be present. These include Al(H2O)63+ and its different hydrolysis products, mainly monomeric species such as [Al(OH)(H2O)5]2+ and [Al(OH)2(H2O)4]+,50,51,56 and small amounts of dimeric species (e.g. [Al2(OH)2(H2O)8]4+).56 Finally, the use of Al2(SO4)3 may result in the presence of [Al(SO4)(H2O)5]+, with a SO42− anion in the coordination sphere instead of H2O.56
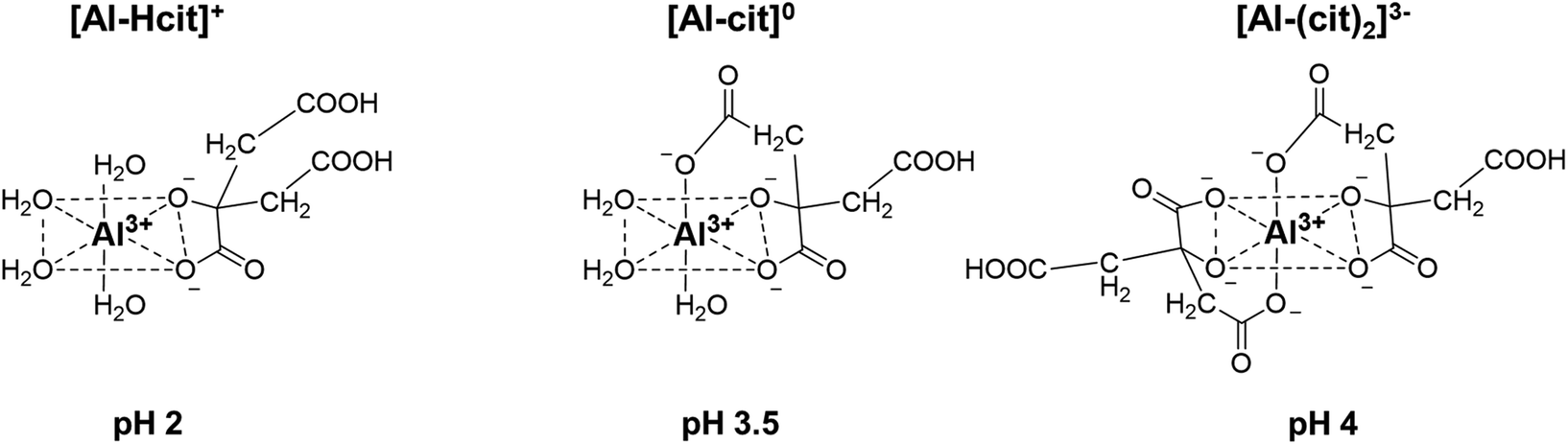 | ||
| Fig. 2 Structures of different Al-citrate complexes and pH values at which their presence is most dominant.50,51 | ||
In an attempt to identify the catalytically relevant aluminum species, liquid 1H- and 27Al-NMR measurements were performed on solutions with the same compositions as the reaction mixtures (i.e. 0.1 M citric acid and 0.05 M Al3+; Table S4, ESI†). The 27Al-NMR spectra (Fig. 3) showed that as the pH increases, the intensity of the signal at 0.9 ppm, corresponding to Al(H2O)63+ (and hydrolysis products [Al(OH)(H2O)5]2+ and [Al(OH)2(H2O)5]+), and −2.4 ppm, corresponding to [Al(SO4)(H2O)5]+ gradually decreases. These signals remain prominent up to a pH of 1.76, but at higher pH their intensity decreases more sharply, and they disappear at a pH around 4. This can be explained by the change in coordination of the Al3+ ion, from octahedral complexes to tetrahedral hydroxy species.51 The chemical shift of the signal at 6–8 ppm corresponds to Al-citrate complexes [Al-Hcit]+, [Al-cit]0 and [Al-(cit)2]3−. Starting at pH 1.76, this peak initially increases, becomes most distinct at a pH value of 2.48, before decreasing and finally almost disappearing at pH 4. Again, this can be understood as an evolution from octahedral compounds to tetrahedral or oligomeric species, like [Al3(OH)(H−1Cit)3]4− (H−1Cit is a fourfold deprotonated citrate anion with charge −4).51 As mentioned earlier, there is little consensus on the speciation above a pH of 4 in the literature. In addition, the linewidth of the peak increased at higher pH values, which can be a result of a fast exchange between citrate and OH− ligands.50 Below a pH value of 1.5, the 27Al-NMR spectra do not give evidence for the formation of Al-citrate complexes.
 | ||
| Fig. 3 27Al-NMR spectra of mixtures containing 0.1 M of citric acid and 0.25 eq. Al2(SO4)3 at different pH levels: pH 1.15 (2 eq. H2SO4), pH 1.30 (1 eq. H2SO4), pH 1.48 (0.5 eq. H2SO4), pH 1.76 (no additive), pH 2.48 (0.5 eq. NaOH), pH 3.15 (1 eq. NaOH) and pH 4.15 (2 eq. NaOH). The signal at 6–8 ppm corresponds to the Al-citrate complexes; the signal at 0.9 ppm corresponds to non-chelated Al.50,51,56 | ||
The complexation of Al3+ by citrate in the reaction mixture was also studied by 1H-NMR (Fig. 4). At a pH ≤ 1.48, no Al-citrate complexes were observed and the peaks corresponded only to non-coordinated citric acid. Increasing the pH resulted in a shift of the characteristic citric acid peaks to higher field. At a pH of 1.76 and 2.48 the most dominant complex is [Al-Hcit]+, while at a pH of 3.15 the [Al-cit]0 complex prevailed.51 Higher pH values resulted in multiple peaks which were difficult to assign to specific complexes.
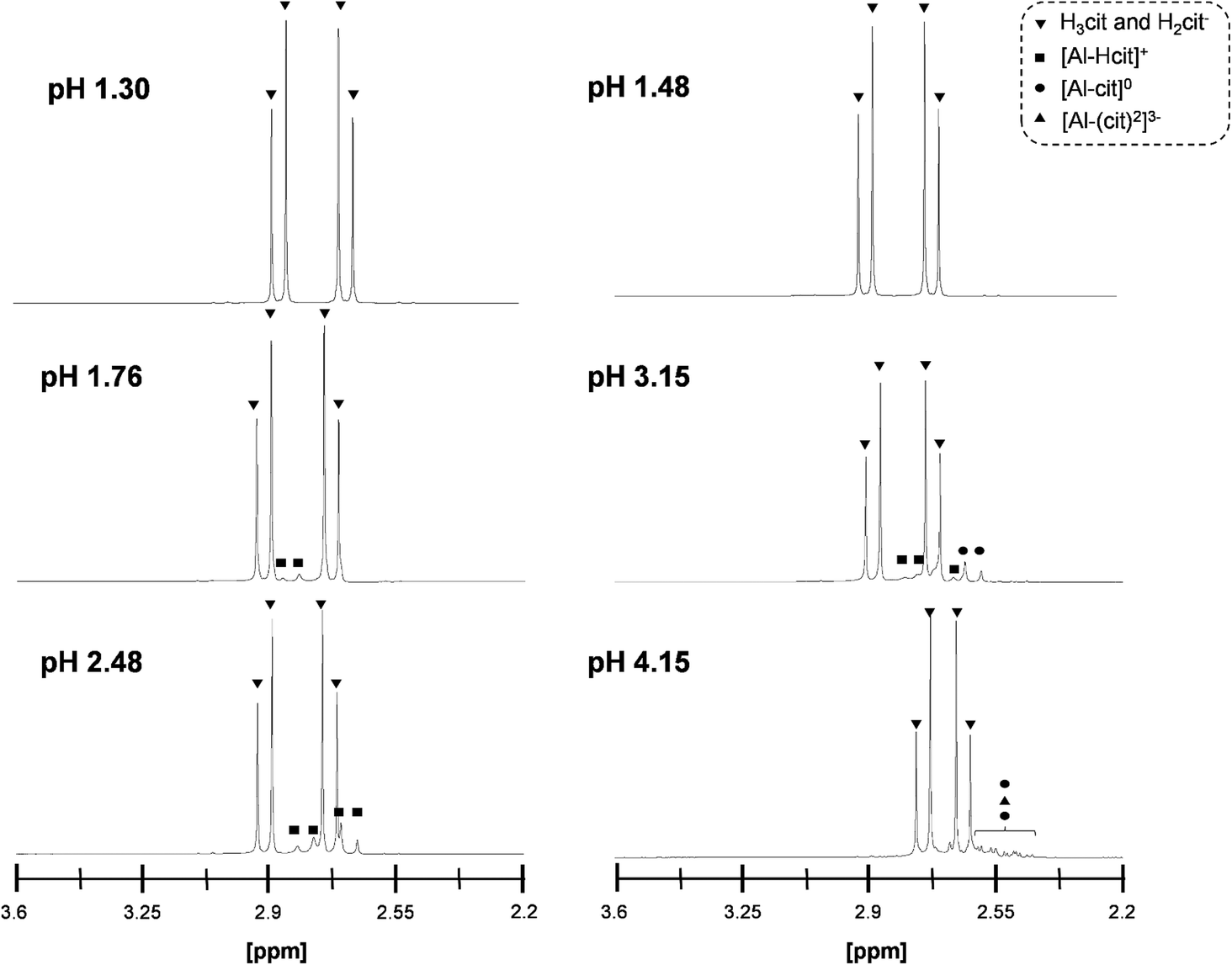 | ||
| Fig. 4 1H-NMR spectra of mixtures containing 0.1 M of citric acid and 0.25 eq. Al2(SO4)3 at different pH levels: pH 1.30 (0.5 eq. H2SO4), pH 1.48 (1 eq. H2SO4), pH 1.76 (no additive), pH 2.48 (0.5 eq. NaOH), pH 3.15 (1 eq. NaOH) and pH 4.15 (2 eq. NaOH).50,51,56 | ||
These results, obtained at 0.05 M [Al]total, may suggest that the availability of free aluminum species (Al(H2O)63+, [Al(OH)(H2O)5]2+, [Al(OH)2(H2O)4]+ and [Al(SO4)(H2O)5]+) is important for the catalytic dehydration of citric acid, since the highest PTA yields were obtained at pH values where seemingly no Al-citrate complexes were observed. It must be remarked however, that the sensitivity of NMR to detect small concentrations of (chelated) Al3+ is not very high, especially if the peaks are broad and the concentration moderate (0.1 M). Moreover, the measured pH values are initial values, and since PTA is a weaker acid than citric acid, the pH is expected to increase during reaction. Therefore, the measured pH values likely underestimate the typical pH values at intermediate conversion. Finally, pH values were measured at room temperature, while the actual reaction proceeds at 150 °C; both acid dissociation and complexation equilibria may be temperature dependent. Therefore, 27Al-NMR measurements were repeated at a 10-fold increased concentration, which is catalytically even more relevant (cfr. infra), and at varying temperatures between 25 and 80 °C (Fig. 5).
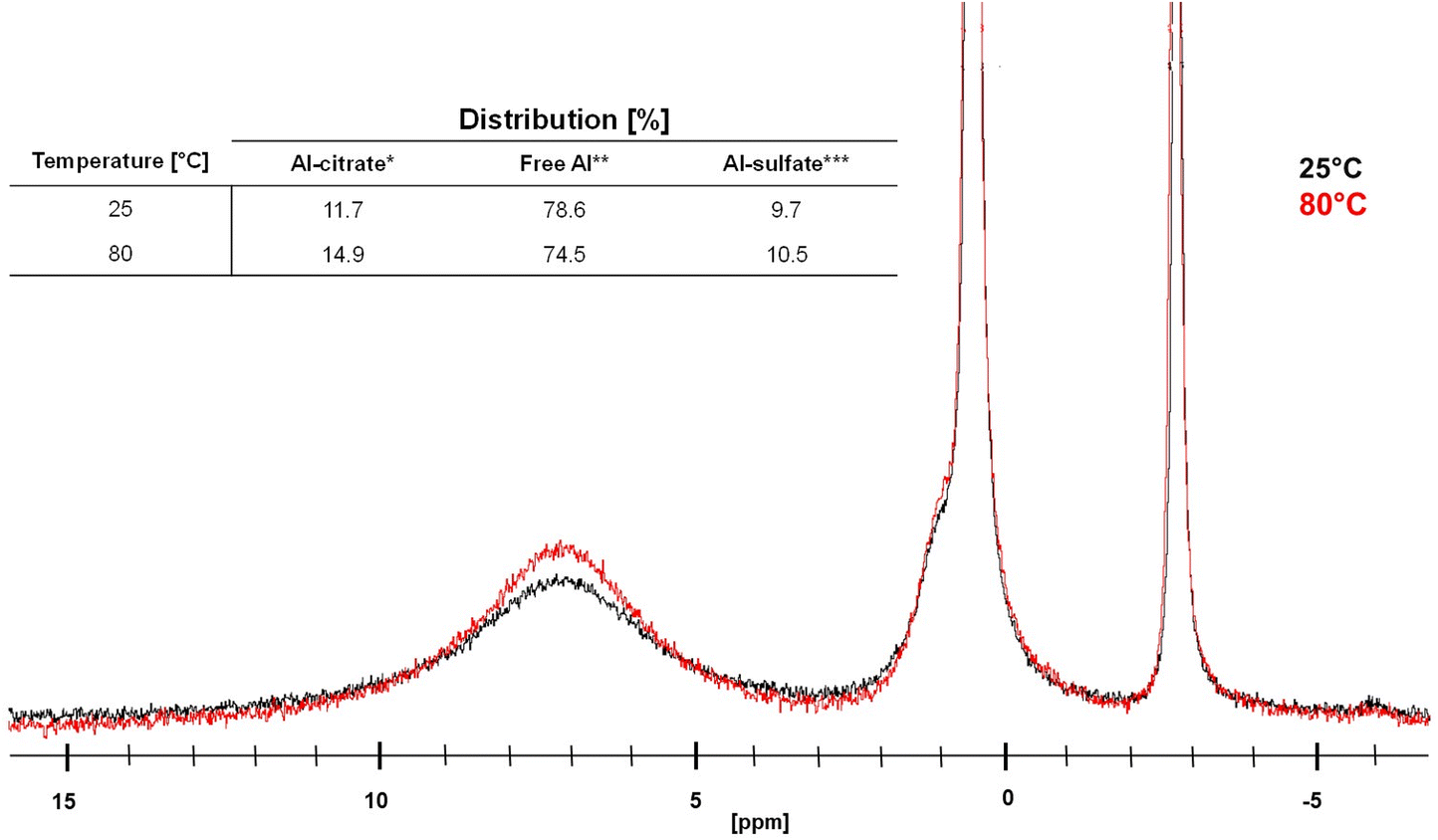 | ||
| Fig. 5 27Al-NMR spectra of mixture containing 1 M of citric acid and 0.125 eq. Al2(SO4)3 at 25 °C (black) and 80 °C (red), with a clear increase of the signal at 6–8 ppm corresponds to the Al-citrate complexes. *Al-citrate includes [Al-Hcit]+, [Al-cit]0, and [Al-(cit)2]3−; **Free Al includes Al(H2O)63+, [Al(OH)(H2O)5]2+ and [Al(OH)2(H2O)5]+ and ***Al-sulfate includes [Al(SO4)(H2O)5]+. | ||
The results show that temperature does have an effect on the complexation between Al3+ ions and citric acid, since from a temperature of 70 °C onwards a clear increase of the amount of Al-citrate in the mixture was observed. Since catalytic reactions are performed at even higher temperatures (i.e. 150 °C), it can be expected that Al-citrate complexes will indeed be formed within the reaction mixture. The majority of these complexes are likely [Al-Hcit]+, which is the predominant speciation at low pH values (vide supra).50,51,56
Based on these findings, a reaction mechanism is proposed for the homogeneously catalyzed dehydration of CA in presence of Al3+ ions (Scheme 2), in which the Al(H2O)63+ coordinates with the citric acid, via two carboxylic groups and the tertiary hydroxy group (1), creating a reactive complex (∼[Al-Hcit]+ or [Al-cit]0). If the pH is too low, only small amounts of active Al-citrate complexes are available, and the reaction is suppressed. In the complex, the tertiary hydroxyl group has been proposed to bind in the deprotonated, alcoholate state. We propose the reaction to start by net transfer of an –OH group from the citric acid backbone to the Al3+ center; obviously this requires that the tertiary hydroxy group is in a protonated state. This explains that the pH must be sufficiently low for the reaction to proceed fast; indeed, upon protonation, a –OH group is formed which is a much better leaving group than the alcoholate (–O−). The loss of the tertiary hydroxyl group weakens the complex,49 most likely leading to its decomposition into free aluminum species (i.e. [Al(OH)(H2O)5]2+) and an intermediate carbocation (2). Next, the hydroxyl group in the coordination sphere of the Al3+ can act as a base and accept a proton from a –CH2– adjacent to the carbocation, leading to formation of aconitic acid (3). The double bond in aconitic acid is highly reactive and is hydrogenated to PTA under the applied conditions (150 °C, 10 bar H2, Pd0/C). The free Al3+ (i.e. Al(H2O)63+) is restored and can in turn interact with a new citric acid molecule repeating the catalytic process (4).
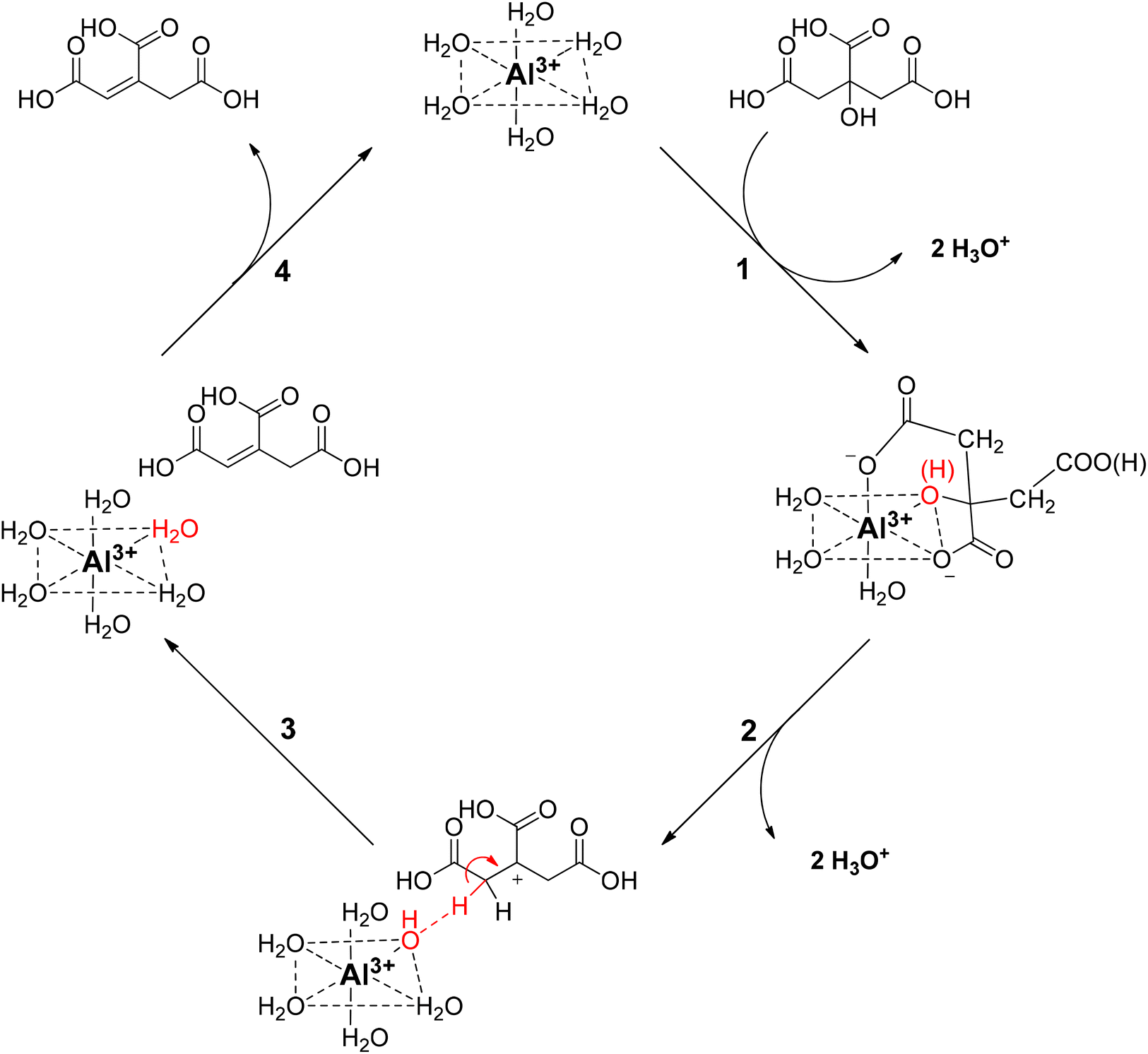 | ||
| Scheme 2 Proposed reaction mechanism for the homogeneously catalyzed dehydration of citric acid with Al3+ ions. | ||
When performing the reaction at a 0.1 M citric acid concentration, lowering the pH by adding sulfuric acid was beneficial to increase the reaction rate. However, since citric acid itself is a Brønsted acid, a higher substrate concentration can be used to lower the pH instead. As mentioned before, a higher concentration of citric acid and Al3+ may also drive the equilibrium of the complexation of Al3+ with citrate to the right, favoring formation of reactive chelates.50,51 Several reactions were performed to optimize the concentration of Al2(SO4)3 in presence of higher concentrations of citric acid (Table S7, ESI†). A carbon yield of 88% PTA was obtained starting from 1 M citric acid, 0.125 eq. Al2(SO4)3, 20 bar H2 at 150 °C after 20 h of reaction time. The initial composition of the reaction mixture resulted in a pH of 1.16 (at 25 °C) which is lower than in the previous experiments. Meanwhile, 27Al-NMR measurements clearly show the presence of Al-citrate complexes (Fig. S4, ESI†), and 1H-NMR confirmed them to be [Al-Hcit]+ (Fig. S5, ESI†). Next, the reaction was scaled-up a tenfold with the use of 60 mL pressure reactor. A carbon yield of 93% of PTA was achieved under the same reaction conditions, confirming the scalability of the dehydration-hydrogenation process. Based on this upscaled result, the reaction time was varied to record a time profile (Fig. 6). After a reaction time of 20 h, a maximum carbon yield of 93% of PTA is achieved. This value remained relatively constant; however, small losses (∼4%) were observed after 48 h which may be the result of fragmentation36 and hydrogenolysis reactions58 (Scheme S1, ESI†). In addition, after 20 h of reaction time, the pH of the solution has increased to 1.46, as a result of PTA being a weaker acid compared to citric acid. Overall, the data indicate a high stability of PTA.
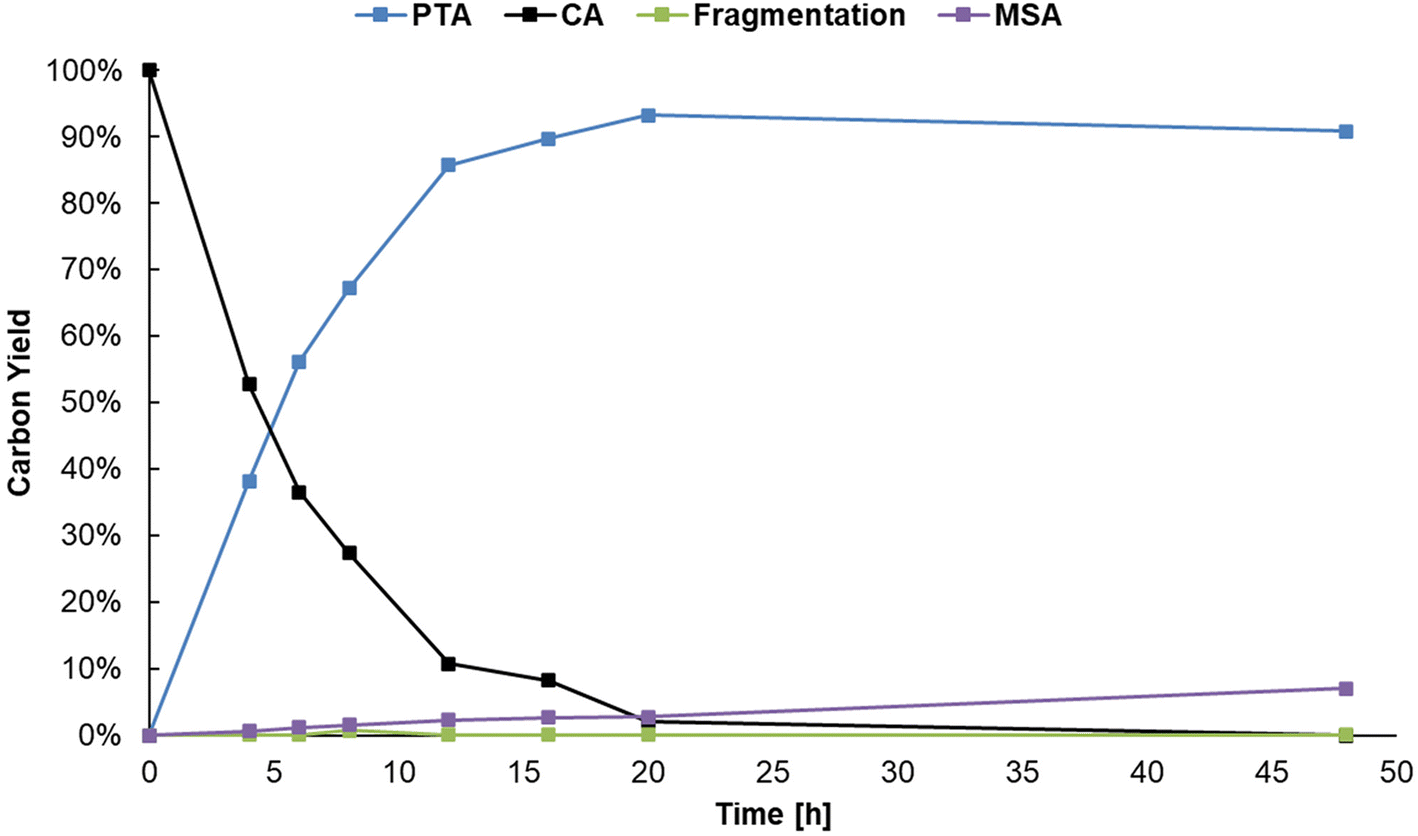 | ||
| Fig. 6 Time profile of the dehydration-hydrogenation of citric acid. Reaction in water (20 mL) with citric acid (20 mmol), Al2(SO4)3 (1.25 mmol), Pd0/C (0.5 mol% Pd0) with 20 bar H2 at 150 °C. | ||
Robustness of the dehydration-hydrogenation system
Next, the robustness of the dehydration-hydrogenation of citric acid to PTA was tested. Since purification can imply a substantial cost during the production of citric acid, a robust system is preferred because it would allow to circumvent at least certain purification steps, hence the use of a less purified stream. During the catalyst screening (Table 1) it was already shown that different inorganic ions (which may even be present in tap water) actually catalyze the dehydration of citric acid; thus proven not to be problematic for the deoxygenation process. Next to inorganic salts, both amino acids and sugar molecules can also be present at some point in the fermentative production of citric acid.59 As such, the influence of S-containing amino acids was evaluated (Fig. 7 and Fig. S10, ESI†). Reactions with different amounts of cysteine and methionine showed that the hydrogenation catalyst (i.e. Pd0/C) is poisoned during the process. The hydrogenation activity was not completely suppressed, since MSA was still present in the reaction mixture, however, the activity of Pd0 decreased to such an extent that the hydrogenation rate was too slow to significantly suppress the spontaneous aconitic acid decarboxylation. This phenomenon was more pronounced with cysteine than with methionine. These results are expected, since sulfur-containing compounds are notorious for poisoning noble metals.60 Secondly, the presence of another organic acid (i.e. glutaric acid) and an alcohol (i.e. threonine) was evaluated. Both components appeared to exert a limited influence on the system, only resulting in small decreases in conversion, compared to the standard reaction (89%): a PTA carbon yield of 70% and 66% were observed at a concentration of 5 mol% glutaric acid and 5 mol% threonine, respectively. Lastly, the influence of a monosaccharide was tested (i.e. glucose). While no major influence on the selectivity was observed, the obtained conversions were significantly lower (39%–61%). These reduced conversions are a result of the added organic components, or derivatives, reacting under the given reaction conditions.52,61,62 In this way the components compete with the citric acid for the Al3+ ions or Pd0/C, effectively slowing down the dehydration-hydrogenation reaction. Additionally, a real industrial post-fermentation process stream was used as substrate. This intermediate process stream underwent only partial purification of citric acid and contained up to 430 g L−1 (∼2.2 M) of citric acid, ∼730 mg L−1 inorganics and ∼1100 mg L−1 other organics (Table S9, ESI†). The stream was diluted to reach a citric acid concentration of around 1 M (see model system) and successfully used as a substrate for dehydration-hydrogenation with a PTA carbon yield of 84% (Fig. S11, ESI†), providing additional evidence for the robustness of the dehydration-hydrogenation reaction.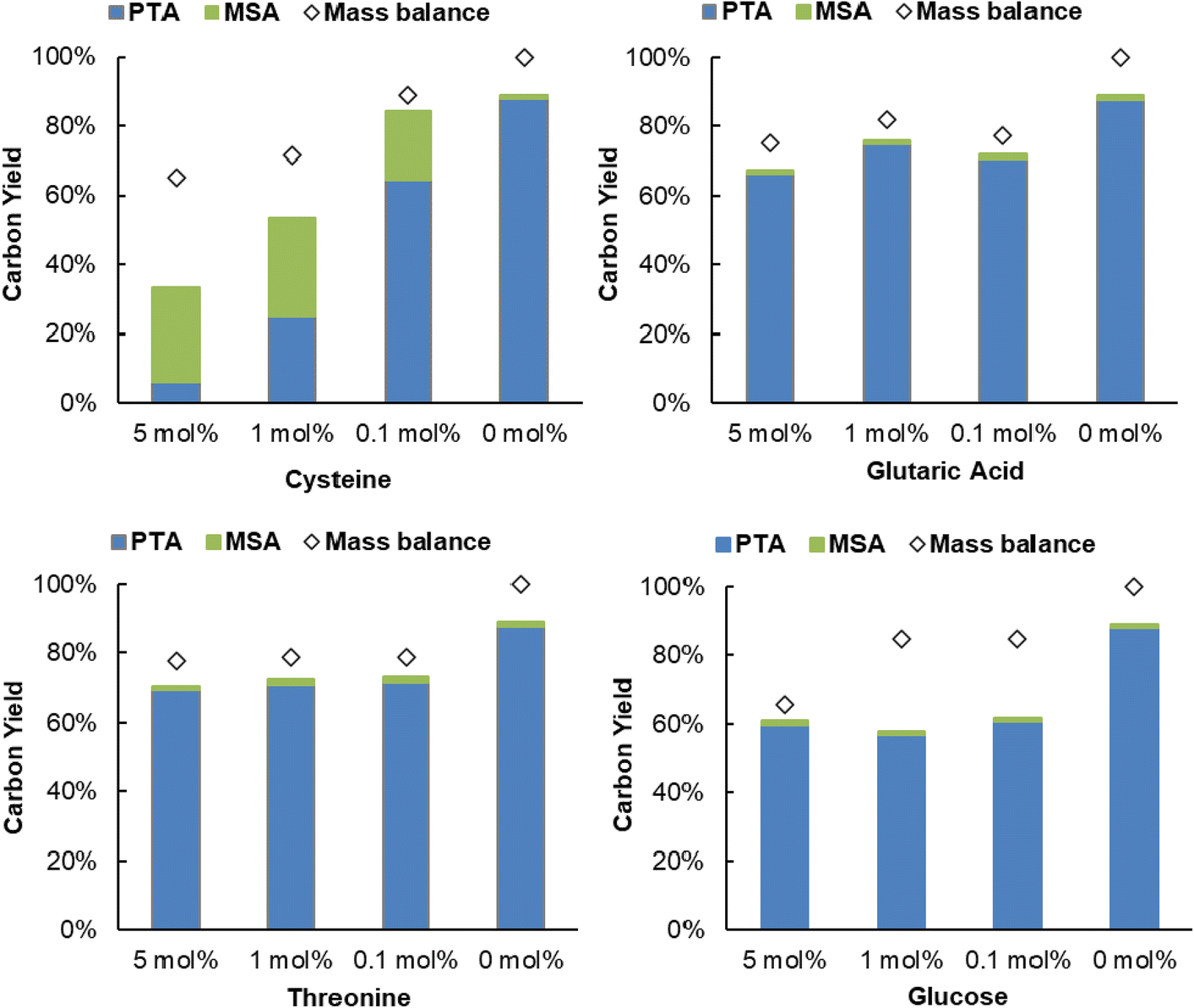 | ||
| Fig. 7 Influence of amino acids with different functional groups (–SH, –OH, –COOH) and glucose on dehydration-hydrogenation of citric acid. Reaction in water (2 mL) with citric acid (2 mmol), Al2(SO4)3 (0.25 mmol), Pd0/C (0.5 mol% Pd0), 20 h with 20 bar H2 at 150 °C. | ||
Fischer esterification of PTA
The dehydration-hydrogenation reaction of citric acid was coupled to a Fischer esterification with n-butanol using a Dean–Stark setup (Fig. 8). In this way, the produced acids were converted to the corresponding butyl esters for their potential application as functional plasticizers in PVC and PLA.46,47 The aqueous 1 M product mixture (20 mL) of the scaled-up reaction (93% PTA, 3% MSA and 2% CA) was transferred (after removing Pd0/C via centrifugation) to a glass flask and n-butanol was added (1:3.4 molar ratio). Since the crude mixture will be used without any purification, the applied Al2(SO4)3 catalyst remains present in the mixture during the Fischer esterification. However, this is not disadvantageous since Al2(SO4)3 can catalyze the esterification reaction. To our delight, as more water is removed, Al2(SO4)3 precipitates as a result of its low solubility in n-butanol. Using this setup, it is therefore possible to esterify the previously produced acids and to recover the precipitated homogeneous catalyst. After 20 h of reaction time at 135 °C, the mixture was purified (see Experimental section), resulting in a pure mixture of 5.06 g esters (93% TBPTC and 7% DBMS), which corresponds to a total yield of 90% directly from the initial citric acid.
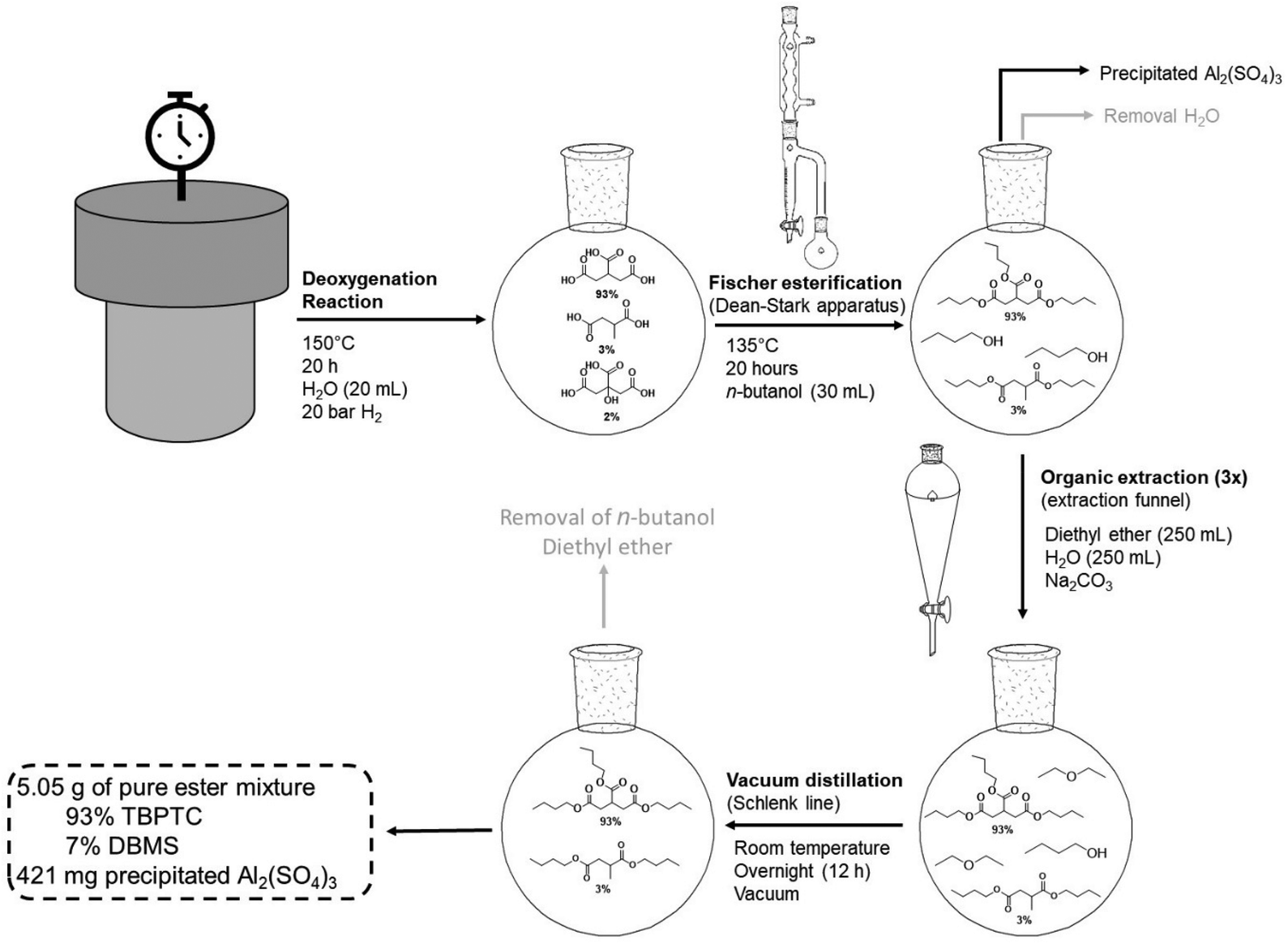 | ||
| Fig. 8 Dean–Stark setup for Fischer esterification of reaction mixtures (scaled-up reaction). Reaction mixture was added to 30 mL of n-butanol stirred at 500 rpm at 135 °C for a period of 20 h. | ||
To investigate whether recovered Al2(SO4)3 could be reused, a scaled-up reaction (1 M CA in 20 mL H2O, 150 °C, 20 bar H2 during 20 h) was performed using the precipitated catalyst (see Experimental section). This resulted in a product mixture of 85% PTA, 4% MSA and 8% CA, which clearly shows that this system allows the recovery and reuse of the homogeneous catalyst, hence showing the catalyst recyclability.
Stability dehydration-hydrogenation system
The recyclability of the catalytic system (i.e. Pd0/C and Al2(SO4)3) was evaluated (Fig. 9). Initially, a dry mass of 421 mg Al2(SO4)3 was recovered, corresponding to a 98% catalyst recovery. After ICP-OES measurements, it was determined that 64% of this mass could be assigned to anhydrous Al2(SO4)3, which corresponds to the fresh commercial Al2(SO4)3 (Table S6, ESI†). It is important to consider that the precipitate most likely consists of different types of common Al-hydrates (i.e. Al2(SO4)3·5H2O, Al2(SO4)3·16H2O and Al2(SO4)3·18H2O). During subsequent runs, Al2(SO4)3 recovery dropped, resulting in slightly lower PTA carbon yields. However, the addition of fresh Al2(SO4)3 after the fourth run to compensate for catalyst losses, resulted in the catalytic system regaining its initial activity (91% PTA carbon yield). The stability of the hydrogenation catalyst was confirmed by CO chemisorption after the first run, showing a similar Pd0 dispersion of 37% before and after reaction (20 h) (Table S5, ESI†). This demonstrates good recyclability of the catalytic system, with the hydrogenation catalyst maintaining its stability although Al2(SO4)3 recovery could be improved.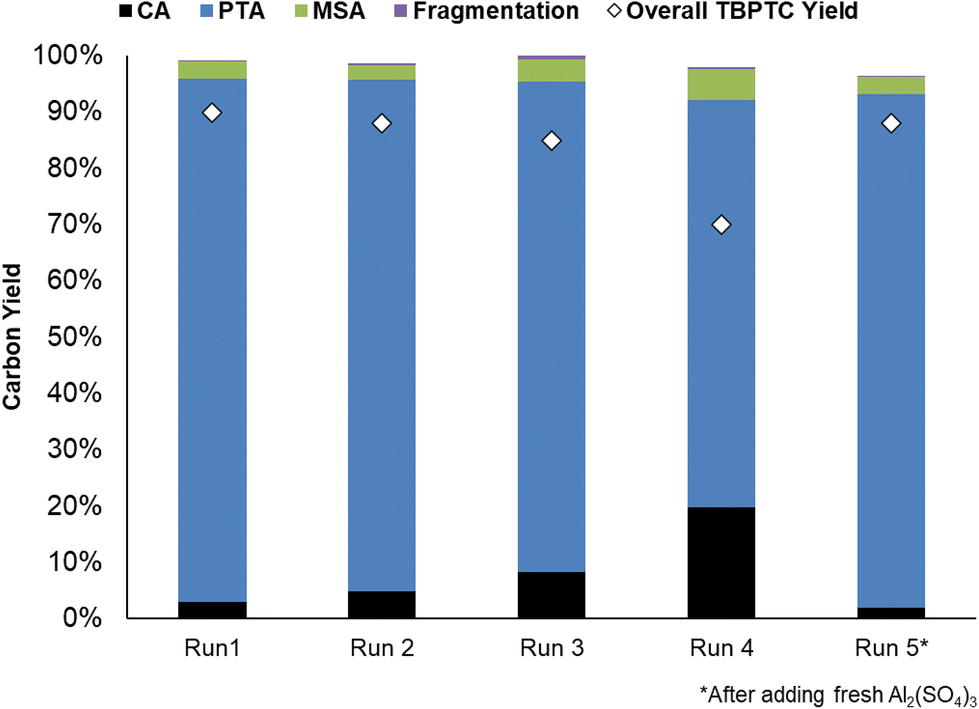 | ||
| Fig. 9 Recycling of the catalytic system (i.e. Pd0/C and Al2(SO4)3) for the two-step process. Reaction in water (20 mL) with citric acid (20 mmol), Al2(SO4)3 (1.25 mmol), Pd0/C (0.5 mol% Pd0), 20 h with 20 bar H2 at 150 °C. | ||
Conclusion
In conclusion, Al2(SO4)3 combined with Pd0/C resulted in an excellent catalytic system for the dehydration-hydrogenation of citric acid towards PTA. Al2(SO4)3 catalyzes the dehydration of citric acid to aconitic acid, through the formation of an active Al-citrate complex at low pH (1.3–1.5), which was thoroughly investigated by liquid 1H- and 27Al-NMR. The system was scaled-up to convert 20 mmol of citric acid (1 M in 20 mL H2O) with 0.125 equivalent of Al2(SO4)3, which resulted in a PTA carbon yield of 93% under optimized conditions. Interestingly, these elevated concentrations of citric acid permitted the omission of additional H2SO4. The robustness and industrial relevance of the system were subsequently verified by the addition of various impurities and the use of an industrial process stream. Considering the impurities, only sulfur containing components, like cysteine and methionine, poisoned the hydrogenation catalyst and suppressed to some extent the formation of PTA. At last, the dehydration-hydrogenation process was coupled to a Fischer esterification, which resulted in a total yield of 90% TBPTC starting from citric acid, with the main side product being the dibutyl ester of MSA (7% DBMS). Both molecules are thought to be potentially high quality biobased plasticizers for PVC and PLA. Interestingly, 98% of the homogeneous dehydration catalyst (i.e. Al2(SO4)3), was recovered and reused successfully. However, after 4 runs, some fresh Al2(SO4)3 needed to be added for the catalytic system to regain its initial activity. As such, a green and total synthesis route of biobased tricarballylate plasticizers directly from citric acid was successfully developed via a two-step process. Furthermore, the high substrate concentrations, scalability, robustness to impurities and recyclability of both catalysts, evidence the high industrial potential of this process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from VLAIO (HBC.2019.2387), in a collaborative project between Citribel and KULeuven. Wouter Stuyck is grateful to the FWO for his SB PhD fellowship (1SC1519N). The authors would like the thank Sam Van Minnebruggen for performing CO chemisorption measurements, Rodrigo de Oliveira Silva for performing the 27Al-NMR experiments.References
- PlasticEurope, Plastics - the facts 2021: An analysis of European plastics production, demand and waste data, 2020, https://plasticseurope.org, (accessed April, 2022) Search PubMed.
- A.L. Andrady and M.A. Neal, Phil. Trans. R. Soc. B. Sci., 2009, 364, 1977–1984 CrossRef CAS PubMed.
- R. Pfaendner, Polym. Degrad. Stab., 2006, 91, 2249–2256 CrossRef CAS.
- B. Pelzl, R. Wolf and B. L. Kaul, Plastic, Additives in Ulmann's Encyclyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2018, pp. 1–56 Search PubMed.
- M. P. Stevens, Polymer Chemistry—An Introduction, Oxford University Press, New York (United States), 1999 Search PubMed.
- K. Pivnenko, M. K. Eriksen, J. A. Martín-Fernández, E. Eriksson and T. F. Astrup, Waste Manage., 2016, 54, 44–52 CrossRef CAS PubMed.
- P. M. Lorz, F. K. Towae, W. Enke, R. Jäckh, N. Bhargava and W. Hillesheim, Phthalic Acid and Derivatives in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2007, pp. 131–180 Search PubMed.
- D.-H. Phan-Vu and C.-S. Tan, RSC Adv., 2017, 7, 18178–18188 RSC.
- S. Windels, T. Diefenhardt, N. Jain, C. Marquez, S. Bals, M. Schlummer and D. E. De Vos, Green Chem., 2022, 24, 754–766 RSC.
- C. Casals-Casas and B. Desvergne, Annu. Rev. Physiol., 2011, 73, 135–162 CrossRef CAS PubMed.
- K. A. Richardson, P. R. Hannon, Y. J. Johnson-Walker, M. S. Myint, J. A. Flaws and R. A. Nowak, Reprod. Toxicol., 2018, 77, 70–79 CrossRef CAS PubMed.
- K. J. Groh, T. Backhaus, B. Carney-Almroth, B. Geueke, P. A. Inostroza, A. Lennquist, H. A. Leslie, M. Maffini, D. Slunge, L. Trasande, A. M. Warhurst and J. Muncke, Sci. Total Environ., 2019, 651, 3253–3268 CrossRef CAS PubMed.
- M. Zarean, M. Keikha, P. Poursafa, P. Khalighinejad, M. Amin and R. Kelishadi, Environ. Sci. Pollut. Res., 2016, 23, 24642–24693 CrossRef CAS PubMed.
- R. M. David, R. D. White, M. J. Larson and J. K, Herman and R. Otter, Toxicol. Lett., 2015, 238, 100–109 CrossRef PubMed.
- European Chemicals Agency, Annex XVII to REACH - Conditions of restriction: Entry 51, 2021, https://echa.europa.eu/substances-restricted-under-reach, (accessed June, 2022) Search PubMed.
- United States Consumer Product Safety Commission, Phthalates Business Guidance & Small Entity Compliance Guide, 2019, https://www.cpsc.gov/Business-Manufacturing/Business-Education/Business-Guidance/Phthalates-Information#, (accessed June, 2022) Search PubMed.
- European Commission, IP/99/829 Ban of phthalates in childcare articles and toys, 1999, https://ec.europa.eu/commission/presscorner/detail/en/IP_99_829, (accessed April, 2022) Search PubMed.
- M. Bocqué, C. Voirin, V. Lapinte, S. Caillol and J.-J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 11–33 CrossRef.
- W. D. Arendt and J. Lang, J. Vinyl Addit. Technol., 1998, 3, 3278–3283 Search PubMed.
- L. Larsson, P. Sandgren, S. Ohlsson, J. Derving, T. Friis-Christensen, F. Daggert, N. Frizi, S. Reichenberg, S. Chatellier, B. Diedrich, J. Antovic, S. Larsson and M. Uhlin, Vox Sang., 2021, 116, 60–70 CrossRef CAS PubMed.
- M. O. Ahmed, S. O. Neill, F. Reid and M. Kinnear, Pharm. World Sci., 2009, 31, 40–141 CrossRef PubMed.
- E. H. Immergut and H. F. Mark, Principles of Plasticization in Plasticization and Plasticizer Processes, Advances in Chemistry, American Chemical Society, United States, 1965, pp. 1–26 Search PubMed.
- P. G. Demertzis, K. A. Riganakos and K. Akrida-Demertzi, Polym. Int., 1991, 25, 229–236 CrossRef CAS.
- H. E. Bair and P. C. Warren, J. Macromol. Sci., Part B: Phys., 1981, 20, 381–402 CrossRef.
- J. I. Kroschwitz, Encyclopedia of Polymer Science and Engineering, John Wiley and Sons, 1990, pp. 739–743 Search PubMed.
- C. E. Wilkes, J. W. Summers and C. A. Daniels, PVC Handbook, Hanser, Munich (Germany), 2005 Search PubMed.
- Y. Wang, C. Zhou, Y. Xiao, S. Zhou, C. Wang, X. Chen, K. Hu, X. Fu and J. Lei, Iran. Polym. J., 2018, 27, 423–432 CrossRef CAS.
- G. Feng, L. Hu, Y. Ma, P. Jia, Y. Hu, M. Zhang, C. Liu and Y. Zhou, J. Cleaner Prod., 2018, 189, 334–343 CrossRef CAS.
- K. Nara, K. Nishiyama, H. Natsugari, A. Takeshita and H. Takahashi, J. Health Sci., 2009, 55, 281–284 Search PubMed.
- S. Ting, CN Pat, 101353305B, 2007 Search PubMed.
- B. L. Song, G. Zhang, Y. Z. Wang and W. J. Yanli, CN Pat, 101402571A, 2008 Search PubMed.
- G. Kia, Z. Yang, Z. Pengwei, H. Wei and L. Xiaolin, CN Pat, 102351696B, 2011 Search PubMed.
- L. Jianzhong, Z. Yuepeng and L. Hiulai, CN Pat, 102633640B, 2012 Search PubMed.
- C. Xingjian, CN Pat, 106928065A, 2017 Search PubMed.
- J. F. Day, US Pat, 20060094894A1, 2004 Search PubMed.
- J. Verduyckt and D. E. De Vos, Chem. Sci., 2017, 8, 2616–2620 RSC.
- J. Verduyckt, A. Geers, B. Claes, S. Eyley, C. Van Goethem, I. Stassen, S. Smolders, R. Ameloot, I. Vankelecom, W. Thielemans and D. E. De Vos, Green Chem., 2017, 19, 4642–4650 RSC.
- A. Stuart, M.-M. McCallum, D. Fan, D. J. LeCaptain, C. Y. Lee and D. K. Mohanty, Polym. Bull., 2010, 65, 589–598 CrossRef CAS.
- H. C. Erythropel, P. Dodd, R. L. Leask, M. Maric and D. G. Cooper, Chemosphere, 2013, 91, 358–365 CrossRef CAS PubMed.
- H. C. Erythropel, S. Shipley, A. Börmann, J. A. Nicell, M. Maric and R. L. Leask, Polymer, 2016, 89, 18–27 CrossRef CAS.
- F. C. Magne and R. R. Mod, Ind. Eng. Chem., 1953, 45, 1546–1547 CrossRef CAS.
- R. J. Reid Jr. W. M. Smith and B. H. Werner, US Pat, 2802802, 1957 Search PubMed.
- W. Stuyck, J. Verduyckt, A. Krajnc, G. Mali and D. E. De Vos, Green Chem., 2020, 22, 7812–7822 RSC.
- Z. Li, X. Wen and H. Liu, Green Chem., 2022, 24, 1650–1658 RSC.
- W. Stuyck, A. L. Bugaev, T. Nelis, R. de Oliveira-Silva, S. Smolders, O. A. Usoltsev, D. A. Esteban, S. Bals, D. Sakellariou and D. E. De Vos, J. Catal., 2022, 408, 88–97 CrossRef CAS.
- P. Canton, G. Fagherazzi, M. Battagliarin, F. Menegazzo, F. Pinna and N. Pernicone, Langmuir, 2002, 18, 6530–6535 CrossRef CAS.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics, CRC Press, Florida (United States), 97th edn, 2016 Search PubMed.
- R. B. Martin, J. Inorg. Biochem., 1986, 28, 181–187 CrossRef CAS PubMed.
- H. K. Wen, M. K. Wang, M. H. Pan, W.-W. Chia, M. C. Chia and L. W. Shan, Water Res., 2005, 39, 3457–3466 CrossRef PubMed.
- M. E. Essington, The Complexity of Aqueous Complexation: The Case of Aluminum- and Iron(III)-Citrate in In Biophysico-Chemical Processes of Heavy Metals and Metalloids in Soil Environments, John Wiley and Sons, Hoboken (United States), 2007, pp. 373–416 Search PubMed.
- T. Wang, J. A. Glasper and B. H. Shanks, Appl. Catal., A, 2015, 498, 214–221 CrossRef CAS.
- G. A. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson Education, Delhi (India), 5th edn, 2007 Search PubMed.
- F. Fringuelli, F. Pizzo and L. Vaccaro, Tetrahedron Lett., 2001, 42, 1131–1133 CrossRef CAS.
- F. Fringuelli, F. Pizzo and L. Vaccaro, J. Org. Chem., 2001, 66, 4719–4722 CrossRef CAS PubMed.
- S. Berger, J. Nolde, T. Yüksell, W. Tremel and M. Mondeshki, Molecules, 2018, 23, 1–16 Search PubMed.
- L. O. Ohman, Inorg. Chem., 1998, 27, 2565–2570 CrossRef.
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307–313 RSC.
- S. Mores, L. P. de S. Vandenberghe Jr., A. I. Magalhães, J. C. de Carvalho, A. F. M. de Mello, A. Pandey and C. R. Soccol, Bioresour. Technol., 2021, 320, 124426 CrossRef CAS PubMed.
- J. A. Moulijn, M. Makkee and A. Van Diepen, Chemical Process Technology, John Wiley and Sons, Hoboken (United States), 2nd edn, 2013 Search PubMed.
- J. Verduyckt, R. Coeck and D. E. De Vos, ACS Sustainable Chem. Eng., 2017, 5, 3290–3295 CrossRef CAS.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04678d |
| This journal is © The Royal Society of Chemistry 2023 |