
Renewed interest in biopolymer composites: incorporation of renewable, plant-sourced fibers
Cody W.
Weyhrich
 ae,
Stella P.
Petrova
d,
Kevin J.
Edgar
bc and
Timothy E.
Long
*ae
ae,
Stella P.
Petrova
d,
Kevin J.
Edgar
bc and
Timothy E.
Long
*ae
aSchool of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA
bMacromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA
cDepartment of Sustainable Biomaterials, College of Natural Resources and Environment, Virginia Tech, Blacksburg, VA 24061, USA
dDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
eBiodesign Center for Sustainable Macromolecular Materials and Manufacturing, Arizona State University, Tempe, AZ 85281, USA
First published on 13th December 2022
Abstract
High-performance polymer-based composites provide materials that display a spectrum of properties, including those required by rapidly developing aerospace and automotive technologies. The composite structure enables optimization of a variety of physical properties – molecular weight, mechanical strength, toughness, and thermal stability. Too often, however, environmental impact and recyclability remain unaddressed. Presently, carbon emissions and climate change constitute great threats to human society, leading us to examine more closely the impact of fossil fuel-based composites. Herein we discuss diverse pathways to achieve the advantageous properties of composites, while remaining attentive to environmental consequences. The planet provides numerous natural fibers with properties that could permit a wide range of performances; however, biosources are currently undervalued and underutilized. The combination of natural fibers with bio-renewable matrices is an end goal for environmentally friendly composites, allowing for the advancement of structural composites with greatly reduced ecological and economical cost. This review highlights the significance and utility of bio-based composites with special attention to the regional resiliency of resources.
 Cody W. Weyhrich | Cody W. Weyhrich received his B.S. in Chemistry from the University of North Carolina at Chapel Hill in May 2018 with a focus on polymer chemistry. He is currently a fifth-year Ph.D. candidate in the School of Molecular Sciences at Arizona State University under the tutelage of Professor Timothy E. Long, expecting to graduate in May 2023. His research interests focus on employing high-performance polymers, such as poly(arylene ether sulfone)s and fully aromatic polyimides, in UV-assisted additive manufacturing processes, including vat photopolymerization and direct ink-write. |
 Stella P. Petrova | Stella P. Petrova is a 5th year Ph.D. Candidate in the Department of Chemistry at Virginia Tech with affiliations at the Macromolecules Innovation Institute, the Department of Sustainable Biomaterials, and the GlycoMIP Scholars at Virginia Tech. She is advised by Dr Kevin Edgar and is expected to graduate in May 2023. She received her B.S. in Biology (Chemistry minor) at Virginia Commonwealth University and worked in industry for Afton Chemical and at DARPA-startup Embody Inc. prior to graduate school. Her research focuses on sustainable biomaterials, synthetic polysaccharide modification strategies, and structure–property relationships of polysaccharide polymers for oral drug delivery and other applications. |
 Kevin J. Edgar | Edgar is a professor in the Virginia Tech Dept. of Sustainable Biomaterials, associate Dean in the Graduate School, and co-directs the VT Infectious Disease Interdisciplinary Graduate Education Program. His interests include synthesis of novel polysaccharide derivatives, unraveling their structure property relationships, and exploiting their useful properties in demanding, high-value applications. He is an ACS Fellow and an editor-in-chief of Carbohydrate Polymers. Edgar received his BS in Chemistry from Bucknell Univ. and his Ph.D. in Organic Chemistry from Duke Univ., then worked for many years for Eastman Chemical Company before his current appointment at VT. |
 Timothy E. Long | Timothy E. Long received his B.S. in Chemistry from St. Bonaventure University in 1983 and his Ph.D. in 1987 at Virginia Tech under Professor James E. McGrath. Following this, he worked in the Corporate Research Laboratories at Eastman Kodak Company and the Polymers Research Division at Eastman Chemical Company before joining the Virginia Tech Department of Chemistry faculty in 1999. His research group's interests included high-performance step-growth polymers, novel 3D printing materials, and hydrogen-bond-containing polymers before moving to Arizona State University in 2020 to head the Biodesign Center for Sustainable Macromolecular Materials and Manufacturing. |
1. Introduction
Effective composite materials combine properties of their components, such as the tensile strength of a single fiber and the toughness of the matrix, making them important contributors to commerce and science. Dispersing a reinforcing element into a compatible polymeric matrix can provide materials that are lighter and stronger than metallic analogs; this review focuses on fiber-reinforced polymeric composites.1–5 Investigations into polymeric composites emerged in the 1940s with World War II driving the need for lighter replacements for heavy metallic components used in helicopters, airplanes, and other military vehicles. The demand for materials fitting specific military needs coupled with advances in polymer science created a focused research effort on composite materials post-WWII, as shown in Fig. 1.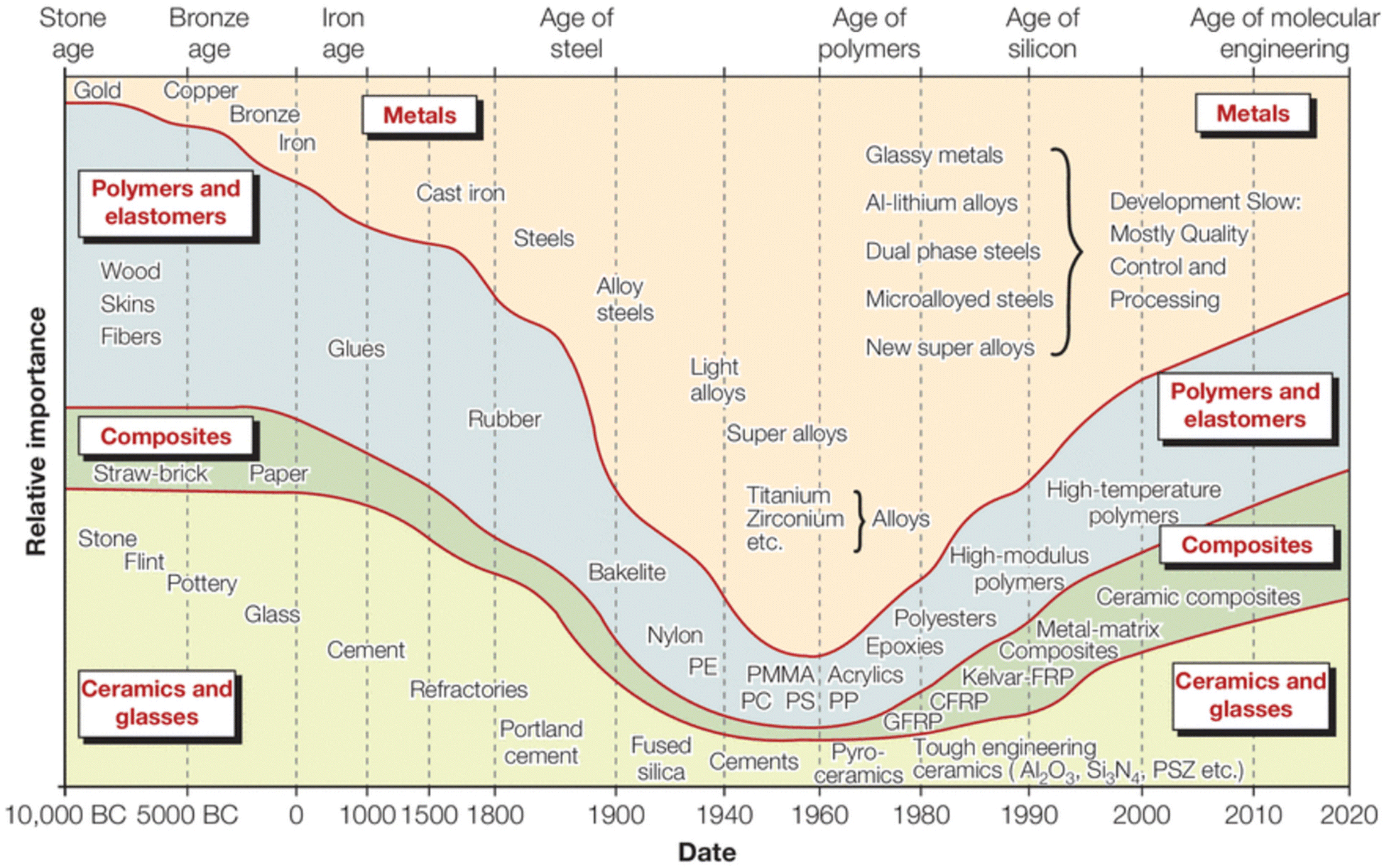 | ||
| Fig. 1 Relative importance of the four main classes of materials as a function of time in engineering applications.6 | ||
Key differences in their philosophy and relative importance of fundamental principles differentiate sustainability and green chemistry. Sustainability evaluates material design through a lens of environmental impact and emphasizes the relationship between the ecosystem and manufacturing. This assessment weighs both the commercial practicality and the ecological impact informing justifiable choices to ensure a healthy ecosystem for future generations. However, green chemistry outlines preferred adjustments for chemical reactions to reduce environmental impact. Twelve principles define green chemistry, including waste prevention, safe alternative use, and the introduction of atom economy. Green chemistry is a pathway that chemists use to promote sustainability with the incorporation of safer, renewable feedstocks, but the two concepts remain independent ideals. However, both offer valuable approaches that are ingrained in the concept of composite materials.
The climate is changing; therefore, utilizing more sustainable chemistry is imperative for reducing negative impacts on the planet in the 21st century. Composite materials are not exempt from this trend, and fiber-reinforced composites research now focuses on more environmentally friendly approaches. Currently, two important ways in which composites are becoming more sustainable are composite material recycling, and the use of renewable components.7–9 This review highlights advances towards more renewable composite materials and the recyclability of composite materials.10,11
Resource regionality refers to the locality where a natural fiber is readily available and produced. Employing readily accessible, local fiber sources when considering green composites is more cost-efficient and creates a smaller carbon footprint. Thus, green and sustainable composites differ depending on their origin.12 Currently, there is a large diversity of fibers available for use in natural fiber composites, illustrated below in Fig. 2.
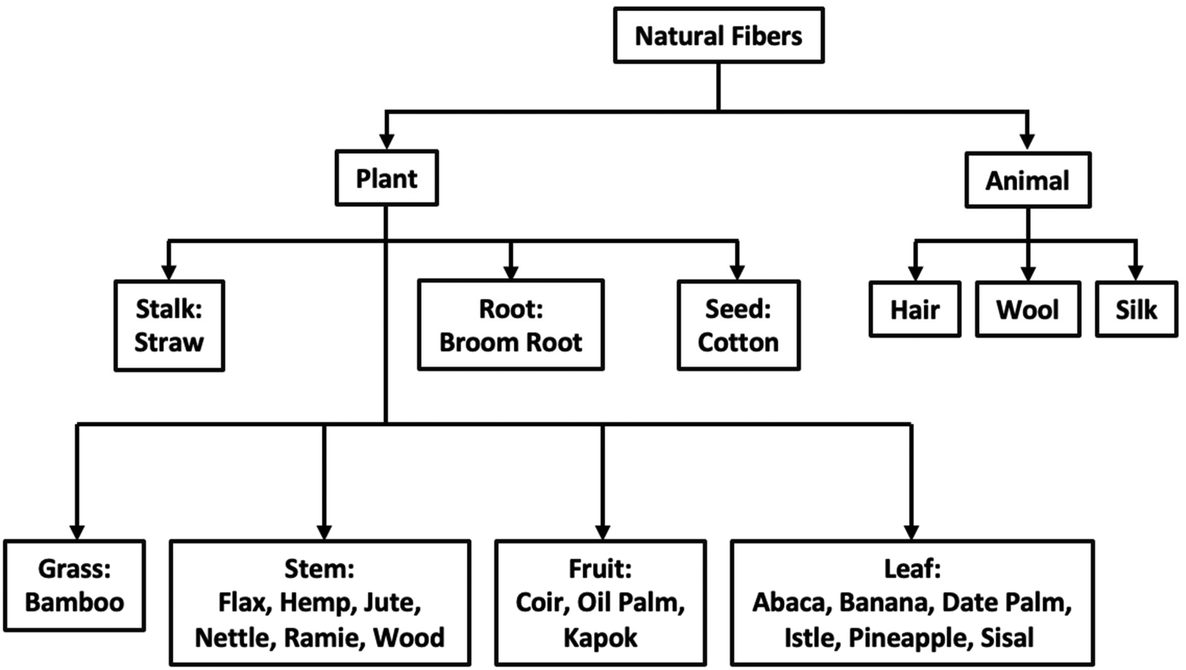 | ||
| Fig. 2 Origin and classification of different natural fibers. Reproduced from ref. 13 with permission from Elsevier, copyright 2013.13 | ||
Applications of high strength, lightweight fibers for composites traditionally use carbon fibers as they exhibit excellent properties, which include high stiffness and high strength. However, their high cost limits potential applications.14 Traditional carbon fiber preparation from the carbonization of poly(acrylonitrile) accounted for 96% of global carbon fiber production in 2012, but this method is not sustainable as acrylonitrile is a non-renewable, petroleum-based monomer.15,16 Furthermore, energy requirements for the manufacture of carbon fibers are intense, typically requiring temperatures exceeding 1000 °C. The non-renewability of carbon fibers prompted investigations of greener and more sustainable carbon fiber production pathways.17 High temperature carbon fiber production requires more energy than production of natural fibers, which are generally produced using mild chemical treatments or short exposures to plasma, as discussed in subsequent sections.
Key potential substitutes for carbon fibers include natural fibers found in a variety of flora such as trees, ferns, and grasses. It is important to note that most natural fibers are unable to fully replicate the excellent properties imparted by carbon fibers. However, natural fibers have unique value in composites, as renewable components with admirable matrix strengthening properties, thus providing numerous benefits when integrated into production. For example, natural fiber usage in the automotive industry reduces fossil fuel consumption (10% reduction in vehicle weight equates to 6–7% reduced fuel consumption) through vehicle weight reduction, thereby increasing overall fuel efficiency.18 In sustainable material composite development (or, for that matter, development of any composite) it is important to quantify environmental impacts, from reagent sourcing through production to disposal. Life cycle engineering, as this is called, provides an additional lens through which these novel materials are analyzed to determine their viability in a sustainable future.19 This review highlights renewable components in fiber-reinforced composite materials. The renewability of fibers and composite matrices is discussed herein with potential applications for green composites in areas where non-renewable, less sustainable composites currently dominate.
2. Abundance and regionality of renewable reinforcing fibers
Natural fiber properties vary considerably depending on variables including the plant species, the portion of the plant where the fiber is found, the time of harvest, and others. The most employed subgroups of natural fibers are derived from the stem/bast, leaf, seed, reed/grass, and/or core of plants. Stem/bast natural fibers are the most promising because they support the weight of the plant and are the source of the plant's rigidity.20 The type of flora that are local to a region influence which green composites are most attractive choices, because utilizing nearby resources reduces transportation costs and is thus more sustainable than sourcing materials from distant locations. Natural fibers have low density and are expensive to transport. Many fibers are sourced from annual crops, which have limited growing seasons, creating the need for storage conditions that optimally preserve fiber stability. In this way the natural fiber can supply the composite manufacturing plant year-round, enabling so-called “fiber regionality”. Many of these natural fibers that are suitable and receive significant consideration for composite materials are found in developing countries with tropical environments, permitting longer growing seasons and reducing the need for post-harvest storage. This is beneficial for the resident economy and is advantageous for local access to high-strength materials that could lower the cost of construction and increase further municipal development. Additionally, it is important to note that some fibers, such as flax, have competing markets, which will lead to fluctuating raw material costs.2.1. Chemical composition of natural fibers
The exact chemical makeup of the various natural fibers discussed herein varies based on the variables noted above, as well as the purification method. However, there are four primary chemical components in natural fibers, i.e., lignin, pectin, hemicellulose, and cellulose.Cellulose provides structural support with its strength derived from its linear, ribbon-like nature and its high degree of crystallinity.25,26 Hemicelluloses lower the interfacial energy between the hydrophobic lignin and the hydrophilic cellulose fibers found in the plant cell wall composite, which assists polymeric cellulose chains in strengthening the plant.27 Reduction of interfacial energy is accomplished by intercalation of the hemicellulose chain into the cellulose fiber bundle, hydrogen bonding to the cellulose fiber, and covalent bonding to lignin. Monosaccharide composition and cell wall hemicellulose structure vary with plant species and the portion of the plant from which it originated. Lignin, the continuous phase component of the plant fiber secondary cell walls (which comprise much of the plant's weight), is a crosslinked polymer network consisting of phenylpropane-derived structural units.28 An oxidative, enzyme-catalyzed radical polymerization mechanism links the molecules depicted in Fig. 3, which produces a network containing an irregular sequence. Lastly, pectin is a complex polysaccharide composed of different monosaccharides and architectures, with galacturonic acid and rhamnose the most abundant. Pectin acts as the continuous phase of the matrix within the natural fiber primary cell walls.29
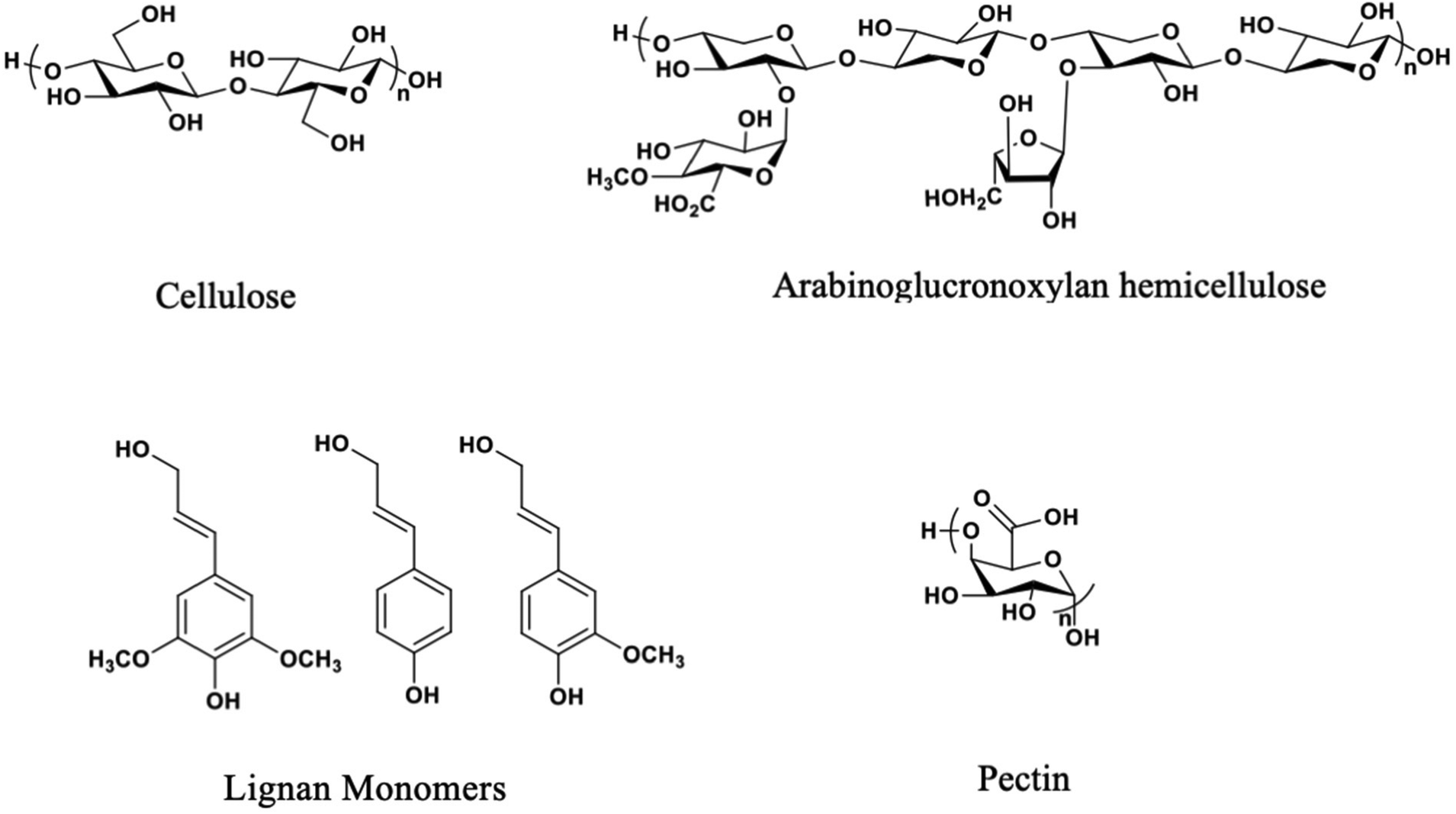 | ||
| Fig. 3 Chemical structures of the four primary components of natural fibers (pectin and arabinoglucuronoxylan structures simplified).21–24 | ||
2.2. Bast fibers
Structural sections of the plant, typically within the stem, host the strong, bast fibers (Fig. 4). The most significant bast fibers for composite materials are found in bamboo, ramie, flax, kenaf, hemp, and jute. Their high cellulose contents, tabulated in Table 1, provide significant strength.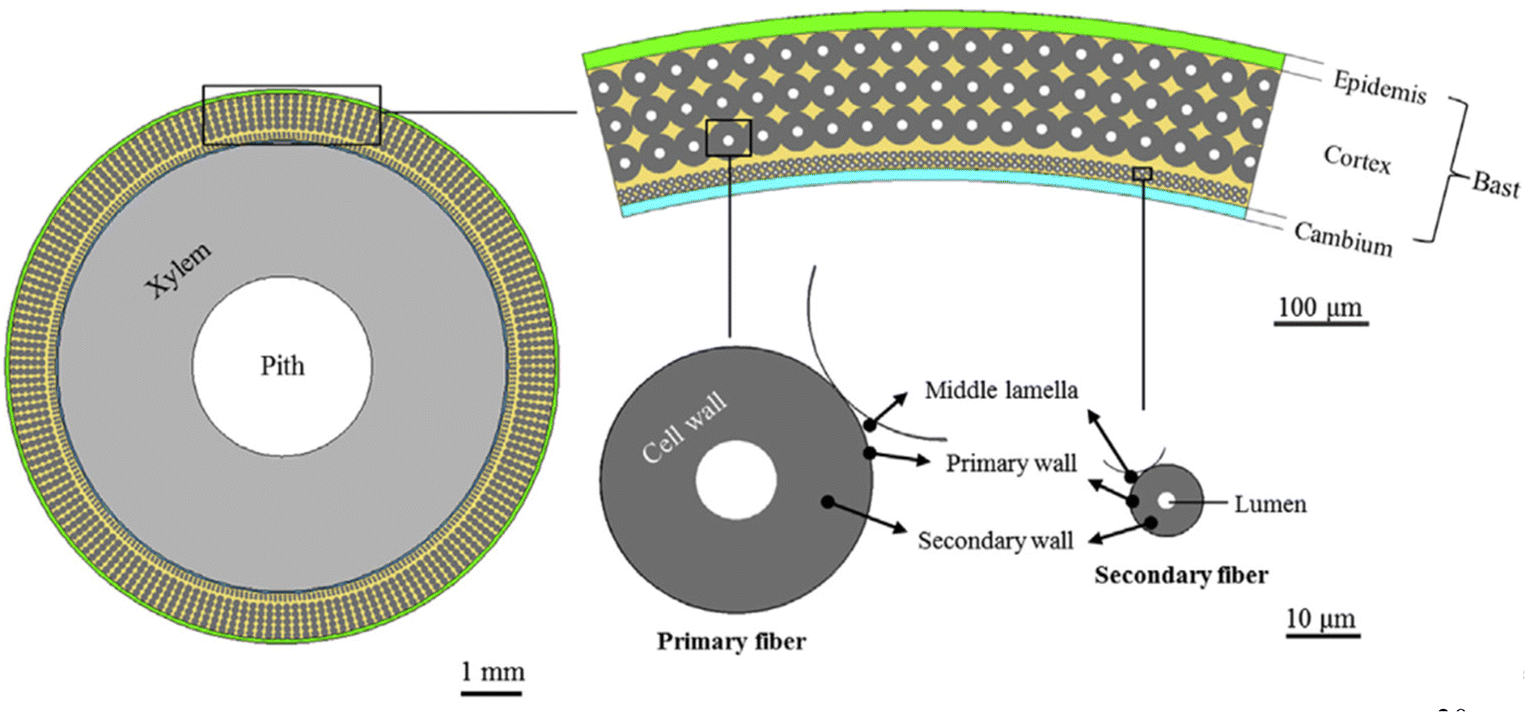 | ||
| Fig. 4 Depiction of the source of bast fibers from their parent plant.30 | ||
| Species | Cellulose (wt%) | Hemicelluloses (wt%) | Lignin (wt%) | Pectin (wt%) |
|---|---|---|---|---|
| Bamboo | 26–43 | 15–26 | 21–31 | — |
| Ramie | 68–76 | 13–15 | 0.6–1 | 1.9–2 |
| Flax | 60–81 | 14–19 | 2–3 | 0.9 |
| Kenaf | 44–57 | 21 | 15–19 | 2 |
| Hemp | 70–92 | 18–22 | 3–5 | 0.9 |
| Jute | 51–84 | 12–20 | 5–13 | 0.2 |
These fibers are of particular interest due to their impressive mechanical properties that are comparable to those of synthetic fibers, as seen in Fig. 5. The specific modulus was calculated using the average reported density and tensile modulus for each material. The high tensile properties of the commercially abundant bast fibers (∼14 million tons per year produced) identify them as robust candidates for modern green composites.32
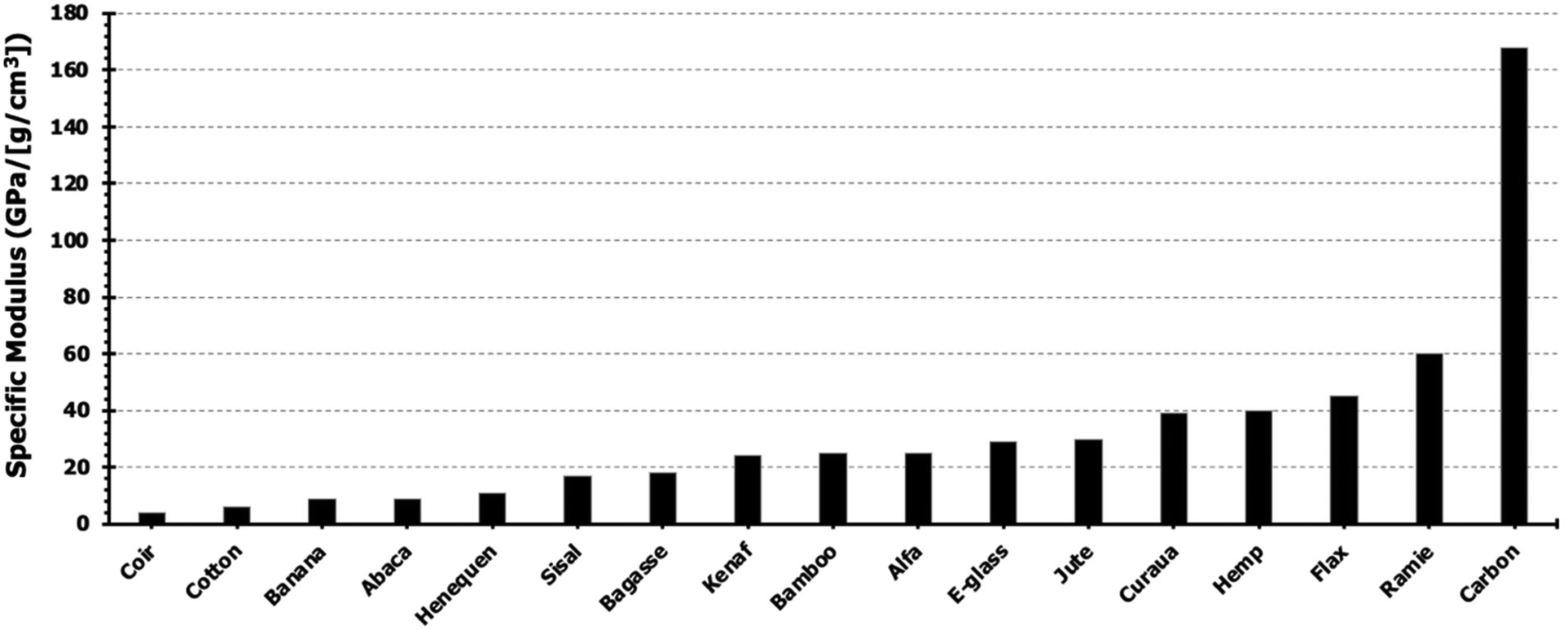 | ||
| Fig. 5 Specific modulus of common natural and synthetic fibers. Reproduced from ref. 33 with permission from Elsevier, copyright 2012.33 | ||
2.3. Leaf fibers
Leaf fibers originate from the stalks of the leaves or the leaves themselves. Leaf the library of available leafy fibers is sizable, but a few stand out, including abaca, pineapple, sisal, and banana. These fibers have wide compositional ranges, which provide advantagous properties, including the high tensile strength of pineapple fibers and the high elongation of abaca fibers (Table 2). However, leaf-based fibers have higher water content than other fibers, which requires more drying and leads to a lower yield of fiber per weight of plant cultivated.| Species | Cellulose (wt%) | Hemicelluloses (wt%) | Lignin (wt%) | Pectin (wt%) |
|---|---|---|---|---|
| Abaca | 61–64 | 21 | 12 | 0.8 |
| Pineapple | 80–81 | 16–19 | 4.6–12 | 2–3 |
| Sisal | 43–78 | 10–13 | 4–12 | 0.8–2 |
| Banana | 60–65 | 6–19 | 5–10 | 3–5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tonnes of dry fiber annually if all leaves were collected and the fibers extracted. The relatively high supply and low demand have driven recent cost of pineapple fibers down to ∼$15 USD per tonne, whereas common softwood fibers are ∼$250 USD per tonne.45
000 metric tonnes of dry fiber annually if all leaves were collected and the fibers extracted. The relatively high supply and low demand have driven recent cost of pineapple fibers down to ∼$15 USD per tonne, whereas common softwood fibers are ∼$250 USD per tonne.45
3. Biodegradable and renewable polymer matrices
Utilizing natural fibers to reinforce composites reduces their environmental footprint, but incorporating a green matrix, which comprises most of a composite's weight and volume, provides more impact. Renewability and biodegradability are both beneficial for moving towards a more environmentally friendly composite future. Ester-containing polymers, particularly poly(hydroxyalkanoates), are ubiquitous throughout the biodegradable material library as they are more hydrophobic than polysaccharides, often are thermally processable (unlike most polysaccharides), and can degrade by two readily available pathways – hydrolytic and enzymatic.52,53 An important differentiation between these two mechanisms is that enzymatic degradation only occurs where the enzyme has access to the material, which is confined to the surface for solid materials (Fig. 6).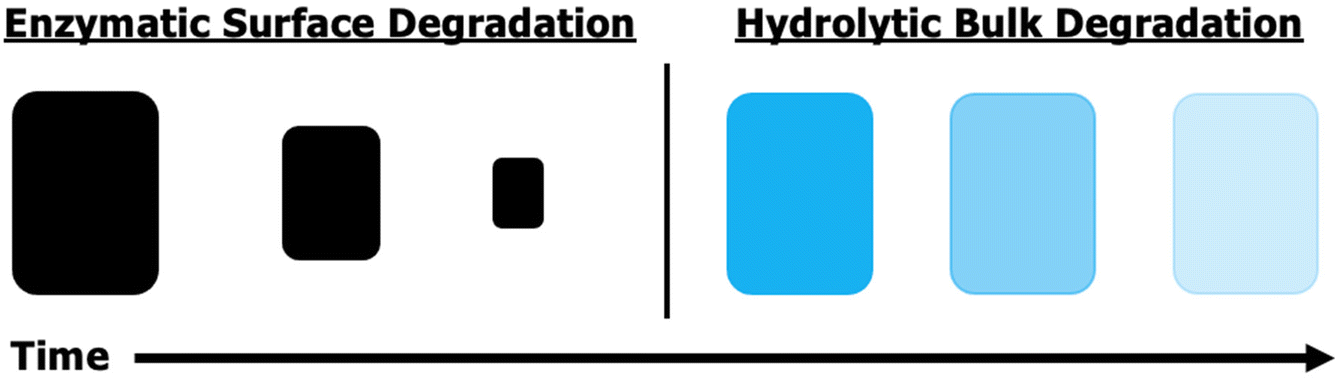 | ||
| Fig. 6 Differences in characteristics of surface active enzymatic vs. bulk hydrolytic degradation. | ||
3.1. Glycerol-based matrices
Use of biofuels as fossil fuel alternatives has recently increased, in part for environmental and energy security reasons. Plant oils extracted from seeds of different types of trees are valuable biodiesel precursors that can be accessed sustainably. This process also provides glycerol as a byproduct; glycerol is a practical renewable feedstock commonly employed in green matrices. The large abundance of trees that produce seeds containing triglycerides means that biodiesel can make a significant contribution to the worldwide fuel market in the future, with a substantial market increase already underway, as seen in Fig. 7.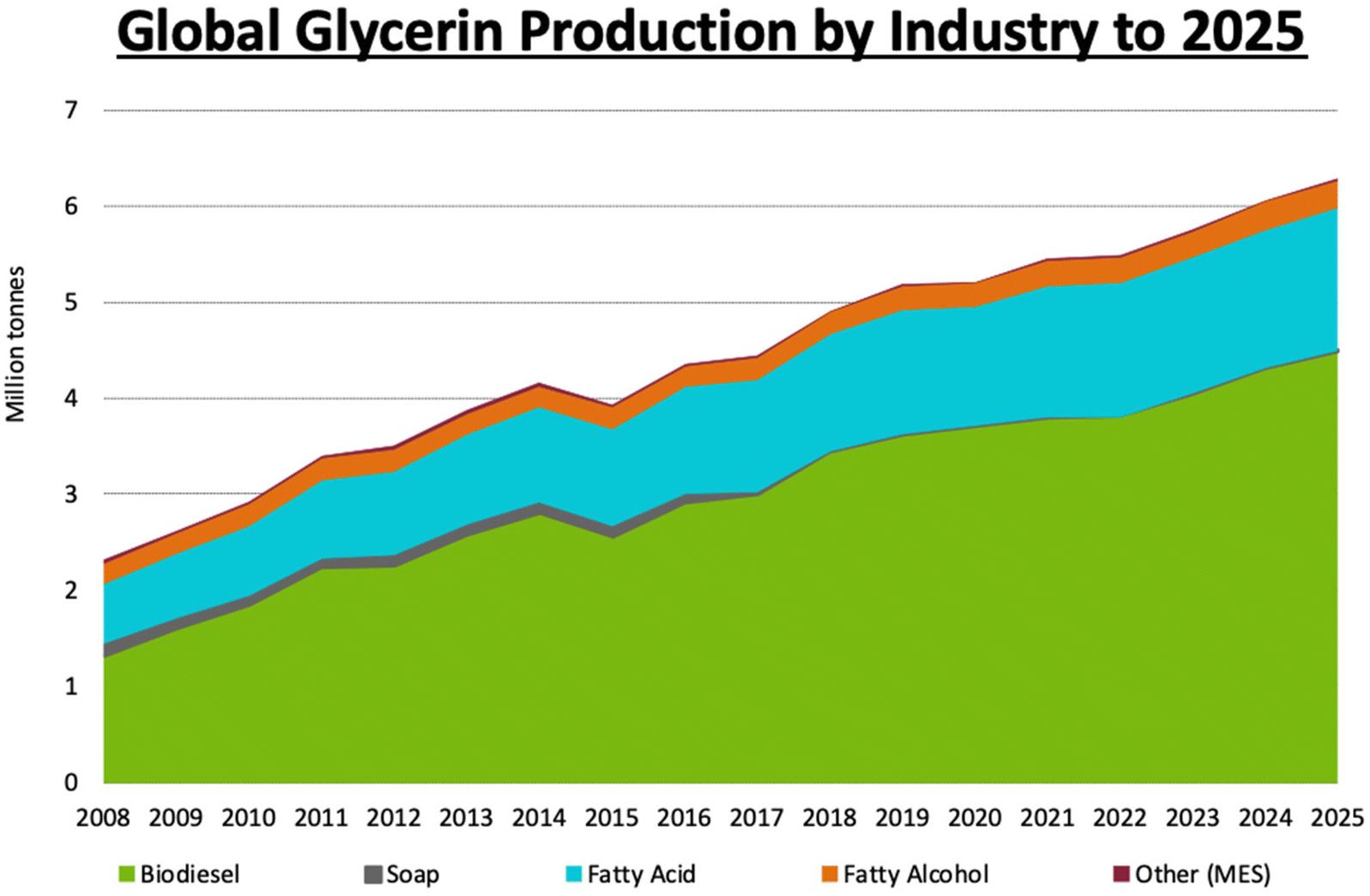 | ||
| Fig. 7 World-wide production of glycerol projected through 2025. Reproduced from ref. 54 with permission from LMC International, copyright 2022.54 | ||
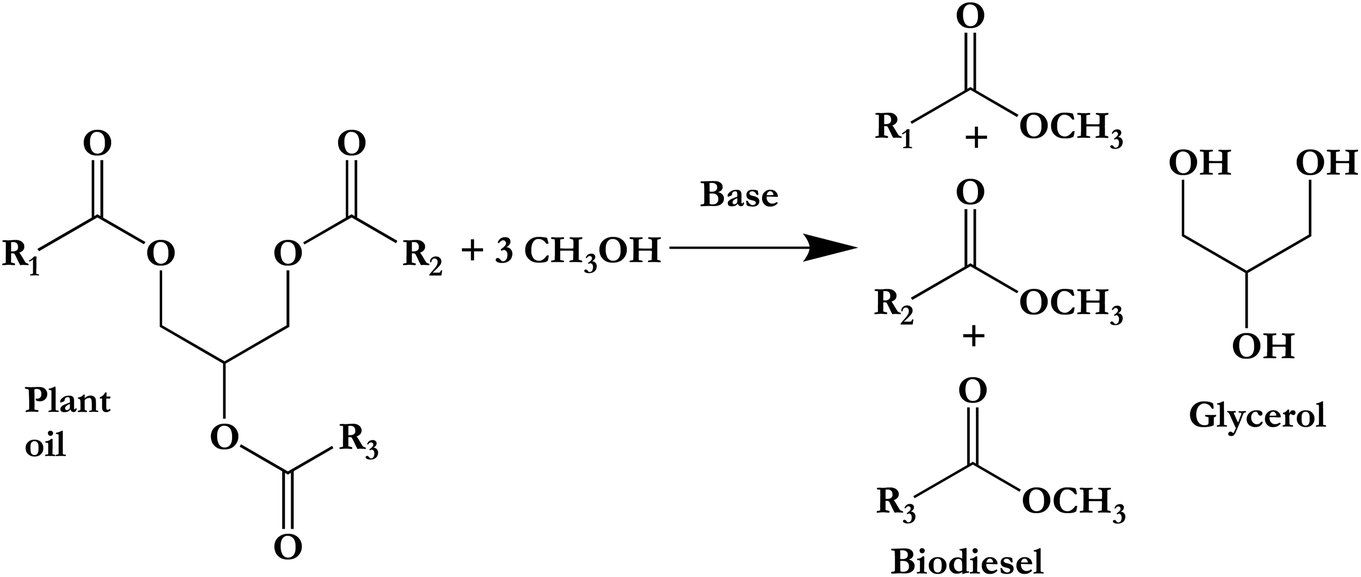 | ||
| Scheme 1 Plant oil transesterification with glycerol by-product.56 | ||
This process exhibits great promise as the alternative supercritical alcohol transesterification method is comparatively expensive.57
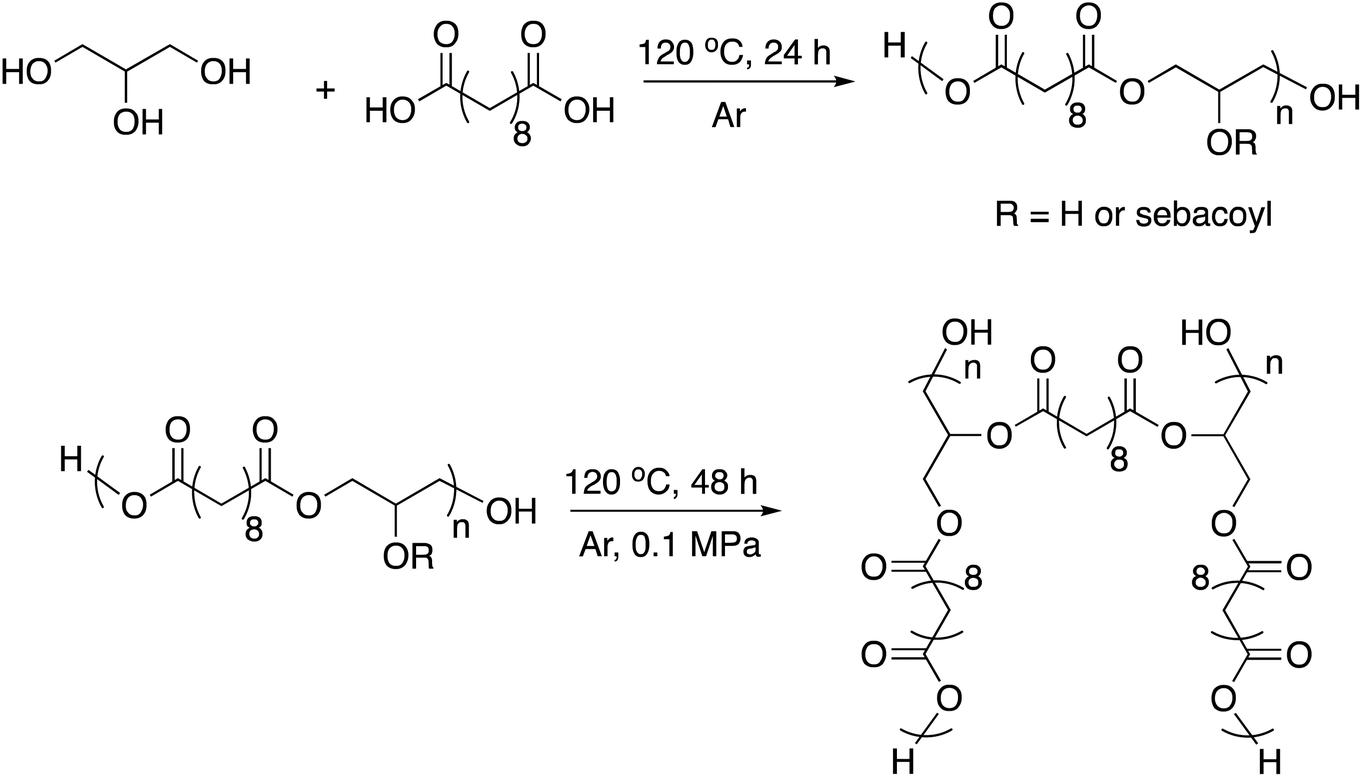 | ||
| Scheme 2 Synthetic route to the PGS thermoset matrix.61,62 | ||
Select biomimetic applications require more robust materials than a neat PGS matrix, which inspired the use of reinforcing glass fibers for cartilage tissue regeneration scaffolds.63 Replacing glass fibers with biodegradable materials, such as silk fibroin or chitosan, for use in skin scaffold engineering enhanced sustainability.64 Kasetaite et al. found that loading a different glycerol-based matrix, crosslinked glycerol diglycidyl ether in this case, with various industrial wastes, such as rapeseed cake and horn meal, produced reinforced composites exhibiting tensile strengths over 120 MPa.65
3.2. Poly(lactic-co-glycolic acid)
The natural sources, high biocompatibility across tissue types, and biodegradability of poly(lactic acid) (PLA) and poly(glycolic acid) (PGA) homopolymers make them particularly useful for medical applications, such as absorbable surgical sutures.66 Like the glycerol-based matrix, these aliphatic polyesters degrade through hydrolytic mechanisms, as illustrated in Scheme 3.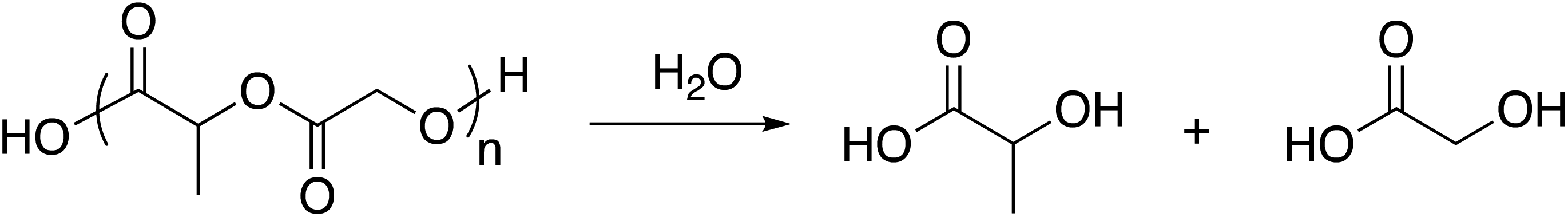 | ||
| Scheme 3 General reaction scheme for the hydrolytic degradation of PLGA.67 | ||
Degradation rates of poly(hydroxyalkanoates) are predictable and controllable by composition. Hydrolytic degradation of PLA is slower than that of PGA due to the wider available approach angles available to PGA carbonyls and relatively more non-polar PLA composition, while the copolymers, PLGA, display degradation rates between those of PLA and PGA.68
3.3. Other polyhydroxyalkanoates
Poly(hydroxyalkanoate)s (PHAs) comprise a broader natural polyester family, that includes PLA and PLGA, many of which are produced by a variety of microorganisms for energy and carbon storage.71 Additionally, PHAs exhibit considerable structural diversity with over 150 different PHA monomers currently identified (Fig. 8). Thus, some degree of material property control can be imparted through monomer selection.72 However, the high cost of production is challenging to overcome when incorporating PHAs into commercial markets to replace much cheaper petroleum-sourced materials.73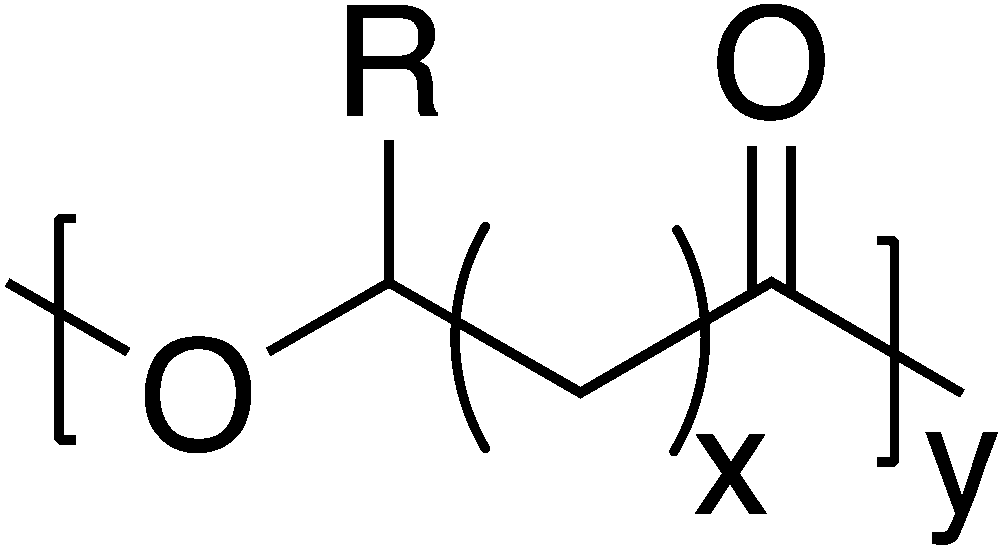 | ||
| Fig. 8 Generalized PHA structure. | ||
| Sample | T g (°C) | T m (°C) | Young's modulus (MPa) | Compressive strength (MPa) | Tensile strength (MPa) |
|---|---|---|---|---|---|
| PLA | 45–60 | 150–162 | 350–3500 | 58–72 | 21–60 |
| PLLA | 55–65 | 170–200 | 2700–4140 | 124 | 16–150 |
| PLGA | 45 | — | 225 | — | 6 |
| PHAs | −53 to −1 | n/a–168 | 200–3500 | — | 1–22 |
| Cortical bone | — | — | 7000–30000 |
130–200 | 50–151 |
| Trabecular bone | — | — | 50–500 | 0.1–16 | 2–19 |
3.4. Poly(butylene succinate)
The ester functionality is common in biodegradable materials because of abundant natural enzymes that catalyze ester hydrolysis, in addition to purely chemical hydrolysis. Poly(butylene succinate) (PBS), for example, exhibits promising material properties in conjunction with degradation rates that can be tuned through clever copolymerization strategies (Fig. 9).81,82 PBS finds use across multiple environmentally friendly applications such as mulch films, catering products, foams, and compostable bags. Unfortunately, PBS suffers from the same faults as other bio-sourced materials – high production cost and low commercial availability. Researchers are investigating more cost-efficient methods for PBS precursor production in hopes of increasing the availability of PBS.83 In addition, PBS and other aliphatic polyesters suffer from characteristically low melt strength, which complicates melt processing techniques. PBS molecular weight offers some control over melt strength but often blends with other polymers are necessary to enable use of PBS in high melt strength processes, e.g. foams.84–86 Consequently, blending PBS with non-biodegradable materials minimizes the applicability and relevance for green applications. However, PBS blends with cellulose esters show good miscibility while maintaining the biodegradability expected from PBS.87,88 PBS/cellulose acetate/dextran membrane separation efficiency and pore size showed a dependence on membrane composition – higher dextran incorporation increased the porosity, which consequently increased the degradation rate.89 PBS also served as an effective matrix for aligned and randomly oriented reinforcing cellulose nanofibers. Through addition of aligned nanofibers, the fracture stress and Young's modulus increased by 1.5 and 2.4 times while the randomly oriented samples saw an increase of 1.3 and 2.0 times, respectively.90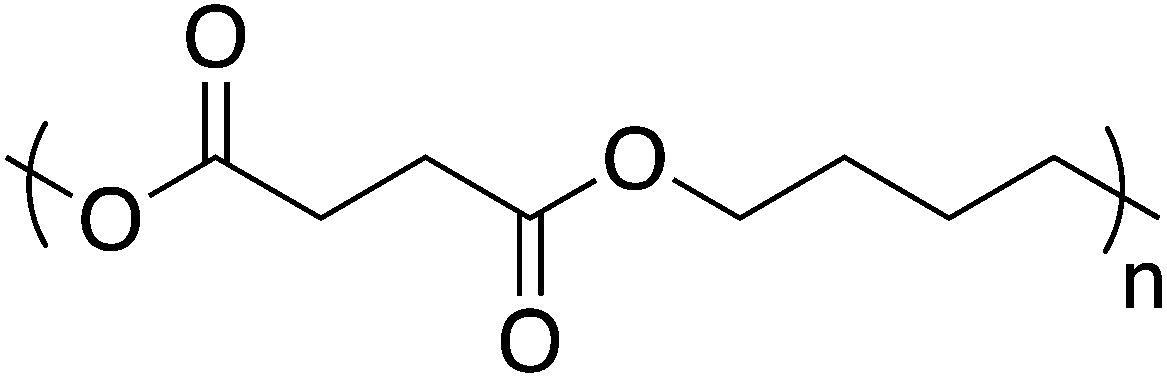 | ||
| Fig. 9 Poly(butylene succinate) structure. | ||
3.5. Polycaprolactone
Like PLA, polycaprolactone (PCL) is synthesized by anionic or cationic ring-opening polymerization (Fig. 10). PCL is a biodegradable, semicrystalline polyester with a Tg of −60 °C and Tm near 60 °C.94 These relatively low thermal transitions ease processability but ultimately complicate practical applications. To overcome this, PCL is often used in blends with other polymers or fillers, similarly to PBS.95 PCL degrades at rates that allow it to persist up to 4 years; rate is influenced by polymer molecular weight, crystallinity, porosity, thickness, and shape. Consequently, PCL often finds use in applications that require significant durability, such as in long-term drug delivery devices.96 Like other aliphatic polyesters, PCL has relatively weak thermomechanical properties, which prevent its use in structural applications, such as in plates or screws. However, PCL has garnered considerable interest since the 1990s due to its superior rheological and viscoelastic properties compared to other resorbable materials, which increases its processability and versatility.97–100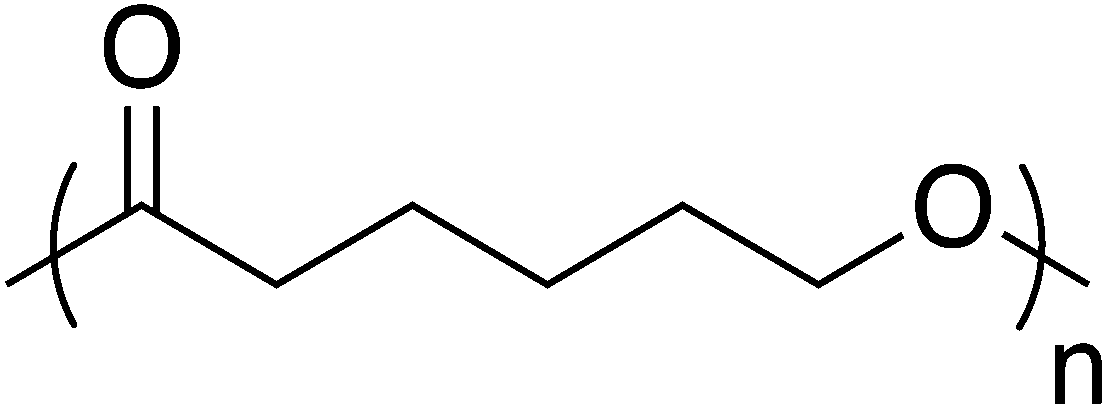 | ||
| Fig. 10 Polycaprolactone structure. | ||
4. Production of fiber reinforced polymer composites
4.1. Choice of fiber for matrix
Favorable interactions between the polymer matrix and reinforcing fiber are essential for a cohesive composite structure. Fiber-reinforced polymer composites are heterogeneous mixtures, whose strength varies with factors such as composition, constituent properties, and matrix-fiber interfacial interactions. The first two factors can be controlled with matrix/fiber selection and relative composition, while the last depends on matrix-fiber interactions, and the choice of processing method.103Eqn (1) estimates the elastic modulus of the resulting fiber-reinforced composite using component weight fractions and moduli.| E = ωfEf + ωmEm | (1) |
Eqn (1). The rule of mixtures equation used to estimate elastic modulus; ωx is the weight fraction of a given component and Ex is the respective elastic modulus.104
Composite processing also contributes to material properties. Employing an incompatible method of processing for a chosen fiber-matrix unintentionally induces defects within the composite, thereby reducing its effectiveness.105 To meet the needs of a particular application, choosing an appropriate matrix-fiber pair is essential.106 The large variety of available matrices and fibers facilitate production of acceptable composites that exhibit the desired properties. The most important aspect of fiber-reinforced composites is the interfacial cohesion between matrix and fiber. In the absence of interfacial cohesion, there is insufficient load transfer, and the fiber is unable to strengthen the surrounding matrix.107Fig. 11 illustrates the structure of a fiber-reinforced matrix, including the interface where cohesion is required.
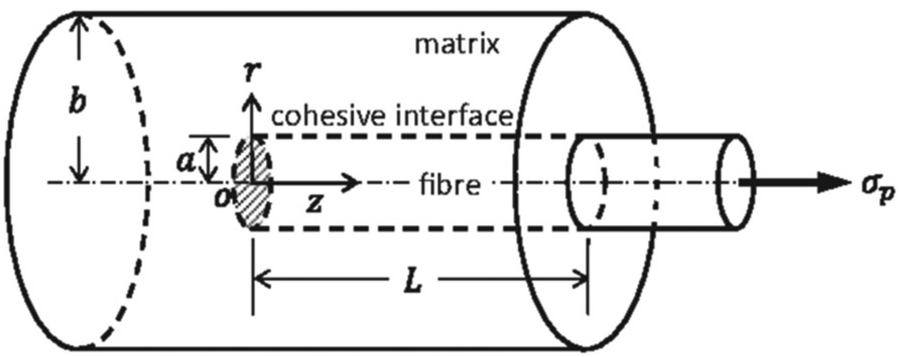 | ||
| Fig. 11 Diagram showcasing a cohesive matrix-fiber composite.108 | ||
4.2. Importance of matrix-fiber adhesion
In the interfacial region of the composite, two types of interactions strengthen the material; the interfacial energy, which describes the chemical interaction of the matrix and the fiber, and the interfacial shear stress, which accounts for the frictional component of the fiber when sliding across the matrix.109 The pull-out test, which measures the energy required to move a fiber from a given matrix, characterizes the strength of the interfacial bond.110,111 Other methods which test the interfacial shear strength are employed in the natural fiber reinforced composite literature – single fiber fragmentation, push-out, and the microdroplet test are common.112–115 Results from these analyses indicate whether the interface requires strengthening; such strengthening is typically achieved through chemical alterations of the fiber's surface. Such modifications may reduce surface energy, and/or increase chemical bonding between components, thus increasing frictional interactions.116Conventionally, carbon fiber surfaces have been oxidized to make the non-polar fiber more suitable with a polar epoxy matrix.117 Duan et al. showcase one method for surface oxidation of carbon fibers, which requires clean carbon fibers heated in a tubular furnace under air between 800–1200 °C for 30 min.118 More complicated oxidation methods exist, such as anodic oxidation, where the carbon fiber acts as an anode, and the potential allows for an electrolytic oxidation process to occur.119 Non-renewable carbon fibers commonly use these methods, but naturally sourced fibers require different strategies.
Natural fibers often require very different modifications than those required for carbon fibers. Whereas carbon fibers are more nonpolar than most composite matrix polymers, natural fibers are typically much more hydrophilic than the matrix. Plasma treatment, chemical modification, or numerous forms of physical alterations are employed to treat fiber surfaces. The hydrophilic polysaccharides of natural fibers contain many hydroxyl groups, leading to high levels of surface water absorption. Both the inherent fiber hydrophilicity and water uptake consequently decrease adhesion to more hydrophobic matrices; the tendency to take up water can also lead to dimensional instability and changing properties in response to, e.g., humidity.120 The following sections describe several common chemical methods used to adjust fiber hydrophilicity.
Immersing fibers in a concentrated sodium hydroxide solution, known as an alkaline treatment, is employed for natural fibers, and has been claimed to interrupt the hydrogen bonding that occurs between the hydroxyl groups located on the fibers. Although natural fibers contain partially crystalline cellulose, other substances, such as hemicellulose, are also present. Hemicelluloses (best described as non-cellulosic plant polysaccharides containing β-1–4-linked, equatorial polypyranose main chains)121 are soluble in aqueous alkali, and therefore will at least partially dissolve under such treatment, creating a rougher fiber surface, increasing frictional interactions.122,123 This treatment is used to reduce fiber-fiber interactions (crystallinity), and thereby, may facilitate fiber-matrix associations.
Hydrophobic chemical modification is another widely practiced method. For example, silylation of natural fibers is commonly employed, involving reaction of free hydroxyl groups on the fiber surface with an electrophilic reagent like a silyl halide. In one method, fibers were submerged in a 3.5 pH, 0.5 wt% acetone solution of a silane coupling agent for 45 min and dried at 65 °C displayed decreased hydrophilicity.124 Kabir et al. compared these methods, reporting that alkaline treatment yielded composites of a more cohesive nature, which exhibited higher strengths and elastic moduli.125 It should also be noted that silylation is relatively expensive, and that the resulting silyl ethers may be hydrolytically labile. Fig. 12 depicts silylation of a natural fiber surface.
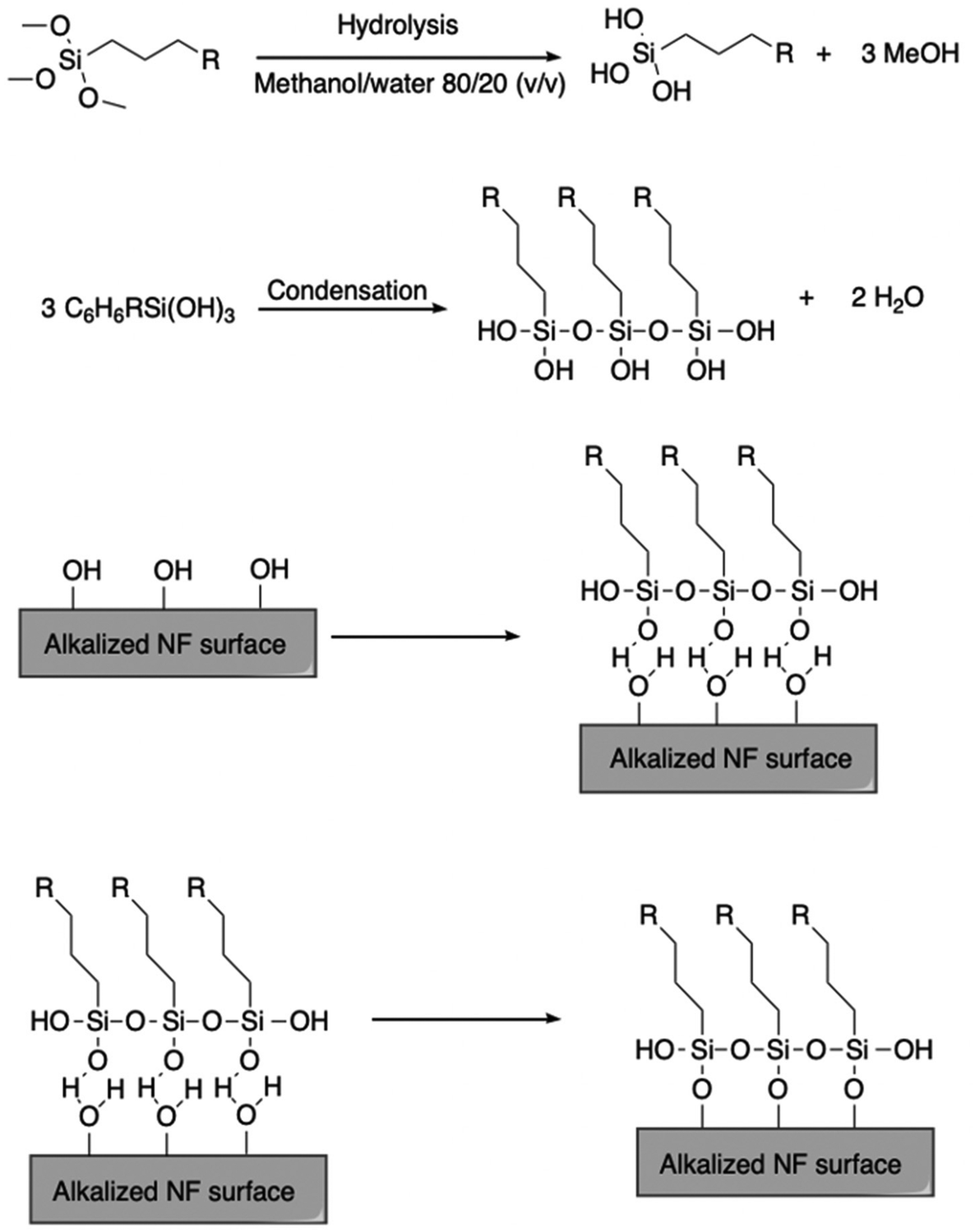 | ||
| Fig. 12 Reaction scheme detailing the synthesis of silane spacers and the natural fiber surface functionalization. Reproduced from ref. 126 with permission from Springer Nature under the Creative Commons Attribution 4.0 international license (https://creativecommons.org/licenses/by/4.0/), copyright 2018.126 | ||
Dispersion of the treated natural fiber into the chosen matrix is then carried out, by methodology depending on the fiber properties and the targeted application. The choice of treated fiber will also depend on the expected matrix-fiber interactions, as previously described, in addition to the required mechanical characteristics needed to meet the needs of the proposed function.
5. Cellulose-based renewable composites and applications
5.1. Physiochemical properties of cellulose
Polymers based on cellulose are highly desirable for composite applications due to cellulose abundance (approximately 1010 to 1011 tons produced annually), and their robust mechanical properties, predictable and tunable biodegradability, sustainability, and utility as lightweight materials. Isolation of cellulose from the lignin, hemicellulose, and pectin present in plant feedstocks is accomplished through methods summarized in Fig. 13. The isolated cellulose polymer consists of anhydroglucose (AGU) units covalently bonded through β-(1→4) glycosidic linkages.127 The process of separating cellulose from other plant components is called pulping, and the corresponding biomass pretreatment methods are divided according to chemical, physical and biological subsets. Furthermore, the selection of the appropriate purification process depends on the requirements of the downstream cellulose application.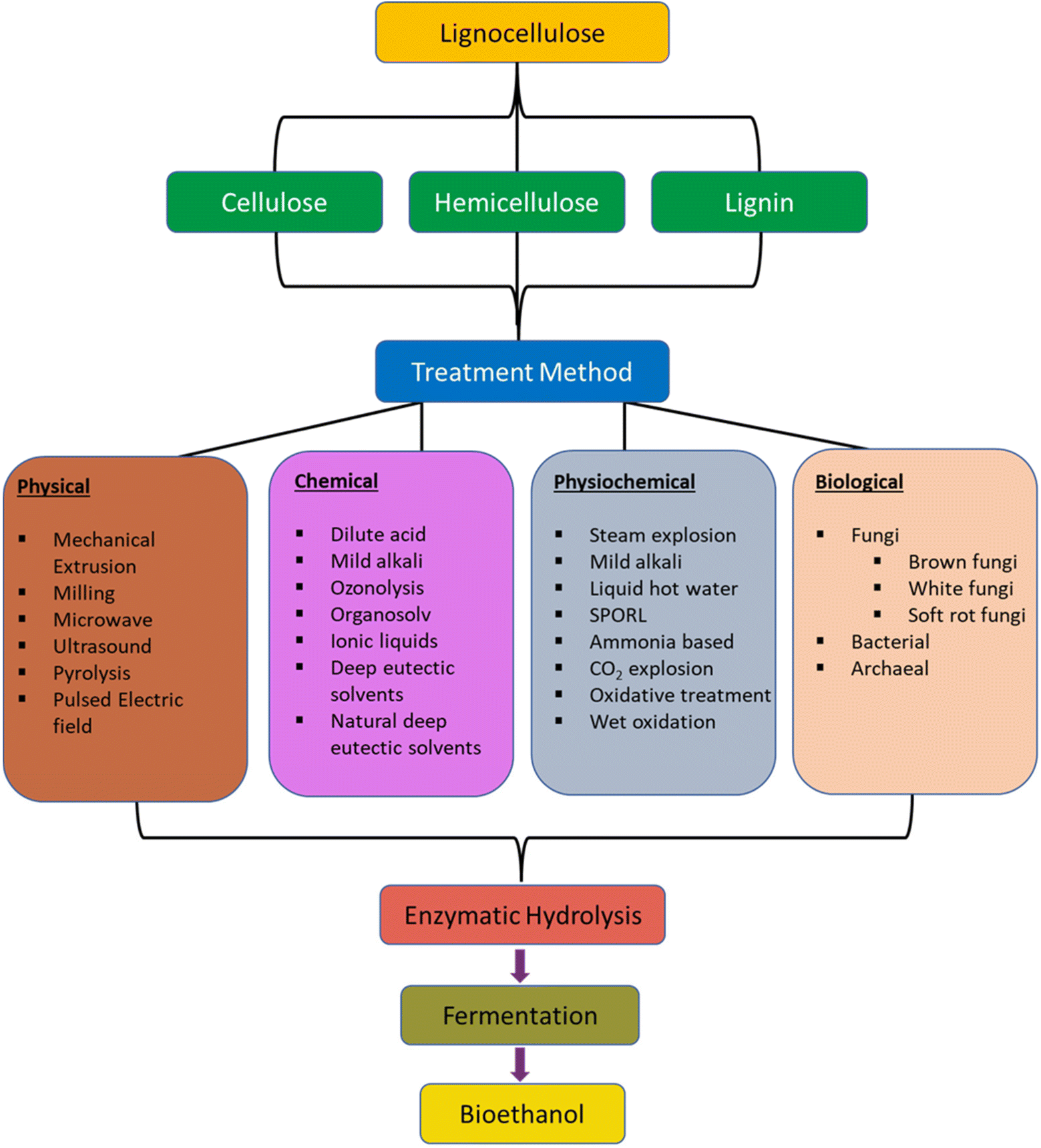 | ||
| Fig. 13 Summary of delignification processes for isolating cellulose and hemicellulose. Reproduced from ref. 127 with permission from Springer Nature under the Creative Commons Attribution 4.0 international license (https://creativecommons.org/licenses/by/4.0/), copyright 2017.127 | ||
Cellobiose can be viewed as the effective structural repeat unit of cellulose because of the nearly 180° twist between each pair of glucose units. Each glucose (chemically cellulose is a homopolymer of glucose) bears three hydroxyl groups, making cellulose a hydrophilic polymer prone to absorbing water. Further, the cellulose hydroxyl groups are in the equatorial position on the glucopyranose ring; this orientation creates hydrophilic and hydrophobic faces of the polymer chain. This permits the cellulose chains to stack on top of each other as long ribbons, forming tightly knit, extensive inter- and intramolecular hydrogen bonding networks.128 While hydrogen-bonding and high alignment of the polymer chains provide the mechanical strength along the fiber axis and reinforcement desired in polymer composites, the high crystallinity of cellulose fibers renders them insoluble in water and all single organic solvents. Cellulose also does not exhibit a Tg or Tm before thermal decomposition (Td), rendering thermal processing infeasible. Cellulose is quite thermally stable, and most cellulose decomposition occurs above 300 °C, but because of the extensive hydrogen bonding of cellulose, its (theoretical) Tg and Tm values are at higher temperatures and are not observed or attainable.129 Such behavior is typical of polysaccharides. Thermal decomposition may also depend on the crystalline content of the cellulose polymer, where increasing crystallinity correlates with higher Td values. Finally, the hygroscopicity and the highly polar nature of the polymer make it difficult to blend with hydrophobic polymers, resulting in poor interface interactions, adhesion issues, and compromised mechanical properties.130,131 Cellulose can readily absorb 10% of its weight or more of water at typical humidity values, which can also lead to weak interfaces, undesirable humidity responsiveness, and dimensional instability.132
5.2. Chemical modification of cellulose
To address issues of low solubility, thermal processability, and polymer compatibility, cellulose is often chemically modified through esterification, etherification, oxidation, and other similar methods. Typical cellulose esterification methods use acid pre-treated cellulose that undergoes subsequent reactions with carboxylic anhydrides and a strong mineral acid catalyst. This direct esterification method produces various degrees of substitution (DS).133 DS refers to the average number of substituted hydroxyl positions per AGU and reaches a maximum value of roughly three per AGU. Esterification with cyclic anhydrides, such as phthalic anhydride, in the presence of organic bases like pyridine and triethylamine in solvent (DMAc/LiCl) or in ionic liquids such as 1-allyl-3-methylimidazolium chloride is possible as well.134–136 Acid-catalyzed reaction with cyclic anhydrides is impractical due to polysaccharide crosslinking caused by the difunctional reagent.137Sulfuric acid catalysis significantly degrades the polymer during esterification; therefore, it is not a viable approach if very high molecular weight polymers are requisite for the target composite application (though it is sufficient for achieving molecular weights of ca. 30–100 K, which are adequate for many plastic applications).138 On the other hand, reacting cellulose fibers with aqueous H2SO4 is the usual preparation method for cellulose nanocrystals (CNCs) used in composites. Acid degrades the more accessible amorphous regions of the cellulose polymer first, leaving behind partially sulfated cellulose nanocrystals.139 TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy)-mediated oxidation occurs on the primary C-6 hydroxy groups; this modification is often used on cellulose nanocrystals to enhance interfacial adhesion.140 Various grafting strategies such as ATRP (atom transfer radical polymerization) and RAFT (reverse addition fragmentation radical polymerization) of cellulose pulp, nanocrystals, and nanofibers can further modify the AGUs and control hydrophilicity, surface compatibility, mechanical strength, and other properties.139–145 A summary of numerous chemical modification strategies for CNCs is presented in Fig. 14.
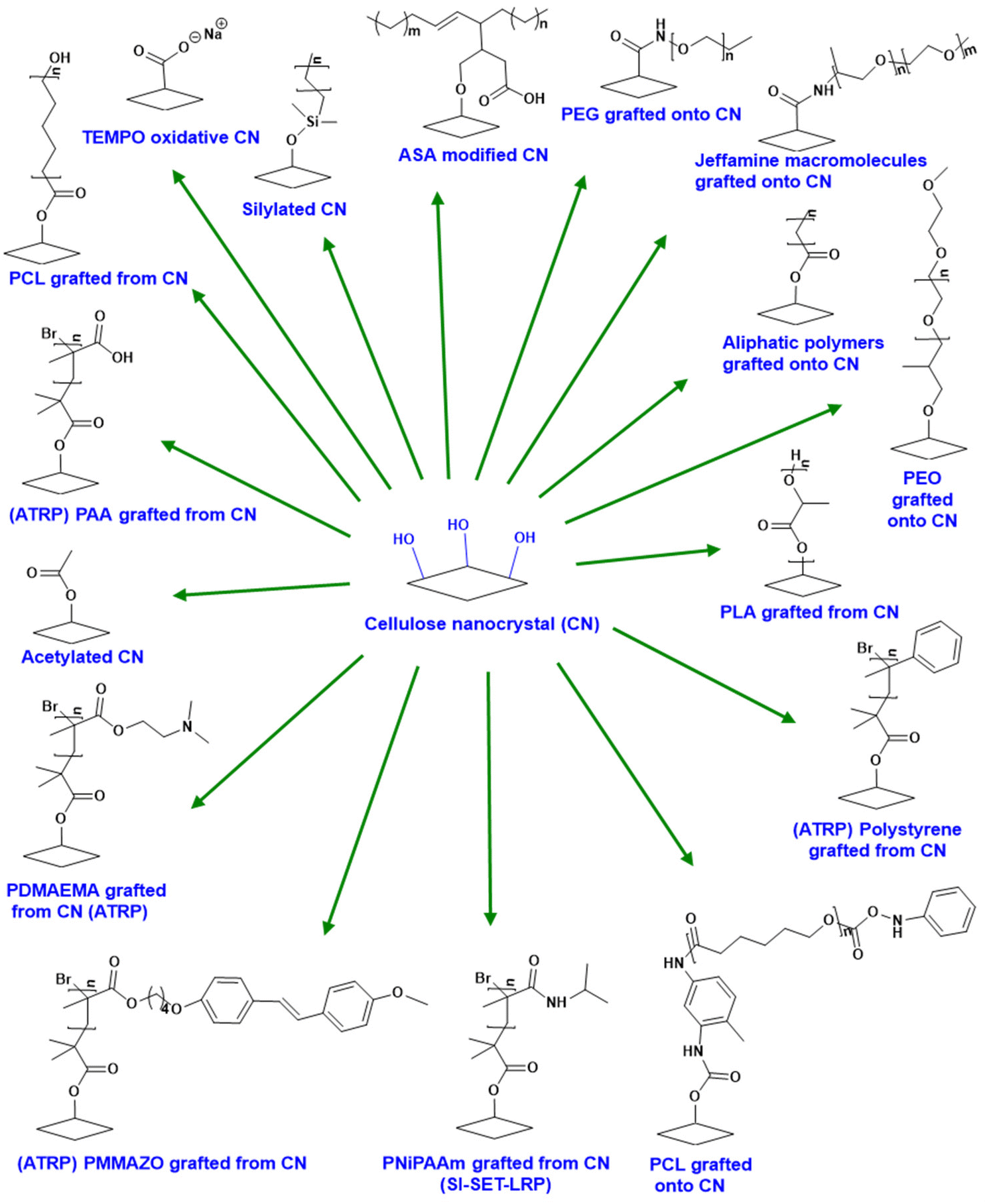 | ||
| Fig. 14 Chemical modifications of nanocellulose. Reproduced from ref. 145 with permission from Elsevier, copyright 2013.145 | ||
5.3. Cellulose nanocrystals for composite reinforcement in green plastics
Cellulosic materials are a valuable resource for satisfying the demand to develop green composite materials that replace petrochemical-based polymers with biodegradable polymers.146,147 One approach to this problem involves blending CNCs with various biodegradable polymers such as poly (lactic acid) (PLA), poly(3-hydroxybutyrate) (P3HB), and polysaccharides like starch, chitosan, and κ-carrageenan.148,149 CNC's highly ordered, crystalline nature (Fig. 15), high Young's modulus, and low density make them desirable for composite reinforcement applications that improve mechanical, thermal, and barrier properties of neat polymers. For example, blending molten PLA with CNC sulfate esters suspended in DMAc/LiCl, where both polymers are biodegradable materials, significantly increased the mechanical tensile strength of the resulting film from 48 MPa (PLA) to 78 MPa (PLA/CNC sulfate ester) while the Young's modulus improved from 2.9 to 3.9 MPa.150 Other reports demonstrate that blending CNCs with chitosan or κ-carrageenan biopolymer films led to tensile strength properties in the range of synthetic polymer-based films such as LDPE, HDPE, and PP. Additionally, they exhibited favorable water and UV barrier properties, thereby showing promise as biodegradable materials for food packaging applications.148,151 Generally speaking, polysaccharide and derivative films have desirable oxygen barrier properties but poor water barrier properties. Hydrophobic polymers such as PLA tend to have a complementary set of properties. | ||
| Fig. 15 Transmission electron micrograph images of the highly crystalline and ordered morphology of cellulose nanocrystals.139 | ||
Ultimately, blends of polysaccharides with hydrophobic polymers like PLA create the potential for materials with acceptable barrier properties towards both water and oxygen. Unfortunately, issues of CNC agglomeration are a plaguing problem in composites, especially during co-extrusion, injection molding, and/or casting, which are typical commercial methods for incorporating CNCs into a polymer matrix to manufacture the resulting composite material. Agglomeration (such as with CNC nanoparticles) occurs when fillers within a composite material attract each other in such a way that the cohesive forces between them are stronger than those with the surrounding matrix material. Consequently, the nanoparticles will begin to aggregate and form a non-uniform dispersion within the matrix. Agglomeration can result from the concentration, type, and shape of the CNC present in the composite resulting in disadvantageous effects on composites such as weakened load bearing, localized stress points within the composite, loss of thermal stability, and reduced interfacial interactions (reduced mechanical and chemical bonding between the filler and the surrounding matrix.152–154 These undesirable physiochemical and mechanical properties emerge in composites and hinder their potential for wider use; therefore, research has focused on using computer modeling, improved material processing, and methods for functionalizing CNCs to address agglomeration and the resulting disadvantageous properties. The most critical research has focused on addressing compatibility issues of the CNC with its surrounding polymer matrix, followed by evaluating the morphology of the matrix and the nanofiller, which influences the behavior of the interfacial interactions and overall characteristics of the composite.155
More recently, starch (comprising amylose and amylopectin biopolymers, Fig. 16) has also garnered interest as a material for green composites. It is abundant, commercially available, and comparatively inexpensive. Although starch typically exhibits weak mechanical properties and brittleness, and pure, dry starch cannot be thermally processed, glycerol or water will plasticize starch granules, permitting convenient thermal processing of the polymer. Various studies have also demonstrated that the addition of CNC fillers improves mechanical properties of the starch/glycerol blends. The resulting films had Youngs moduli drastically improved from 30 to 500 MPa, and tensile strength improved from 4 to 12 MPa. These values were on par with carbon-nanotube fillers in starch/glycerol blends yet were fabricated from completely renewable and truly green composites.149,156
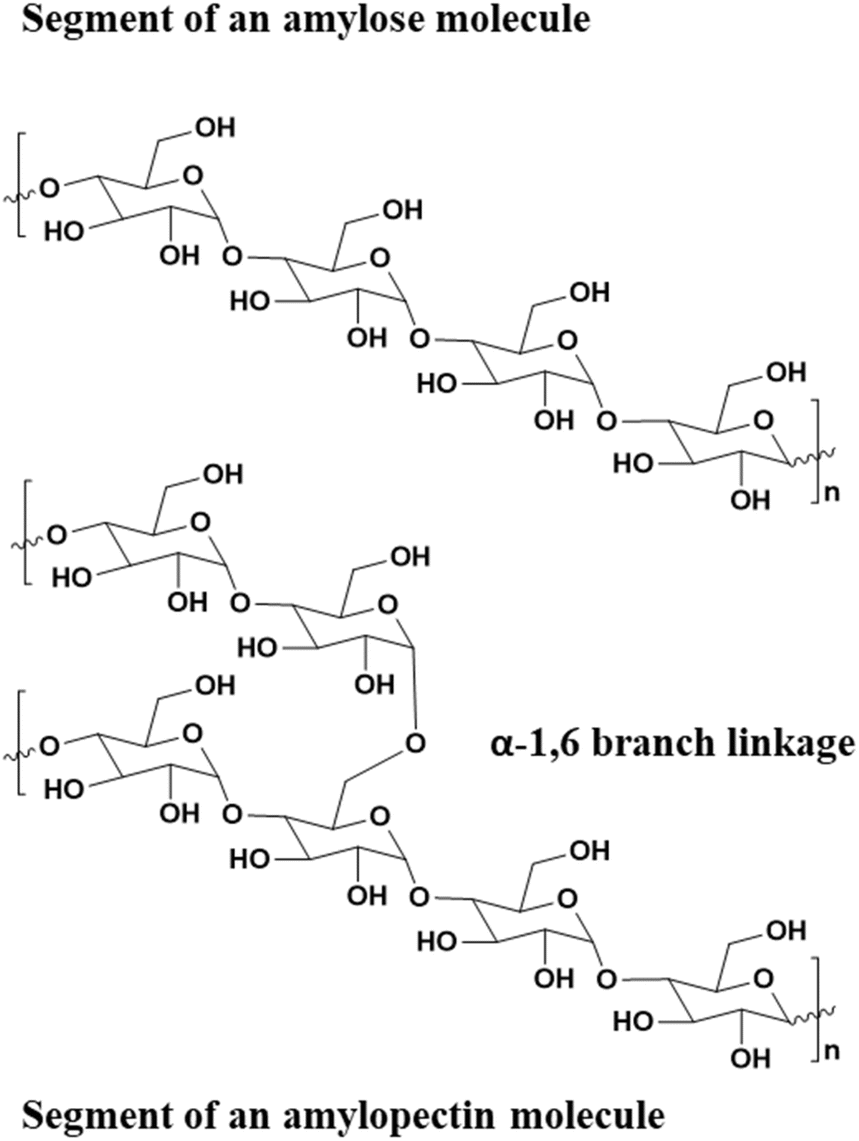 | ||
| Fig. 16 Structure of amylose and amylopectin biopolymers comprising starch.149 | ||
Although glycerol and water are known to improve the processability of starch, starch by-itself is hygroscopic, and the resulting materials tend to have high water permeability, low tensile strength, and overall low reinforcing mechanical properties which prevents their use in many composites applications.149,157 Other plasticizers such a urea, formamide, and polyols can be used to plasticize and process starch although they don't address the issues of poor mechanical strength and/or water permeability. However, reports demonstrating the use of CNC fillers in starch show that you can improve the water vapor sensitivity issue compared to starch films alone while still maintaining an environmentally friendly resulting material. CNC/starch films are highly compatible together due to their strong hydrogen bond network which prevents water molecules from overcoming the hydrogen bond network and solubilizing the composite compared to starch films alone.158–160
Another approach to addressing the water vapor permeability issue and improving mechanical properties of starch is through the esterification of starch producing various degrees of substitution (DS) of starch acetates. Numerous reports have demonstrated that achieving DS (acetyl) values between 1.2–1.7 imparted both optimal hydrophobicity resulting in desirable water barrier properties while also balancing out mechanical properties.161,162 Starch acetate films between these DS values have also shown to be biodegradable when placed under compostable environments. Furthermore, regioselectivity esterifying starch can also affect is biodegradability and water-barrier properties. For example, regioselective 2-O-acetyl starch was shown to only swell in water while non-regioselectively esterified starch was completely soluble even when the DS (acetyl) values were similar.163 These findings suggest that when choosing a matrix and a filler, especially for completely green composites, factors such as the type of biopolymer, impact of regioselectivity of the biopolymer (if functionalized), as well as the type of filler are all important factors to consider for the final desired properties and the downstream application of the material.
5.4. Cellulose nanofibers and composite applications
Cellulose nanofibers (CNFs) are another important class of nanocellulose-based materials. They are typically extracted from plant cell wall biomass or made from bacterial sources producing two different classes of fibrils, plant-sourced and bacteria sourced, although CNFs can also be obtained from marine animal tissues such as tunicates.164–166 A major morphological difference between CNCs and CNFs is in their dimensions. Whereas CNCs resemble rod-like structures and are nanometric in all dimensions, ranging from 100–200 nm in length and 5–20 nm in diameter, CNFs are micron-sized in length, on the order of > 1 μm, while having diameters in the nanoscale range of 5–200 nm.166,167 Extraction methods for CNFs involve homogenization, grinding, cryo-crushing, high-intensity ultrasonic treatment, enzymatic methods, alkaline treatment, and mild acid hydrolysis.165,167,168 As cellulose nanofibril extraction methods do not employ the same strong acid extraction treatment (H2SO4) used to isolate CNCs, CNFs contain both amorphous and crystalline regions comprising of entangled, flexible cellulose chains. The elastic moduli of highly crystalline cellulose nanofibrils can be on the order 150 GPa in the longitudinal direction and between 18–50 GPa in the transverse direction, making them highly desirable for imparting both mechanical strength and sustainability in composite materials.166,169Cellulose nanofibers have been incorporated into a variety of composites (e.g. electronic devices, aerospace materials) due to their impressive thermal expansion coefficients of 5 × 10−6 K−1 (parallel to layer) and 7 × 10−6 K−1 (perpendicular to layer) at low temperatures (−120 °C to −150 °C). These values are close to those of ceramics but much lower than the thermal expansion coefficients of typical polymers and metals used in membranes for purification and water filtration applications.164,170–172 Wang, et al., used in situ polymerization of pyrrole and catechol-containing tannic acid (low-cost plant polyphenol) on the surface of CNFs with the addition of FeCl3 as an oxidant to create interconnected nanohybrid network composites for potential applications in energy storage. The CNF TA/polypyrrole networks were embedded into commercial, inexpensive soy protein isolate matrices, and the resulting flexible composites showed high conductivity (0.018 S m−1) with excellent mechanical properties (tensile strength of 14.5 MPa and toughness of 3.5 MJ m−3) as compared to other hybrid film, rigid-filler-reinforced SPI composites.173
Use of nanocellulose-based composites, such as CNFs, provides significant advantages with regard to cost, sustainability, and low environmental impact relative to petrol-based polymers, especially in energy-based applications such as manufacturing of solar cells and batteries.174 Plastics and inorganic materials such as those from group III–V elements are typically used in solar cells. However, nanocellulose can provide a green alternative to plastics in solar cells due to thermal stability, tunable mechanical properties, and optical properties (such as transparency) that rival and in some cases are better than those of petroleum-based plastics making CNCs potentially viable replacements in those industries.175 One could envision utilizing recyclable nanocellulose for the production of transparent films used in solar cells for example, to both reduce plastic waste and the environmental impact of nanocellulose extraction itself, making the entire manufacturing process greener and more sustainable.
5.5. Cellulose composites in biomedical applications
Benign cellulose is also of value in biomedical composite applications. In addition to beneficial mechanical properties, cellulose fibers and cellulose nanocrystals exhibit low toxicity, low immunogenicity, and positive stability against degradation in vivo, proving useful in the development of novel drug delivery vehicles, orthopedic devices, biosensors, and vascular grafts.176–178 Recent studies developed composites with CNC, poly (ethylene glycol) diacrylate (PEGDA), and a compatibilizer (1,3-diglycerolate diacrylate (DiGlyDA)). The resultant resins were 3D printed into various shapes for possible orthopedic applications.179The CNC composite resin was cured using digital light processing (DLP) and exhibited improved mechanical properties relative to the PEGDA/DiGlyDA polymer blends without CNCs. Furthermore, CNC incorporation did not interfere with the fidelity of the printed structures, closely matching the original SolidWorks CAD designs (Fig. 17).179 They demonstrated that varying CNC concentration affects water uptake of the resulting composites, providing a useful avenue for controlled swelling behavior of printed CNC-composites.
 | ||
| Fig. 17 Image of CAD designs (column one), corresponding DLP 3D printed resins without CNC (second column), and with 1 wt% CNC filler in the resin (third column).179 | ||
Hydrogels are utilized in numerous biomedical applications such as drug delivery, tissue engineering, and wound dressings. Natural polymer-based hydrogels garner attention for biomedical applications due to their compatibility with many tissues, and in many situations, tissue-mimicking consistency, porosity, and ability to absorb fluids, such as water or cellular media.180–182
Other investigators have tried to combine the high stiffness modulus and reinforcement properties of CNCs with softer, self-healing polymer matrices to form nanocomposite hydrogels.183–185 Supramolecular, 3-component CNC hydrogels were developed for this purpose.185 For example, CNCs were functionalized with copolymers dimethylaminoethyl methacrylate (DMAEMA) and napththyl-functionalized methacrylate (NpMA) repeat units. The soft matrix was prepared from poly (vinyl alcohol) (PVA) modified with pendant methyl viologen groups (PVA-MV). Hydrogel composites processing utilized the physical crosslinking between the CNC-g-P(DMAEMA-r-NpMA) and PVA-MV, which was facilitated by host–guest interactions with cucurbit [8] (CB [8]), a macrocyclic molecule that physically entrapped “guests” in the CB [8] pocket. The CNC nanocomposite hydrogels exhibited excellent self-healing properties, high storage modulus resulting from the CNC-reinforcing component, and good storage stability. The hydrogels maintained their self-healing properties after several months of ambient storage.185
Demand for renewable and sustainable materials is based on societal trends leading us to believe that it will continue to increase; therefore, incorporating renewable polymers such as cellulose-based nanocrystals, fibrils, and polymers into existing and new composites is a viable route to improving biodegradability. Herein several examples of technological advancements and applications for cellulose-based renewable composites are presented. Further developments in 3D printing, microfluidics, and other fabrication technologies are also needed to enhance the scalability of novel green materials, thereby creating manufacturing-friendly processes for large-scale production of future candidate renewable composites. Immiscibility of naturally hydrophilic polysaccharides with hydrophobic polymers has obstructed progress towards such polymer blends. Immiscibility of polymer blends is problematic as the interfaces between two immiscible polymers are weak points, encouraging material fracture. Compatibilizers blended with the polymer system of interest may overcome this problem; however, creating such compatibilizers is not a trivial task, and the required compatibilizer structure depends on the specific polymer system.186
The hygroscopic nature of polysaccharides and their poor water barrier properties may also hinder their use in industrial applications, depending on the end-use of the material and its potential exposure to water.187 These barrier and water absorption challenges can be addressed through polysaccharide functionalization.138 Currently, important research topics include functionalizing cellulose for improved water vapor barrier properties, thermal processing, and lowered polysaccharide aggregation within composites.188,189 In addition, surface functionalization is often required to facilitate favorable interfacial interactions between hydrophilic natural materials, such as cellulose, and hydrophobic synthetic polymers.
6. Potential applications for green composite integration
While all facets of society require greener materials, it may be more feasible to integrate green composites into new technologies under development, rather than trying to supplant materials used in current products and technologies. The wide variety of properties available in the diverse population of natural fibers, detailed in Table 4, will enable a wide range of performance and application. Additionally, it is important to acknowledge the financial benefit of natural fiber composites as they are generally available at a lower cost than synthetic fibers, though prices will be influenced by weather and other agricultural market forces.190,191 Along with clear financial benefit, life cycle analyses have shown that natural fibers consume considerably less energy during production than do synthetic materials (reed fiber: 3.64 MJ kg−1, glass fiber: 48.33 MJ kg−1, polypropylene: 77.19 MJ kg−1).192,193 One caveat is that natural fibers may negatively impact aquifers due to fertilizer run-off, leading to higher concentrations of pollutants like nitrates and phosphates. Such impacts must be balanced against reduced carbon emissions from synthetic material sourcing and synthesis.194 A life cycle assessment determined that an automotive component constructed from a natural fiber/synthetic fiber hybrid composite generated 33% lower environmental impact compared to spring steel.195 Similarly, Boland et al. reported that natural fiber composite replacements for glass fiber composites resulted in 6.5–7.4% energy savings through the material lifetime.196| Fiber | Density | Young's modulus (GPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|---|
| Flax | 1.54 | 27.5–85 | 345–2000 | 1–4 |
| Ramie | 1.5–1.56 | 27–128 | 400–1000 | 1.2–3.8 |
| Hemp | 1.47 | 17–70 | 368–800 | 1.6 |
| Jute | 1.44 | 10–30 | 393–773 | 1.5–1.8 |
| Sisal | 1.45–1.5 | 9–22 | 350–700 | 2–7 |
| Coconut | 1.15 | 4–6 | 131–175 | 15–40 |
| Cotton | 1.5–1.6 | 5.5–12.6 | 287–597 | 7–8 |
| Nettle | 1.51 | 24.5–87 | 560–1600 | 2.1–2.5 |
| Kenaf | 1.2 | 14–53 | 240–930 | 1.6 |
| Bamboo | 0.6–1.1 | 11–17 | 140–230 | — |
| E-glass | 2.5 | 70 | 2000–3500 | 2.5 |
| Carbon | 1.4 | 230–240 | 4000 | 1.4–1.8 |
6.1. Carbon fiber based prosthetics
Carbon fiber composites are frequently used in the healthcare industry, e.g., in prosthetics and orthotics. Incorporating composites into the prosthetic industry has been revolutionary as amputees, especially those who lost lower limbs, were shown to expend more energy performing the same tasks as non-amputees.198 In this rapidly changing field, lightweight composites not only reduce the energy expenditure needed, but may afford more support and assistance to the wearer than previous non-composite materials. The current carbon fiber/epoxy composites found in prosthetics, such as running blades, are candidates for replacement with biodegradable components that would allow the materials to return to the environment through decomposition after their lifetime. Such improved devices would allow patients to gain function while increasing device sustainability and renewability.6.2. High performance drones
Technological advances in the 21st century have relied heavily upon robotics and electronics. Contemporary applications of unmanned aerial vehicles (UAVs) are growing tremendously, not only within the military, and in surveying and surveillance, but also in the relatively recent commercialization of the UAV format for personal use drones.199 Composites are advantageous in UAVs due to their high strength-to-weight ratio, which is ideal for aeronautical applications. For UAVs, the fuselage is a primary area of concern as it contains key components, thus manufacturers aim to protect high-cost equipment using carbon fiber/epoxy composites.200 The drone market is projected to reach $36.9 billion in 2022, further highlighting the opportunity for composites and their protective qualities within UAVs.2016.3. All-electric aircraft
Air travel revolutionized the world and ushered in a new age where once unimaginable places were relatively easily accessible with planes. However, climate change was unheard of and the resulting impact of this high emission mode of transportation was yet to be understood. Currently, jet fuel combustion accounts for nearly 3% of annual energy use related CO2 emissions.202 Multiple viable methods for reducing air travel's impact on climate change exist, but the idea of all-electric aircraft has garnered major interest as increased electrification of modern jets is already underway and has contributed to increased fuel efficiency.203 Technology enabling electric flights up to 600 nautical miles would reduce fossil fuel consumption and CO2 emissions by 15% and flight capabilities up to 1200 nautical miles would provide a reduction up to 40%.204 While all-electric aircraft remain an idyllic situation of the future, research encompassing green electricity generation, energy storage, and electric propulsion provides steps towards these ideals.6.4. Composites in wind turbines
Capturing renewable energy, such as wind, requires high-strength, durable materials currently obtained from primarily non-renewable sources. Energy capture structures such as wind turbines reduce the carbon footprint of energy use, but the construction of such structures in and of itself produces waste. Investigators have demonstrated using life cycle energy assessments that natural fiber reinforced composites require 40–60% less energy compared to glass composites.205 While some wind turbines boast 80% recyclability of materials, ample room for improvement exists, achievable using green composites. For example, some glass fiber composites are incinerated after the life cycle of the turbine.206 Wind energy is becoming an increasingly important source of renewable energy, as shown in Fig. 18, and even minor increases to overall turbine recyclability will lead to significant amounts of carbon waste reduction.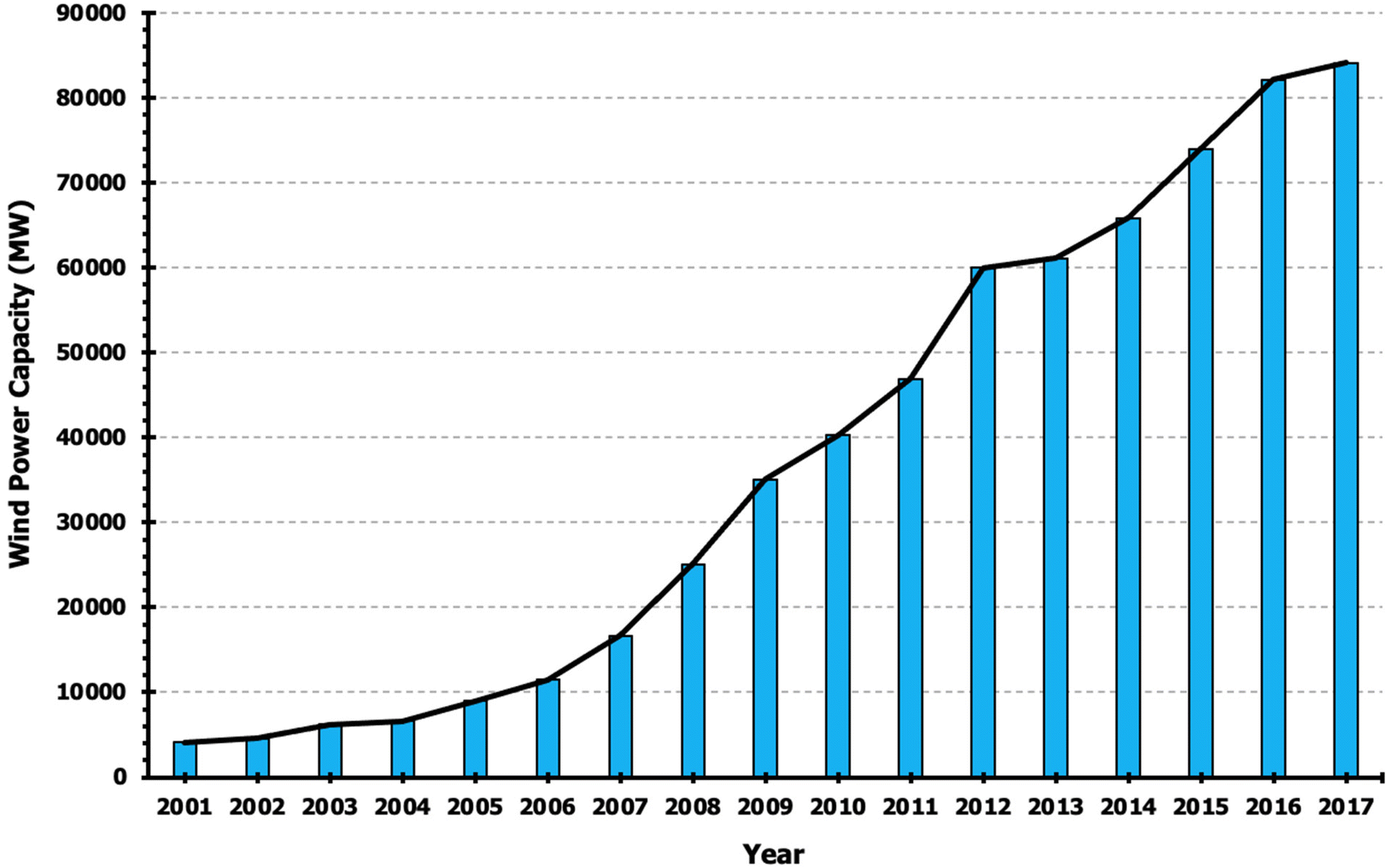 | ||
| Fig. 18 Annual wind energy production in the United States.207 | ||
Typically, E-glass fibers find use in wind turbine blades due to their high tensile strength, which is required to maintain structural integrity while experiencing rotational and wind-induced forces. Recently, Batu et al. determined through various mechanical tests that natural fiber-reinforced composites perform nearly identically to the industry standard E-glass/epoxy networks, while delivering 28–38% weight reduction.208 Natural fiber-reinforced composites are limited to non-load-bearing portions within turbines, for reasons that highlight the aspects of natural-fiber based composites that are still in need of research and improvement. The tendency of natural fibers to absorb moisture and consequently their relative dimensional instability, potentially combined with mismatched interfacial energies, can lead to softening and loss of strength over time.209
7. Conclusions
Composites are complex materials comprising multiple components, leading to complexities not faced by traditional single component materials, such as interfacial adhesion. Composites are more complicated compared to conventional metals or plastics as their properties depend on composition, matrix/fiber match, fiber orientation, fiber length, and additional variables. While such factors can increase development time and cost for composites, their ability to combine desirable material properties has enabled immense growth across various fields, most notably aerospace, infrastructure, and automotive. Natural biopolymer-based composites may be expected to fuel further growth as they enable materials that better meet societal sustainability and greenhouse gas reduction goals. Key points of our review include:• Natural fibers are inherently hydrophilic and often require chemical modification to facilitate strong interfacial interactions between matrix and reinforcing fibers.
• Natural fibers are available at a lower cost than synthetic fibers and have reduced energy requirements compared with synthetic materials.
• Natural fibers, specifically leaf fibers, are often waste products, potentially enhancing supply and reducing cost of the materials.
• Available natural fibers exhibit a wide array of properties that enable production of composite materials with excellent mechanical properties.
• Many markets currently dominated by unsustainable materials show promise for replacement with green composites including packaging, automotive, electronics, and biomedical industries.
• Current biodegradable polymer matrices comprised primarily poly(hydroxyalkanoates), due to their natural abundance or sourcing, and natural degradation pathways (hydrolytic and enzymatic).
• Biodegradable polymer matrices are currently used primarily in resorbable biomedical applications.
• Incorporating nanocellulose into composites can reduce the use of fossil resource-based plastics without compromising physicochemical and mechanical properties in many applications.
• Important ongoing research goals must include enhancing compatibility between natural reinforcing agents and matrices, economically reducing water sensitivity of biopolymer-based components, and ensuring that use of biopolymers in composites does not negatively impact food availability.
8. Future directions
Natural fibers show great promise as potential replacements for synthetic, petroleum-dependent fibers, such as carbon fiber. The diversity of commercially available, natural fibers permits virtually endless fiber/matrix combinations, allowing for tunable mechanical properties. Bast fibers with increased cellulose content lend themselves to high strength, low flexibility applications, while those with increased pectin content, like coconut fibers, allow for more dynamic and flexible materials. The principal issue for entirely green composites is the relatively constrained current set of available natural polymer matrix materials. In addition, attention must be paid to mitigating disadvantages inherent in natural fibers, including their hydrophilicity, dimensional instability, and the challenges of natural fiber processability. Natural fiber composites are often overly sensitive to environmental conditions, which is impeding green composites displacing traditional materials. These sensitivities are detrimental to green composite integration in particular applications; for example, a highly water unstable structural component, such as PLGA, is not suitable for applications with exposure to moisture, rain, and snow. Much current research is aimed at increasing the strength of natural fiber composites, and thereby widening the scope of their usage. Successful assimilation of green composites will significantly reduce CO2 emissions, both because additional cultivation of source crops sequesters more CO2 and because of reduced fossil fuel-based material production.Conflicts of interest
There are no conflicts to declare.References
- E. J. Babero, Smart composites: mechanics and design, CRC Press, Boca Raton, 1st edn, 2013 Search PubMed.
- R. Rahman and S. Z. F. S. Putra, in Mechanical and Physical Testing of Biocomposites, Fibre-Reinforced Composites and Hybrid Composites, 2018, pp. 81–102 Search PubMed.
- T. Ozbakkaloglu, J. F. Chen, S. T. Smith and J. G. Dai, Int. J. Polym. Sci., 2016, 5804145 Search PubMed.
- V. Dhand, G. Mittal, K. Y. Rhee, S. J. Park and D. Hui, Composites, Part B, 2015, 73, 166–180 CrossRef CAS.
- B. D. Agarwal, L. J. Broutman and C. W. Bert, Analysis and Performance of Fiber Composites, 2009, vol. 48 Search PubMed.
- M. F. Ashby, Materials Selection in Mechanical Design, Elsevier, 4th edn, 2011 Search PubMed.
- R. Naghdi, Int. Wood Prod. J., 2021, 12, 178–193 CrossRef.
- A. A. Pérez-Fonseca, H. Teymoorzadeh, J. R. Robledo-Ortíz, R. González-Nuñez and D. Rodrigue, in Handbook of Composites from Renewable Materials, 2017, vol. 1–8, pp. 25–59 Search PubMed.
- Y. Huang, S. Kormakov, X. He, X. Gao, X. Zheng, Y. Liu, J. Sun and D. Wu, Polymers, 2019, 11(2), 187 CrossRef.
- M. C. S. Ribeiro, A. Fiúza, A. Ferreira, M. de L. Dinis, A. C. M. Castro, J. P. Meixedo and M. R. Alvim, Recycling, 2016, 1, 178–193 CrossRef.
- A. E. Krauklis, C. W. Karl, A. I. Gagani and J. K. Jørgensen, J. Compos. Sci., 2021, 5, 28 CrossRef CAS.
- S. Taj, M. A. Munawar and S. Khan, Proc. Pak. Acad. Sci., 2007, 44, 129–144 CAS.
- Z. N. Azwa, B. F. Yousif, A. C. Manalo and W. Karunasena, Mater. Des., 2013, 47, 424–442 CrossRef CAS.
- N. Hiremath, S. Young, H. Ghossein, D. Penumadu, U. Vaidya and M. Theodore, Composites, Part B, 2020, 198, 108156 CrossRef CAS.
- X. Li, X. Q. Zhu, K. Okuda, Z. Zhang, R. Ashida, H. Yao and K. Miura, Xinxing Tan Cailiao/New Carbon Mater., 2017, 32, 41–47 CrossRef CAS.
- S. Das, J. Warren, D. West and S. M. Schexnayder, Global Carbon Fiber Composites Supply Chain Competitiveness Analysis, 2016 Search PubMed.
- J. Wei, S. Geng, O. Pitkänen, T. Järvinen, K. Kordas and K. Oksman, ACS Appl. Energy Mater., 2020, 3, 3530–3540 CrossRef CAS.
- S. Chandgude and S. Salunkhe, Polym. Compos., 2021, 42, 2678–2703 CrossRef CAS.
- A. Fitzgerald, W. Proud, A. Kandemir, R. J. Murphy, D. A. Jesson, R. S. Trask, I. Hamerton and M. L. Longana, Sustainability, 2021, 13, 1–25 CrossRef.
- H. Pingulkar, A. Mache, Y. Munde and I. Siva, Mater. Today: Proc., 2021, 44, 3872–3880 CAS.
- H. P. S. Abdul Khalil, A. S. Adnan, E. B. Yahya, N. G. Olaiya, S. Safrida, M. S. Hossain, V. Balakrishnan, D. A. Gopakumar, C. K. Abdullah, A. A. Oyekanmi and D. Pasquini, Polymers, 2020, 12(8), 1759 CrossRef PubMed.
- A. A. Houfani, N. Anders, A. C. Spiess, P. Baldrian and S. Benallaoua, Biomass Bioenergy, 2020, 134, 105481 CrossRef CAS.
- P. Sainvitu, K. Nott, G. Richard, C. Blecker, C. Jérôme, J. Wathelet, M. Paquot and M. Deleu, Biotechnol., Agron., Soc. Environ., 2012, 16, 115–124 Search PubMed.
- L. Cao, W. Lu, A. Mata, K. Nishinari and Y. Fang, Carbohydr. Polym., 2020, 242, 116389 CrossRef CAS.
- S. Yamanaka and J. Sugiyama, Cellulose, 2000, 7, 213–225 CrossRef CAS.
- T. Hayashi, M. P. Marsden and D. P. Delmer, Plant Physiol., 1987, 83, 384–389 CrossRef CAS PubMed.
- J. Lucenius, J. J. Valle-Delgado, K. Parikka and M. Österberg, J. Colloid Interface Sci., 2019, 555, 104–114 CrossRef CAS.
- D. Rico-García, L. Ruiz-Rubio, L. Pérez-Alvarez, S. L. Hernández-Olmos, G. L. Guerrero-Ramírez and J. L. Vilas-Vilela, Polymers, 2020, 12, 1–23 Search PubMed.
- S. Cui, B. Yao, M. Gao, X. Sun, D. Gou, J. Hu, Y. Zhou and Y. Liu, Carbohydr. Polym., 2017, 157, 766–774 CrossRef CAS PubMed.
- M. Liu, A. Thygesen, J. Summerscales and A. S. Meyer, Ind. Crops Prod., 2017, 108, 660–683 CrossRef CAS.
- L. Y. Mwaikambo, Afr. J. Sci. Technol., 2006, 7, 120–133 Search PubMed.
- A. Athijayamani, M. Thiruchitrambalam, U. Natarajan and B. Pazhanivel, Polym. Compos., 2009, 16, 344–353 Search PubMed.
- D. B. Dittenber and H. V. S. Gangarao, Composites, Part A, 2012, 43, 1419–1429 CrossRef.
- P. M. Tahir, A. B. Ahmed, S. O. A. Saifulazry and Z. Ahmed, BioResources, 2011, 6, 5260–5281 CAS.
- H. T. Sreenivas, N. Krishnamurthy and G. R. Arpitha, Int. J. Lightweight Mater. Manuf., 2020, 3, 328–337 Search PubMed.
- T.-D. Ngo, Nat. Artif. Fiber-Reinf. Compos. Renewable Sources, 2017 DOI:10.5772/intechopen.71012.
- L. Mohammed, M. N. M. Ansari, G. Pua, M. Jawaid and M. S. Islam, Int. J. Polym. Sci., 2015, 2015, 1–15 CrossRef.
- A. Amiri, T. Krosbakken, W. Schoen, D. Theisen and C. A. Ulven, Proc. Inst. Mech. Eng., Part P, 2018, 232, 28–38 Search PubMed.
- M. L. Longana, V. Ondra, H. Yu, K. D. Potter and I. Hamerton, Recycling, 2018, 3(4), 52 CrossRef.
- A. M. Hashim, K. E. Tanner and J. K. Oleiwi, ZANCO J. Pure Appl. Sci., 2016, 28, 379–389 Search PubMed.
- R. D. Gupta and N. Raghav, Int. J. Biol. Macromol., 2020, 147, 921–930 CrossRef CAS.
- B. J. D. Barba, J. F. Madrid and D. P. Penaloza, J. Chil. Chem. Soc., 2020, 65(3), 4919–4924 CrossRef CAS.
- Future Fibres: Abaca, https://www.fao.org/economic/futurefibres/fibres/abaca0/en/, (accessed 23 September 2019).
- M. F. Hossain, Afr. J. Food, Agric., Nutr. Dev., 2016, 16, 11443–11456 Search PubMed.
- A. L. Leão, B. M. Cherian, S. Narine, S. F. Souza, M. Sain and S. Thomas, Biofiber Reinf. Compos. Mater, 2015, pp. 211–235 Search PubMed.
- M. K. Huda and I. Widiastuti, IOP Conf. Ser. Earth Environ. Sci., DOI:10.1088/1742-6596/1808/1/012015.
- F. Unal, O. Avinc and A. Yavas, Sustainable Textile Designs Made from Renewable Biodegradable Sustainable Natural Abaca Fibers, 2020, pp. 1–30.
- S. Senthil Kumaran, K. Srinivasan, M. Ponmariappan, S. Yashwhanth, S. Akshay and Y. C. Hu, Mater. Res. Express, 2020, 7, 55102 CrossRef.
- M. A. Paglicawan, B. A. Basilia and B. S. Kim, Compos. Res., 2013, 26, 165–169 CrossRef.
- F. Berzin, T. Amornsakchai, A. Lemaitre, E. Di Giuseppe and B. Vergnes, Polym. Compos., 2018, 39, 4115–4122 CrossRef CAS.
- Pineapple Leaf Fibers, ed. M. Jawaid, M. Asim, P. M. Tahir and M. Nasir, Springer Singapore, Singapore, 2020 Search PubMed.
- H. Shirahama, Y. Kawaguchi, M. S. Aludin and H. Yasuda, J. Appl. Polym. Sci., 2001, 80, 340–347 CrossRef CAS.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS.
- L. International, Global Glycerin Production by Industry to 2025, 2022 Search PubMed.
- H. Rassem, A. Nour and R. M. Yunus, Aust. J. Basic Appl. Sci., 2016, 10, 117–127 CAS.
- A. B. Leoneti, V. Aragão-Leoneti and S. V. W. B. de Oliveira, Renewable Energy, 2012, 45, 138–145 CrossRef CAS.
- C. S. Singh, N. Kumar and R. Gautam, Environ. Prog. Sustainable Energy, 2021, 40, 1–13 Search PubMed.
- Z. Mao, X. Wen, J. Wang and D. Zhu, Eighth Int. Conf. Photonics Imaging Biol. Med. (PIBM 2009), 2009 Search PubMed, vol. 7519, pp. 75191N.
- W. S. Ao-Ieong, S. T. Chien, W. C. Jiang, S. F. Yet and J. Wang, Polymers, 2021, 13(12), 1960 CrossRef CAS PubMed.
- D. Lin, B. Cai, L. Wang, L. Cai, Z. Wang, J. Xie, Q. X. Lv, Y. Yuan, C. Liu and S. G. Shen, Biomaterials, 2020, 253, 120095 CrossRef CAS.
- A. Gadomska-Gajadhur, M. Wrzecionek, G. Matyszczak, P. Pietowski, M. Wiecław and P. Ruśkowski, Org. Process Res. Dev., 2018, 22, 1793–1800 CrossRef CAS.
- X. J. Loh, A. Abdul Karim and C. Owh, J. Mater. Chem. B, 2015, 3, 7641–7652 RSC.
- M. T. Souza, S. Tansaz, E. D. Zanotto and A. R. Boccaccini, Materials, 2017, 10, 83 CrossRef PubMed.
- X. Zhang, C. Jia, X. Qiao, T. Liu and K. Sun, Polym. Test., 2017, 62, 88–95 CrossRef CAS.
- S. Kasetaite, J. Ostrauskaite, V. Grazuleviciene, D. Bridziuviene and E. Rainosalo, J. Compos. Mater., 2017, 51, 4029–4039 CrossRef CAS.
- X. Deng, M. Qasim and A. Ali, J. Biomed. Mater. Res., Part A, 2021, 109, 2065–2081 CrossRef CAS.
- M. L. Houchin and E. M. Topp, J. Pharm. Sci., 2007, 97, 2395–2404 CrossRef.
- A. C. R. Grayson, G. Voskerician, A. Lynn, J. M. Anderson, M. J. Cima and R. Langer, J. Biomater. Sci., Polym. Ed., 2004, 15, 1281–1304 CrossRef CAS PubMed.
- R. C. Thomson, M. J. Yaszemski, J. M. Powers and A. G. Mikos, Biomaterials, 1998, 19, 1935–1943 CrossRef CAS.
- R. L. Simpson, S. N. Nazhat, J. J. Blaker, A. Bismarck, R. Hill, A. R. Boccaccini, U. N. Hansen and A. A. Amis, J. Mech. Behav. Biomed. Mater., 2015, 50, 277–289 CrossRef CAS PubMed.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, 1–20 CrossRef CAS.
- G. Q. Chen, X. Y. Chen, F. Q. Wu and J. C. Chen, Adv. Ind. Eng. Polym. Res., 2020, 3, 1–7 Search PubMed.
- M. Gallardo-Cervantes, Y. González-García, A. A. Pérez-Fonseca, M. E. González-López, R. Manríquez-González, D. Rodrigue and J. R. Robledo-Ortíz, J. Appl. Polym. Sci., 2021, 138, 50182 CrossRef CAS.
- G. Mármol, C. Gauss and R. Fangueiro, Molecules, 2020, 25(20), 4653 CrossRef PubMed.
- P. Cinelli, M. Seggiani, N. Mallegni, V. Gigante and A. Lazzeri, Int. J. Mol. Sci., 2019, 20(2), 284 CrossRef PubMed.
- D. Puppi and F. Chiellini, Appl. Mater. Today, 2020, 20, 100700 CrossRef.
- Ł. Kaniuk and U. Stachewicz, ACS Biomater. Sci. Eng., 2021, 7, 5339–5362 CrossRef.
- C. Sanhueza, F. Acevedo, S. Rocha, P. Villegas, M. Seeger and R. Navia, Int. J. Biol. Macromol., 2019, 124, 102–110 CrossRef CAS PubMed.
- D. Puppi, G. Pecorini and F. Chiellini, Bioengineering, 2019, 6(4), 108 CrossRef CAS PubMed.
- S. Bi, B. Tan, J. L. Soule and M. J. Sobkowicz, Polym. Degrad. Stab., 2018, 155, 9–14 CrossRef CAS.
- S. H. Lee and S. Wang, Composites, Part A, 2006, 37, 80–91 CrossRef CAS.
- S. A. Rafiqah, A. Khalina, A. S. Harmaen, I. A. Tawakkal, K. Zaman, M. Asim, M. N. Nurrazi and C. H. Lee, Polymers, 2021, 13, 1–28 CrossRef PubMed.
- O. Platnieks, S. Gaidukovs, V. Kumar Thakur, A. Barkane and S. Beluns, Eur. Polym. J., 2021, 161, 110855 CrossRef CAS.
- J. Yang, J.-Z. Liang and F.-J. Li, J. Macromol. Sci., Part B: Phys., 2012, 51, 1715–1730 CrossRef CAS.
- K. Ru, S. Zhang, X. Peng, J. Wang and H. Peng, Polymer, 2019, 185, 121967 CrossRef CAS.
- V. Ghaffarian, S. M. Mousavi and M. Bahreini, Desalin. Water Treat., 2015, 54, 473–480 CrossRef CAS.
- T. Uesaka, Polymer, 2000, 41, 8449–8454 CrossRef CAS.
- A. H. Bahremand, S. M. Mousavi, A. Ahmadpour and M. Taherian, Carbohydr. Polym., 2017, 173, 497–507 CrossRef CAS PubMed.
- N. Kurokawa, S. Kimura and A. Hotta, J. Appl. Polym. Sci., 2018, 135, 45429 CrossRef.
- J. Xu and B. H. Guo, Biotechnol. J., 2010, 5, 1149–1163 CrossRef CAS PubMed.
- M. Zhou, J. Yan, Y. Li, C. Geng, C. He, K. Wang and Q. Fu, RSC Adv., 2013, 3, 26418 RSC.
- O. Platnieks, A. Barkane, N. Ijudina, G. Gaidukova, V. K. Thakur and S. Gaidukovs, J. Cleaner Prod., 2020, 270, 122321 CrossRef CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- P. Matzinos, V. Tserki, C. Gianikouris, E. Pavlidou and C. Panayiotou, Eur. Polym. J., 2002, 38, 1713–1720 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- K. H. Lee, H. Y. Kim, M. S. Khil, Y. M. Ra and D. R. Lee, Polymer, 2003, 44, 1287–1294 CrossRef CAS.
- A. Luciani, V. Coccoli, S. Orsi, L. Ambrosio and P. A. Netti, Biomaterials, 2008, 29, 4800–4807 CrossRef CAS PubMed.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- F. Serio, N. Silvestri, S. Kumar Avugadda, G. E. P. Nucci, S. Nitti, V. Onesto, F. Catalano, E. D'Amone, G. Gigli, L. L. del Mercato and T. Pellegrino, J. Colloid Interface Sci., 2022, 607, 34–44 CrossRef CAS PubMed.
- T. Theeranattapong, T. Luangwattanawilai, J. Suwanprateeb, W. Suvannapruk, S. Chumnanvej and W. Hemstapat, Key Eng. Mater., 2015, 659, 19–23 Search PubMed.
- F. Sarasini, J. Tirillò, D. Puglia, F. Dominici, C. Santulli, K. Boimau, T. Valente and L. Torre, Compos. Struct., 2017, 167, 20–29 CrossRef.
- S. Khandelwal and K. Y. Rhee, Composites, Part B, 2020, 192, 108011 CrossRef CAS.
- S. K. Golushko, J. Phys.: Conf. Ser., 2017, 894, 12021 CrossRef.
- M. Kawai, S. Yajima, A. Hachinohe and Y. Kawase, Compos. Sci. Technol., 2001, 61, 1285–1302 CrossRef CAS.
- S. S. R. Raj, J. E. R. Dhas and C. P. Jesuthanam, J. Reinf. Plast. Compos., 2021, 40, 41–69 CrossRef CAS.
- P. Robinson, E. S. Greenhalgh and S. Pinho, Failure mechanisms in polymer matrix composites: criteria, testing and industrial applications, Woodhead Publishing, 2012 Search PubMed.
- Z. Chen and W. Yan, Mech. Mater., 2015, 91, 119–135 CrossRef.
- H. Lilholt and B. F. Sorensen, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 219, 012030 Search PubMed.
- X. Hao, H. Zhou, B. Mu, L. Chen, Q. Guo, X. Yi, L. Sun, Q. Wang and R. Ou, Composites, Part B, 2020, 185, 107778 CrossRef CAS.
- C. Yang, R. Han, M. Nie and Q. Wang, Mater. Des., 2020, 186, 108332 CrossRef CAS.
- A. Kandemir, T. R. Pozegic, I. Hamerton, S. J. Eichhorn and M. L. Longana, Materials, 2020, 13(9), 2129 CrossRef CAS.
- D. Tripathi and F. R. Jones, J. Mater. Sci., 1998, 33, 1–16 CrossRef CAS.
- J. Jäger, M. G. R. Sause, F. Burkert, J. Moosburger-Will, M. Greisel and S. Horn, Composites, Part A, 2015, 71, 157–167 CrossRef.
- S. Réquilé, A. Le Duigou, A. Bourmaud and C. Baley, Compos. Sci. Technol., 2019, 181, 107694 CrossRef.
- S. Tiwari and J. Bijwe, Procedia Technol., 2014, 14, 505–512 CrossRef.
- J. Qin, C. Wang, R. Lu, S. Su, Z. Yao, L. Zheng, Q. Gao, Y. Wang, Q. Wang and H. Wei, Compos. Sci. Technol., 2020, 195, 108198 CrossRef CAS.
- L. Duan, X. Zhao and Y. Wang, J. Alloys Compd., 2020, 827, 154277 CrossRef CAS.
- L. Peng, H. Liu, W. L. Wang, Z.-B. Xu, X. Y. Ni, Y. H. Wu, Q. Y. Wu and H. Y. Hu, Chem. Eng. J., 2020, 384, 123239 CrossRef CAS.
- T. Srinivasan, G. Suresh, P. Ramu, V. G. Ram, M. Giresh and K. Arjun, Mater. Today: Proc., 2021, 45, 1334–1337 CAS.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- A. Benyahia, A. Merrouche, Z. E. A. Rahmouni, M. Rokbi, W. Serge and Z. Kouadri, 21 Congr. Fr. Macanique, 2013, 1–6.
- K. R. Sumesh, K. Kanthavel and V. Kavimani, J. Compos. Mater., 2020, 54, 3655–3671 CrossRef.
- M. A. Sawpan, K. L. Pickering and A. Fernyhough, Composites, Part A, 2012, 43, 519–526 CrossRef CAS.
- M. M. Kabir, H. Wang, K. T. Lau, F. Cardona and T. Aravinthan, Composites, Part B, 2012, 43, 159–169 CrossRef CAS.
- A. Oushabi, F. Oudrhiri Hassani, Y. Abboud, S. Sair, O. Tanane and A. El Bouari, Int. J. Ind. Chem., 2018, 9, 335–343 CrossRef CAS.
- A. K. Kumar and S. Sharma, Bioresour. Bioprocess, 2017, 4, 1–19 CrossRef PubMed.
- K. H. Gardner and J. Blackwell, Biochim. Biophys. Acta, Gen. Subj., 1974, 343, 232–237 CrossRef CAS PubMed.
- L. Szcześniak, A. Rachocki and J. Tritt-Goc, Cellulose, 2008, 15, 445–451 CrossRef.
- S. Kalia, B. S. Kaith and I. Kaur, Polym. Eng. Sci., 2009, 49, 1253–1272 CrossRef CAS.
- G. Díaz-Ramírez, F. Maradei and G. Vargas-Linares, Rev. UIS Ing., 2019, 18, 117–130 Search PubMed.
- P. Denton, J. Text. Inst., Trans., 1956, 47, T570–T585 CrossRef CAS.
- C. R. Fordyce, Cellulose Esters of Organic Acids, 1945, pp. 309–327 Search PubMed.
- C. L. McCormick and T. R. Dawsey, Macromolecules, 1990, 23, 3606–3610 CrossRef CAS.
- C. F. Liu, R. C. Sun, A. P. Zhang, J. L. Ren, X. A. Wang, M. H. Qin, Z. N. Chao and W. Luo, Carbohydr. Res., 2007, 342, 919–926 CrossRef CAS PubMed.
- K. J. Edgar, K. M. Arnold, W. W. Blount, J. E. Lawniczak and D. W. Lowman, Macromolecules, 1995, 28, 4122–4128 CrossRef CAS.
- T. Heinze, O. A. El Seoud and A. Koschella, Cellulose Derivatives: Synthesis, Structure, and Properties, 2018, pp. 259–292 Search PubMed.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- M. Mariano, N. El Kissi and A. Dufresne, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 791–806 CrossRef CAS.
- A. Isogai, T. Hänninen, S. Fujisawa and T. Saito, Prog. Polym. Sci., 2018, 86, 122–148 CrossRef CAS.
- R. Kato, J. H. Lettow, S. N. Patel and S. J. Rowan, ACS Appl. Mater. Interfaces, 2020, 12, 54083–54093 CrossRef CAS.
- J. H. Lettow, H. Yang, P. F. Nealey and S. J. Rowan, Macromolecules, 2021, 54, 10594–10604 CrossRef CAS.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- K. Missoum, M. N. Belgacem and J. Bras, Materials, 2013, 6, 1745–1766 CrossRef.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- P. P. Das and V. Chaudhary, Cleaner Eng. Technol., 2021, 4, 100182 CrossRef.
- K. N. Ganesh, D. Zhang, S. J. Miller, K. Rossen, P. J. Chirik, M. C. Kozlowski, J. B. Zimmerman, B. W. Brooks, P. E. Savage, D. T. Allen and A. M. Voutchkova-Kostal, Environ. Sci. Technol., 2021, 55, 8459–8463 CrossRef CAS PubMed.
- M. Yadav, K. Behera, Y. H. Chang and F. C. Chiu, Polymers, 2020, 12(1), 202 CrossRef CAS PubMed.
- C. Calvino, N. Macke, R. Kato and S. J. Rowan, Prog. Polym. Sci., 2020, 103, 101221 CrossRef CAS.
- K. Oksman, A. P. Mathew, D. Bondeson and I. Kvien, Compos. Sci. Technol., 2006, 66, 2776–2784 CrossRef CAS.
- M. Yadav and F. C. Chiu, Carbohydr. Polym., 2019, 211, 181–194 CrossRef CAS PubMed.
- E. C. Demir, A. Benkaddour, D. R. Aldrich, M. T. McDermott, C. Il Kim and C. Ayranci, J. Compos. Mater., 2022, 56, 1591–1604 CrossRef PubMed.
- R. F. Gibson, Principles of Composite Material Mechanics, CRC Press, 2016 Search PubMed.
- A. C. Corrêa, K. B. R. Teodoro, J. A. Simão, P. I. C. Claro, E. de Morais Teixeira, L. H. C. Mattoso and J. M. Marconcini, Polym. Compos., 2020, 41, 1736–1748 CrossRef.
- Z. Miao, J. Seo and M. A. Hickner, Polymer, 2018, 152, 18–24 CrossRef CAS.
- Z. W. Abdullah and Y. Dong, J. Mater. Sci., 2018, 53, 15319–15339 CrossRef CAS.
- X. Ge, L. Yu, Z. Liu, H. Liu, Y. Chen and L. Chen, Int. J. Biol. Macromol., 2019, 125, 370–375 CrossRef CAS PubMed.
- A. M. Slavutsky and M. A. Bertuzzi, Carbohydr. Polym., 2014, 110, 53–61 CrossRef CAS PubMed.
- S. Tunc, H. Angellier, Y. Cahyana, P. Chalier, N. Gontard and E. Gastaldi, J. Membr. Sci., 2007, 289, 159–168 CrossRef CAS.
- M. Abdollahi, M. Alboofetileh, M. Rezaei and R. Behrooz, Food Hydrocolloids, 2013, 32, 416–424 CrossRef CAS.
- R. Narayan, S. Bloembergen, A. L. Narayan, R. Bloembergen and A. S. Lathia, US5869647A, 1999.
- F. D. S. Larotonda, K. N. Matsui, P. J. A. Sobral and J. B. Laurindo, J. Food Eng., 2005, 71, 394–402 CrossRef.
- T. Liebert, W. M. Kulicke and T. Heinze, React. Funct. Polym., 2008, 68, 1–11 CrossRef CAS.
- Q. F. Guan, H.-B. Yang, Z. M. Han, L. C. Zhou, Y. B. Zhu, Z. C. Ling, H.-B. Jiang, P. F. Wang, T. Ma, H. A. Wu and S. H. Yu, Sci. Adv., 2020, 6, 1–9 Search PubMed.
- B. Thomas, M. C. Raj, B. K. Athira, H. M. Rubiyah, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS.
- K. Li, C. M. Clarkson, L. Wang, Y. Liu, M. Lamm, Z. Pang, Y. Zhou, J. Qian, M. Tajvidi, D. J. Gardner, H. Tekinalp, L. Hu, T. Li, A. J. Ragauskas, J. P. Youngblood and S. Ozcan, ACS Nano, 2021, 15, 3646–3673 CrossRef CAS PubMed.
- D. A. Gopakumar, S. Thomas and Y. Grohens, Nanocelluloses as Innovative Polymers for Membrane Applications, Elsevier Inc., 2016 Search PubMed.
- N. Carter, I. Grant, M. Dewey, M. Bourque and D. J. Neivandt, Front. Nanotechnol., 2021, 3, 1–11 Search PubMed.
- S. Iwamoto, A. Isogai and T. Iwata, Biomacromolecules, 2011, 12, 831–836 CrossRef CAS PubMed.
- O. A. T. Dias, S. Konar, A. L. Leão, W. Yang, J. Tjong and M. Sain, Front. Chem., 2020, 8, 420 CrossRef CAS PubMed.
- M. L. Hassan, S. M. Fadel, R. E. Abouzeid, W. S. Abou Elseoud, E. A. Hassan, L. Berglund and K. Oksman, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed.
- M. Lay, I. González, J. A. Tarrés, N. Pellicer, K. N. Bun and F. Vilaseca, Eur. Polym. J., 2017, 91, 1–9 CrossRef CAS.
- Z. Wang, S. Zhao, A. Huang, S. Zhang and J. Li, Carbohydr. Polym., 2019, 205, 72–82 CrossRef CAS.
- D. Lasrado, S. Ahankari and K. Kar, J. Appl. Polym. Sci., 2020, 137, 1–14 CrossRef.
- P. Tayeb and A. H. Tayeb, Carbohydr. Polym., 2019, 224, 115149 CrossRef CAS.
- J. Rojas, M. Bedoya and Y. Ciro, in Cellulose – Fundamental Aspects and Current Trends, InTech, 2015 Search PubMed.
- M. Vasudevan, M. J. Y. Tai, V. Perumal, S. C. B. Gopinath, S. S. Murthe, M. Ovinis, N. M. Mohamed and N. Joshi, J. Taiwan Inst. Chem. Eng., 2021, 118, 245–253 CrossRef CAS.
- M. A. C. M. Haniffa, K. Munawar, C. Y. Chee, S. Pramanik, A. Halilu, H. A. Illias, M. Rizwan, R. Senthilnithy, K. R. R. Mahanama, A. Tripathy and M. F. Azman, Carbohydr. Polym., 2021, 267, 118136 CrossRef CAS PubMed.
- V. C. F. Li, X. Kuang, A. Mulyadi, C. M. Hamel, Y. Deng and H. J. Qi, Cellulose, 2019, 26, 3973–3985 CrossRef CAS.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- Q. F. Guan, H.-B. Yang, Z. M. Han, Z. C. Ling, C. H. Yin, K. P. Yang, Y. X. Zhao and S. H. Yu, ACS Nano, 2021, 15, 7889–7898 CrossRef CAS PubMed.
- A. Zennifer, P. Senthilvelan, S. Sethuraman and D. Sundaramurthi, Carbohydr. Polym., 2021, 256, 117561 CrossRef CAS PubMed.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- L. Bai, X. Jiang, Z. Sun, Z. Pei, A. Ma, W. Wang, H. Chen, H. Yang, L. Yang and D. Wei, Cellulose, 2019, 26, 5305–5319 CrossRef CAS.
- J. R. McKee, E. A. Appel, J. Seitsonen, E. Kontturi, O. A. Scherman and O. Ikkala, Adv. Funct. Mater., 2014, 24, 2706–2713 CrossRef CAS.
- K. J. Arrington, J. V. Haag, E. V. French, M. Murayama, K. J. Edgar and J. B. Matson, ACS Macro Lett., 2019, 8, 447–453 CrossRef CAS PubMed.
- A. Nešić, G. Cabrera-Barjas, S. Dimitrijević-Branković, S. Davidović, N. Radovanović and C. Delattre, Molecules, 2020, 25(1), 135 CrossRef PubMed.
- N. Chopin, X. Guillory, P. Weiss, J. Bideau and S. Colliec-Jouault, Curr. Org. Chem., 2014, 18, 867–895 CrossRef CAS.
- Q. Liu, B. Duan, X. Xu and L. Zhang, J. Mater. Chem. B, 2017, 5, 5690–5713 RSC.
- F. G. Torres and M. L. Cubillas, Polym. Test., 2005, 24, 694–698 CrossRef CAS.
- F. I. Mahir, K. N. Keya, B. Sarker, K. M. Nahiun and R. A. Khan, Mater. Eng. Res., 2019, 1, 88–99 CrossRef.
- S. V. Joshi, L. T. Drzal, A. K. Mohanty and S. Arora, Composites, Part A, 2004, 35, 371–376 CrossRef.
- F. Ahmad, H. S. Choi and M. K. Park, Macromol. Mater. Eng., 2015, 300, 10–24 CrossRef CAS.
- T. Corbière-Nicollier, B. G. Laban, L. Lundquist, Y. Leterrier, J. A. E. Månson and O. Jolliet, Resour., Conserv. Recycl., 2001, 33, 267–287 CrossRef.
- M. R. Mansor, M. T. Mastura, S. M. Sapuan and A. Z. Zainudin, The environmental impact of natural fiber composites through life cycle assessment analysis, Elsevier Ltd, 2018 Search PubMed.
- C. S. Boland, R. De Kleine, G. A. Keoleian, E. C. Lee, H. C. Kim and T. J. Wallington, J. Ind. Ecol., 2016, 20, 179–189 CrossRef CAS.
- A. Célino, S. Fréour, F. Jacquemin and P. Casari, Front. Chem., 2014, 1, 43 Search PubMed.
- R. L. Nelham, J. Biomed. Eng., 1981, 3, 305–314 CrossRef CAS PubMed.
- M. K. Idris, J. Qiu, G. W. Melenka and G. Grau, Eng. Res. Express, 2020, 2, 025022 CrossRef.
- A. Rane and R. Kabra, Int. J. Curr. Eng. Technol., 2016, 6, 765–767 Search PubMed.
- P. M. N. Araujo, T. R. Costa and E. C. Silva, in Composite Materials. Rio de Janeiro, 2018, pp. 562–569.
- International Energy Agency, CO2 Emissions from Fuel Combustion 2019, OECD, 2019 Search PubMed.
- P. Wheeler, in 2016 IEEE International Conference on Automatica (ICA-ACCA), IEEE, 2016, pp. 1–5.
- A. W. Schäfer, S. R. H. Barrett, K. Doyme, L. M. Dray, A. R. Gnadt, R. Self, A. O'Sullivan, A. P. Synodinos and A. J. Torija, Nat. Energy, 2019, 4, 160–166 CrossRef.
- B. Mutnuri, C. Aktas, J. Marriott, M. Bilec and H. GangaRao, in Proceedings of COMPOSITES 2010, Las Vegas, 2010, pp. 9–11.
- A. S. Rahman, Reinf. Plast., 2019, 63, 21–25 CrossRef.
- Following Recent Surge, Wind Now Generates 5.5 Percent of U.S. Electricity, https://e360.yale.edu/digest/following-recent-surge-wind-now-generates-5-5-percent-of-u-s-electricity.
- T. Batu, H. Lemu and B. Sirhabizuh, Adv. Sci. Technol. Res. J., 2020, 14, 67–75 CrossRef.
- D. Sundar, S. Narasimalu, Y. Yang and S. Sharma, in 2017 Asian Conference on Energy, Power and Transportation Electrification, ACEPT 2017, 2017, vol. 2017-Decem, pp. 1–7.
| This journal is © The Royal Society of Chemistry 2023 |