
History repeats itself again: Will the mistakes of the past for ILs be repeated for DESs? From being considered ionic liquids to becoming their alternative: the unbalanced turn of deep eutectic solvents
J.
Afonso†
 a,
A.
Mezzetta†
b,
I. M.
Marrucho
*a and
L.
Guazzelli
*b
a,
A.
Mezzetta†
b,
I. M.
Marrucho
*a and
L.
Guazzelli
*b
aCentro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal. E-mail: isabel.marrucho@tecnico.ulisboa.pt
bDepartment of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy. E-mail: lorenzo.guazzelli@unipi.it
First published on 29th November 2022
Abstract
The establishment of the place of Deep Eutectic Solvents (DESs) as a new class of green solvents was essentially grounded on naïve comparisons with Ionic Liquids (ILs), since both are composed of charged compounds. The easiness of DESs’ preparation afforded the quick preparation and utilization of a massive number of solvents and their use in wide variety of applications with minimal fundamental knowledge of their thermophysical properties and phase equilibria studies. As time went on, the need to define DESs from the thermodynamic point of view and to differentiate them from other classes of solvents was imperative. This perspective review aims at dispelling some myths about DESs through the use of experimental data and computational chemical calculations and establishing fair comparisons with other classes of solvents, ILs, eutectic solvents (ESs) and volatile organic compounds (VOCs), so that clear and sound conclusions can be drawn. Several important parameters typically used to characterize solvents and that have been much used to justify DESs’ wide range of applications, such as vapor pressure, thermal stability, polarity, toxicity and water miscibility, were accessed for these different solvents and comparisons were established. Moreover, a comparative analysis in a selected research area, biopolymer dissolution and treatment, was chosen to illustrate the unique potential of ILs and DESs and the challenges that still need to be addressed. Literature available for the diverse polysaccharides selected (cellulose, hemicellulose and chitin) and lignin highlighted pros and cons and the different level of knowledge gained to date for both ILs and DESs. This part is complemented by recycling and techno-economic considerations for the two classes of solvents, which are additional key aspects to consider for the development of an effective integrated biorefinery process. The conclusion is obvious: DESs are a new class of solvents, with distinct properties from other classes of solvents which are essentially dependent on the properties of their constituent compounds. Therefore, when starting compounds are wisely selected, DESs become an additional and promising pathway in the pursuit of environmentally friendly solvents to replace traditional VOCs for a given application. However, some fundamental studies are still needed to fully understand these systems and define their most effective areas of application.
1. DESs’ definition over time
Eutectic mixtures date back to 1884, when Guthrie defined them as mixtures in such proportion that a minimum temperature of liquefaction could be obtained.1 Nowadays, eutectic solvents (ESs) are a well-known class of multicomponent systems, presenting more or less pronounced melting point temperature depressions relative to the starting compounds. Many examples, in the pharmaceutical field for solubilizing or liquefying specific compounds,2–4 as phase change materials,5–7 as liquid crystals8–10etc., can be found in the open literature. In 2003, Abbott et al.11 showed that it is possible to obtain remarkably large melting point depressions using one salt, that acts as hydrogen bond donor (HBD), and one hydrogen bond acceptor (HBA) to obtain liquid mixtures at room temperature, presenting for the first time the so-called deep eutectic solvents (DESs). DESs were then defined as supramolecular complexes formed between one hydrogen bond donor (HBD) and one hydrogen bond acceptor (HBA), and the large decrease in the mixture melting point was attributed to charge delocalization and the extensive hydrogen bond network that occurs between the HBD and the HBA. The first reported DES was a mixture of cholinium chloride (ChCl) and urea (U), with a melting temperature of 12 °C, much lower than those of the starting materials, 302 °C and 133 °C, respectively, and thus the designation of deep. Since then, a large amount of work has been carried out under the DES flagship, most of it regarding applications of DESs as tunable solvents, and only a handful addressing the fundamentals of these mixtures. Knowledge of the solid–liquid phase diagram, defining the phase behavior at a given temperature and composition and thus the understanding of the phase coexistence, the depression of the melting point through the intersection of the solubility lines and the liquid window available at a specific temperature, is vital to the design of chemical processes and products using DESs.Since the dawn of DESs, many authors have referred to them as ionic liquid (IL) analogues.12–14 This was mainly due to the presence of salts or solid at room temperature ILs, especially those based on the cholinium cation. Contrary to what happened in research into ILs, which were very well studied and characterized from the fundamental point of view prior to their use, DESs started to be used in applications with little fundamental knowledge and molecular understanding. This is probably a consequence of their easy preparation, most often just by mixing and heating the pure components. However, some authors pointed out the unsuitability of this preparation method,15,16 especially for DESs composed of carboxylic acids, since their lack of thermal stability might lead to unwanted reactions and thus impurities. Consequently, despite the simple and easy preparation method, the chemical structure of the final DESs should always be verified.
In 2011, Choi and co-workers17,18 reported many natural deep eutectic solvents (NADESs) based on natural compounds only, particularly primary metabolites, such as organic acids, amino acids, sugars etc. Soon, the attractive features of NADESs were widely extrapolated to the whole DES family, which became widely known as being easy to prepare, inexpensive, environmentally benign, green, sustainable etc. In 2019, Coutinho and co-workers19 brought some order into to the field by defining DESs as a mixture of pure compounds for which the eutectic point temperature is considerably below that of the ideal mixture description, thus presenting significant negative deviations from ideality. This not only clearly sets DESs apart from ESs or eutectic mixtures, but also emphasizes the importance of the knowledge of solid–liquid phase equilibria diagrams, which sometimes are not easy to measure and most times are not available. Although most DESs that fall under this definition are ionic, meaning they have salts in their constitution, non-ideal mixtures of fully neutral compounds, such as D,L-menthol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thymol,20,21 can also exhibit non-ideal behavior and thus also need to be considered as DESs. Conversely, not all ESs containing salts present large deviations from the ideal behavior.19,22 This distinction between DESs and ESs implies the recognition of the latter as another platform for solvent development. If both DESs and ESs are to be used as solvents, the temperature depression should be such that a liquid mixture needs to be attained at the operating temperature for a certain composition range. The acknowledgement that DESs’ composition does not have to be fixed at the eutectic point but can be another variable in the preparation of DESs and ESs, just like in any other mixture, adds further flexibility to these platforms of designer solvents.
:thymol,20,21 can also exhibit non-ideal behavior and thus also need to be considered as DESs. Conversely, not all ESs containing salts present large deviations from the ideal behavior.19,22 This distinction between DESs and ESs implies the recognition of the latter as another platform for solvent development. If both DESs and ESs are to be used as solvents, the temperature depression should be such that a liquid mixture needs to be attained at the operating temperature for a certain composition range. The acknowledgement that DESs’ composition does not have to be fixed at the eutectic point but can be another variable in the preparation of DESs and ESs, just like in any other mixture, adds further flexibility to these platforms of designer solvents.
2. ILs and DESs – properties and myths
The boom in designer solvents occurred with the discovery of stable molten salts that are liquid at room temperature or temperatures below room temperature – ILs.23 Besides the commonly accepted definition for an IL, a salt with a melting temperature below the boiling point of water, this new class of solvents, composed solely of cations and anions, soon attracted the attention of the scientific community. ILs usually consist of a large, asymmetric organic cation coupled with a generally smaller, weakly coordinating anion. Due to the large number of possible cation and anion combinations, that could reach 106 according to some authors,24 their physicochemical properties can be easily tuned, justifying the designation of designer solvents. However, the complex synthetic and purification steps that are often needed to prepare an IL with a purity equivalent to commercial organic solvents was rarely mentioned.Another important property of (aprotic) ILs is their negligible vapor pressure,25,26 typically in the order of units of Pa,27 which eliminated the major problems associated with volatile organic solvents, damage to the ozone layer, human health hazards and the loss of solvent throughout a process due to its high volatility. This property alone granted ILs the supposedly green solvent label, without considering the different aspects through which ILs might enter the terrestrial ecosystem. The very rich and complex interactions present in ILs and through which they solvate solutes, either gases, liquids, or solids, have also been seen by most authors as a true advantage over other solvents. Nevertheless, there are many examples in the literature where ILs act as sponges, dissolving very high amounts of solutes, which can hardly ever be recovered, thus bringing additional challenges from the sustainability and circular economy point of view.
One of the greatest challenges in the design of ILs was, and still is, the preparation of liquid-at-room-temperature ILs. Despite the ubiquitous place that the imidazolium cation has in the IL portfolio, the introduction of the cholinium cation and, in particular, the ChCl salt, was a game changer since it provided a framework to design more benign ILs. However, the fact that the ChCl is a solid at room temperature hindered its application as a solvent, until it was used in the preparation of DESs. Thus, the “birth” of DESs from solid ILs prompted many erroneous generalizations. To promote the “beneficial” properties of ILs and DESs, the scientific community made wide generalizations about their properties, instead of recognizing that both these classes of solvents have their independent classes of solvents with different properties. Even common volatile organic compounds (VOCs) have very different chemical and biological properties among themselves. However, some VOCs can have similar properties to each other, as recognized by the pharmaceutical industry where scales have been created to replace some solvents by others with similar chemical properties but different toxicity.
The most frequent generalizations are about DESs’ toxicity and volatility, without paying attention to DESs’ constituents. Just as the properties of ILs are intimately connected to the properties of their ions, those of DESs are also dependent on their constituents. Another important point is that unlike ILs, DESs are not pure fluids. Consequently, the properties of DESs depend also on the composition (HBA/HBD molar ratio), and thus the DESs’ constituents can have a different impact on each property. On the other hand, as non-ideal mixtures, DESs might have properties that are not in between those of their parent compounds, depending on the interactions playing a dominant role in their bulk or at the surface, according to the property under consideration.
Table 1 depicts the evolution of the scientific community's perception of the properties of ILs in early 2000 and today, along with the current perception of DESs. Will the mistakes of the past for ILs be repeated for DESs? By critically analyzing the single entries, it can be easily stated that perceptions of both ILs and DESs are not correct. As soon as ionic and non-ionic DESs found their own space outside the ILs family, a race to praise their properties against those of ILs started, instead of recognizing them as another interesting class of media alternative to VOCs. It is evident that errors of the past, experienced previously in the IL research area, are being repeated nowadays in the DES research area (overestimating the properties of DESs, often to the detriment of ILs). In conclusion, DESs are not IL analogues or complexes at a fixed stoichiometric ratio of components, but non-ideal mixtures of two or more compounds. DESs are not always green, not always cheap, not always nontoxic: their properties depend on their components and their interactions within the DES mixture.
| Property | Ils early 2000 | Ils Today | DESs Today |
|---|---|---|---|
| Tunability | ✓ | ✓ | ✓ |
| Thermal stability | ✓ | X | ✓ |
| Low cost | ✓ | X | ✓ |
| Safety (toxicity/biodegradability) | ✓ | X | ✓ |
| Green character | ✓ | X | ✓ |
| Ease of preparation | ✓ | X | ✓ |
3. Is there any relationship between ILs and DESs?
The seminal work of Abbott in 200311 opened the door to the preparation of DESs using ILs. In fact, a large amount of DESs are based on ammonium salts, especially ChCl, as HBA, and a wide variety of HBDs such as acids, amino acids, alcohols, glycols etc.28,29 More recently, water has also been proposed as a HBD and thus mixtures of DESs and water have been investigated to understand the composition up to which the DES's structure is maintained.30–34 On the other hand, mixtures of imidazole-based ILs, [C2C1im]Cl, [C4C1im]Cl, [C6C1im]Cl, with a large variety of HBDs have been widely prepared and used in several applications.35–41 The relevant role of the chloride anion, present in the IL, in the establishment of hydrogen bond donors in DESs has also been addressed.42–50 Even more, mixtures of ILs could give rise to new DESs. Nevertheless, solid–liquid phase diagrams are reported only for a few systems. As shown in Fig. 1, although it is possible to convert ILs to DESs, the reverse cannot be attained. | ||
| Fig. 1 IL and DES relationship (ring dimensions are arbitrary). | ||
Recently this type of transition was made by adding carboxylic acids, namely acetic acid, to the IL [C2C1im]Cl, in various molar ratios, leading to the formation of DESs with a melting point below −78 °C. Additionally, NMR and solvatochromic parameters were used to prove that the acid does not ionize and lead to the formation of the IL [C2C1im]OAc as might be expected; in fact, the eutectic character of this mixture was thoughtfully explored and its capacity to solubilize cellulose was found to be negligible, in contrast to [C2C1im]Cl and [C2C1im]OAc, proving once again the possibility and applicability of transitioning from an IL to a (D)ES.51
4. On the properties of DESs, ESs, ILs and VOCs
Although much research has been conducted on the properties of DESs, their properties are rarely compared with those of other classes of solvents.52,53 In the following sections some of the most important properties of DESs that are often freely extrapolated from those of ILs will be discussed and illustrated. Vapor pressure, thermal stability, polarity and toxicity are the properties selected to illustrate and discuss the difference between ILs and DESs, and we also introduce ESs’ and VOCs’ properties and how their properties compare with the former classes of solvents.4.1. Do DESs and ESs have negligible vapor pressure?
With the objective of rebutting the common claim that DESs and ESs have negligible vapor pressure, this section presents a comparison between DESs, ESs, ILs and VOCs. It should be noted that DESs and ESs are mixtures of compounds, and thus the difference in volatility of each one of the constituent compounds as well as the nature of their interactions may lead to the preferential evaporation of one of the compounds to the detriment of the other. This implies that the composition of the DESs in the vapor phase can thus be quite different from that of the liquid phase, especially for DESs and ESs composed of salts or ILs and other neutral compounds. The isoteniscope is the most commonly used method to measure vapor pressure.54 Despite the recent redesigns of the apparatus,55 this procedure is very demanding regarding the manipulation of the apparatus and requires large volumes of the sample under study. Thus, recently, this method has been replaced by head-space gas chromatography mass spectrometry (HS-GC-MS), for DESs and ESs, and thermogravimetric analysis (TGA) mainly for ILs. HC-GC-MS has the advantage of clearly identifying DES and ES vapor phase composition. Despite the scarcity of data regarding DESs and ESs, some conclusions can be already drawn.The truth is that DESs and ESs present a much lower vapor pressure than common solvents. For example, comparing with the vapor pressure of VOCs such as dichloromethane and diethyl ether, which exhibit values of 102 kPa and 123 kPa at 40 °C,56 respectively, the highest vapor pressure found in the literature for ESs is that of 1-tetradecanol:D,L-menthol (1:2), which does not surpass 16 Pa at the same temperature. This difference is even higher when comparing VOCs with ionic DESs/ESs that have a salt as one of the components, 4 Pa being the highest value of vapor pressure observed for [P4444]Br:sulfolane (1:4), at the same temperature. This difference in vapor pressure is still several orders of magnitude different compared with less volatile VOCs, like ethyl acetate or toluene, which exhibit a vapor pressure of 25 kPa and 8 kPa, at 40 °C,56 respectively. Moreover, other common environmentally friendlier solvents, such as ethanol or even water, still have a much higher vapor pressure, 19 kPa and 7 kPa, respectively, at the same temperature.56
In fact, eutectic mixtures usually have a lower vapor pressure than their pure components as result of the favorable interactions, mostly hydrogen bonds, established between the two compounds. This is true for both ionic and neutral DESs and ESs, even when their components have a very low vapor pressure. For example, considering U-based eutectics, the vapor pressure of ChCl:U (1:2)57 is 1.33 Pa at 100 °C, contrasting with 1.97 Pa for pure U at the same temperature.58 Additionally, the proportion of each compound in the eutectic mixture plays a decisive role in the difference between the vapor pressure values for the pure components and the DESs or ESs. This can be easily understood considering, for example, the eutectic mixtures of thymol:D,L-menthol at 100 °C, for which the vapor pressure is 975 Pa at the molar ratio (1:2), while at a molar ratio of (1:1) it decreases to 680 Pa.57 In this case, the lowest vapor pressure corresponds to the eutectic point (1:1) of the eutectic mixture, which can be interpreted as, at this molar ratio, the two compounds having more favorable interactions, and therefore being less volatile. On the other hand, mixtures of decanoic acid:D,L-menthol have, at 100 °C, a vapor pressure of 540.9 Pa at (1:1) molar ratio59 and 895.8 Pa at a (1:2) molar ratio.60 Since (1:2) is closer to the eutectic point (2:3) and it presents a higher vapor pressure, it can be concluded that the vapor pressure of each neat component impacts the tunability of the vapor pressure of the mixture. Another point to consider is how deep the melting temperature depression of the DES/ES is; that is, how much the system deviates from ideality. Similarly, a mixture of thymol:lidocaine, at 100 °C, at a molar ratio of (1:1) has a vapor pressure of 53.9 Pa,60 while the (1:2) ratio has a vapor pressure of 329.4 Pa (ref. 59) (Fig. 2).
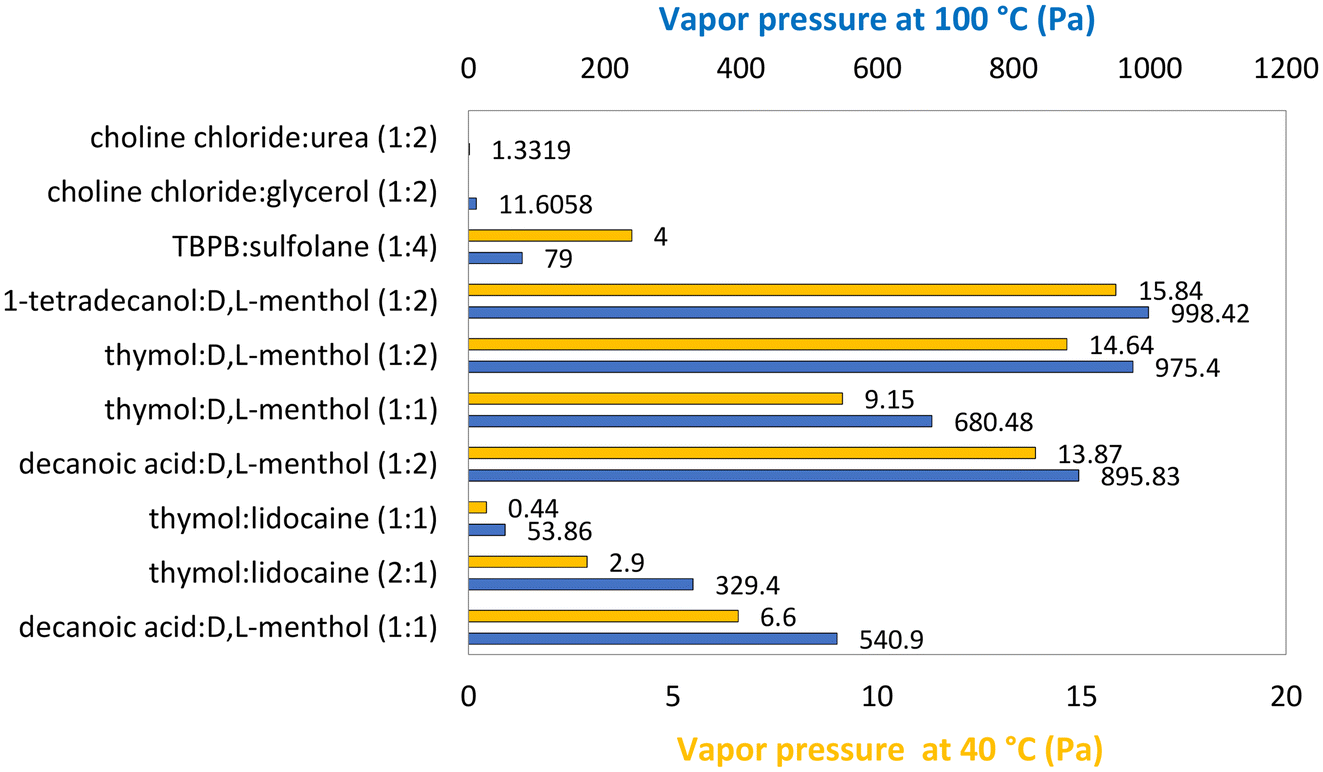 | ||
| Fig. 2 Vapor pressure of various eutectic solvents at 40 °C (yellow) and 100 °C (blue).57,59,60,64 | ||
Aprotic ILs (AILs) present a much lower vapor pressure than all the above-mentioned DESs/ESs. The fact that ionic DESs/ESs contain a salt or an ionic liquid in their constitution has led to incorrect generalizations about their properties. When compared with ionic DESs/ESs, ILs have a much lower vapor pressure, below 1 Pa at 200 °C, in most cases.61 Also, it should be mentioned that ionic DESs/ESs usually have lower vapor pressure than neutral DESs/ESs. For example, thymol:lidocaine (1:1) has a vapor pressure of 7.28 Pa at 70 °C, while ChCl:Gly (1:2) presents a vapor pressure of 2.14 Pa at the same temperature.59,60 Still, both these vapor pressure values are much higher than those of ILs.
In the case of protic ILs (PILs), as they are a mixture of the neat constituents and their respective ionic species, the vaporization phenomenon can be understood through the concept of a reactive azeotrope.62 In this scenario, it is possible to define an equilibrium between 3 species, namely the two neutral species (Brønsted acid and base) and the formed PIL. The vapor pressure is led by the volatility of the molecular compounds as their respective ionic forms have negligible vapor pressure. This implies that PILs have higher vapor pressure than aprotic ones. Indeed, this vapor pressure can be calculated for various molar ratios of the two components considering vapor–liquid equilibrium conditions and a reaction constant K for the previously stated equilibrium. In this regard, the pKa and pKb of the acid and base are a measurement of the proton exchange process efficiency,62,63 and therefore influence the stoichiometry of the azeotropic point. Nevertheless, the volatility of the molecular components also plays an essential role in the vapor pressure shown by PILs.62 Furthermore, evidence suggests that PILs also display an equilibrium between ionic and neutral species in the gas phase.63
In conclusion, the negligible vapor pressure of ILs, even at high temperatures, is an achievement that DESs/ESs cannot match, despite their relatively low vapor pressure when compared with commonly used organic solvents. Of course that DESs prepared by mixing two ILs will have vapor pressure as low as ILs.
4.2. Is the thermal stability of DESs, ESs and ILs on the same level?
DESs/ESs are usually wrongly labeled as being as thermostable as ILs. It is important to highlight the subjectiveness of what can be labeled as thermostable, as it is based not only on the capacity of other similar compounds to remain active, without decomposition or deactivation, but is also connected to its function and often linked to a specific application. Although usually both DESs/ESs and ILs are more thermostable than VOCs, this does not mean that they are all the same in this regard.The thermostability of solvents is typically evaluated by TGA, in which the sample is gradually heated, the weight monitored, and the onset temperature (Tonset) determined. Although the onset temperature is usually used as measure of the thermal stability of compounds, it is usually obtained over short periods of time, which leads to divergence between the onset temperature and the real degradation temperature when the compound is exposed to temperature for long periods. Furthermore, reported values of Tonset have many fluctuations, as measurement conditions such as sample mass, inert gas flow and heating rate significantly influence the results. For this reason, Tz/y is usually calculated, where z and y correspond to the degree of decomposition and time of utilization, respectively.65
A plot of the distribution of Tonset of 51 DESs/ESs and 165 ILs is shown in Fig. 3. It is easy to see that most DESs/ESs clearly have a lower thermal stability than ILs. In fact, almost all DESs/ESs have a Tonset between 30 °C and 250 °C,66–69 while only about one-quarter of the considered ILs have a Tonset in this range.65 In fact, eutectic mixtures display Tonset as low as 34.3 °C for ChCl:acetic acid (1:2),66 up to 265.1 °C for ChCl:pentaerythritol (2:1).66 For eutectic mixtures composed of ChCl and organic diacids at (1:1) molar ratio, the Tonset increases with chain length of the organic acid, from 128.3 °C for malonic acid, 229.1 °C for succinic acid, up to 232.3 °C for adipic acid.66 Additionally, different counter ions of cholinium salt also have an impact on the thermal stability of the corresponding eutectic mixtures, since larger ions lead to lower Tonset, for example, Tonset of 128.3 °C for ChCl:malonic acid (1:1), 126.2 °C for ChBr:malonic acid (1:1) and 125.2 °C for ChI:malonic acid (1:1).66 Moreover, worth mentioning is also the capacity of easy tuning of the thermal stability of these solvents. For example, ChCl:D-fructose (2:1) has a Tonset of 161.5 °C, while ChCl:xylitol (2:1) has Tonset of 261.1 °C,66 despite the fact that both HDBs are sugars that only differ in the oxidation state of one functional group. The scarcity of data on this subject does not allow further conclusions to be made, and thus it is very important to measure onset temperature for DESs and ESs, as well as decomposition temperatures for long exposure periods, for instance T0.01/10 h, conditions which are commonly used for ILs.65
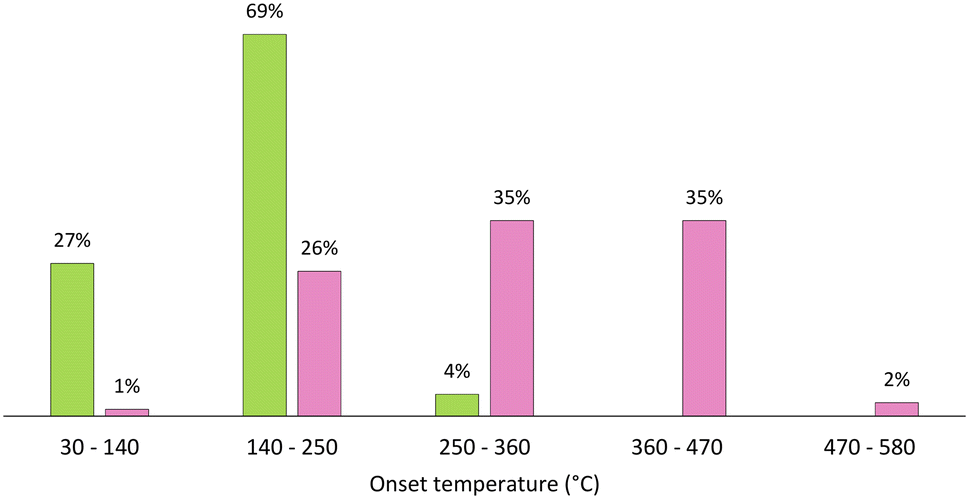 | ||
| Fig. 3 Distribution of ESs (green) and ILs (pink) based on their Tonset (°C), considering intervals of 110 °C.65–69 | ||
Another aspect of the TGA methodology worth mentioning is the lack of knowledge about transformations that may occur on the sample that do not result in mass loss. For example, although ChCl:oxalic acid (1:1) has a reported Tonset of 162.1 °C,66 this same eutectic solvent undergoes esterification with a 34% yield at a temperature of 100 °C after 2 hours.16 Actually, this eutectic solvent shows considerable esterification (10%) after 2 hours at 60 °C,16 and these esterification reactions were also reported in this range of temperatures for other mixtures of ChCl and diacids at diverse molar ratios.16 Within this line of thought, techniques like NMR, FTIR and MS that are already used to understand the mechanisms of degradation are also recommended in order to verify the integrity of the sample. In another vein, in some cases TGA methodology might underestimate DES/ES thermal stability. Indeed, as eutectic mixtures do present vapor pressure, evaporation of the neutral species under gas flow has been observed. This means that DESs/ESs may be more thermally stable than what is determined by TGA, since the observed mass loss might be due to evaporation and not degradation, as the gas flow in TGA helps the evaporation process.
Focusing on ILs, their Tonset ranges from 100 °C ([P4440]NO3) up to 575.2 °C for ([C9(C1im)2][NTf2]2),65 although nearly 97% of them present a Tonset in the range of 140 °C to 470 °C. Nevertheless, they can reach much higher temperatures than most DESs/ESs without degradation. The degradation mechanism and the effect of different anions and cations have already been discussed at length, with the order of stability reported over various series of anions for a given cation as well as the other way around.65 Regardless of this, some effects are too complex to be reported over a series of ions, as happens in the case of chain length effect. For instance, the Tonset of [C2C1im]Phe is 156 °C, [C4C1im]Phe is 158 °C, [C6C1im]Phe is 162 °C, [C8C1im]Phe is 168 °C and [C10C1im]Phe is 180 °C; one could conclude that the Tonset increases as chain length grows.65 In contrast, [C4C1im]OAc and [C8C1im]OAc have a Tonset of 216 °C and [C6C1im]OAc of 220 °C, indicating that chain length has no effect on the thermal stability of these ILs.65 Finally, predictions of these temperatures and mechanisms of degradation can be made by various models such as quantitative structure–property relationship (QSPR), ab initio quantum chemical calculation, and COSMO-RS as well as correlations with physical properties or spectroscopic parameters, for example peak position on the IR spectra.70
In the case of PILs, the principal decomposition mechanism is reverse proton transfer, because in this type of IL the difference between the value of pKa of both the acid and the base represents a major factor in thermal stability.65 For instance, taking into account the ILs [P4440]NO3, [P4440]MsO, [P4440]TfO and [P4440]NTf2, the Tonset displayed is, respectively, 100, 210, 220 and 240 °C.65 This is also in accordance with the superior thermal stability of superbase-derived PILs, between 300 ([BEMPH]NTf2) and 380 °C ([mTBDH]NTf2), as the ΔpKa is higher than for other PILs. Furthermore, considering phosphonium and ammonium-based PILs the thermal stability increases as the alkylic chain grows; for instance, the Tonset for [P4440]TfO is 220 °C, while for [P8880]TfO it is 371 °C. The presence of electron-withdrawing groups has the opposite effect; for instance taking into account [N8880]TfO and [Ph3NH]TfO, the respective Tonset values are 362 and 208 °C.65
4.3. Are DESs and ESs less polar than ILs? What about common solvents?
As a complex parameter to evaluate in a one-dimensional scale, polarity is often evaluated using empirical polarity scales. The most widely used method to describe multiple polarity properties of a solvent is the Kamlet–Taft polarity scale. In this scale, polarity is unfolded over 3 factors: α, which accounts for hydrogen bond donor capability, β, which accounts for hydrogen bond acceptor capability and π*, which accounts for polarizability/dipolarity.As stated before, the polarity measurement is too complex for a one-dimensional scale as this property is the result of different intermolecular interactions between the solvent and the solute, namely hydrogen bonding, van der Waals interactions and π-interactions. The complexity of this property is even greater when the solvent is composed of a mixture of substances, like in eutectic mixtures, as each component has its own capacity to establish interactions with the probes.71 Typically, polarity is measured through the analysis of the UV-vis spectra of a set of solvatochromic probes in the desired solvent. Hence, the interaction of the solvent with solvatochromic probes is evaluated through the changes in the probes’ absorption spectrum.72 The most used sets of solvatochromic probes are those that allow the determination of the so-called Kamlet–Taft parameters, using 4-nitroaniline, N,N-diethyl-4-nitroaniline and Reichardt's dye 30 as probes. Usually, the solvatochromic shift of the absorption bands of these probes is measured as positive bathochromic shift (shift to the red), or negative hypsochromic shift (shift to the blue), and then related to the Kamlet–Taft parameters.73 Is also important to mention that Reichardt's dye ground state is more stabilized by solvents with a high hydrogen bonding capability than the excited state; this is why in case of these solvents alternative probes are used, namely, Reichardt's betaine dye 33 is commonly used for eutectic solvents71 and Nile red for ILs.74 Although this approach enables a better understanding of the polarity of a solvent, especially DESs, ESs and ILs, these classes of solvents have an added difficulty in the determination of the α parameter due to their hygroscopic nature, as the water concentration affects the measurement.75 To overcome this problem, 13C NMR measurements of this parameter were developed and reported, based on the carbon's chemical shifts of the pyridine-N-oxide probe.76 As a matter of fact, some of the α values used in this critical review for comparisons were measured using this method, for example those for eutectic solvents comprising tetraalkylammonium salts;77 as each one of the Kamlet–Taft parameters is weighted by coefficients (a, b and s) for a given probe, these values of α can be directly compared with the ones obtained by other methodologies as they represent the same scale.78
It is important to emphasize that these parameters’ values may change if a different probe is used, making it hard to fairly compare different sets of data. Additionally, this method represents an economically and time-demanding step, making room for in silico prediction of these parameters, a much cheaper and quicker method. COSMO-RS (conductor like screening model for real solvents) is a quantum chemical-based model that calculates thermodynamic properties of molecules.75 This model has already been applied to predict Kamlet–Taft parameters based on the equilibrium constant of the tautomerism between a β-keto ester and a β-diketone with their respective enolic forms in various molecular solvents.79 A different method was proposed for hydrophilic and hydrophobic eutectic mixtures, by establishing a linear correlation between three calculated energies, namely, hydrogen bonding, van der Waals, and misfit energy and the α and β parameters. These two correlations were then used to predict the values of α and β for other eutectic solvents.75
In order to compare and distinguish each one of these parameters for DESs, ESs, ILs, and VOCs, each of these parameters will be analyzed separately. It is important to note that this type of comparison between these classes of solvents is scarce, and usually reported taking into account only a small amount of data.71,74
:glycolic acid (1:1)) up to 1.79 (menthol:dodecanoic acid (2:1)).71 This parameter is very sensitive to each component of the DES/ES. For example, menthol:octanoic acid (1:1) has α a value of 1.77,71 while [N4444]Br:octanoic acid (1:2) has α a value of 0.98. In fact, just changing the counter ion of the tetraalkylammonium salts also has a large impact, as can be concluded from the α value (0.84) of [N4444]Cl:octanoic acid (1:2). The length of the alkyl chains of the tetraalkylammonium salts also shows significant influence on the α value. For example, [N2222]Cl:octanoic acid (1:2) and [N3333]Cl:octanoic acid (1:2) have an α value of 0.96 and 0.90, respectively, allowing the conclusion that this parameter decreases with the increase in length of the alkyl chain of the ammonium salt.77 This is probably due to the fact that smaller alkyl chains have a lower electronic contribution to the nitrogen atom, thus increasing its capability to be a hydrogen bonding acceptor and therefore increasing the HBD capacity (α) of the other component. This influence of the alkyl chains can be appreciated by comparing the α values of eutectic mixtures with ChCl, where a decrease of the value of α as the alkyl chain length of the carboxylic acid increases is also noticeable for different tetraalkylammonium salts.77 Changing the proportion of the components also has an impact on the value of α. For instance, [N4444]Cl:decanoic acid (1:2) has an α value of 0.85, while for the molar ratio of (1:1) a value of 0.91 was reported.77
Similarly, ILs also show a wide range of α values, between 0.38 for [C4C1im]NTf2 and 1.55 for [Ch]Mal. Nevertheless, the majority of ILs (typically based on imidazolium and pyridinium cations) usually present an average value around 0.63, while ILs containing [HOC2C1im] or cholinium cations present higher values of α. This is probably due to the presence of the OH group, corroborating the importance of hydrogen bond interactions within ILs.81 The influence of the IL's anion in the α value can be appreciated by considering the ILs [HOC2C1im]Cl, [HOC2C1im]NO3, [HOC2C1im]DCA, [HOC2C1im] ClO4, [HOC2C1im]PF6 and [HOC2C1im]NTf2, which display α values of 0.73, 0.77, 0.80, 1.06, 1.17 and 1.17, respectively.74 It is also interesting to note that, similarly to what happens with eutectic mixtures composed of carboxylic acids, comparing ILs composed of cholinium and two distinct acids, the value of α decreases with the increase of the alkyl chain length of the acids. For example, [Ch]Mal has an α value of 1.55, while [Ch]Lev has an α value of 1.07.71 Another interesting fact is that the α values obtained for ILs are usually higher than those obtained for eutectic mixtures composed of the same compounds; for example, [Ch]Gl has α value of 1.29, while ChCl:glycolic acid (1:1) shows a value of 0.49.71 Nevertheless, in general, the eutectic mixtures have higher values of α than ILs, which can be explained by the different types of compounds present in each one of these classes of solvents. For example, eutectic mixtures containing carboxylic acids present a much higher acidity than the most common IL cations, imidazolium, pyridinium and pyrrolidinium, combined with the corresponding carboxylic basis.
Protic ILs display α values between −0.06 for [HC1im]OEt and 1.415 for [HC4im]HSO4. Just like AILs, for protic ILs this parameter is also mainly controlled by the anion. For example, considering PILs based on the nitrate anion, the α values are 1.09 for ethylammonium nitrate, 1.1 for ethanolammonium nitrate, 1.09 for propylammonium nitrate and 1.08 for butylammonium nitrate.82 Changing the anion has a much more pronounced effect on the α value, which increases with the strength of the conjugated acid of the anion; for example, [HC1im]HSO4 has an α value of 1.225, [HC1im]OF 0.812 and [HC1im]OPr 0.506.83 In comparison with AILs, PILs have usually a higher α as expected since they are based on a proton transfer mechanism.
On the other hand, VOCs usually have an α value very close or equal to zero, apart from alcohols that usually display an α between 0.76, for isopropanol, and 0.93, for methanol, given their known capability of acting as HBD as well as HBA to some extent. VOCs that have a labile hydrogen atom also have a high value of α, like chloroform (0.44) and dichloromethane (0.30), as α is a measure of acidity of hydrogen bond.84,85
However, there is a wider distribution of the β parameter values compared with the α parameter, even reaching values near zero. Overall, it can be observed that eutectic mixtures and ILs both present similar values of α and β. Nonetheless, tetraalkylammonium salts eutectic mixtures show higher values of β than ILs and other DESs and ESs, showing increasingly high values of β with the increase of the alkyl chains length of the tetraalkylammonium salts. For instance, (2:1) eutectic mixtures of octanoic acid with [N2222]Cl, [N3333]Cl and [N4444]Cl have β values of 0.87, 0.96 and 1.19,77 respectively. Other eutectic mixtures (based on D,L-menthol or ChCl) have similar β values, around 0.53, while ILs exhibit a wider variety of values.
It is also important to compare the β parameter of eutectic mixtures and ILs composed of the same compounds. It has been shown that eutectic mixtures of ChCl with levulinic, malonic or glycolic acid present a lower β value than their respective ILs. For example, ChCl:glycolic acid (1:1) shows a β value of 0.50, while [Ch]Gl has a β value of 0.79, thus supporting the conclusion that the formation of eutectic mixtures by the establishment of hydrogen bonds decreases their capacity to form additional hydrogen bonds.71
Concerning PILs, they also present a wide range of β values which are slightly above the average of AILs. Again this parameter is mainly controlled by the anion, as can be seen from the β values of [N2200]OF, [N4000]OF and [N5000]OF, which are 0.91, 0.93 and 0.93, respectively.82 In contrast to what happens with the α parameter, the β parameter values increase with the decrease in acidity; for example, for [HC1im]HSO4, [HC1im]OF, [HC1im]OAc the β value respectively displayed is 0.61, 0.81 and 0.85.83
Concerning VOCs, the alcohols present values of β values between 0.62, for methanol, and 0.95, for isopropanol. The remaining VOCs present a different distribution over the β axis, as amides, ethers, esters, and ketones have β values distinct from the more apolar solvents like alkanes, presenting β values between 0.37 for 1,4-dioxane and 0.69 for dimethylformamide.84,85
:hexanoic acid (1:2).77 This fact was expected since ILs are composed of ions, whose charges confer a strong dipole moment and thus a larger polarizability. On the other hand, the π* value of neutral eutectic mixtures is usually lower than those of ionic eutectic mixtures, as there is no charged species on the former. For example, the highest π* value (0.66) for neutral eutectic solvents was obtained for D,L-menthol:levulinic acid (1:1), while the lowest π* value (0.69) for ionic eutectic mixtures was observed for [N4444]Cl:decanoic acid (1:2).71,77 In fact, these mixtures have π* values comparable to those of VOCs such as ethers, esters, ketones, and alcohols, as the presence of heteroatoms forms a dipole on these molecules, thus showing higher π* when compared with other organic apolar solvents, like alkanes. VOCs also exhibit a larger π* parameter values distribution, when compared with the other classes of solvents, between 0.18 exhibited by butyl ether and 1.00 by dimethyl sulfoxide.84,85
Focusing on PILs, the values of π* are similar to those displayed by AILs. In contrast with the other parameters, the π* parameter values vary when the cation of the PIL is changed. For instance, [N5000]OF has a π *value of 0.66, [N4000]OF 0.7 and [N2200]OF 1.06,82 indicating that for PILs as the alkyl chain length of the amine increases, the value of π* decreases. The anion has also an important effect on the π* parameter, as stronger acids lead to the formation of PILs with higher π* values. For instance, [HC1im]OAc, [HC1im]OF and [HC1im]HSO4 have π* values of 1.03, 1.10 and 1.17, respectively.83
An overall analysis of the 3 solvation parameters is now due. Exchanging one of the components from a salt and an organic compound has a huge influence on α and β. For instance, eutectic mixtures of D,L-menthol and carboxylic acids have a higher α value and lower β value than eutectic mixtures of the same acids with tetra alkyl ammonium salts, as they are better hydrogen bond acceptors but worse donors compared with D,L-menthol. For example, the α and β values of D,L-menthol:dodecanoic acid (2:1) and [N4444]Br:dodecanoic acid (1:2) are, respectively, 1.79, 0.57 and 1.45, 1.04.71 Even the presence of a different counterion has influence on these parameters, as eutectic mixtures with ammonium chloride salts have lower values of α and higher values for β than those with bromide ammonium salts. For example, the α and β values for eutectic mixture (2:1) of decanoic acid and [N4444]Br and [N4444]Cl are, respectively, 0.95, 1.05 and 0.85, 1.28.77 Furthermore, changes in the HBD also have a great impact on these parameters. For example, ChCl eutectic mixtures with levulinic acid have less than half the value of α parameter (0.51) than mixtures of ChCl with better hydrogen bond donors like malonic acid (1.39) or EG (1.47).71
In Fig. 4, a 3-dimensional representation of the Kamlet–Taft parameters for the classes of solvents, described above, VOCs, ILs, eutectic mixtures, is shown. A quick glance shows that there are no overlapped points, meaning that no IL or DESs/ESs, from those here considered, could fully replace a VOC.
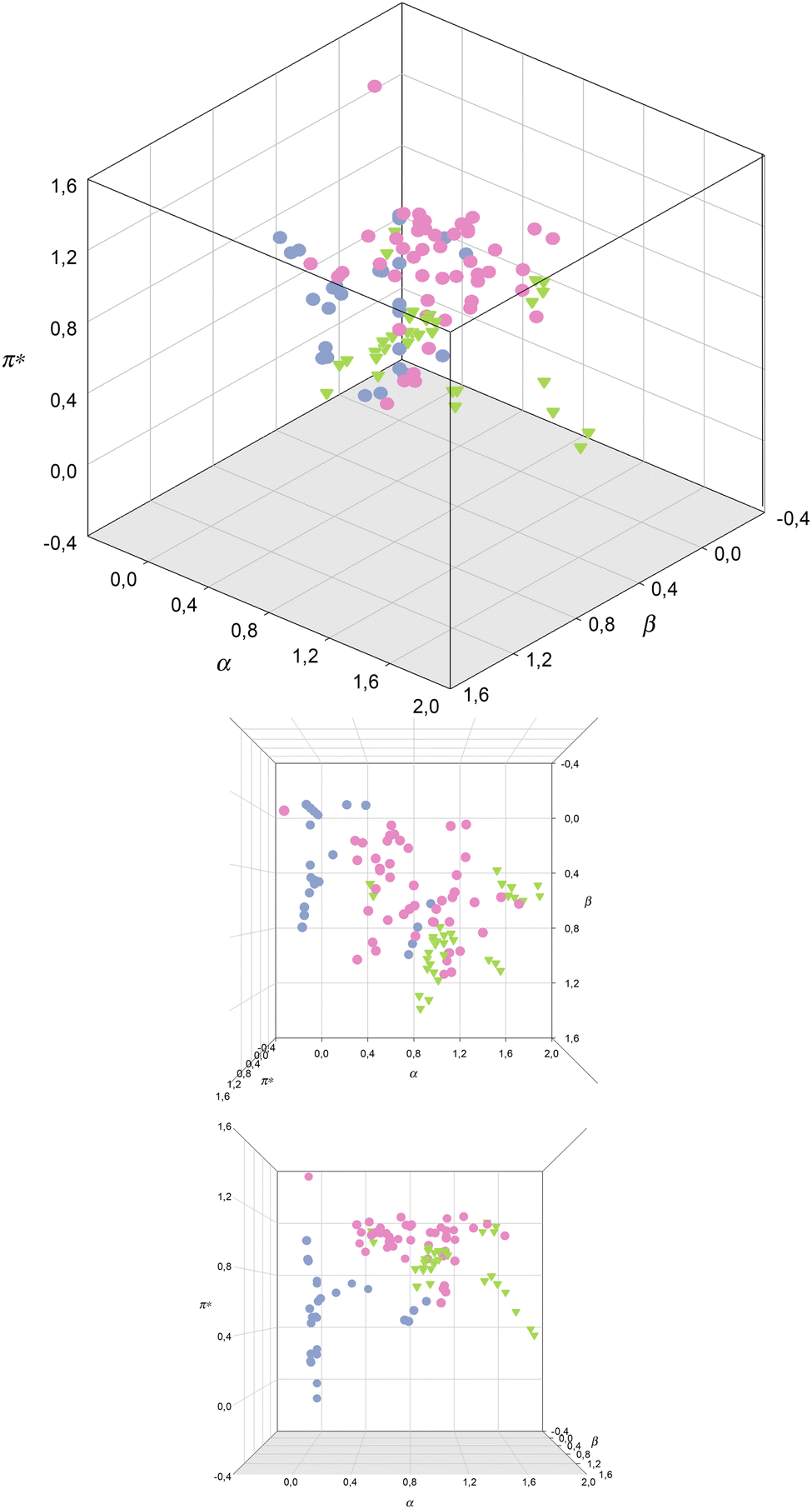 | ||
| Fig. 4 Kamlet–Taft parameters for VOCs (blue), ILs (pink) and DESs and ESs (green).71,74,77,86 | ||
As mentioned before, the difference in the values of α parameters for these classes of solvents can clearly be appreciated, with VOCs displaying very low α, apart from acetonitrile and alcohols, followed by ILs and eutectic mixtures, which show the highest values for this parameter. Nevertheless, these families of solvents have members with intermediate values of α (around unity), meaning that it is possible to replace VOCs with ILs and eutectic mixtures in terms of the α values.
The difference between β values for the 3 classes of solvents is more subtle than for the α values, as there are members of the 3 classes that can have similar values of this parameter. Nevertheless, eutectic solvents reach higher β values than the other two classes of solvents, meaning that in terms of this parameter it is easier to design an IL to replace a VOC than DES/ES. Nevertheless, and just like for the α values, it is possible to find members of the 3 classes of solvents with intermediate β values.
Regarding the π* values, it can be observed that ILs present the highest π* values amongst the 3 studied families, while VOCs and eutectic solvents share similar π* values. In this case, it would be difficult to replace a common solvent with an IL, but it would be possible to find a eutectic mixture as a replacement of a VOC, since they share the same π* values. Nevertheless, only VOCs (hexane, cyclohexane, butyl ether, isopropyl ether, carbon tetrachloride, ethyl ether and tetrachloroethylene) display very low π* values.84,85
Also, aqueous solutions of eutectic mixtures have Kamlet–Taft parameters distinct from their respective neat solvents, since water molecules interfere in the hydrogen bonding network between the constituents of the eutectic mixture, establishing stronger interactions with them and leading to a decrease in the capability of the solvent to establish new hydrogen bonds, which translates into lower values of α and β as the water content increases. Consequently, these hydrogen bond interactions with water molecules reduce ionic and dipole interactions within eutectic mixtures, leading to an increase of π* value with the water content in the mixture. It is important to note that although the β value of pure water is the limit value for these diluted solutions, the other two parameters do not follow this tendency. This provides new insights into the interaction between water and these solvents, as they do not act as simple aqueous solutions of the eutectic mixture constituents but have a more complex mechanism of interaction, at least up to a water content of 50%.87,88 To illustrate this, Table 2 compiles the Kamlet–Taft parameters of two eutectic mixtures of ChCl:U and ChCl:EG, both at (2:1) molar ratio, with different content of water as well as pure water.
:U (2:1) and ChCl:EG (2:1) as function of water content87,88
| Water content % (w/w) | ChCl:U (2:1) |
ChCl:EG (2:1) |
Water | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 10 | 30 | 50 | 0 | 10 | 30 | 50 | ||
| A | 0.79 | 0.69 | 0.62 | 0.60 | 0.67 | 0.63 | 0.45 | 0.40 | 1.17 |
| B | 0.84 | 0.79 | 0.71 | 0.66 | 0.69 | 0.67 | 0.53 | 0.47 | 0.47 |
| π* | 1.14 | 1.18 | 1.25 | 1.27 | 1.08 | 1.10 | 1.39 | 1.55 | 1.09 |
It can be observed that the Kamlet–Taft parameters for water are not the lower/upper limiting values of these eutectic mixtures, thus indicating the evolution of complex solvation phenomena as the water content changes.
It should be highlighted that although ILs and eutectic mixtures can both have their polarity tuned, it is possible for the latter to have slight variations of α, β and π* with simple changes in the proportion of the constituents.
4.4. Are DESs and ESs nontoxic?
The ecotoxicological profile of a solvent is of extreme importance as it can be a restriction on its applicability. The toxicity of a given solvent can be measured over different scales, such as minimum inhibitory concentration (MIC), half maximal inhibitory concentration (IC50), half maximal effective concentration (EC50), median lethal dose (LD50) and many others that can be more or less specific to a group of model living beings. In fact, ecotoxicity evaluation of solvents is rarely reported against more than one model organism, namely zebrafish, Vibrio fischeri, Escherichia coli, Daphnia magna, Lemna minor and even human cellular lines. This diversity of models to evaluate ecotoxicity brings unfair and imprecise comparisons between different datasets, which can lead to erroneous conclusions. Some correlations for given scales between different organisms have been developed, although they are only accurate for highly similar species.89 This unevenness of the reported ecotoxicological data has led to attempts to make uniform different values on a single scale, to enable predictions on the toxicity of a given solvent not yet studied.90Toxicity studies on the Gram-negative bacteria Vibrio fischeri are rather common, as these bioluminescent bacteria are trademarked as a toxicological test under the name of Microtox®; the effects of compounds can be visualized with the decrease of the luminescence produced by the bacterial cellular respiration.91 In this scenario, it is usual to report the toxicity as EC50, the half maximal effective concentration, which is the concentration at which the toxicant induces a response of 50% of the effect, in this case the reduction of bioluminescence. For this test, the bacteria are exposed to a known concentration of each pollutant and after an incubation time (usually 5, 15 and 30 minutes) the luminescence produced is measured and compared with the control. This procedure is repeated for different concentrations of the pollutant under study, and the concentration of 50% inhibition is calculated.92 As there are reports about the ecotoxic effects of eutectic mixtures,92–95 ILs,96 and VOCs97 for this system, it was chosen for comparison here. Fig. 5 shows the EC50 (mM) value for several solvents belonging to the 3 different classes of solvents under study.
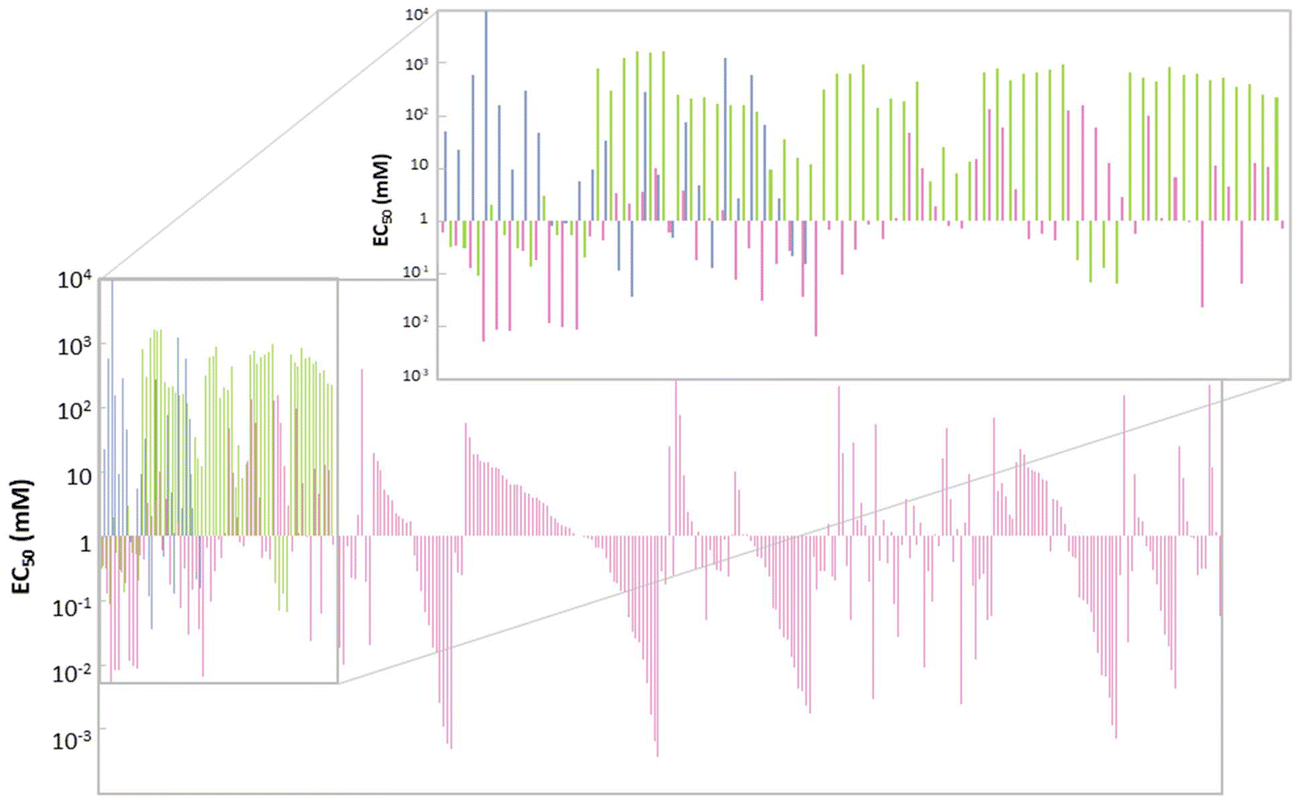 | ||
| Fig. 5 Ecotoxicity in EC50 (mM), against Vibrio fischeri, of each one of the considered solvents.92–97 VOCs (blue), ILs (pink) and DESs and ESs (green). | ||
In general, ILs seem to have lower values of EC50 than VOCs and DESs/ESs. In fact, 56% of the ILs considered have EC50 < 1 mM (Fig. 6), which means that lower concentrations are required to attain 50% of inhibition of the bioluminescence and, therefore, they are the most ecotoxic of these 3 classes of solvents. However, ILs also display a wider range of EC50 values, from 0.4 μM ([C10C1IM]FeCl4) to 398.11 mM ([C2DABCO]Br),96 the last one having a value of EC50 similar to the majority of eutectic mixtures and VOCs. This huge variety of ecotoxicity values is also present in ILs of the same family. For instance, [C2DABCO]Br, [C4DABCO]Br, [C6DABCO]Br, [C8DABCO]Br, and [C10DABCO]Br have EC50 values of 398.11 mM, 70.79 mM, 5.01 mM, 0.19 mM and 0.02 mM, respectively. The anion also has a high impact on IL ecotoxicity. For example, the ILs [C1C4mor]NTf2, [C1C4mor]CF3SO3, [C1C4mor]N(CN)2 and [C1C4mor]Br exhibit EC50 of 0.31 mM, 25.12 mM, 77.62 mM and 281.84 mM, respectively.96 As both cation and anion are very impactful on the toxicity of the final IL, it is not possible to assign a given family of ILs as more or less ecotoxic than the majority.
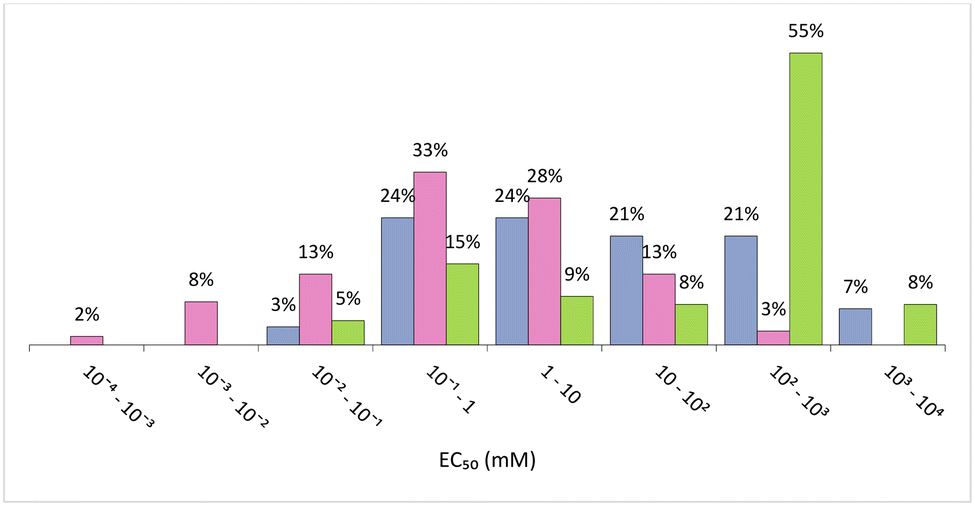 | ||
| Fig. 6 Distribution of EC50 (mM) against Vibrio fischeri considering intervals of 1 order of magnitude.92–97 Organic solvents (blue), ILs (pink) and eutectic solvents (green). | ||
Considering PILs, they present a narrower range of EC50 values, between 1.69 mM for [H2N(CH2CH2OH)2]OEt and 15.13 mM for [H3N(CH2CH2OH)]OPr.96 Similar to what happens for AILs, the ecotoxicity of PILs also varies based on changes in both ions, for instance considering the PILs [HN(CH2CH2OH)3]OBut, [H2N(CH2CH2OH)2]OBut and [H3N(CH2CH2OH)]OBut the values of EC50 are, respectively, 2.11, 4.15 and 15.13 mM. This can also lead us to the conclusion that ethanolamine-based PILs are less ecotoxic than di and triethanolamine-based ones considering the same anion, and therefore, the ecotoxicity of these PILs increases with the number of hydroxy-ethyl chains. The anion also plays an important role in the ecotoxicity, and for the same cation the ecotoxicity of PILs increases as the carboxylic aliphatic chain length increases. For instance, considering the PILs [H2N(CH2CH2OH)2]OAc, [H2N(CH2CH2OH)2]OBut and [H2N(CH2CH2OH)2]OPent, the EC50 values are, respectively, 10.61 mM, 4.15 mM and 1.69 mM. Ramifications on the side chain of the acids seem to slightly decrease the ecotoxicity of these PILs, as [H2N(CH2CH2OH)2]OiBut has an EC50 of 4.40 mM.96
Similarly, for VOCs, the range of values for EC50 is very wide, from 0.04 mM for carbon tetrachloride to 10 M for methanol. Nevertheless, this value for methanol is almost ten times the EC50 of the second less ecotoxic VOC, dimethyl sulfoxide, which has a EC50 value of 1.259 M. In general, alcohols up to three carbons, acetone, acetonitrile, dimethyl sulfoxide, dimethylformamide have EC50 values above 150 mM, while chlorinated solvents, aromatic ether compounds, other alcohols and ketones have much lower values, below 75.86 mM for ether.97 Considering linear alcohols, the ecotoxicity seems to increase with the length of the chain, as methanol has a EC50 of 10 M while the value of this parameter for ethanol is 758.58 mM, for propanol 154.88 mM and for butanol 50.12 mM. This increase of ecotoxicity with longer alkyl chains also occurs in ethers as, for instance, ethyl ether has an EC50 of 75.86 mM, while butyl ether 0.48 mM. Also, for chlorinated compounds the ecotoxicity increases as more atoms of chlorine are present. For example, considering dichloromethane, chloroform, and carbon tetrachloride, the EC50 values are 33.88 mM, 5.62 mM and 0.04 mM, respectively.97
Eutectic mixtures have a narrower range of values for EC50, ranging from 0.07 mM for ChCl:propionic acid (1:4)95 up to 1681.41 mM for ChCl:Gly:water (1:2:1).93 However, and despite the wide range of EC50 values, with several orders of magnitude, more than 60% of these values are above 0.1 M, while for VOCs less 30% have EC50 above this value.
It is worth underlining the importance of reporting the water content of eutectic mixtures in this type of measurement, as two of the datasets considered here report different values of EC50 for ChCl:U (1:2) and ChCl:Gly (1:2), as eutectics with a high water content present low ecotoxicity. For instance, ChCl:Gly (1:2) presents EC50 values of 803.50 mM (ref. 93) and 969.20 mM (ref. 95) for 0.0275% and 0.9% (w/w) of water. In fact, the effect of the presence of water on the ecotoxicity of these eutectic mixtures was already reported93 and corroborated this ecotoxicity trend. For example, EC50 values of 304.30 mM and 1592.89 mM were obtained for ChCl:U (1:2) and ChCl:U:water (1:2:1.7),93 respectively. Similarly, the EC50 values for ChCl:Gly (1:2) and ChCl:Gly:water (1:2:1) are, respectively, 803.50 mM and 1681.41 mM, and for ChCl:EG (1:2) and ChCl:EG:water (1:2:1) the EC50 values are 1234.37 mM and 1638.89 mM, respectively.93
In general, it can be concluded that eutectic mixtures have higher EC50 values than that of their most ecotoxic components, which allows the comparison of eutectic mixtures that have one component in common. For instance, eutectic mixtures of ChCl:propanol, that have EC50 values between 228.33 mM for the molar ratio of (1:4) and 393.31 mM for (2:1) 30, are less ecotoxic than eutectic mixtures of this alcohol with tetraalkylammonium chloride salts, that show values ranging from 9.26 mM ([N3333]Cl:propanol (2:1)) to 245.98 mM ([N1111]Cl:propanol (1:1)).94 However, ChCl has a lower EC50 value (2.89 mM)95 than [N1111]Cl (207.03 mM) and [N2222]Cl (116.11 mM).94 Thus, the ecotoxicity of a eutectic solvent is different from the ecotoxicity of each one of its neat components and a more complex mechanism needs to be considered. To rationalize these results, the mixture toxicity theory was proposed. This is based on two different toxicity action models working simultaneously, namely the Concentration Addition (CA), where the mixture of toxicants targets the exact same site of action, and the Independent Action (IA), where instead different sites of action are targeted.94 This approach is also supported by the evidence of the dissociation of the components of a eutectic mixture above a specific concentration of water, that is easily surpassed in this type of assay.94,95 Consequently, the ecotoxicity of a given eutectic mixture is studied as a mixture of compounds and enabling the application of these models. In the case of Vibrio fischeri, it is suggested that these individual compounds act as membrane disruptors in these organisms,95 which is in agreement with the superior data description of the CA model, in which each component of the eutectic mixture acts as diluter of the other, the reason why it is unusual to have a trend of values linear with the molar fraction of one of the components. For instance, mixtures of [N3333]Cl with EG have EC50 values of 5.76 mM, 25.82 mM, 8.20 mM and 13.67 mM for molar ratios of 1:2, 1:1, 2:1 and 4:1, respectively.94
When the components of the eutectic mixture have identical targets, they can act as synergists or antagonist of each other; this means that each of them can boost or attenuate the ecotoxicity of the other. Eutectic mixtures based on ChCl, with different HBD (EG, U, Gly, 1,2-propanodiol, propanol) all presented an antagonistic character,95 meaning that these DESs/ESs display lower ecotoxicity than their neat compounds. ChCl:propionic acid was the exception, displaying a synergistic character for some molar ratios, in agreement with the theory that the HBD determines the ecotoxicity of eutectic mixtures.95 For these eutectic mixtures based on ChCl, a ecotoxicity sequence can be proposed, from less to more ecotoxic, Gly < 1,2-propanediol and EG < U < propanol < propionic acid < acetic acid < lactic acid < glycolic acid < citric acid.95 In fact, with the exception of EG, this series is identical to that of neat compounds. Tetraalkylammonium chloride-based eutectic mixtures composed of EG presented lower ecotoxicity than propanol-containing ones; for example, [N1111]Cl:EG (1:4) exhibits an EC50 of 916.79 mM, while [N1111]Cl:propanol (1:4) displays an EC50 of 219.45 mM.95
On a final note, it is important to keep in mind that even though eutectic mixtures presented the lowest ecotoxicity among the three families of solvents here considered, Vibrio fischeri represents only a small fraction of the biodiversity present in the Earth's environment, demanding deeper knowledge about the mode of action and potency of alternative solvents like ILs and eutectic mixtures as pollutants to ensure their “greenness”. On the other hand, it should be kept in mind that eutectic mixtures can be quite ecotoxic, depending on their constituents.
Switching to a human-focused perspective, information about the cytotoxicity of alternative solvents is very scarce and mostly only available against a specific skin cell line (HaCaT). As these solvents are a “greener” option than VOCs, it is important to address this lack of knowledge. Despite the diversity of the data found in the literature, this work presents a comparison between the reported values of IC50 against HaCaT cells displayed by VOCs98 and eutectic mixtures,99 with an exposure time of 72 hours, as well as PILs100 and AILs,101 with an exposure time of 24 hours.
According to Fig. 7, eutectic mixtures are clearly less cytotoxic than VOCs and ILs, and the difference between the reported values of IC50 may be even larger considering the lower period of exposure of ILs relatively to DESs and ESs. In fact, these are the only class of solvents with values of IC50 in the two upper categories (10 to 1000 mM). PILs and VOCs share the middle categories (0.1 to 10 mM) although, as stated before, this is not a fair comparison due to the different time of exposure. Finally, AILs are the only representative of the two lowest categories (0.001 to 0.1 mM) and therefore are the most cytotoxic to the HaCaT cell line of the solvents considered.
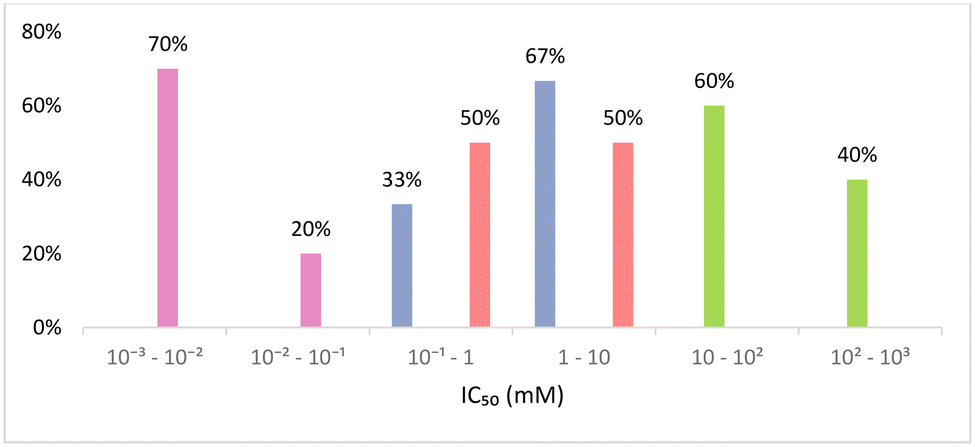 | ||
| Fig. 7 Distribution of IC50 (mM) against HaCaT cell line considering intervals of 1 order of magnitude.98–101 VOCs (blue), AILs (pink), PILs (red) and DESs and ESs (green). | ||
Focusing on eutectic mixtures, the cytotoxicity of [N4444]Cl-based eutectic seems higher than that of ChCl or [N1111]Cl-based ones, as all eutectic mixtures based on these latter HBAs increased cell viability, even at the highest concentrations tested for most cases.99 Thus, it is impossible to calculate a reasonable IC50 value for these eutectic mixtures, while meaningful information can be obtained from the [N4444]Cl based eutectics. For instance, the cytotoxicity of mixtures of this tetraalkylammonium salt and carboxylic acids seems to decrease as the length of the alkyl chain of the acid increases. For example, [N4444]Cl:butanoic acid (1:1) and [N4444]Cl:hexanoic acid (1:1) have an IC50 of 108.7 and 112 mM, respectively. These two eutectic mixtures are also the least toxic to the HaCaT cell line, displaying the highest values of IC50, in contrast to [N4444]Cl:EG (1:1), which showed the lowest IC50 of 34.1 mM.99 Moving on to VOCs, the reported values of IC50 are between 0.14 mM for dimethylformamide and 2.27 mM for 2-methoxyethanol.98 However, due to the reduced number of VOCs tested and the heterogeneity in IC50 values, it is impossible to take conclusions from this set of data. Additionally, the scarcity of information about the cytotoxicity of VOCs is frightening, as they are widely and intensively used all over the world.
Focusing on ILs, as mentioned before PILs showed higher values of IC50 than AILs. AILs have IC50 values ranging from as low as 0.001 mM, for [C14Qn]Br, up to 0.035 mM, for [C8C1im]Cl. Both the cation and anion seem to have influence on the cytotoxicity of aprotic ILs against the HaCaT cell line. For instance, imidazolium-based ILs showed a lower IC50 for long alkyl side chains, namely [C8C1im]Cl, [C14C1im]Cl and [C16C1im]Cl, which have an IC50 of 0.035, 0.005 and 0.004 mM, respectively. The effect of the anion is evident when the IC50 of [C16C1im]Cl (0.004 mM) is compared with that of [C16C1im]NTf2 (0.017 mM).101 PILs have an IC50 value ranging from around 0.4 mM for [H3N(CH2CH2OH)]OF and 2.5 mM for [H3N(CH2CH2OH)]OF, and similarly to AILs their cytotoxicity is influenced by both ions. For example, both [N2000]OF and [N2000]NO3 display an IC50 below 0.4 mM, while [N2000]Gl has a value of 1.3 mM and [N2000]OAc shows the highest value of 2.4 mM. The role played by the cation can also be illustrated by considering the case of [H3N(CH2CH2OH)]OAc with an IC50 of 2.5 mM, [H2N(CH2CH2OH)2]OAc with an IC50 of 0.9 mM and [HN(CH2CH2OH)3]OAc with an IC50 of 1.6 mM.100
Switching to a more realistic model to analyze the toxicity of DESs, a mixture of Bet and Gly at a molar ratio of 1:2 and with 10% (v/v) water was used to extract antioxidant phenolic compounds from green coffee beans; this extract, besides being composed of only natural occurring compounds, proved to be lethal to rats. Even though Bet and the extracted phenolic compounds have very positive effects in rats, namely antioxidative effects, Gly is known to be harmful to these mammals, leading to renal failure, and therefore the prepared DES had a similar effect. This is also in accordance with the dissociation of the DES at high percentage of water, possibly in the stomach.102
4.5. Water in DESs and DESs in water
Trace amounts of water are often unavoidable as an impurity in ILs and DESs/ESs, as these solvents have proved to be very hygroscopic. Nonetheless, the addition of specific amounts of water has been a strategy followed by many researchers to overcome two major drawbacks of both these classes of fluids: their high viscosity and high price.103 Consequently, understanding the effects of different amounts of water on the liquid structure of ILs and DESs/ESs at the molecular level is highly important, since it allows control of their macroscopic properties. Although the effect of water on ILs’ structure is well understood and a substantial amount of work has been published,104 the same cannot be said for DESs/ESs, which have been studied to a considerably lesser extent and with a greater emphasis on mixtures of ChCl with U.105,106 Chunyan published a comprehensive review107 of similarities and differences of water in ILs/ionic DESs and ILs/ionic DESs in water. The mechanisms of solubilization of both these classes of solvents in water as well as inorganic salts are summarized in Fig. 8.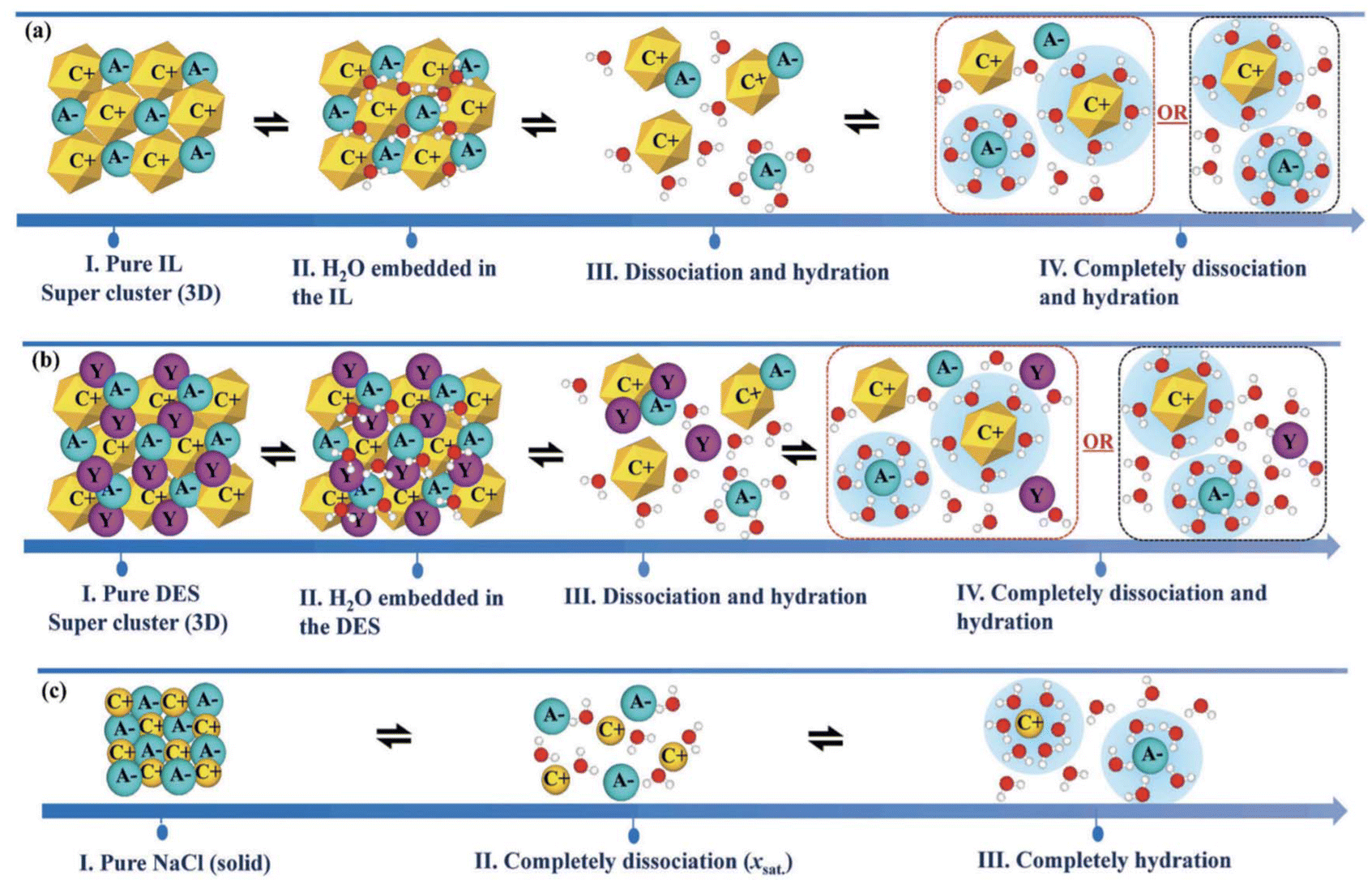 | ||
| Fig. 8 Structural effects of the addition of water to ILs (a), DESs (b) and NaCl (c).107 | ||
A given IL exposed to a small quantity of water can embed water molecules without breaking the liquid structure of the pure IL. As the addition of water increases, the cluster starts to dissociate and the anions begin to strongly hydrate, and finally the addition of more water molecules completely dissociates the IL ions and fully hydrates them. Even though this is true for many ILs, some larger anions, like BF4−, do not get fully hydrated, and hydrophobic anions, such as NTf2−, do not even get hydrated. This feature is another proof of ILs’ tunability; when switching from smaller and hydrophilic ions to larger and more hydrophobic anions the resulting IL will be less capable of inserting water molecules into its structure, but also more stable in highly diluted solutions.107 For example, MD simulations show that in [C4C1im]Cl when mixed with water, ion pairs can be found even at an IL molar fraction of 0.001. Even though these simulation studies lack experimental validation, they are helpful in understanding the difference between the effect of water in ILs and in salts.108 As for this last case, a solution of a given salt at its saturation point will result in the complete dissociation of the ions that compose the salt, and finally the addition of more water will carry the full hydration of these ions.107
It is also important to point out that the stability of a given IL cluster depends on both the structure of the cation and the anion, meaning that it is not possible to make a general rule, as two distinct ILs may have different concentrations of water that results in dissociation and hydration, and as explained above this may even not happen in some ILs.107 In fact, MD simulations of aqueous solutions of ILs based on [C10C1IM] and different anions, all at the concentration of 1 M, show very distinct arrangements. For instance, [C10C1IM]NTf2 at this concentration still remains as a cluster with few water molecules embedded in it, while [C10C1IM]PF6 at the same concentration is already dissociating and hydrating.109
The structural modifications of pure DES clusters due to the addition of water are similar to those of ILs, starting with the embedding of water molecules in the cluster, going through dissociation and hydration as more water is added, until full dissociation and hydration is achieved. Also, once again it is not possible to make a general rule, as different combinations of HBA and HBD can result in DESs with different affinity for water and, therefore, more or less stable clusters at high water content.107 Either way, the concepts of water in DES and DES in water are already established, as water in DES refers to the embedding of water molecules into the DES structure while DES in water corresponds to the dissociation of the cluster.
The solubility of DESs in water is complex because DESs are mixtures and thus the DES components are going to solubilize in water according to their individual solubility in water. Considering a binary DES, if it is composed of two hydrophilic compounds, a one-phase solution is obtained, but if one of the components is hydrophobic a two-phase equilibrium will be obtained, with one aqueous phase and one hydrophobic phase. Note that in this last case, both the hydrophilic and the hydrophobic components will solubilize in the water phase according to their solubility.110 Thus, the hydrophobic phase will be mainly composed of the hydrophobic component since the hydrophilic components will leach to the water phase. In fact, this was proved by bringing eutectic solvents into contact with water and analyzing both phases through 1H-NMR; for DESs composed of [N4444]Cl and various carboxylic acids, the ammonium salt was always present in water and the carboxylic acids with short alkyl chain, for example, acetic acid also leached to the aqueous phase, to some extent. Switching from the ammonium salt to neutral compounds, such as D,L-menthol, only the DESs composed of short carboxylic acids showed the presence of these acids in the water phase, while for acids with long alkyl chains like octanoic, decanoic and dodecanoic acids, none of the components were identified in the water phase. Of course, this does not mean that these DESs are not soluble in water, but that their concentration in the aqueous phase is below the limit of detection of the 1H-NMR technique.111 Even for a hydrophobic DES composed of two hydrophobic compounds, the final DES composition will always slightly change, since the two compounds will not have the same solubility in water. This does not hinder the use of hydrophobic DESs as extractants of compounds from aqueous phases as the quantity of leached compounds will be, most times, negligible when compared with the extracted compounds. For example, taking into account two DESs composed of decanoic acid and either D,L-menthol or [N4444]Cl in the proportion 2:1, for the latter one the water solubility into DES is 6.9 wt% at 298 K and the solubility of the ammonium salt in water is 13.5 wt% at 298 K, while for the former DES the solubility of water in DES is 2.1 wt% at 295 K and the total solubility of both compounds in water is 0.04 wt% at 295 K.112 Consequently, it is possible to conclude that even hydrophobic DESs have some solubility in water that should not be ignored. Moreover, various authors present the solubility of a DES in water as if it is an individual compound, or only measure the water solubility for one of the compounds (typically the hydrophilic or charged compound), instead of presenting the water solubility of all the individual compounds of the eutectic mixture. It is also important to point out that changing one of the constituents of the DES can lead to significant changes in the solubility of water in DES and vice versa. Finally, it is worth noticing that when in contact with water the remaining DES phase will have a different molar ratio compared with the initial one, as each compound solubilizes to a different extent from the other.110
5. Comparative analysis in a selected research area: is there a clear winner and a clear loser between ILs and DESs?
When reading the general statements found in the introduction of the vast majority of papers, one has the impression that DESs are by nature greener and better performing systems than ILs, not to mention other solvents.As mentioned in the previous sections, DESs and ILs share several common features which make them suited for sustainable development. For instance, they can be made up of natural molecules that originate from renewable feedstock such as sugars and sugar-based compounds,113–116 amino acids,117–119 fatty acids120–123 and lignin monomers.124,125 This peculiarity presents for the first time the option of using largely available, non-petroleum-derived materials for the preparation of a wide range of liquid media. In an attempt to answer the heading question of this article, we focused the present analysis on the research area which, in our opinion, will likely play the lion's share in sustainable chemistry, namely the exploitation of lignocellulose materials. From this point onwards, this comparative analysis is directed towards selected examples concerning the dissolution and treatment of the most available biopolymers: cellulose, hemicellulose, lignin and chitin. In this chosen area of research, new media are always sought given the limited applicability of most of the traditional organic solvents.
Before starting this analysis, a word of caution is needed when considering the results reported by different authors, even considering the same kind of biopolymer. Indeed, different publications are based on polymers with different degrees of polymerization, crystallinity, composition (for hemicellulose) and polymorphism, as well as various degrees of acetylation (for chitin). Also, it is not always trivial to organize this information, which hinders the correct comparison and evaluation of the proposed systems’ performances. Moreover, when considering complex biomasses, factors like the natural heterogeneity of lignocellulosic material, the size of the material and possible additional pretreatments all render critical comparisons less reliable.
5.1. Lignocellulosic biomass
Cellulose is the most abundant biopolymer in nature, being present in 30–50 wt% of terrestrial biomass and even more prominent in marine biomass. The exploitation of this resource is essential in the current scenario of sustainable development and circular economy, and requires the development of solvents to extract, purify, dissolve and process it effectively.126 Although cellulose has been used to make yarns and paper for centuries, most of the protocols used in these processes are not environmentally sustainable.127 For example, the cultivation of cotton produces high-purity cellulose but at the same time it is very expensive (2.7 € per kg,128 price for 1 ton) and above all has a marked environmental impact in terms of water footprint (10000 L of water per 1 kg of cotton) and massive use of pesticides.129 Pulp for paper production is obtained more economically (0.4 € per kg,128 price for 1 ton) using the Kraft process, which, however, needs harsh conditions (high temperature as well as pressure in addition to the use of reagents harmful to the environment) and employs only cellulose-rich biomass. This latter aspect raises serious concerns as some areas of the planet are being deforested and certain types of trees are preferred, to the detriment of biodiversity. In addition, cellulose from the Kraft process typically contains 15–25 wt% of hemicelluloses and 5–10 wt% of lignin and therefore needs to be purified to make it suitable for other applications.130,131 These purification processes are called “pulping” and also involve the use of hazardous chemicals and severe conditions.
Nowadays, the Kraft process accounts for about 80% of cellulose pulp production. As mentioned above, this protocol operates under harsh conditions (150–180 °C) and utilises hazardous sodium hydroxide (NaOH) and sodium sulfate (Na2SO4) as reagents.132 Although the optimised Kraft process has improved energy efficiency, serious issues remain, firstly related to corrosion due to the alkaline environment, but also to the disposal of process water. In addition, the resulting cellulose is intensely coloured due to the degradation products of lignin and hemicellulose, and therefore requires many bleaching treatments. Lastly, the Kraft process breaks down the hemicellulose almost completely and the obtained lignin is of very poor quality due to its strongly degraded and sulfur-rich content.133 In view of an integrated biorefinery process, these issues are critical and need to be addressed.
The sulfite pulping process has been developed as an alternative to the Kraft process, its main advantage being the flexibility to treat a wide variety of biomasses. In fact, by varying the dosage and the composition of the chemicals, the entire pH range can be covered.133 Sulfite pulping enables the valorisation of the hemicellulose and the lignin fractions through their transformation into chemicals such as lignosulfonates, ethanol, fodder yeast, soda, vanillin, acetic acid and furfural. However, the quality of the cellulose fibres obtained is worse than that obtained in the Kraft process.133
Overall, the best industrial process is the Organosolv pulping process, which makes it possible to obtain good quality cellulose fibres while preserving the structure of the other components. For example, the lignin obtained from the Organosolv process is sulfur-free and presents a structure more similar to the native one in comparison with that obtained with the processes described above. This allows for an effective further exploitation and facilitates the valorisation of lignin as high-value material. The Organosolv process patented in 1971 used water or water/volatile organic solvent mixtures of different nature in the presence of a catalyst.134 The most widely used industrial mixtures are methanol/water 50–100 wt% and ethanol/water 40–60 wt% in the presence of mineral acids or calcium and magnesium salts as catalysts.135 Although the Organosolv process represents a significant improvement over the Kraft and sulfite processes, the poor solubility of lignin still gives rise to major issues: the significant quantities of volatile and flammable organic solvents needed, combined with the high energy consumption for their recovery, makes it environmentally but also economically unsustainable.133,136 For these reasons, further improvement in the form of new processes that are economically as well as environmentally sustainable, is very much needed.
5.2. Pretreatment of biomass
The main challenge is to develop systems that yield cellulose from all sorts of biomasses in a sustainable way, while simultaneously allowing the valorisation of all biomass components without altering their structure and properties. This advanced biorefinery concept must be implemented in harmony with the principles of green chemistry in order to be fully sustainable. In this context, ILs and DESs can play a key role, separately or even synergistically, as sustainable media capable of selectively solubilising each individual biomass component and thereby resulting in an “integrated biorefinery”.Over the years, many biomass pretreatment processes aimed at obtaining raw cellulose for use in saccharification processes have been based on the use of ILs and more recently of DESs. These processes, commonly referred to as pretreatment processes, afford cellulose suitable for enzymatic hydrolysis into glucose which is eventually transformed into ethanol using second-generation (2G) protocols.
Given the amount of knowledge in the area, the use of ILs affords a more mature technology compared with DES-based methods. There are two different approaches in the pretreatment of biomass with ILs: the dissolution process and the Ionosolv process.137 In the dissolution process, the entire biomass is completely dissolved and the individual components are obtained by precipitation. This enables a more efficient separation of lignin and cellulose. Furthermore, the recovered cellulose is completely amorphous and thus more readily attacked in hydrolysis processes.137 These processes employ aprotic ionic liquids (AILs) with imidazolium cation, which, besides their high cost ($50 per kg)138 and low water tolerance, have a strong depressing effect on hydrolysis and fermentation processes even if present in traces (as little as 0.25% residual [C2C1im]OAc in the fermentation broth will completely inhibit yeast growth and biofuel production).139 The limited numbers of papers in the literature that use the dissolution process suggest that it has not yet been fully optimised. Conversely, numerous examples of Ionosolv processes have been reported. The Ionosolv process, whose name is deliberately similar to the Organosolv process, involves the use of ILs in pure form or in a mixture with organic solvents (or water) for the dissolution of lignin and hemicellulose, while cellulose remains undissolved and is not appreciably decrystallised. This way, the raw cellulosic material obtained remains suitable for use in applications where the crystalline cellulose fraction is needed, such as for nano-crystalline cellulose production or as a polymer additive. In addition, ILs can be used in a mixture with water or alcohol. This feature, in spite of a significant reduction of solvent cost, reduces contamination due to entrapment in the cellulose fibres obtained after the re-precipitation step. Thus, the use of ILs has a dual benefit: it allows for a higher percentage of recycled solvents and produces pure cellulose suitable for enzymatic hydrolysis and fermentation processes.
The effectiveness of this approach is confirmed by the Ionosolv process marketed by Imperial College London under the name BioFlex process.140 This pretreatment process, which uses aqueous solutions of PIL triethylammonium hydrogensulfate ([N2220]HSO4), demonstrated high feedstock flexibility. Indeed, by modifying the operating conditions, it proved to be efficient with biomasses such as hardwood,141 softwood,142 forest residues and switchgrass,143 agricultural wastes such as rice straw, rice husk, wheat straw, sugarcane bagasse and coconuts waste.144–146 This process has been scaled-up to pilot plants of 200 L capacity and plans for a unit able to produce ca. 200 t per year of pulp have been drafted.140 The selection of PILs has been the best in economic terms.147 In addition to [N2220]HSO4, other PILs with hydrogen sulfate as anion such as tetramethylguanidinium, monoethanolammonium, diethanolammonium, triethanolammonium, and diisopropylammonium have also performed excellently.148 Apart from the type of solvent, the effect of water as well as pretreatment conditions such as biomass loading, acidity, particle size, time, and temperature have been studied extensively.149,150 The impact of different kinds of treatment on hemicellulose and lignin will be discussed in sections 5.3 and 5.5. The cellulose obtained is characterised by partial hydrolysis of the amorphous portion and consequent decrease in the molecular weight, as a result of the severe conditions used in these processes. Although this is desirable for some downstream uses such as saccharification and nanocellulose production, it should be avoided for applications such as fibre manufacturing. For this reason, both the type of solvent and the operating conditions must be appropriately selected in order to match with the target use of the cellulosic material.
The hybrid Ionosolv–Organosolv process developed by Chen et al. is certainly noteworthy.151 In this hybrid fractionation process, a mixture of organic solvents (ethanol, butanol or acetone) and [N2220]HSO4 shows very good fractionation efficiency both on Miscanthus and on recalcitrant biomass such as pine wood. Pretreatments using butanol 60 wt% or ethanol 40 wt% with [N2220]HSO4 show a glucose yield of 85%, which is 10% higher than that obtained with the Ionosolv process. This could be explained by the improved delignification of the biomass during fractionation, confirmed by compositional analysis of the pulps. Moreover, the Ionosolv–Organosolv pretreatment demonstrates the ability to operate at high biomass loading (up to 50 wt%) and to simultaneously generate a highly enzyme-accessible cellulose fraction and high-quality lignin, with excellent potential for high value-added uses.151 The effect of pretreatments on lignin will be discussed in the dedicated section (section 5.3).
PILs with hydrogen sulphate anion are not the only ones that show great promise. Ferrari et al.152 reported a Ionosolv pretreatment process followed by enzymatic hydrolysis based on a mixture of [H3N(CH2CH2OH)]OAc/[H3N(CH2CH2OH)]OHex PILs in a 1:1 molar ratio. For this process, integrated within a 2G ethanol production process, an in-depth techno-economic analysis was carried out,153 showing its effectiveness despite the fact that acetate is a proven metabolic inhibitor of most yeast systems.137
In biomass pretreatment processes, DESs are extremely competitive compared with ILs due to their low cost and their low toxicity. The great number of publications in this area is tangible evidence of their effectiveness in this application. Similarly to ILs, the majority of papers focus on the pretreatment of different types of biomass, from forestry and agricultural sources, and its impact on cellulose saccharification and ethanol production. Various kinds of DESs and NADESs, with HBD and HBA of different nature and molar ratio, have been employed in pretreatment processes. Very often, the pretreatment processes were Ionosolv-like, where cellulose was extracted by reactive and non-reactive solubilisation of lignin and hemicellulose. The solubilisation of hemicellulose and lignin by DESs and NADESs will be discussed in section 5.5. Investigating the contribution of the HBD portion in the pretreatment process reveals that mono- and di-carboxylic acids have superior efficiencies compared with polyols and U.154 A comparative study was carried out on DESs with the same HBD moiety (lactic acid, LA) and different HBA parts, which showed that hydrogen bond acidity (α of K–T parameter) had a positive linear relationship with pretreatment efficacy.155 Among the various carboxylic acids used as HBDs, LA is by far the most popular despite its known inhibitory effect on enzymatic hydrolysis processes.156 Finally, the effect of the molar ratio on the pretreatment efficiency was evaluated for the DES ChCl:LA. The best results were obtained with ChCl:LA (1:4) at 130 °C. The cellulosic material obtained had a high degree of crystallinity and a low degree of polymerisation due to the hydrolysis of the amorphous cellulose portion.157
Although DESs with carboxylic acids as HBDs are apparently the best-performing systems, they present problems during recycling. In fact, it is well known that for cholinium-based DESs, an esterification reaction between the acid and the hydroxyl group of the cholinium occurs. However, it is also true that this phenomenon only affects a DES fraction and remains constant over time. Furthermore, Morais et al.158 investigated the effect of DES ChCl:LA (1:10) on the polysaccharide portion during the pretreatment process at 130 °C at different time points (0.5–24 h). Surprisingly, the DES had a negative impact already at pretreatment times beyond 4 h, resulting in variety of undesired events such as the esterification of cellulose by LA, the shortening of fiber length and their agglomeration. Moreover, the authors noted side reactions of the hemicelluloses fraction such as furfural and humin formation as well as LA grafting. In addition, this DES also undergoes transformations due to lactide formation and polymerisation. For these reasons, treatments with lactic acid-based DESs are recommended only for a short period of time.158 Based on this DES, Kumar et al. carried out a technical–economic analysis for a biorefinery process that utilises different types of biomass for the production of cellulose, lignin, and silicates in a plant- and economically viable way.159
Although the inhibition effect of the LA component of DES was easily overcome by optimising one-pot saccharification pretreatment processes,156 the remaining problems were more difficult to solve. Replacing LA with other organic acids only partially solved the difficulties described above. From a circular economy perspective, the use of lignin-derived DESs reported by Kim et al.160 is of particular interest. These DESs, which are based on ChCl and 4-hydroxybenzyl alcohol, catechol, vanillin, and p-coumaric acid, showed good results and high recyclability as solvents for biomass pretreatment in terms of lignin removal and sugar release after enzymatic saccharification. A viable alternative to acid HBDs is represented by DES based on Gly, a natural product widely available for large-scale applications and a well-known enzyme-stabilizing agent.161 The performance of glycerol-based DES, ChCl:Gly (1:1) and Bet:Gly (1:1), was compared with IL [C2C1im]OAc.162 The pretreatment efficiency of the different media was evaluated by comparing the enzymatic digestibility of the cellulose obtained. Pretreatment tests were carried out using 90 wt% DES in water or [C2C1im]OAc at 80 °C for 24 h with a 5 wt% loading of different cellulose types. The enzymatic digestibility of the pretreated substrates was evaluated in buffer and in the presence of 30 wt% and 80 wt% of DES or [C2C1im]OAc.162 Although the stability analysis identified glycerol-based DESs as stabilising agents for cellulases, the overall efficiency of the process appeared to be in favour of [C2C1im]OAc. This can mainly be attributed to a lower pretreatment efficiency of the DES compared with the IL. Pretreatment efficiency can be, however, improved by coupling DES treatment with ultrasonic irradiation or mechanical manipulation. Sharma et al.163 reported excellent results in the pretreatment of sugarcane bagasse using ChCl:Gly (1:10) coupled with ultrasonication. Under these conditions, the neutral DES was found to be the best choice even when compared with DESs containing acids as HBDs.163 Of great interest is the pretreatment procedure devised by Ai et al.164 that combines ChCl:Gly (1:2) with a twin-screw extruder. The use of ChCl:Gly-mediated extrusion preserved the basic lignin structural characteristics with no significant differences between extruded biomasses at a solid loading of 30% and 50%.164 Besides the excellent results obtained, it is important to point out that the use of an acidic DESs in pretreatment would lead to major corrosion concerns.
5.3. ILs and DESs for the ‘lignin first’ biorefinery process
When it comes to biomass utilisation processes, lignin is regarded as the “ugly duckling” when compared with the various other components.165 The recalcitrant character of biomass materials is in fact mostly due to the properties of this very component. The origin of lignin's bad reputation goes back to well-known problems in both papermaking and ethanol production processes, where the presence of lignin and its derivatives leads to the darkening of the final product and enzyme inhibition, respectively. Lignin derived from traditional pulping and pretreatment processes is quite different from native lignin. In fact, of the various components of lignocellulosic biomass, lignin is the one that undergoes the largest structural changes. The resulting material, which is often named after the process employed (e.g. Kraft lignin), only partially retains its pristine structure and loses many of its original properties, with a dramatic reduction in applicability. Therefore, it comes as no surprise that only 2% of the extracted lignin is used in industry and agriculture while the rest is discarded as waste.166Lignin cannot yet be isolated in its native unaltered state and for this reason the native structure has not yet been completely elucidated. Despite this, this peculiar chemical species is thought to be a complex amorphous long-chain heterogeneous polymer of phenylpropane linked by ether bonds with a molecular weight in the range of 2500–10000 g mol−1.167 The polymer is composed of aromatic subunits called monolignols like guaiacyl (G), syringyl (S) and p-hydroxyphenyl (H) subunits derived by sinapyl (3,5-dimethoxy-4-hydroxycinnamyl), coniferyl (3-methoxy-4-hydroxycinnamyl) and p-coumaryl (4-hydroxycinnamyl) alcohols, respectively (Fig. 9).
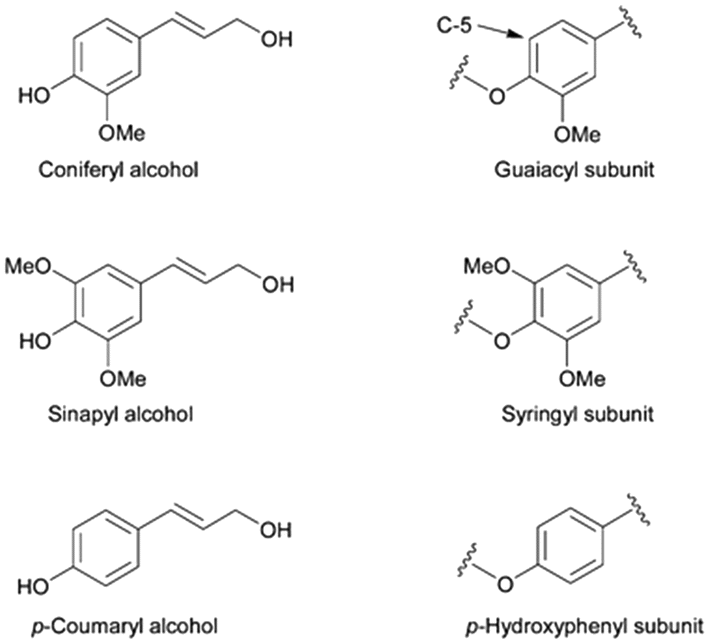 | ||
| Fig. 9 The three monolignols from which lignin is synthesised and derived aromatic subunit. Reproduced from ref. 168 with permission from The Royal Society of Chemistry, copyright 2013. | ||
Non-canonical subunits that have been identified include ferulates (which form linkages between hemicellulose and lignin), coniferaldehyde, sinapaldehyde, and 5-hydroxyconiferyl.169 Lignin monolignols are predominantly linked either through ether or C–C bonds. In native lignin, two-thirds or more of the total linkages are ether bonds, while the other linkages are C–C bonds. The predominant linkages between the structural units of lignin are β-O-4′ (β-arylether), β–β′ (resinol), and β-5′ (phenylcoumaran), but β–5, β–1, dibenzodioxocin, 5–5′, and α-O-4′ are also present (Fig. 10). Roughly half of the total lignin linking is represented by the β-O-4′ ether type. The latter kind are the bonds that allow the lignin chains to elongate through the formation of linear strands from which branches are connected through ether and C–C bonds.169
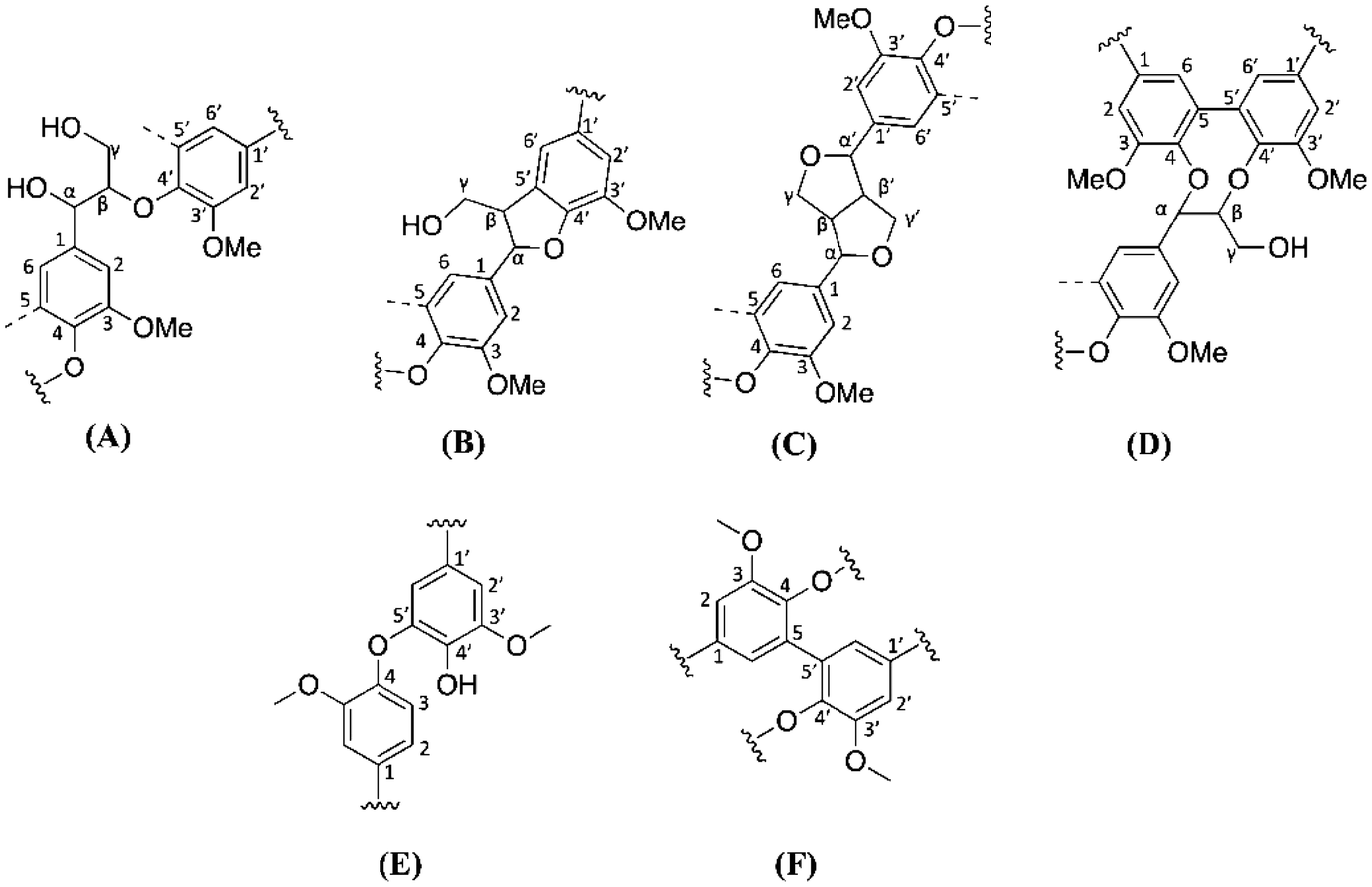 | ||
| Fig. 10 Common lignin interunit linkages: (A) β-O-4′, (B) β-5′, (C) β–β′, (D) dibenzodioxocin, (E) 4-O-5′, and (F) 5–5′ linkage. Adapted with permission from Dutta et al.169 Copyright 2017 American Chemical Society. | ||
The amount, structure and composition of the aromatic subunits are highly dependent on plant taxonomy and whether the source of lignin is softwood (gymnosperm), hardwood (angiosperm) or grasses. Softwoods contain between 27 and 33 wt% lignin, consisting almost exclusively of G units (90–95%) while hardwoods contain 18–25 wt% of lignin, which is rich in S units (45–75%) with a lower amount of G units (25–50%). Grasses contain a low amount of lignin (17–24 wt%) and a higher amount of H units (5–35%), which is negligible in hardwood and softwood.170 The composition of lignin has a strong impact on both its potential utilisation and its stability to the biomass deconstruction processes. In fact, during classical delignification processes, lignin is solubilised via the breaking of the β-O-4′ bond with concomitant lowering of the molecular weight. However, under the same conditions the guaiacyl groups tend to form stable C–C bonds with other units at the C-5 position that are stable to hydrolysis. This leads to an increase in molecular weight and to the formation of a stable lignin-like solid. This material is often insoluble and can deposit on other lignocellulosic components, lowering their quality.168 Lignin with a high number of C–C bonds is of poor quality and cannot be used in the production of chemicals given its resistance to degradation. This phenomenon makes the delignification of softwood biomass more difficult than hardwood. This is essentially due to hardwood's lower amount of G groups together with the fact that in S groups the C-5 position is unreactive to crosslinking due to the presence of the methoxyl group.168
The myth of lignin's recalcitrance is attributable not to pristine lignin, but to the material obtained by classical pretreatment processes. The so-called technical lignin has a low number of ether bonds, a high sulfur content and a high number of C–C bonds, which makes dissolution and transformations difficult. Fig. 11 shows the characteristics of three common types of technical lignin.
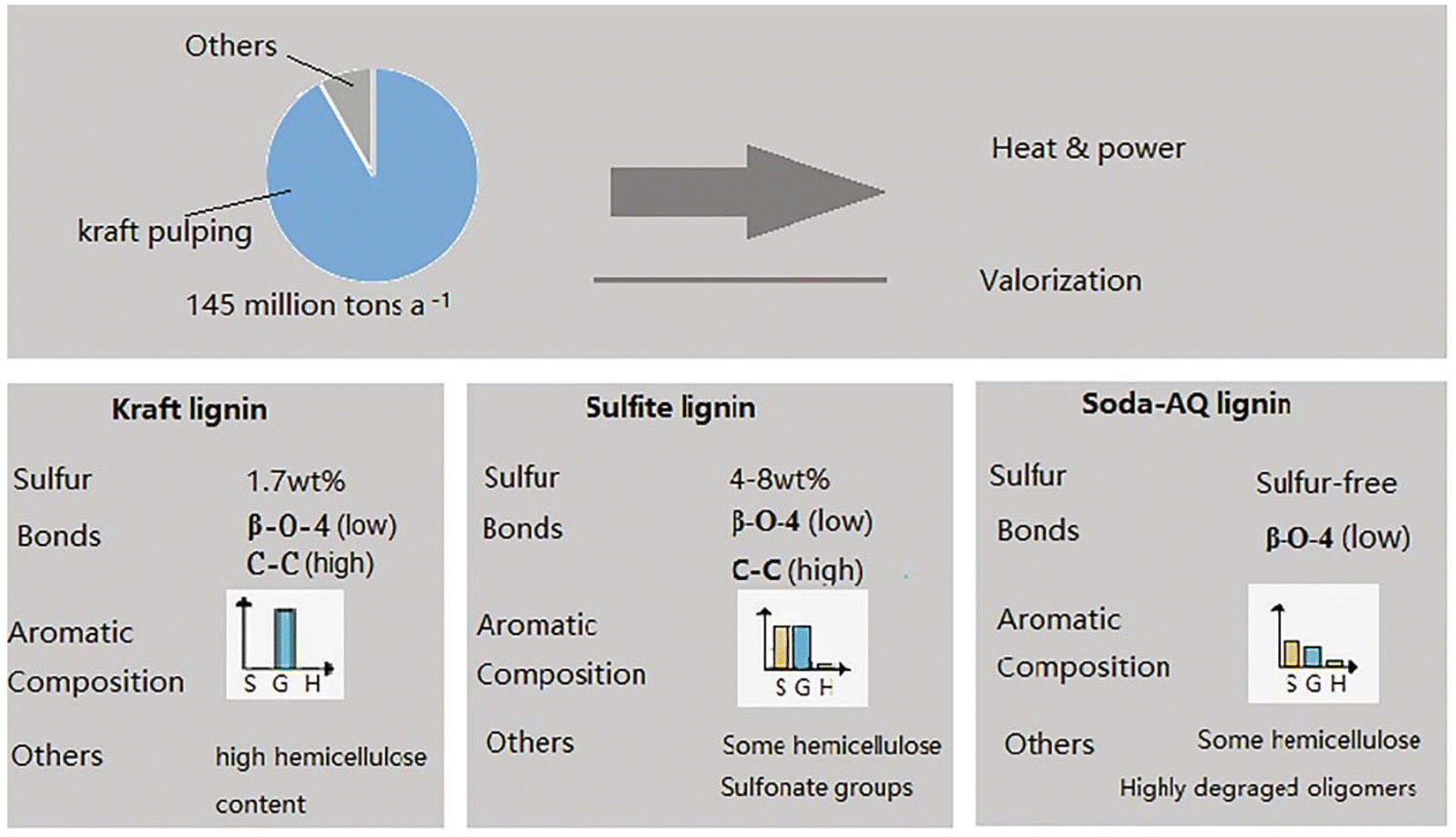 | ||
| Fig. 11 Structural characteristics of Kraft, Sulfite and Soda-AQ technical lignin. Reproduced from ref. 166 with permission from Elsevier, copyright 2020. | ||
In order to exploit the full potential of lignin, the spotlight has shifted in recent years to the so-called ‘lignin first’ biorefinery processes. According to the protocols here used, the removal of lignin takes place through a solubilisation process that limits the modification of the primary structure and thereby restricts recondensation phenomena.171 The price of lignin can vary between 250 and 750 USD per ton for low- and medium-quality lignin, respectively, depending on the degree of purity. This price can rise significantly in the case of low molecular weight native and/or high-purity lignin, thus showing the economic relevance of the above described ‘lignin first’ processes.172
Of the industrial-scale pretreatment processes, Organosolv processes are the mildest towards the structure of lignin and thus the most suitable for the ‘lignin-first’ biorefinery.173 Although the limitations of such processes have previously been pointed out both economically and environmentally, lignin obtained by Organosolv processes is nowadays the reference material in the lignin field.174 In this context, it can be argued that the use of ILs and DESs in this field has great potential. Numerous papers and reviews on the processing of lignin have been reported in recent years with a focus on the amount of solubilised lignin, the mechanism of solubilisation and structural modification.
The most interesting ILs in lignin removal are AILs with acetate anion and PILs with hydrogen sulfate anion. In addition, cholinium ILs with amino acidic anion are also a viable alternative to AILs with imidazole cations as they are cheaper, biobased, and biodegradable. Given the high structural variability of lignin and the fact that many solubilisation studies have been carried out with different technical lignins, a comparison of the solubilisation capacity of different ILs is unrealistic and of little interest. Instead, the solubilisation mechanism and the modification of the lignin structure by ILs are certainly topics of interest, especially in view of employing this valuable resource in biorefinery processes.
AILs were the first ILs to be used for lignin processing, and the material obtained from such processes is often used as a reference. The lignin-removing capabilities of [C2C1im]OAc were tested on hardwood (Eucalyptus globulus) and switchgrass (Panicum virgatum) biomasses under different pretreatment conditions and the effect on the structure of the obtained material was evaluated.175 In particular, it was observed that temperature has the greatest effect on structure, with a preferential breakdown of S-lignin at high pretreatment temperature (160 °C) in both hardwood and switchgrass while breakdown of G-lignin for hardwood was observed at 120 °C. In contrast, no preferential breakdown of either S- or G-lignin was observed in switchgrass at low temperature.175
Exploiting this system, Sathitsuksanoh et al.176 developed a versatile system for the pretreatment and subsequent enzymatic hydrolysis of wheat straw, Miscanthus, and Loblolly pine. This protocol has been used successfully in the treatment of diverse biomasses so that lignins with different structures can be obtained by varying the reaction conditions. Lignin partitioning into the diverse streams can be controlled by altering the pretreatment parameters. Processing with [C2C1im]OAc produces lignins with various molecular masses that can be isolated in the different streams (Fig. 12).
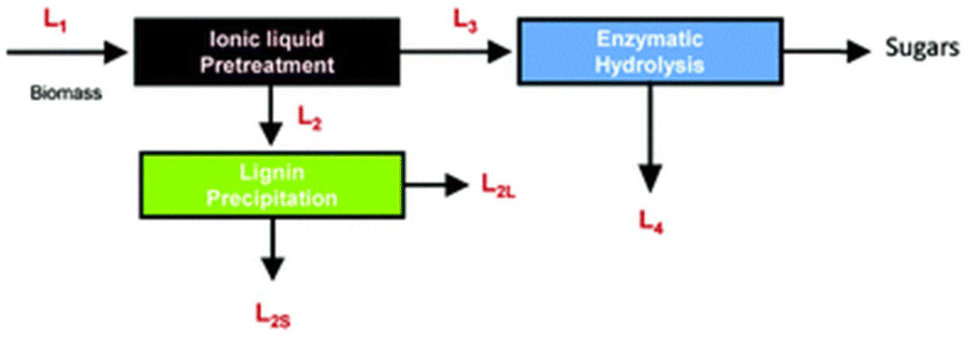 | ||
| Fig. 12 Diagram of extracted lignin from different streams during IL pretreatment and enzymatic hydrolysis. L1: lignin from untreated biomass, L2: solubilized lignin in [C2C1im]OAc, L2S: precipitated solid lignin from L2, L2L: remaining lignin in the supernatant after L2S precipitation, L3: lignin in pretreated biomass, and L4: lignin remaining after enzymatic hydrolysis. Reproduced from ref. 176 with permission from The Royal Society of Chemistry, copyright 2014. | ||
Indeed, at the end of the treatment process, the higher molecular weight lignin is precipitated from IL (L2S) by the addition of 1:1 acetone–water (v/v) while the low molecular weight fraction (L2L) remains in solution. In addition, a high molecular weight and chemically unmodified lignin (L4) is recovered at the end of the enzymatic hydrolysis reaction. Lignin extraction can be tuned by changes to the pretreatment conditions. With milder conditions the amount of lignin removed is low and the recovered lignin is of high molecular weight and has its structure largely unmodified. In contrast, harsher conditions favour the formation of lower molecular mass lignin products in the IL. The major benefit of using [C2C1im]OAc in these protocols is that the formation of lignin condensation products is not observed even under the most extreme conditions.176 Although the cost of [C2C1im]OAc is rather high, the inherent flexibility of the process is an overwhelmingly positive feature, especially in view of bulk-scale production of high value-added products.
To overcome the issue of [C2C1im]OAc's cost drawback, Sun and Xue177 studied the effect of water on the dissolution performance of this IL against two different materials: Alcell lignin and Eucalyptus urophylla lignin (obtained by an enzymatic process). The study shows that, in addition to lowering the cost of solvent medium, the addition of water also has a beneficial effect on the solvent capacity. The optimal amount of water is dependent on the type of lignin. Furthermore, a different lignin removal mechanism was observed. Indeed, for lignin from enzymatic hydrolysis, an increase in the syringyl (S) to guaiacyl (G) ratio (S/G) indicated the preferential breakdown of G-unit lignin. In contrast, for Alcell lignin, the S-unit lignin was easily removed from the mixture, indicating a different mechanism of lignin depolymerisation.177
AILs with cholinium cations and amino acid anions [Ch]AA provide a viable alternative to [C2C1im]OAc. Since their discovery, there has been much attention paid to their use for the treatment of biomass.178 The main advantage of these ILs is related to their fully biobased nature and associated biodegradability.179 The appeal of using biobased solvents for the treatment of biomass immediately fascinated researchers. [Ch]AA ILs have been successfully employed in the treatment of various biomasses such as rice straw, sugarcane bagasse, eucalyptus, and pine. Amongst many, cholinium arginate and cholinium lysinate ([Ch]Arg and [Ch]Lys, respectively) were found to be the best-performing ILs.180 [Ch]Lys was also used for the pretreatment of switchgrass and eucalyptus as well as pine, and the respective lignins obtained were analysed in depth.181 Pine delignification was found to be the most difficult process in comparison with eucalyptus and switchgrass, as can be seen from the amount of residual lignin after the hydrolysis process (Fig. 13).
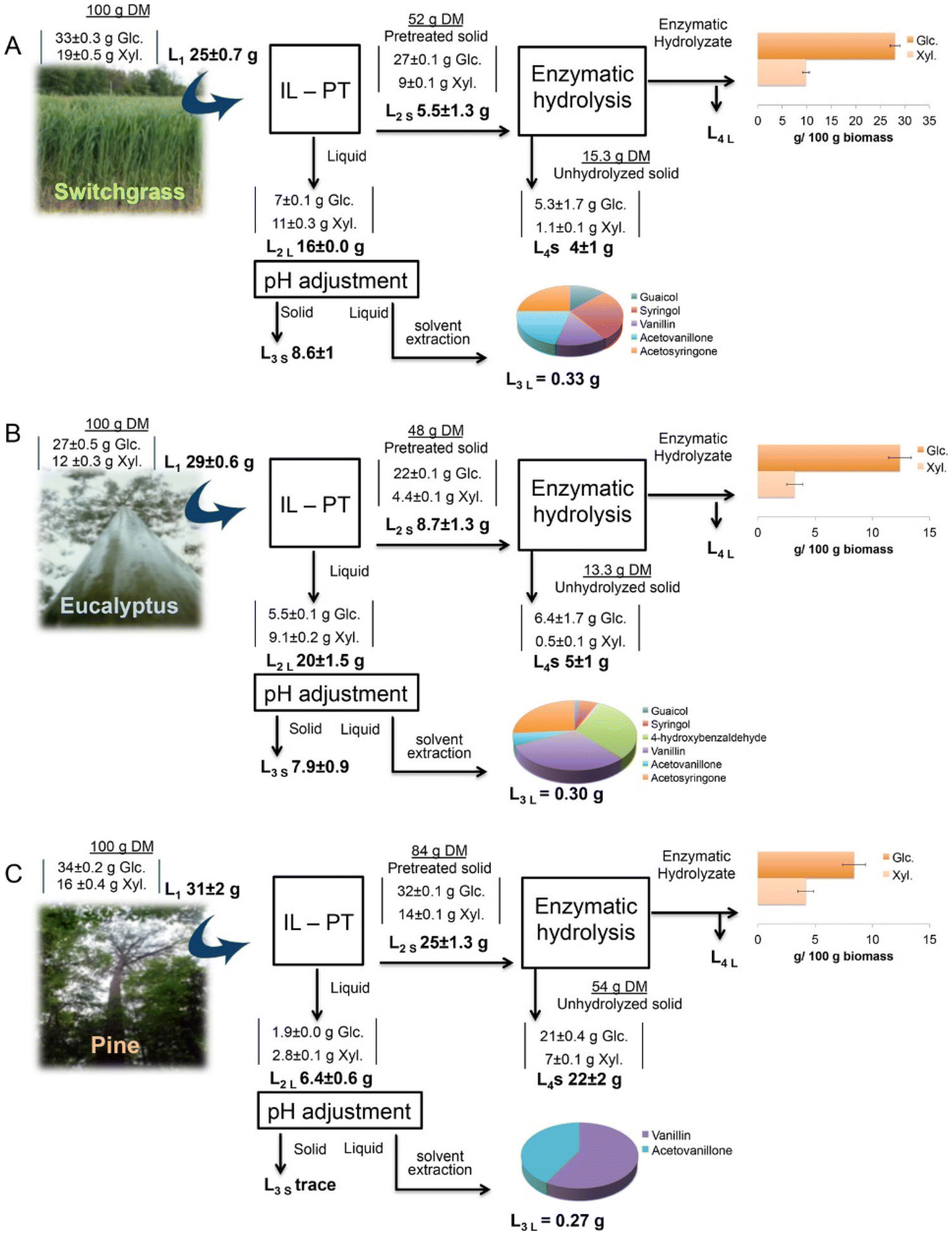 | ||
| Fig. 13 Schematic representation of the pretreatment of switchgrass (A), hardwood (B), and softwood (C) with [Ch]Lys at 140 °C for 1 h. Reprinted with permission from Dutta et al.181 Copyright 2018 American Chemical Society. | ||
Furthermore, from the analysis of the soluble fraction present in the ILs, the pine treatment shows a limited variety of hydrolysis compounds. Subsequently, in a study conducted on Kraft lignin treated with different [Ch]AA, [Ch]Lys was found to be the gentler option in terms of depolymerisation and of reduction of β-O-4′ as well as β–β′ bonds.182
As mentioned in the previous section, the use of PILs in pretreatment processes is currently the most promising in the development of Ionosolv-type industrial processes. For this reason, study of the effects of this pretreatment on lignin is prominent in the recent literature.
Brandt et al.183 described the mechanism of lignin solubilisation through the use of 1-butylimidazolium hydrogen sulfate [HC4im]HSO4 as solvent in a Ionosolv process. This particular PIL breaks lignin–polysaccharide linkages and depolymerizes the lignin through the cleavage of glycosidic, ester and β-O-4 ether bonds. The partially depolymerized lignin then becomes soluble and is separated from the rest of the biomass. By increasing the pretreatment time, the lignin fragments tend to condense through the formation of C–C bonds. The process is flexible with the possibility of obtaining low molecular weight aromatic molecules or alternatively high molecular weight lignin with a low sulfur content.
The Ionosolv BioFlex process is predominantly based on an aqueous solution of [N2220]HSO4 at 80 wt%. Weigand et al.184 analysed the lignin obtained from BioFlex pretreatment of willow hardwood as a function of the severity of the process. In particular, the effect of temperature (120–170 °C), time (0.3–8 h) and acid/base ratio (1.02 and 0.98) was studied. The acid/base ratio of PILs in solution appears to have little impact on the amount of linkages present in the recovered lignin. At low temperatures (150 °C), the prevailing linkage remains the β-O-4′ ether even after 8 h of treatment, while increasing the temperature causes the abundance of this linkage to decrease quickly (Fig. 14).184
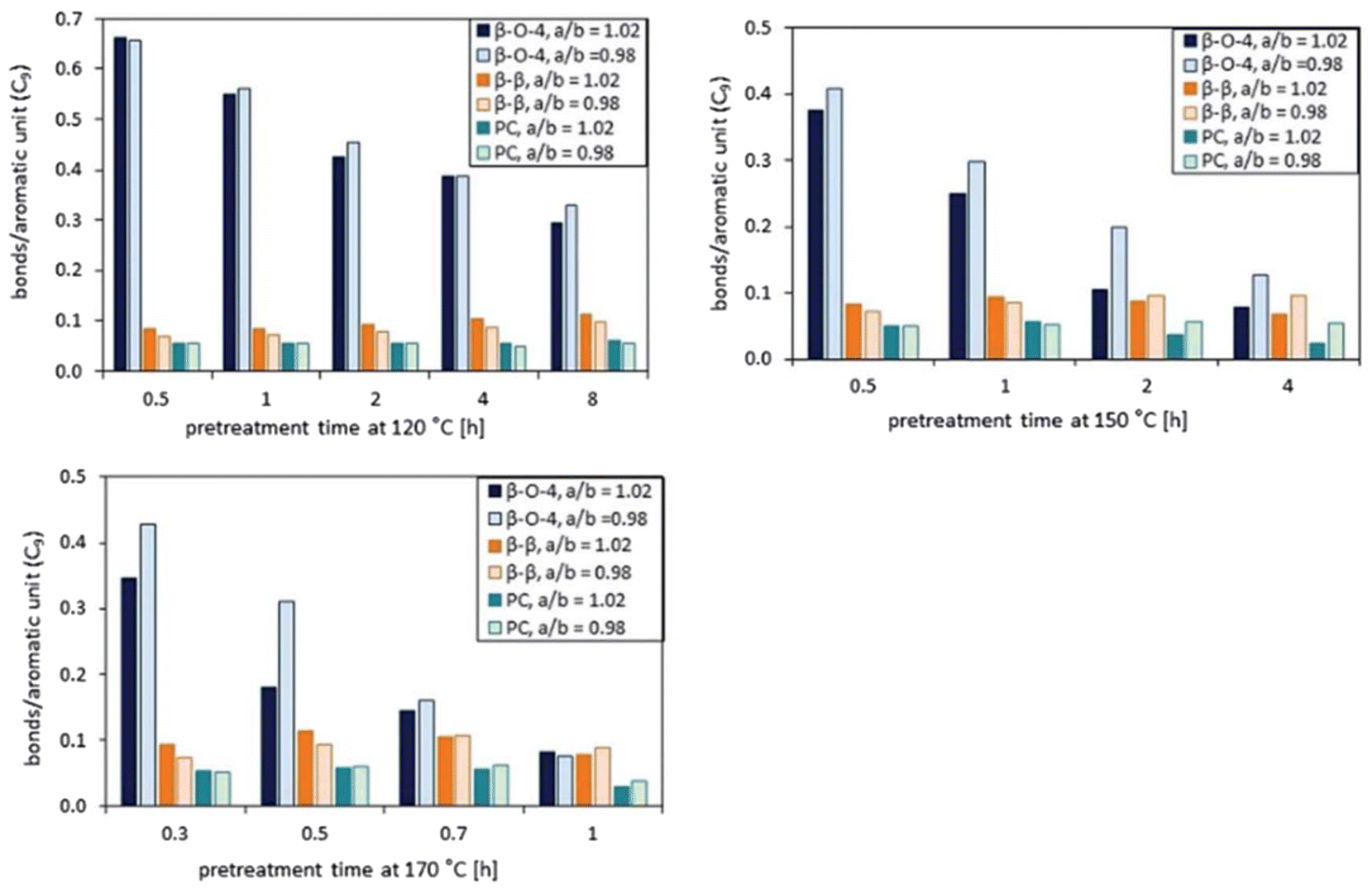 | ||
| Fig. 14 Analysis of linkages (β-O-4′ ether linkage, β–β′ (resinol) linkage and phenylcoumaran (PC) linkage) in lignin isolated after pretreatment with [N2220]HSO4 at different acid/base ratios (a/b = 1.02 and 0.98) and different temperatures. Top left: 120 °C, top right: 150 °C and bottom left: 170 °C. Reproduced from ref. 184 with permission from The Royal Society of Chemistry, copyright 2017. | ||
An acid/base ratio of 0.98 tends to preserve the β-O-4 bond, especially for more harsh pretreatment conditions, long reaction times or high temperatures. The molar weight (MW) of the recovered lignin varied depending on the conditions employed: a strong decrease was detected with increasing pretreatment times, from approx. 25000 g mol−1 after 30 min to approx. 13000 g mol−1 after 2 hours at 120 °C. Under these conditions, differences in the acid/base ratio (1.02 and 0.98) had no significant effects. In contrast, at 170 °C and using short pretreatment times, a significant increase of MW was observed with the 0.98 acid/base ratio medium, leading to a higher molecular weight lignin if compared with the 1.02 ratio.184 This study highlighted how the process is highly tunable, although recondensation of the recovered lignin can easily occur, in contrast to what was observed for [C2C1im]OAc.
Recently, the performance of the Ionosolv process based on 1-methylimidazolium chloride ([HC1im]Cl) and of the Organosolv process based on a 60% (w/w) ethanol/water mixture were compared.185 Although the Ionosolv process was carried out under milder conditions than the Organosolv process (120–150 °C vs. 185–215 °C, respectively) the former was found to be more effective in terms of lignin removal. On the other hand, the lignin recovered after the Ionosolv process showed a high number of C–C bonds indicating a high degree of recondensation. In contrast, the lignin produced by the Organosolv process was characterised by a higher content of C–O linkages with no signs of recondensation.185
A valuable solution to lignin condensation problems is the mixed Ionosolv–Organosolv process based on the N,N-dimethyl-N-butylammonium hydrogen sulfate [N1140]HSO4–ethanol mixture.186 The [N1140]HSO4 system proved to be more efficient than [N2220]HSO4 in the treatment of softwood.187 This process allows for high delignification yields, typical of the Ionosolv process, while at the same time obtaining high-quality lignin. Indeed, studies conducted on the yielded lignin showed that the presence of alcohol induced α-alkoxylation during lignin fractionation and turned β-O-4′ ether units into α-alkoxylated ether units. This modification not only increased the solubility of lignin, but also inhibited the onset of condensation reactions. This hybrid process makes it possible to obtain both high-quality cellulosic and lignin material that is optimal for a biorefinery process.186
In order to compare the three main classes of ILs capable of extracting lignin, Dutta et al.169 reported a comparative study focused on dissolving Kraft lignin with [C2C1im]OAc, [N2220]HSO4, and [Ch]Lys. The [C2C1im]OAc pretreatment showed little change in β-aryl ether content together with a strong decrease in molecular weight due to reduced recondensation phenomena. In contrast, PIL [N2220]HSO4 showed the highest degree of β-O-4′ chain breakage and the highest number of dehydration and recondensation reactions, resulting in the smallest decrease in molecular weight. Additionally, [Ch]Lys showed an intermediate effect on the treated lignin, with a moderate depolymerization, dehydration, and relative recondensation observed on the produced material.169
The type of IL also influences the composition of the soluble fraction. An analysis of the composition after treatment at 140 °C for 1 h showed that [C2C1im]OAc exhibited the highest yield of monomeric lignin depolymerization products. Some products such as vanillin and acetovanillinone, which are commonly present in the [Ch]Lys and [N2220]HSO4 fractions, were only observed in low concentration (Fig. 15). Under severe pretreatment conditions (160 °C for 1, 2, and 3 h), treatment with [Ch]Lys produced mainly guaiacol, while treatment with [N2220]HSO4 produced guaiacylacetone as the main product. Finally, pretreatment with [C2C1im]OAc furnished almost equivalent amounts of guaiacylacetone and guaiacol.169
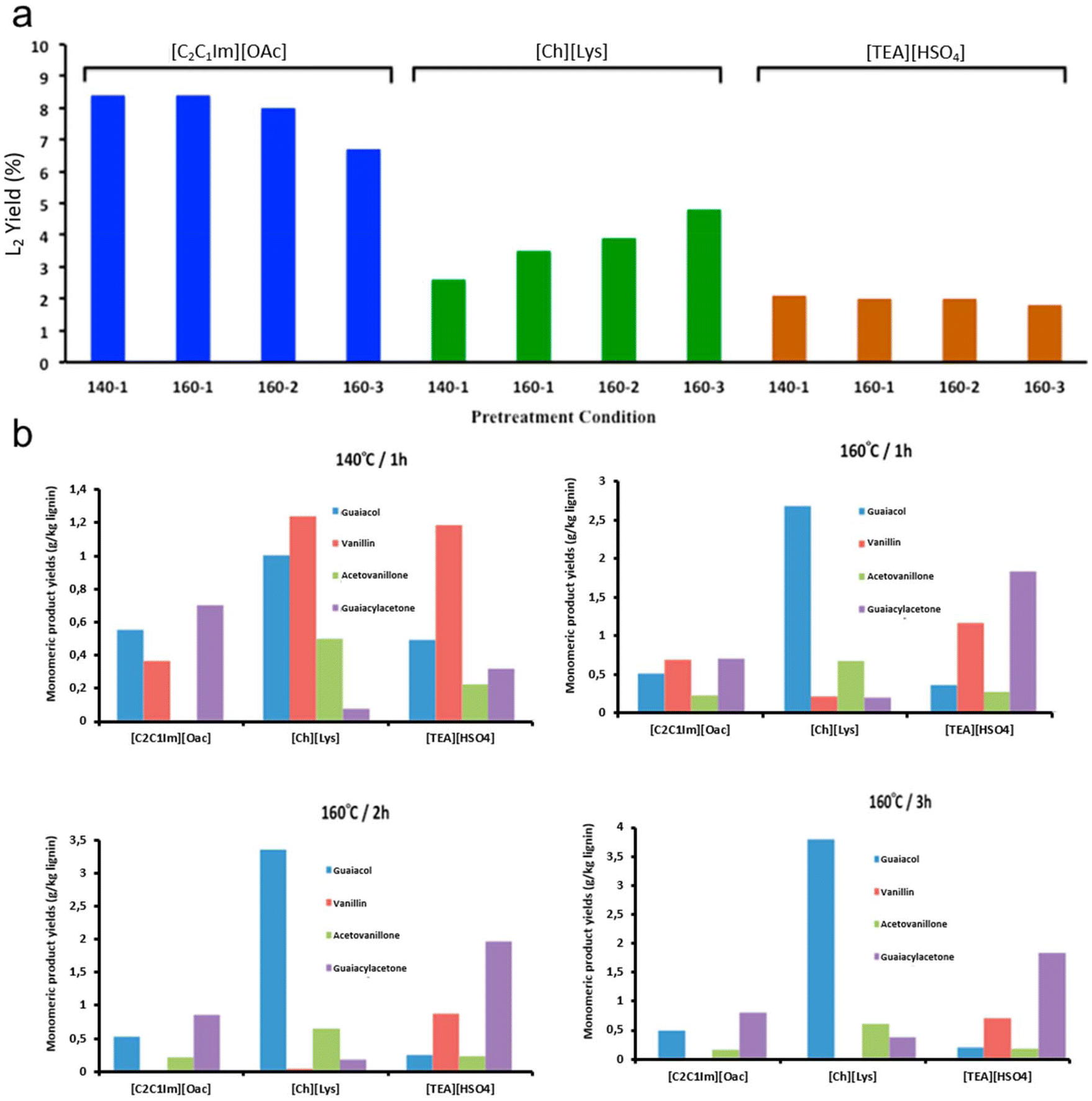 | ||
| Fig. 15 (a) Overall yield (%) of soluble depolymerized lignin products as a function of pretreatment conditions and IL categories. (b) Composition of lignin depolymerization fraction expressed in g kg−1 lignin as a function of pretreatment conditions and IL categories. Adapted with permission from Dutta et al.169 Copyright 2017 American Chemical Society. | ||
DESs are certainly a viable alternative to ILs for the dissolution of lignin. This is reflected by a large number of publications where DESs are used profitably in the extraction of lignin from biomass. Recently, Yuan et al. reported a valuable review on structural changes of lignin after extraction with different DESs.188 Amongst this body of work, the systems that use metals as additives/catalysts,189,190 ILs or synthetic ammonium salts191,192 as HBAs components are of minor interest. Indeed, the use of these materials greatly reduces the usually acknowledged benefits of DESs with a frequent increase in costs.
As mentioned in the previous section, DESs with acidic HBDs are the most studied and those that showed the best lignin removal performance (Fig. 16).
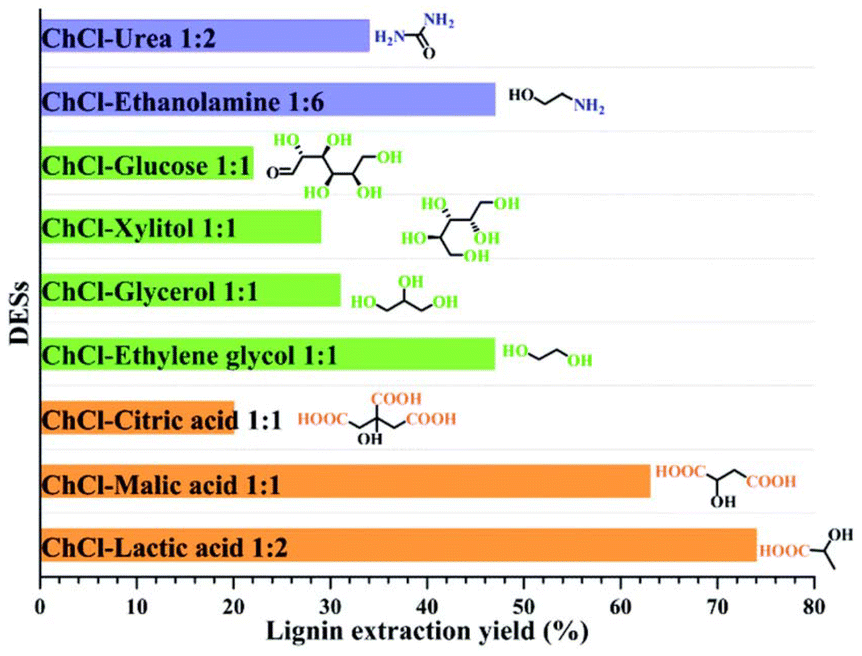 | ||
| Fig. 16 Effect of HBD type on lignin extraction yield. Reproduced from ref. 188 with permission from The Royal Society of Chemistry, copyright 2020. | ||
From the perspective of an integrated biorefinery process, the efficiency of lignin removal cannot any longer be the only parameter taken into consideration. In the evaluation of DESs as tools in biomass valorization, the effect of the pretreatment on lignin structure is also of primary importance. In a seminal paper, Alvarez-Vasco et al.193 described how the quality of lignin extracted from hardwood (poplar) and softwood (D. fir) changes depending on which DES is employed in the process. The system investigated was based on ChCl combined with a selection of four HBDs: acetic acid, LA, levulinic acid and Gly. Since then, numerous studies have been carried out on the mechanism of lignin dissolution in acidic, basic and neutral environments. The findings described in the literature reveal that basic DESs lead to a less pure and highly recondensed lignin with small particle size compared with the material recovered with acidic DESs.194 Among the acidic HBDs, LA is certainly the most commonly used, and its effect on lignin structure was evaluated under different conditions and on different biomasses. The techno-economic benefits of this medium have already been described and highlighted.172 Shen et al.195 studied the extraction capacity of ChCl:LA (1:10) in the temperature range 90–130 °C and with a reaction time of 6 h. Under the optimised conditions of 110 °C for 3 h, the delignification yield increased to 80% and the recovery of the removed lignin to 44%. When employing this protocol, the obtained lignin showed good purity, homogeneous molecular weight and the β-O-4′ and β–β linkages were preserved. These results were partly confirmed by Shen et al.196 in a following work. Indeed, an in-depth analysis of the recovered lignin provided interesting data not only on the depolymerization reaction products but also on the structural modification caused by dehydration and subsequent condensation reactions, consistent with a mechanism similar to that previously observed with [N2220]HSO4. In addition, the LA component of the DES was found to react with the –OH groups of lignin, forming the corresponding esters (Fig. 17).
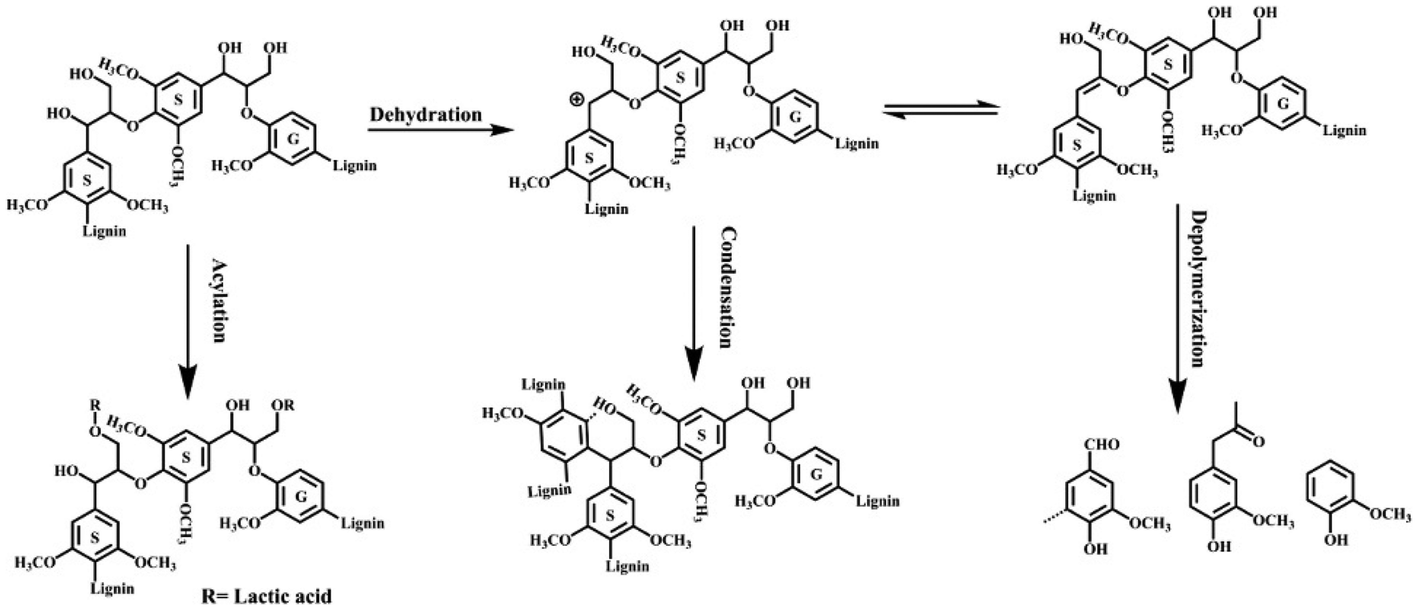 | ||
| Fig. 17 Reaction pathways during the pretreatment process with ChCl:LA (1:10). Reprinted with permission from Shen et al.196 Copyright 2020 American Chemical Society. | ||
Some of the same authors of the previous reference confirmed the presence of γ-acetylated groups in the regenerated alkali lignin (AL) isolated from poplar also at low temperatures.197 However, the presence of the esters was not observed after treatment with cholinium chloride:oxalic acid (ChCl:OA, 1:1). This is attributed to the larger steric hindrance of OA compared with LA. However, treatment with ChCl:OA (1:1) seems to favor condensation reactions. In addition, the presence of S- and G-derived diketones on the depolymerized lignin fraction (lignin oil) in ChCl:LA (1:10) was observed for the first time. The mechanism proposed to account for this reactivity pattern involves the cleavage of β-O-4′ linkages, the removal of Cα hydroxy groups to form benzylic carbocations and finally oxidation of the alcoholic groups to obtain Cα ketones.197
The effect of ChCl:LA (1:10) was compared with that of propionic acid:U (PA:U, 2:1) and cholinium chloride:p-toluenesulfonic acid (ChCl:pTSA, 1:1) using 2-phenoxy-1-phenylethanol (PPE) as a model substrate for the more complex lignin.198 The tests confirmed that ChCl:LA was capable of breaking down PPE and at the same time producing PPE lactic acid ester and lactic acid oligomers. In contrast, PA:U (2:1) was not able to degrade the PPE bonds despite it being able to dissolve lignin. Finally, the high acidity of ChCl:pTSA (1:1) made it the most effective system in breaking down PPE, leading, however, to the condensation of cleaved products with prolonged reaction times. This result highlights the important role played by the acidity of the HBD partner, both in the dissolution and in the occurrence of side reactions of lignin. In the same work, it was observed that the addition of water decreased the cleavage of PPE bonds and strongly suppressed undesired side reactions.198
The presence and the nature of the halide anion also play an important role in the dissolution process. da Costa Lopes et al. evaluated the properties of cholinium bromide (ChBr) by replacing ChCl in the ChCl:pTSA system. The kinetic study performed with ChCl:pTSA (1:1) and ChBr:pTSA (1:1) demonstrated that PPE-halogenide intermediates are important in increasing the rate of cleavage reactions. The results obtained are in line with DFT calculations attributing to the β-O-4 ether bond cleavage a pivotal role.198 Previously, the role of the chloride anion had been investigated in an industrial application concerning milled wood from Eucalyptus globulus, in this case by following lignin cleaving reactions carried out in a ChCl:LA (1:10).199 The DES was compared with the NaCl:LA mixture and showed similar results in terms of lignin dissolution.199 This behaviour highlights the importance of the chloride anion in the dissolution process but raises at the same time some concern as to whether the cholinium portion is actually needed. However, further studies are certainly required to further elucidate this aspect.
As noted in the previous section, the use of ChCl:LA has serious limitations in terms of recyclability. In addition to the cholinium ester formation, lactide formation and LA oligomerization as well as formation of LA esters on both the cellulosic and lignin portions are all encountered issues. These side reactions actually lead to the consumption of the LA HBD component, raising serious doubts as to the real applicability of this DES in the pretreatment of lignocellulosic biomass. Hence, the identification of DESs alternative to ChCl:LA and with fewer limitations is of the highest interest, especially in view of the full utilization of lignin in an economically sustainable manner.
The use of low melting mixtures with boric acid (BA) as HBD component is a viable alternative. Cholinium chloride:boric acid (ChCl:BA) showed good delignification performance of softwood sawdust at 90 °C while avoiding condensation of the solubilized lignin.200 The observed results are attributed to the formation of a cyclic ester between boric acid and the hydroxy functionalities situated at position α and γ. Similarly, the DES composed of ChCl, BA, and polyethylene glycol-200 (PEG-200) in a 1:1:1.5 ratio showed excellent lignin and hemicellulose dissolution capabilities when used on wheat straw biomass.201 The recovered lignin displayed a low molecular weight, a narrow polydispersity index and good retention of the β-O-4′, β–β′, β-5′ bonds present in pristine lignin. Another possible option is the use of DESs with polyalcoholic HBD partners which would overcome the problems associated with the use of carboxylic acids. However, their effectiveness is reduced due to the reduced acidity. For this reason, Chen et al.202 employed cholinium chloride:ethylene glycol (ChCl:EG, 1:2 and 3:2) with the addition of 0.5 or 1.0 wt% H2SO4 for switchgrass fractionation. The lignin properties such as molecular weight, number of ether bonds as well as degree of condensation were modulated by controlling the DES composition and the pretreatment conditions. In general, the obtained lignin exhibited low condensation degree, well-preserved β-O-4′ linkages, and high volatility similar to that of cellulolytic enzyme lignin (CEL). Similar results were obtained using ChCl:Gly (1:2) with 0.9 wt% H2SO4.203 Although the lignin obtained is undoubtedly of excellent quality, the use of H2SO4 represents a limitation, due to corrosion issues, that can be eliminated by mechanical means. As described in the previous section, ChCl:Gly (1:2) was successfully used in the pretreatment of sorghum bagasse using co-rotating twin-screw extruders. The analysis of the recovered lignin after pretreatment with two different biomass loadings (30 and 50 wt%) showed a decrease in molecular weight by 50% and a slight increase in the polydispersity index.204 The decrease in the ratio of S/G units suggested that some condensation reactions, albeit moderately, occurred even during the extrusion process. Furthermore, the data highlighted the decrease of H units in the lignin after extrusion, while analysis of the ether linkages showed that only limited cleavage occurred during the extrusion that followed pretreatment. The quality of the recovered lignin, together with the excellent delignification performance, make this pretreatment one of the most innovative and interesting reported in the literature.
A comparison of ILs and DESs in lignin solubilisation processes does not reveal a clear winner. The choice of the most suitable system depends on the biomass of interest and the type of lignin to be obtained. The use of DESs requires on average longer pretreatment times (6 h vs. 1–2 h for DESs and ILs, respectively) while a wide variety of different lignins can be obtained with both media. The only direct comparison between ILs and DESs present in the literature is between ChCl:LA (1:9) and 1-allyl-3-methylimidazolium chloride [AC1im]Cl.205 However, the unfortunate choice of a poorly efficient IL for lignin dissolution such as [AC1im]Cl makes this analysis undeservedly unfair for ILs. Anyway, even the comparison of the best-performing ILs and DESs, although of interest, would remain confined to the specific case selected, as each process must be evaluated according to many parameters, and possible effects on other biomass components must also be considered.
5.4. Cellulose purification
Full utilisation of cellulose relies on its dissolution, processing and transformation into new materials or chemicals. Transformation of cellulose into low molecular weight products has been reviewed elsewhere and will only be partially discussed in this critical review.206–208 Cellulose solubilisation is at the basis of many of its industrial manipulations. Indeed, unlike commonly used polymers, this polysaccharide cannot be melted prior to processing. For this reason, interest in cellulose solubilisation has been longstanding and many unsolved challenges still remain in this area, which hamper the use of this material. One well-known process for dissolving cellulose is based on the LiCl/N,N-dimethylacetamide (DMAc) system,209 although many other systems have been developed such as molten salt hydrates, aqueous NaOH,210 NaOH/U211 and lithium hydroxide (LiOH)/U solutions.212 Although the LiCl/DMAc system is capable of dissolving up to 15 wt% of cellulose, it requires high dissolution times and temperatures (up to 150 °C and 48 h) and is extremely water-sensitive.213 On top of these aspects, the reduced availability of lithium due to its limited nature and increasing use in battery manufacture has led to a sharp increase in price.214 Base/U systems can also solubilise up to 4 wt% of cellulose at −10 °C and in the presence of water.215 However, the need to work with cryogenic systems increases energy demand and plant costs considerably.In the industrial context, the most used process is the so-called Lyocell process, which uses N-methylmorpholine-N-oxide monohydrate (NMMO·H2O) as the solvent and is capable of solubilising up to 30 wt% of cellulose (depending on the type of cellulose used).216,217 However, the process shows significant limitations due to the occurrence of side reactions. These phenomena have detrimental effects on the structure of the cellulose with consequent loss of mechanical properties and a temporary or permanent discoloration of the fibers. In parallel, a pronounced decomposition of NMMO is observed, leading to an increased need for stabilisers like isopropyl gallate.218
As a way to respond to the shortages of the technologies described above, ILs were proposed as an alternative medium for performing cellulose-based transformations. Since 2002, after the first work by Swatloski et al.219 on cellulose dissolution with ILs, the number of publications and patents in this area has grown exponentially and the field has matured very quickly.216,220,221
Nowadays, there are three pilot-scale processes and one commercial-scale process which use ILs for cellulose transformation. Natural Fiber Welding, Inc. has developed a commercial-scale process that uses [C2C1im]OAc to turn natural fibers into welded materials.222 Thanks to this technology, it is possible to recycle and even upcycle existing natural materials to create new high-performance textiles.
Metsä Spring, a corporate venturing arm of the Metsä Group, is building an IL-based, closed-loop pilot plant capable of producing 40 kg of fiber per hour for a total budget of EUR 40 million.223 Similarly, the German Institutes of Textile and fiber Research Denkendorf (DITF) has developed a spinning process, called HighPerCell®, for the production of hemp-based cellulose fibers. In the process, the cellulose pulp from hemp is dissolved in ILs and then spun in a special wet spinning system.224 Sticking to cellulose fibers, Aalto University in Finland has developed the closed-loop Ioncell® process, a new dry-jet wet spinning process for cellulose fibers production based on a PIL, 1,5-diaza-bicyclo[4.3.0]non-5-enium [DBNH]OAc (Fig. 18).225,226
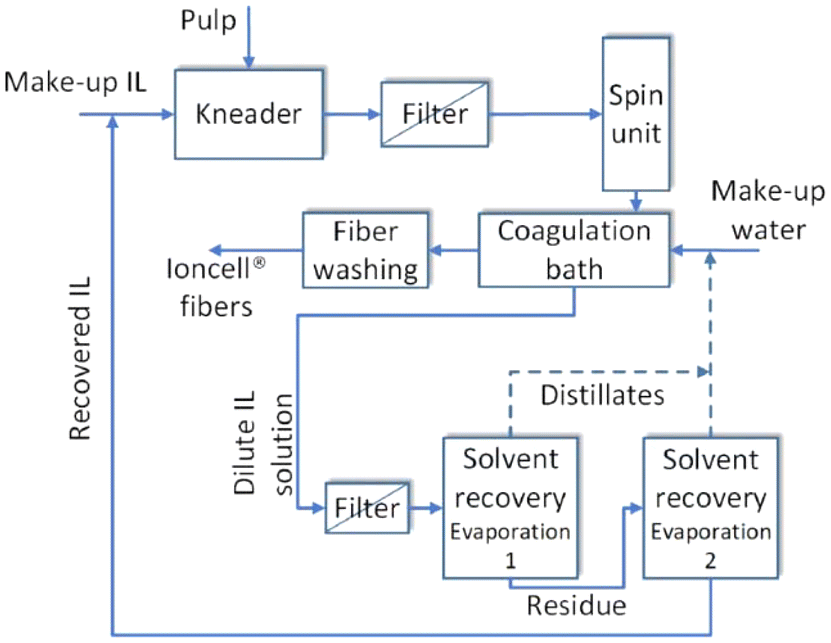 | ||
| Fig. 18 Scheme of the Ioncell-Lyocell process revealing dissolution, fiber spinning, and solvent recovery steps. Reprinted with permission from Elsayed et al.227 Copyright 2020 American Chemical Society. | ||
The use of PILs results in highly oriented cellulosic fibers with a higher tenacity than commercial viscose and NMMO-based Lyocell fibers.225 In 2020, a pilot plant capable of producing 10 kg of fiber per day was launched and is estimated to reach the commercial scale by 2025.228 All these processes have a Technology Readiness Level (TRL)229 in the range from 7 to 9, clearly demonstrating how this field of application is technologically advanced.223
In the last twenty years, a huge number of ILs capable of solubilising cellulose have been developed by varying the structure of the cation and the type of anion.216,230 Alongside, the mechanism of dissolution has been studied and the effects of the ILs’ structural changes on their dissolving power have been documented.221,230,231 A valuable tool for describing and predicting the solvent ability of ILs towards cellulose is based on the Kamlet–Taft parameters. At first, the solvent capacity was attributed mainly to the HBA strength of the anion, hence to the β parameter. In particular, it was reasoned that ILs with β values greater than 0.8 would be the most suitable for dissolving cellulose (acetate and chloride anions).232 Later, the role of the cation was reconsidered thanks to some key observations concerning the behaviour of imidazolium-based ILs. In these systems, the IL establishes hydrogen bonds with cellulose hydroxyl groups via the C-2 of the imidazole ring and at the same time interacts with the hydrophobic portions of the cellulose chains.233 Consequently, ILs with high α-values, which are typical for imidazole cations without electronegative groups (oxygen and/or nitrogen) and poorly hindered (short alkyl chains) around the C-2 position, perform best in cellulose dissolution.234 The effect of the π* parameter is less pronounced than that of α and β parameters. A high π* value denotes a high cation–anion interaction, thus reducing the possibility of ions to interact with the hydroxyl groups of cellulose. In summary, high α and β values and low π* values promote cellulose dissolution.235 In this context, Hauru et al.236 introduced the concept of net basicity, β–α. This parameter more reliably describes both the dissolution and the regeneration of cellulose by ILs as well as by NMMO·H2O. By plotting the β–α and β parameters of neat ILs, the ranges 0.35 < β–α < 0.9 and 0.80 < β < 1.20 were identified as the optimal ranges for cellulose dissolution. This type of correlation between Kamlet–Taft parameters and solvent capacity proves effective not only for neat ILs, but also for their mixtures with polar co-solvents.236 Indeed, polar molecular solvents are widely used in combination with ILs in cellulose dissolution processes. The use of co-solvents expands the portfolio of possible solvent systems for cellulose, lowering the overall cost of the medium and at the same time modulating the viscosity of ILs and IL/cellulose solutions. In this framework, Rinaldi et al.237 introduced the concept of organic electrolyte solutions (OES), composed of mixtures of ionic species and molecular solvents, and showed their ability to instantaneously dissolve cellulose. In particular, low mole fractions of [C2C1im]Cl or [C2C1im]OAc (χIL = 0.08–0.4) in polar solvents (e.g. dimethyl sulfoxide (DMSO), sulfolane, 1,3-dimethyl-2-imidazolidinone, and γ-valerolactone (GVL)) were able to solubilize up to 10 wt% of cellulose in far less time (few minutes) than when used in neat form.237 As OES, even ILs not capable of dissolving cellulose in pure form become potential new options to be tested.238–240 In addition, Holding et al.241 showed how acetate amphiphilic phosphonium ILs, which are poor solvents for the polysaccharide, become suitable solvents for cellulose when mixed with DMSO, dissolving up to 19 wt% cellulose. Furthermore, an additional benefit of this latter series of ILs stems from their poor solubility in water, which allows for their recovery at the end of the cellulose water-based coagulation process, with a clear energy and material-saving advantage.239
PILs are also a valuable option for dissolving cellulose, as they can be easily and inexpensively prepared (please see section 5.7.2). Moreover, one of the most promising processes in the scale-up phase is based on amidinium PILs. The originating Ioncell process employs a [DBNH]OAc IL, which performs very well as it can dissolve up to 17 wt% of cellulose at temperatures around 80 °C in a few minutes.225 [DBNH]OAc possesses a net basicity β–α of 0.57 and a β of 1.1, which makes for an excellent cellulose solvent.242 Similar net basicity parameters are also observed for other guanidium and amidinium acetate PILs, β–α of 0.75 for 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-enium acetate ([mTBDH]OAc) and β–α of 0.41 for 1,5-diaza-bicyclo[4.3.0]undec-5-enium acetate ([DBUH]OAc), all effective solvents for cellulose.243 Unfortunately, many works promoting the use of ILs in processing tend to underestimate potential issues arising from their use in a continuous process. Although PILs have a lower cost than AILs, a key feature for their use in a commercial setting is the ability to efficiently undergo recycling. However, the latter process is sometimes hindered by the presence of by-products in the raw pulp, while this accumulation of impurities can lower the solvent capacity and change the viscosity of the solution. For example, in a continuous spinning process such as Ioncell, these aspects result in a continuous modification of the operating conditions in an attempt to keep the properties of the resulting fibres constant.227 Furthermore, for PILs the recovery process from the coagulation bath is hampered by the fact that the [DBNH] and the [mTBDH] acetate ILs form with water non-stoichiometric azeotropic mixtures with acid–base ratios of 5:3 (acetic acid:[DBN]) and 3:2 (acetic acid:[mTBD]), respectively.244 In addition, amidinium and guanidinium ILs have been shown to be subject to hydrolysis when used in media with water contents higher than 20 wt%.243,245 The presence of varying amounts of these impurities reduces the dissolving power of ILs and is the main hurdle to the industrial development of this process. Based on these considerations, the recyclability of [DBNH]OAc from aqueous media was analysed and an average recovery rate of 95.6 wt% was observed.246 With water content of 4.1–5.4 wt% (6.0–13.6 mol%) per cycle, up to a maximum of 41.5–54.9 mol% of hydrolysis product was observed. The recycling of the IL did not change the chemical composition or degree of polymerisation of the recovered cellulose, but the colour of the regenerated material gradually darkened as the number of recycling cycles increased.246 Replacing [DBNH]OAc with [mTBDH]OAc allowed the number of recycles to be increased to five without a refill of fresh IL.242 This is attributable to the higher stability towards hydrolysis of mTBD compared with DBN.243,245 In addition, the higher basicity of mTBD compared with DBN (pKa = 25.4 and 23.4, respectively, in acetonitrile)227 leads to a higher ΔpKa value and thus to an increased thermal stability of the PIL.247 From the perspective of increasing the hydrolytic stability of [mTBDH]OAc, it was observed that small stoichiometric excesses of acetic acid significantly reduced the kinetics of hydrolysis.243 At the same time, the effect of this addition on the ability to dissolve 12 wt% cellulose was tested for [mTBDH]OAc.227 In a direct dissolution comparison with 3 wt% of water, [mTBDH]OAc could withstand the presence of 20 wt% of hydrolysis products and an acid/base ratio of 1.3, whereas the dissolution capacity of [DBNH]OAc was already compromised at 5 wt% of hydrolysis products and an acid/base ratio of 1.1 (Fig. 19).227
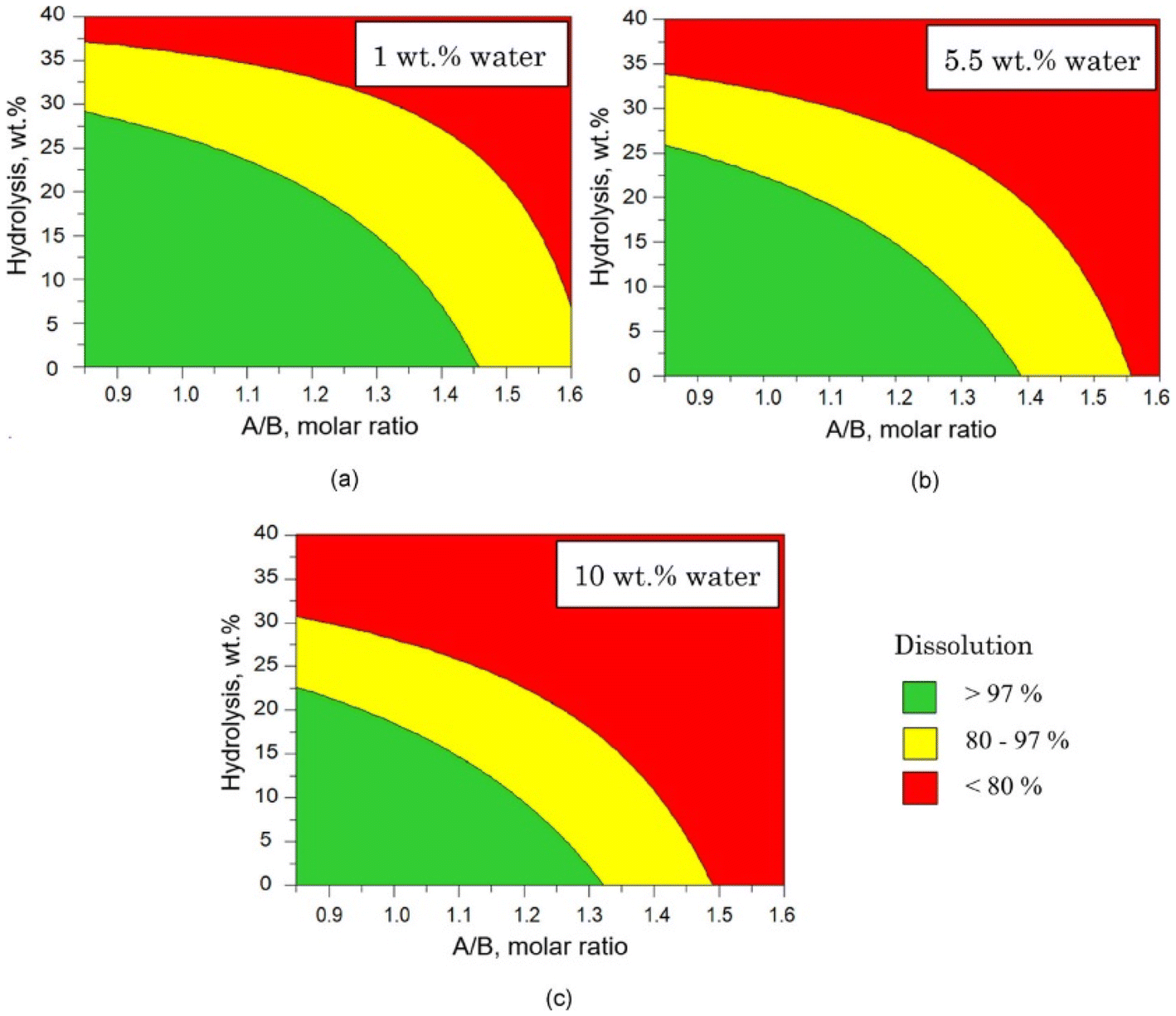 | ||
| Fig. 19 Dissolution contours for 13 wt% cellulose in [mTBDH]OAc in different A/B ratios and hydrolysis product compositions at (a) 1 wt% water, (b) 5.5 wt% water, and (c) 10 wt% water. Reprinted with permission from Elsayed et al.227 Copyright 2020 American Chemical Society. | ||
These data show a higher tolerance of [mTBDH]OAc compared with [DBNH]OAc towards impurities, making the process based on [mTBDH]OAc much more robust in view of its commercialisation.
However, a question arises concerning the effective nature of this solvent system. Indeed, from a formal point of view it is no longer a PIL, but a mixture of mTBD:acetic acid in a molar ratio of 1:1.3. This type of solvent system together with the OESs, described above, could be considered as the “transition zone” between ILs and DESs. It is worth stressing that the potential “deep” nature of the above mixtures has never been verified. Likewise, there are systems capable of dissolving cellulose and defined as “DES” in the literature whose phase diagrams have not been reported, nor do they possess the often vaunted DES properties of low toxicity, low volatility, and ease of preparation. This is the case, for instance, of “DES” systems whose HBAs are imidazole zwitterions248 or allylammonium chlorides,249 which require a preparation process similar to that of ILs. Another controversial example is represented by the DBU/methylthiourea mixture reported by Fu et al.,250 a system capable of dissolving cellulose and whose dissolution mechanism was investigated by quantum mechanical calculations. Apart from the nature of the mixture, which was not investigated, it is important to emphasise that both the strong basicity of DBU and even more the toxicity of methylthiourea are far from the concept of sustainable solvent design.
The first example of cellulose dissolution with solvents with recognised DES nature was reported by Sharma et al. and dates back to 2013.251 In this work, different combinations of ChCl, ChBr, chlorocholinium chloride (ClChCl), and betaine hydrochloride (BetHCl) mixed with U, Gly, and EG in a 1:2 molar ratio were tested in cellulose dissolution processes using microwaves and ultrasound as alternative heating sources. The study showed that DES ChCl:U (1:2) was the best-performing system as it dissolved up to 8 wt% of microcrystalline cellulose (MW 3.12 × 105 Da) at 100 °C.251 The other U-based DESs, ClChCl:U (1:2) and BetHCl:U (1:4), were also able to dissolve cellulose although in reduced amounts, while performances of DESs with alcoholic HBDs were reported to be much less promising.251 A further DES capable of dissolving cellulose was cholinium chloride:imidazole (Im) ChCl:Im (3:7), which was capable of dissolving 2.48 wt% of cellulose in 1 h.252 Conversely, the solvent properties of DESs with an acidic HBD such as OA, citric acid and malic acid (MA) were proved not satisfactory and the effect of the acidity on the structure of the treated cellulose was often even not investigated.253 However, to confirm the potential of these materials, it is worth stressing that acid-based DESs are widely used with high efficiency in the preparation of nanocellulose by erosion of the amorphous part of the cellulose.254–256
The application of U-based DESs as media for cellulose processing has been studied by different research groups.251,257,258 However, these efforts resulted in conflicting data as some papers mention their reaction media as solutions,251,258 while in the majority of cases the obtained mixtures are described as swollen cellulose suspensions.259,260 These discrepancies are likely due to the different types of cellulose processed in terms of molecular weight and crystallinity. However, a detailed analysis of the literature shows that cellulose often undergoes preliminary treatments before being subjected to DES, such as sonication in saturated CaCl2 solutions252 or exposure to solution containing Na+ ions and subsequent washing with acetone.260 These pretreatments often place a significant burden on the sustainability of the whole protocol and partly compromise the benefits of using DESs.
Conversely, an application of DESs in cellulose processing that can lead to interesting industrial developments was proposed by Nguyen et al.261 The NADES ChCl:MA was used as an additive in the Lyocell process with NMMO·H2O as the main solvent. In the study, the effect of different molar ratios of the NADES components (1:1, 1.5:1, 2:1, 1:1.5, 1:2) and of different additive percentages in NMMO·H2O were investigated. The mixture consisting of NMMO·H2O + 15 wt% ChCl:MA (1.5:1) was found to be the best-performing system as it showed a higher dissolving power than the pure solvent (12 wt% versus 17 wt%).261
Furthermore, the use of the additive allowed expansion of the working window of the Lyocell process to room temperature, well below the melting temperature of NMMO (70 °C). The reduction of the working temperature has the dual benefit of lowering energy consumption and reducing solvent and cellulose degradation reactions. In a preliminary recyclability study, under laboratory conditions, the system was found to be recyclable five times, with a recovery rate of more than 95%.261 As noted above for the PIL [mTBDH]OAc, this system also falls outside the formal definitions of DESs. Indeed, giving the intimate interactions between all components, this system is likely better described as a ternary mixture of NMMO/ChCl/MA with peculiar properties rather than NMMO·H2O in combination with NADES as additive. The exact nature of the ternary mixture has not been investigated, but this does not detract from the quality of the whole system which showed great potential for industrial application.
Overall, the use of DESs in the solubilisation and processing of cellulose has severe limitations as the maximum solubilities achieved are rather low. Furthermore, the different data found in the literature for the same DES indicate a strong variability, which is due to the type of cellulose employed and to the pretreatment carried out on the starting materials. Not surprisingly, most of the applications of DES in cellulose technology are related to those areas where actual dissolution is not required.262–265 Prominent among these is the preparation of cellulose nanocrystalline (CNC)255,266–269 and cellulose nanofiber (CNF),256,270–272 where the lower cost of DESs compared with ILs plays in their favour.
From the point of view of sustainability, the synergistic use of ILs and DESs would be certainly desirable. Indeed, this would expand the fields of application and reduce potential drawbacks by exploiting the best features of each medium. In this context, the work reported by Wang et al.,273 where ILs and DESs were used for the preparation of plasticized cellulosic films, represents an inspiring example. [C4C1im]Cl was used to solubilise cotton linters and subsequently form a cellulosic gel. The IL was then washed away with water and recovered, while the DES ChCl:U (1:2) was added to the cellulosic gel. The cellulosic film obtained after drying at 55 °C was completely transparent and colorless, with an improvement in mechanical properties in terms of increase in elongation and decrease in tensile strength.273 The effect of the DES on the quality of the end product was compared with that of Gly and sorbitol, both already used as plasticizers, and showed a similar effect as additive, albeit at lower dosages. Furthermore, with ageing, the sorbitol-plasticized film became opaque and weakened due to crystallisation of the plasticiser. Conversely, this phenomenon was not observed when ChCl:U (1:2) was used.261 This example combines the advantages of both classes of solvents, namely the excellent solvent and gel-forming capabilities of [C4C1im]Cl and the low cost and low toxicity of ChCl:U (1:2) for the preparation of plasticized cellulose films.
5.5. Hemicellulose dissolution
The most undervalued component of lignocellulosic biomass is hemicellulose. Although it represents 15–35 wt% of biomass, its exploitation at industrial level remains very limited.274 Therefore, within the perspective of an environmentally and economically sustainable biorefinery, this fraction must be better exploited and valorised. Currently, hemicellulose is mainly employed as additive in the food and pharmaceutical industries,275,276 while its use as barrier enhancement in polymer films is of potential interest.277–279 A further field of application for hemicellulose is in the tissue paper industry, where it can function as an additive to increase the biomechanical properties of cellulose products.280 The factors limiting the use of hemicellulose are its both heterogeneous and amorphous nature. The first aspect is related to the high variety of forms that this material can have: hemicellulose is a branched polysaccharide composed of hexose sugars (mannose, galactose) and pentose sugars (xylose and arabinose). The structure of hemicellulose varies depending on its source in terms of the type of saccharide units, degree of polymerisation, and presence of functionalities such as acetyl and methyl groups, cinnamic, glucuronic and galacturonic acids.137 Hardwood hemicellulose consists mainly of O-acetyl-4-O-methylglucuronoxylan backbone chains with 8–17% acetyl groups and side groups such as 4-O-methylglucuronic and galacturonic acids. The latter increase the stability of hemicellulose in acidic and basic environments. The average degree of polymerisation is in the range of 100–200.274 Softwood hemicellulose is composed of partially acetylated O-acetyl-galactoglucomannans linear chain (around 6%) with an average degree of polymerisation between 40 and 100. The lower degree of polymerisation of softwood hemicellulose makes it more soluble in water.274 The non-woody or switchgrass hemicellulose is similar to hardwood hemicellulose but with the presence of arabinose, lower degree of acetylation and low degree of polymerisation (DP 50–185).274 Thus, the high structural variability requires the development of very flexible industrial processes, which limits its diffusion and applicability. The second factor that limits the valorisation of hemicellulose is related to its amorphous nature which reduces its stability against hydrolysis.137 For this reason, under the pulping and pretreatment conditions mentioned above, hemicellulose is mainly depolymerised and degraded into the respective sugars and furan derivatives such as furfural.281 Although the process stream resulting from the removal/degradation of the hemicellulose is already recovered and exploited, the sugars obtained are poorly fermentable and the by-products obtained, mainly furans, strongly inhibit enzymatic processes.274 Research aimed at finding fractionation processes that eliminate or at least reduce the degradation of hemicellulose is therefore of primary importance. Once more the utilisation of DESs and ILs represent a viable alternative to conventional industrial processes of hemicellulose removal, which are based on drastic treatments with dilute acids, bases, or alternatively hydrothermal conditions, supercritical fluid, and steam explosion.274,282As mentioned above, the excellent dissolution capacity of halide- and acetate-based AILs towards cellulose and hemicellulose enables extremely efficient processes. Using this technology, it is possible to recover the cellulose by reprecipitation with water and further the hemicellulose with the addition of ethanol.283 However, the primary limitation of this approach remains the associated loss of crystallinity of the resulting cellulose, the high sensitivity to water, and the cost of the medium. The use of AILs/water mixtures has proved successful and widespread, as it allows the selective solubilisation of the hemicellulose while keeping the cellulose intact. Unlike Ionosolv processes that commonly employ acidic AILs or PILs, the use of ILs with basic acetate anion avoids degradation of the hemicellulose during the extraction processes. Among the various systems reported in the literature, ILs/water mixtures are the most widely used. Based on this knowledge, Froschauer et al.284 developed the IONCELL-P(ulp) process using [C2C1im]OAc/water, which proved efficient in the selective removal of hemicellulose. The same authors also investigated the effect of water in the interaction between five different ILs and wood polymers.285 Regarding hemicellulose removal, 1-ethyl-3-methylimidazolium dimethyl phosphate [C2C1im]DMP was found to be superior to its chloride and acetate counterparts but also to PILs 5-methyl-1,5-diazabicyclo[4.3.0]non-5-enium dimethylphosphate [mDBN]DMP, [DBNH]OAc, and 1,5-diazabicyclo[4.3.0]non-5-enium propionate [DBNH]EtCOO. However, PILs showed greater removal of hemicellulose than AILs at pulp loadings above 10 wt% (Fig. 20).
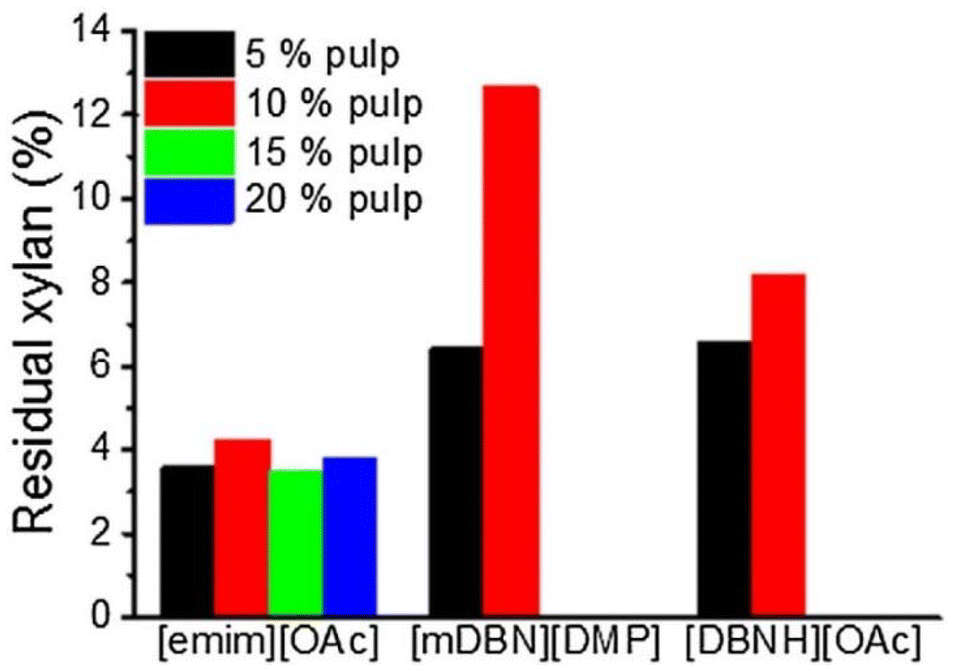 | ||
| Fig. 20 The effect of pulp load on the xylan removal efficiency. Reproduced from ref. 285 with permission from Elsevier, copyright 2017. | ||
The IONCELL-P process has been widely used for the separation of Kraft pulp (originated from different biomasses) from hemicellulose, with the aim of increasing its quality to dissolving grade.286,287
A further interesting system is the organic electrolyte solution (OES) consisting of [C2C1im]OAc and the biobased solvent GVL, in turn derived from cellulose processing. This system performs well in the presence of water, as the GVL/[C2C1im]OAc/water mixture in a 2:1:1 ratio transforms hardwood Kraft pulp into dissolving-grade material under mild conditions.288 In addition, GVL was used to develop a hybrid Organosolv/IONCELL-P process for the fractionation of birch wood and subsequent purification of the resulting cellulose.289 The first step involved the use of the GVL/water mixture to obtain paper-grade pulp, while the IONCELL-P process was used to remove the hemicelluloses and to obtain a GVL-IONCELL pulp. This material was of a significantly higher quality compared with pulp obtained with the more impacting commercial processes.289
A biobased alternative to [C2C1im]OAc is cholinium acetate ([Ch]OAc). In fact, Cheng et al.290 reported the benefits of treating ground bagasse and Southern yellow pine with [Ch]OAc, which can solubilize the hemicellulose and the lignin while retaining the cellulose unchanged. In this study, the authors evaluated the solvent capability of [Ch]OAc and [Ch]OAc/water (5:1 in mass) for single biomass components using MCC for cellulose, xylan for hemicellulose, and Kraft lignin for lignin. Neat [Ch]OAc and [Ch]OAc/water are both able to solubilize up to 13 wt% of xylan.290 This system is appealing because the IL used is derived from renewable sources and is highly recyclable compared with the more expensive [C2C1im]Cl and [C2C1im]OAc.291 A similar system was used by Sun et al.292 for switchgrass fractionation, but the information on the hemicellulosic fraction was not disclosed.
[Ch]OAc has been successfully used as HBD component in the preparation of DESs. Dugoni et al.293 reported the preparation of three different DESs with [Ch]OAc as HBA and levulinic acid, glycolic acid, and imidazole (Im) as HBD moieties, respectively. The three DESs showed very good solvent capabilities towards xylan from beechwood, which was used as a model for hemicellulose. The addition of water to the DESs increased the solvent capacity, which reached an optimum balance with the DES ChOAc:Im (1:1) + 15 wt% water being able to dissolve a considerable 45 wt%. The use of [Ch]OAc in DES systems brought a significant increase in performance compared with pure [Ch]OAc (Fig. 21).293
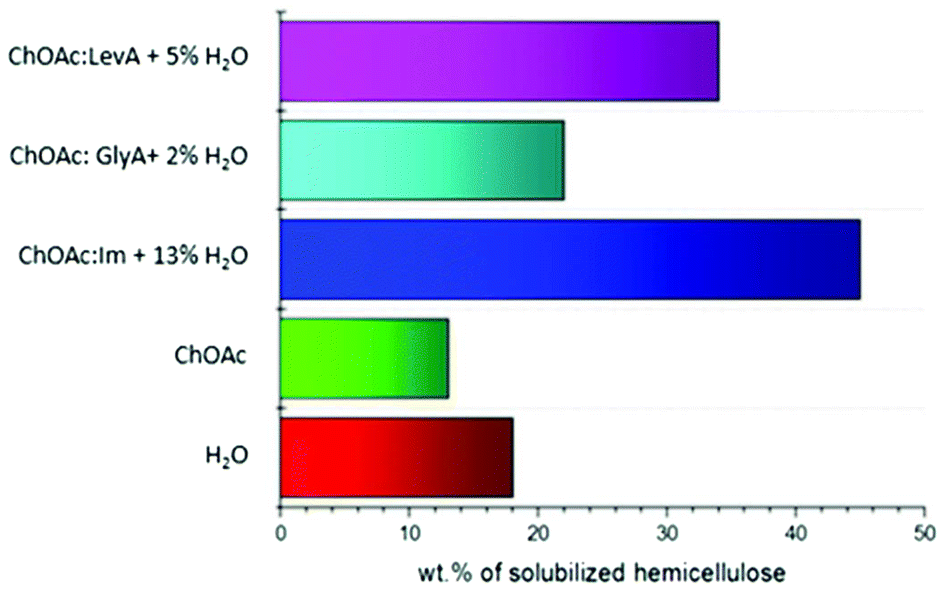 | ||
| Fig. 21 Comparison of the maximum values of hemicellulose solubility obtained in [Ch]OAc-based DESs + wt% water, [Ch]OAcIL and water reproduced from ref. 293 with permission from The Royal Society of Chemistry, copyright 2017. | ||
In addition, [Ch]OAc-based DESs have also proved to be markedly better performing than their ChCl-based analogues. Furthermore, a cost analysis showed that the high price of [Ch]OAc-based DESs is purely due to the limited commercialization of [Ch]OAc compared with the more widespread component ChCl (3.44–1.39 euro per kg for ChCl-based DES vs. 3.58–1.67 euro per kg for [Ch]OAc-based DES).293
The number of papers dealing with hemicellulose solubilization using DESs is very limited. These include works that use DESs comprising toxic chemicals, such as thiourea,294 or acid DESs295–297 that degrade the hemicellulose structure with similar issues to those reported for ILs.
More interesting is the work of Morais et al.298 where DES ChCl:U was used for the removal of hemicellulose from Eucalyptus globulus hardwood. The dissolving power of the DES at different molar ratios, neat and in the presence of water, was evaluated in the solubilization of xylan from beechwood (Fig. 22a). The remarkable value of 310 mg of xylan per g of DES was obtained with ChCl:U 1:2 + 50 wt% water at 90 °C. Under these conditions, the DES is able to solubilize the hemicellulose without significant changes in the degree of polymerization (Fig. 22b).298
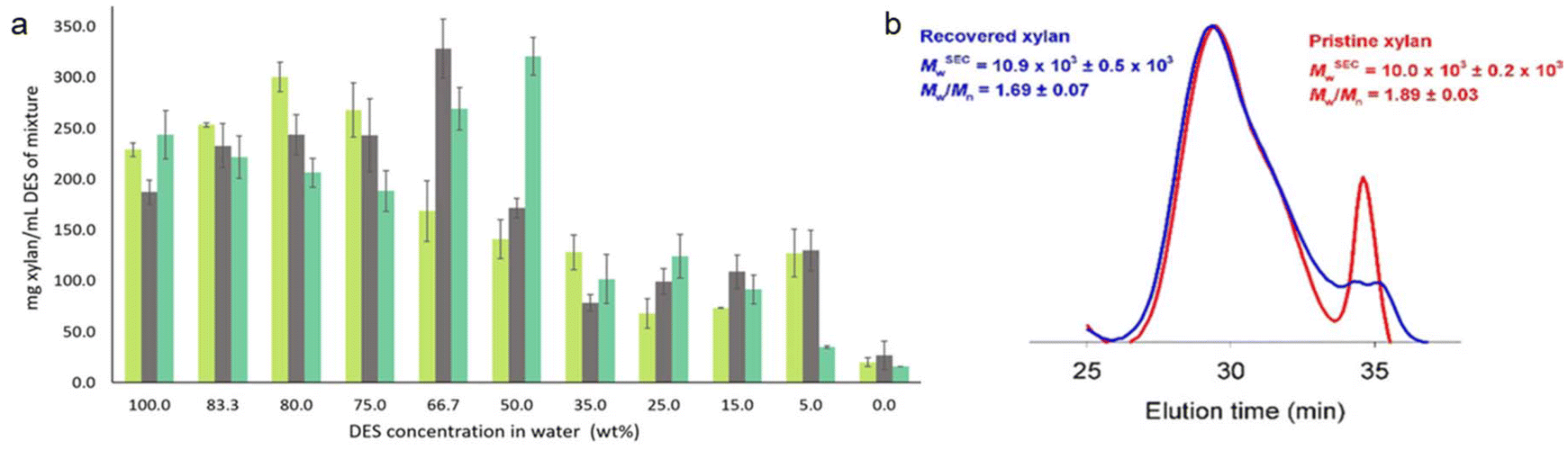 | ||
Fig. 22 (a) Solubility of xylan in aqueous ChCl/U (1:2) at different concentration and different temperatures (70  , 80 , 80  and 90 °C and 90 °C  ). (b) SEC profiles of the pristine and recovered xylans. Reproduced from ref. 298 with permission from Wiley, copyright 2018. ). (b) SEC profiles of the pristine and recovered xylans. Reproduced from ref. 298 with permission from Wiley, copyright 2018. | ||
Considering the excellent results obtained, the use of DES for the solubilisation of hemicellulose seems to be superior to the use of ILs in terms of efficiency, cost and residual toxicity. In particular, the systems developed by Morais et al. and Dugoni et al. are complementary. The first one, based on ChCl:U (1:2, at 50 wt% of water) demonstrated excellent results in the pretreatment of Eucalyptus globulus while the second one, based on ChOAc:levulinic acid, exhibited a very good purification capacity on Kraft cellulose. For both systems, it is important to note the excellent DES recyclability after hemicellulose extraction.
5.6. ILs and DESs toward chitin
Recently, the use of ocean biomass as a source of raw material has attracted enormous interest both at academic and industrial level.299 Among the various marine biopolymers, chitin is undoubtedly the most interesting in terms of its potential applications, but primarily because it is biologically produced in huge quantities. Indeed, chitin is used as a structural material by the majority of invertebrate organisms and is thus the second most abundant polysaccharide biopolymer in nature after cellulose. It has been estimated that between 1012 and 1014 tons of this compound are annually produced in nature as the main component of marine shells (for example α-chitin in the shell of crustaceans and β-chitin in squid pens),300 but it is also found in mushrooms and insects.Chemically, chitin is a polysaccharide only composed of β-1,4-linked 2-(acetylamino)-2-deoxy-D-glucose units. The structure of this polysaccharide is more complex than those reported above for other polysaccharides because, as well as different molecular weights and crystallinity degrees, chitin may present three polymorphic forms (α, β, and γ chitin) and different degrees of acetylation (DA).301 Each of these structural parameters has a strong effect on the mechanism and the difficulty of chitin dissolution. On the other hand, this structural variability makes of this natural polymer an exceptional material with unique properties302,303 like biocompatibility, non-toxicity and antimicrobial power. These features ensure that chitin and its derivative chitosan are considered excellent materials for use in the biomedical field.304 In this sector alone, chitin market size was valued at $42.29 billion in 2020 and is projected to reach $69.29 billion in 2028.305 In addition, chitin is one of the few nitrogen-containing biopolymers, making it an excellent precursor for the synthesis of organonitrogen biochemicals.306,307
Despite this potential, chitin remains underutilized at industrial level, mainly on account of its low solubility in common solvents. Indeed, chitin dissolves at low concentrations in LiCl/DMAc, CaCl2/methanol and hexafluoroisopropanol (HFIP), but these treatments cause considerable changes in the biopolymer structure such as degree of crystallinity and crystal form, molecular weight, and DA.308,309 In addition to these media, solvent systems based on aqueous solutions of acids, bases or salts such as phosphoric acid, LiSCN, NaOH/U, and KOH/U were reported to dissolve chitin.301 Although the base/U systems show great solvent capabilities, they require energy-consuming freezing/thawing cycles, which make them less suitable for industrial applications. For chitin, as for other polysaccharides, the development of solvent systems capable of solubilising a high load without changing the structural characteristics is essential for efficient and cost-effective use.
In this context, ILs are the main tool for the industrial exploitation of chitin. In fact, they are able to solubilise large quantities of chitin while minimising structural changes. For this reason, since the first report by Rogers et al. in 2001, numerous research papers, technical reports and reviews on this subject have appeared in the literature. Rogers and co-workers founded the company 525 Solution and more recently Mari Signum with the aim of obtaining and using chitin for industrial applications. However, which IL works best with respect to chitin dissolution is not entirely clear. In fact, even the literature values for the same ILs present inconsistencies. This discrepancy is undoubtedly due to the different types of chitin used and to the different operating conditions. As a result, although it is clear that dissolution of chitin in ILs is possible, an in-depth comparison is difficult. Nevertheless, some general considerations about the dissolution of chitin in ILs can be advanced.
AILs are undoubtedly those capable of dissolving larger quantities of chitin, and among these, the imidazolium-based ones are the best-performing media.310 The best result reported in the literature was obtained using [C2C1im]OAc, which solubilises 16 wt% of pure chitin at 100 °C in 19 h.311 Differently from what was observed for cellulose dissolution, where only the anion strongly influences the performance, for chitin the combined effect of cation and anion seems to be crucial and predominant over the contribution of the individual ion. For example, for [AC1im]-based ILs, bromide shows higher solvent capacity than chloride (4.8–5 wt% for Br312 compared with 0.5 wt% for Cl313), whereas for the [C2C1im]-based ILs, only chloride is able to dissolve chitin (10 wt%).314 It is also interesting to note that ILs containing a dimethylphosphate anion, which have a high solvent capacity towards cellulose, do not act as solvent in the case of chitin.313 By analysing the solubilisation performance of various ILs for different types of chitin, Shamshina identified and ranked by increasing ability 5 ILs: [AC1im]Cl < [C4C1im]Cl < [C2C1im]Cl < [C4mim]OAc < [C2C1im][OAc].315 In selecting the appropriate IL to be used in a process scale-up, the solvent power is certainly important, but the cost of the medium and its recyclability are likewise relevant. Acetate ILs are very efficient in the dissolution of chitin, but they are also costly and raise some concerns about their recyclability due to the low thermal stability. Nevertheless, Sun et al. reported that [C2C1im][OAc] can be efficiently recycled by using pervaporation systems without significant loss due to degradation.316
Optimising the cost of ILs remains an open issue and the use of “statistical mixtures” of ILs OAc instead of pure ILs has been proposed as a way to considerably reduce their cost and synthetic impact. In fact, the 2:1:1 mixture of [C2C1im]OAc:[C2C2im]OAc:[C1C1im]OAc can be sourced at low cost, without passing through halide intermediates, from the reaction between glyoxal, formaldehyde, a combination of ethyl- and methylamine and acetic acid at 70 °C for a total time of 13 h.317 This mixture was found to have a solvent power toward lignin 1.6 times higher than the corresponding pure ILs.315 Interestingly, [HN(CH2CH2OH)3]OAc and [HN(CH2CH2OH)3]MeSO3, which were unable to dissolve chitin when tested in pure form, became working solvents for this polysaccharide even at room temperature when mixed with ethylenediamine (EDA) at different molar ratios (between 1:0.5 and 1:2).318 These two latter systems presumably represent examples of eutectic mixtures capable of dissolving chitin.
The literature about the use of DESs for chitin dissolution is far less vast than that for ILs. The first work in this area was published by Prasad et al. in 2013319 and describes that cholinium chloride:thiourea (ChCl:thiourea, 1:2) dissolves 9 wt% of α-chitin in 6 h of heating at 100 °C. In this study, the dissolution capability of ChCl:U (1:2), cholinium bromide:U (ChBr:urea, 1:2), ClChCl:U (1:2), and BetHCl:U (1:4) using conventional heating, microwave and ultrasound were tested. The authors claim that the use of DES was favourable compared with ILs due to their lower cost and lower toxicity.319 Although DESs are often less expensive than the majority of ILs, any generalisation regarding their low toxicity should be avoided. Thiourea is indeed a toxic compound (category 4) displaying carcinogenicity (category 2), reproductive toxicity (category 2) as well as long-term aquatic hazard (category 2)320 and therefore DESs containing thiourea are likely toxic. Thiourea was used as HBD also in the preparation of DESs with ILs as HBA ([AC1im]Cl, [C4C1im]Br, [C2C1im]Cl, [C2C1im]Br).321 Although this class of solvents is capable of solubilising chitin (2–5 wt%), their real “deep eutectic nature” was not verified. Therefore, the work of Prasad et al.319 is the only study concerning the dissolution of chitin in DES, which however does not investigate the dissolution mechanism nor the rheological properties of the solution. Moreover, their investigation only highlights the decrease of the molecular weight of the regenerated chitin, while no effect on DA was observed. In contrast, the literature available on chitin dissolution in ILs provides information on the solubilisation mechanism, the characteristics of the polymer in solution and the effects of the dissolution process on chitin structure. These studies, which have been reported in several papers322–324 and reviews,301,325 will not be discussed in detail in this critical review.
As a general rule, inter- and intramolecular hydrogen bond networks need to be destroyed for chitin dissolution (Fig. 23). For ILs, as in the case of cellulose dissolution, a correlation between the α and β Kamlet–Taft parameters and the ability to solubilise chitin was observed. Shimo et al.318 found that only ILs with β values >0.5, typically attributable to basic anions, were able to dissolve chitin. Conversely, the α value, which is ascribable to the cation nature, seems to be less important. These findings, which are surely valid for the ILs under consideration, were in conflict with what was previously reported for ILs and especially with what was experimentally observed for DESs. Indeed, ChCl:EG and ChCl:Gly, which display β values >0.5 (0.57 and 0.52 respectively),326 were unable to solubilise chitin. These inconsistencies indicate that the dissolution mechanism is a rather complex process that cannot be correlated solely to acidity of basicity of the IL's ions.
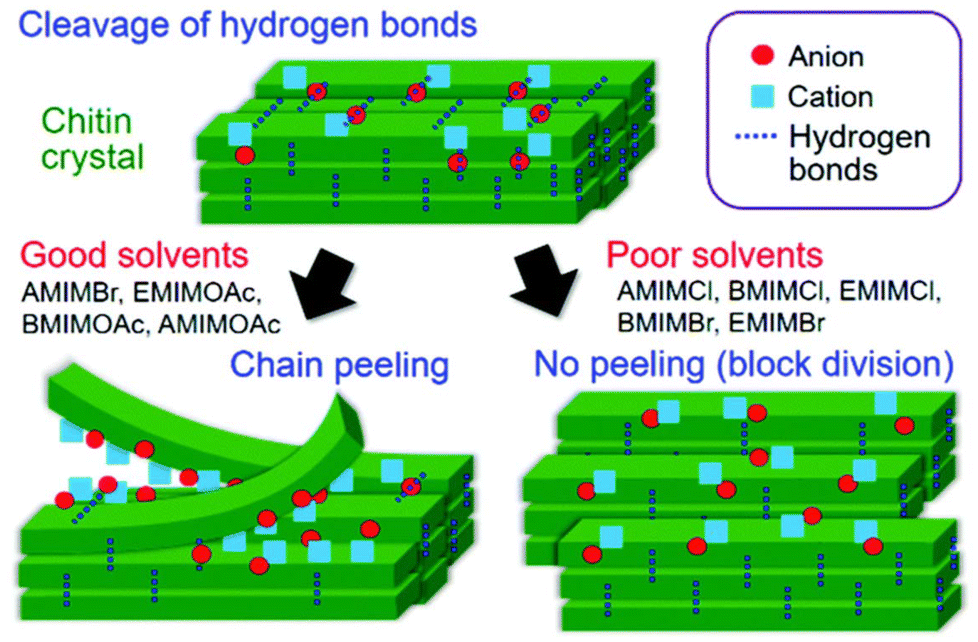 | ||
| Fig. 23 Dissolution mechanism of chitin crystal models in imidazolium-based ILs. Reproduced from ref. 322 with permission from The Royal Society of Chemistry, copyright 2018. | ||
The number of publications describing the use of ILs in chitin technology is substantially larger than that concerning DESs. The main reason for this trend is time-related, as the first reports involving ILs were from 2001327 whereas, as mentioned above, the first study about the same use of DESs was published 12 years later.319 In addition, the limited number of DESs capable of dissolving chitin and the rather low maximum load reached strongly limited the use of DESs in all applications that require the solubilisation of this polysaccharide. Therefore, applications such as wet- and electrospinning,328–330 preparation of hydro and ionogels,331,332 production of porous chitin microbeads,333 membrane fabrication334 as well as enzymatic and chemical transformation of chitin in homogeneous phase335–339 are at the moment prerogative only of ILs. On the other hand, DESs are mainly used in the preparation of nanofibres340 and nanocrystals341,342 through the exploitation of the acidity of the HBD component, which leads to the degradation of the amorphous part of chitin and leaves the crystalline portions intact.
Essentially, ILs are used in processes where chitin requires solubilisation, whereas DESs are used in processes where chitin merely needs to be swelled but not solubilised. This is in line with a similar trend already observed for cellulose, despite DESs possessing a higher solvent capability towards chitin. The process wherein this different behaviour is most evident is the “pulping” of chitin, where ILs and DESs act in an opposite fashion. Indeed, chitin can be extracted through two different approaches: (i) selective dissolution of the biopolymer, which allows for its separation from the inorganics and protein fractions by filtration, and (ii) chemical “erosion” of the inorganic fraction and simultaneous solubilisation/degradation of the protein portion, which permits recovery of the chitin without prominent modification of the chemical structure. Both alternatives are valid and can be followed depending on the end use of the extracted chitin. By using ILs, it is possible to selectively solubilise chitin and then recover it by precipitation, spinning or directly by conversion into the product of interest. Alternatively, the use of DESs is more suitable for the second approach since, by judicious selection of HBA and HBD, it is possible to decompose the inorganic and protein fraction, which are consequently removed. Using DESs would allow raw chitin to be obtained at an overall lower cost. An interesting approach that has not been investigated to date is the combined use of DESs and ILs in chitin pulping. Indeed, the use of acid DESs in the pretreatment would allow for the inorganic part (mainly CaCO3) to be effectively removed and the protein part to be degraded. This approach would be particularly interesting in the treatment of ‘hard’, highly mineralised biomasses (e.g. crab shells) with a high CaCO3 content. The resulting material, highly enriched in chitin, would contain part of the degraded protein fraction and could only be considered ‘technical-grade chitin’. To further purify this chitin, making it suitable for instance for biomedical use, the consecutive selective dissolution in ILs followed by filtration could be pursued. This process would be particularly suitable in terms of efficiency and cost-effectiveness, as it exploits the synergistic effects of DESs and ILs while maximising their benefits and minimising their drawbacks.
5.7. Beyond dissolution and fractionation performances
Most works which deal with this subject both for ILs and DESs describe these solvents’ performances on different cycles, while in depth analysis of energy costs and quality of the medium recovered are less studied. In the case of ILs, a few different recycling options have been analysed and the results are summarized in dedicated reviews.343,344 Conversely, limited data have been gathered thus far for DESs, which have been summarized by Isci and Kaltschmitt,345 and further investigations are needed. It is also worth stressing that some reports on DES recycling could be hidden in the literature on account of misleading titles, as is the case for the pressure-driven membrane processes, nanofiltration, reverse osmosis and pervaporation studies performed on ChCl:EG (1:2) by Haerens et al.346 In the case of biomass treatment, the addition of antisolvents to DES–lignin/hemicellulose mixtures to precipitate the polysaccharides and recover the DES after evaporation of water/ethanol/acetone is the strategy described in most reports. The DES thus obtained is often used in following runs and performances are evaluated without an in-depth study of the DES composition, possible degradation and of the presence of contaminants. Indeed, low molecular weight molecules soluble in the DES are likely to be extracted from the biomass and contaminate the DES.
Overall, it is not trivial to state which of the two media (ILs or DESs) is the most amenable to recycling. This question is further complicated by the fact that the application of interest plays an important part in assessing the medium's performance. Within the biomass treatment research area, a comparison could be attempted according to the works by Chang et al., who reported the recovery both of an IL ([AC1im]Cl)347 and a DES (ChCl:EG, 1:2)348 by electrodialysis. The authors treated the same biomass (Eucalyptus globulus wood) and then studied the recovery with the two media. The schematic electrodialysis process both for IL and DES is reported in Fig. 24.
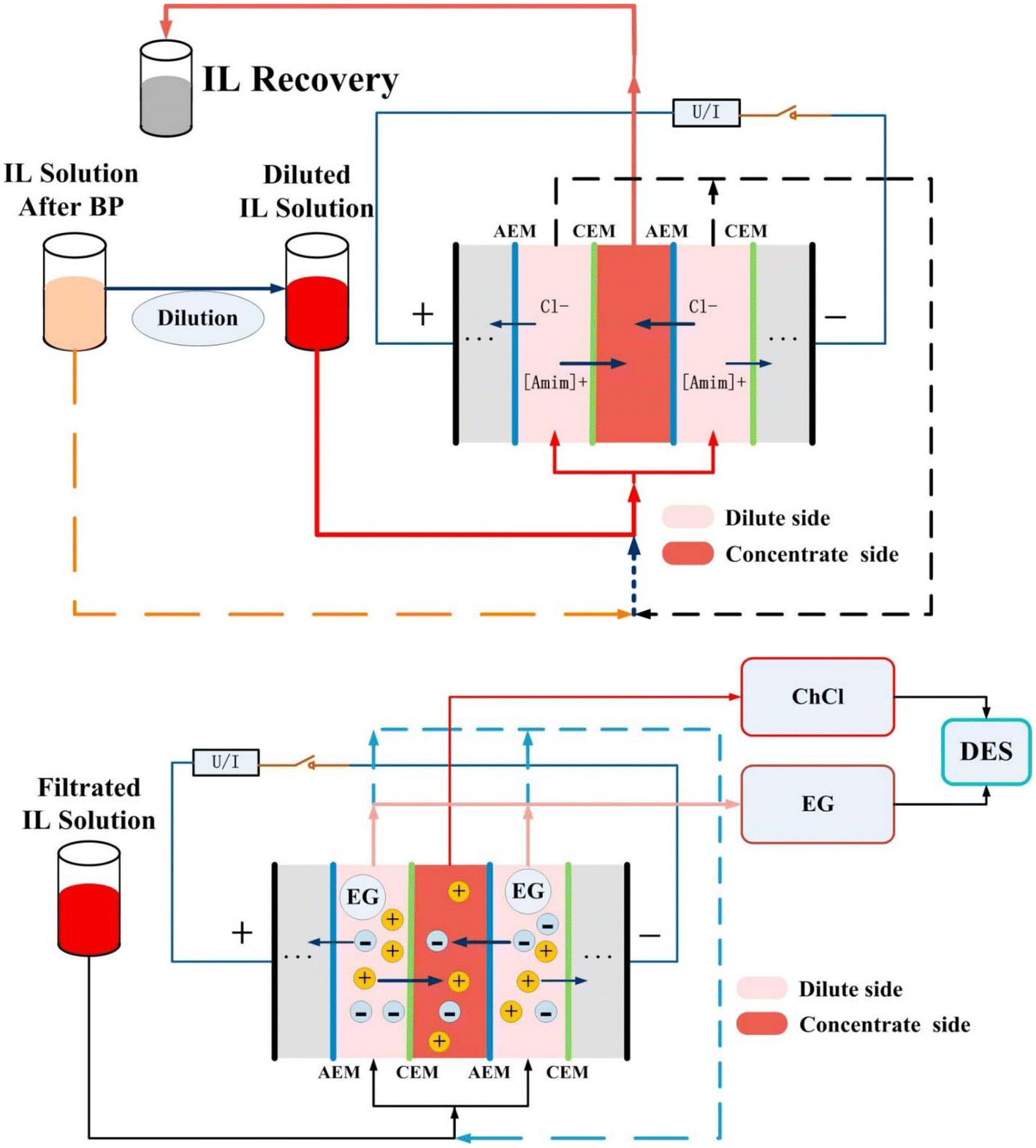 | ||
| Fig. 24 IL (top)347 and DES (bottom)348 recovery process with electrodialysis treatment. Adapted from ref. 347 and 348 with permission from Elsevier, copyright 2017 and 2019. | ||
The main difference between the two schemes stems from the nature of the two media. In particular, the presence of the non-charged species (EG) in the DES case determines the separation of the HBA and HBD during the electrodialysis process and the need to reconstruct the DES system at the end of the process before its further use.
For [AC1im]Cl, the recovery ratio was 66–71% with a specific energy consumption as high as 429–467 g KW−1 h−1 in a single electrodialysis treatment (at 15 V applied voltage and 5 L min−1 solution flow rate). An improvement of performances was observed with the semi-continuous process, where the recovery ratio increased to 93% with a slight drop of the specific energy consumption in the case studied. For ChCl:EG (1:2), under the same applied voltage and solution flow rate, recovery ratios of 84% for ChCl and of 96% for EG with the specific energy consumption lower than 300 g KW−1 h−1 were obtained in a single electrodialysis treatment. In the semi-continuous process, enhancement of ChCl recovery was observed (up to 92%) together with a slight decrease of the EG recovery and beneficial effects on the energy consumption when compared with the single recovery process. On the basis of these results, one could be tempted to claim ChCl:EG (1:2) as a more convenient solvent than [AC1im]Cl in the biomass treatment, at least from the recovery perspective.
However, it is worth stressing again that focusing on a partial picture could bring wrong conclusions. Indeed, the two biomass processes are substantially different: the DES treatment is carried out on a lower biomass loading and, more importantly, it is combined with a previous hydrothermal treatment. This latter removes hemicellulose and low molecular weight molecules, thus preventing DES contamination and making its recycling easier (especially in the case of the non-charged EG). Even more worrying is the observation that the recovered IL and DES act differently in repeated runs. While recycled [AC1im]Cl provided almost the same efficiency as the fresh IL, the reconstituted ChCl:EG (1:2) displayed a worse efficiency than the fresh DES.
In conclusion, it is difficult to have a clear-cut indication of which system is more amenable to efficient recycling, even when the same technique and biomass is employed. Further investigations and more innovative approaches in the recycling of both ILs and DESs are needed for designing industrially feasible processes based on these non-conventional media.
It is also worth stressing that in the case of PILs, the synthetic strategy is as simple as the one commonly employed for DESs and requires a simple mixing step, as mentioned above. An illustrative case is that reported by Hallett et al.,351 who carried out a techno-economic evaluation of PILs used in biomass processing, namely [N2220]HSO4 and 1-methylimidazolium hydrogen sulfate [HC1im]HSO4. The cost prices were estimated as $1.24 per kg and $2.96–5.88 per kg, respectively, with the cost of the former comparing positively with the price of acetone and ethyl acetate. This comparative analysis was further refined by also taking into account the indirect costs such as human health, resources and environmental impact.352 [N2220]HSO4 was found to have the lowest total cost followed by acetone, as an example of a traditional solvent from fossil resources, and Gly, as a representative of solvents from renewable resources. Gly displayed the highest production cost due to the cost of its precursors (rapeseed oil and soybean oil) and considerably higher indirect costs than the other solvents analyzed in terms of human health and environmental impact. This work clearly emphasizes the importance of evaluating the cost of any chemical species from a wider perspective, where direct production costs, although fundamental, are just one of many factors.352 Furthermore, in the context of the present IL/DES comparison, it is of interest to stress that Gly, which is one of the most commonly used HBD of NADESs, presents the highest total cost within the series.
Of the few papers dealing with techno-economic evaluations of processes involving DESs, three are related to the subject of this analysis.353–355 Specifically, these reports describe the treatment of rice straw and switchgrass either with ChCl:LA or ChCl:EG with 1 wt% of H2SO4 for the production of low molecular weight products. Two major conclusions can be drawn from these studies: (a) the development of multi-product biorefineries should be targeted in view of profitable and sustainable processes instead of a single, high-volume–low-value compound, and (b) the biomass/DES ratio should be as high as possible to reduce the costs of solvents required for the recovery of the biomass fractions from the DES system and for the overall DES recycling. In the case studied, solid loadings were in the region of 10–27 wt%, which differ from the usual amounts tested in the vast majority of lab-scale studies reported in the literature to date for DESs. To further highlight the importance of the biomass/DES ratio on the overall reactor sizes and process costs of a DES-based biorefinery, and thus recommend paying attention to this parameter even at an early stage in an investigation, the comparison of the pretreatment process flow steps at 5 wt% and 10 wt% solid loading reported by Kumar et al.356 is summarized in Fig. 25. The amounts of water and acetonitrile required for treating 1 kg of rice straw increased from 53 to 123 liters and from 110 to 210 liters, respectively, by halving the biomass loading.
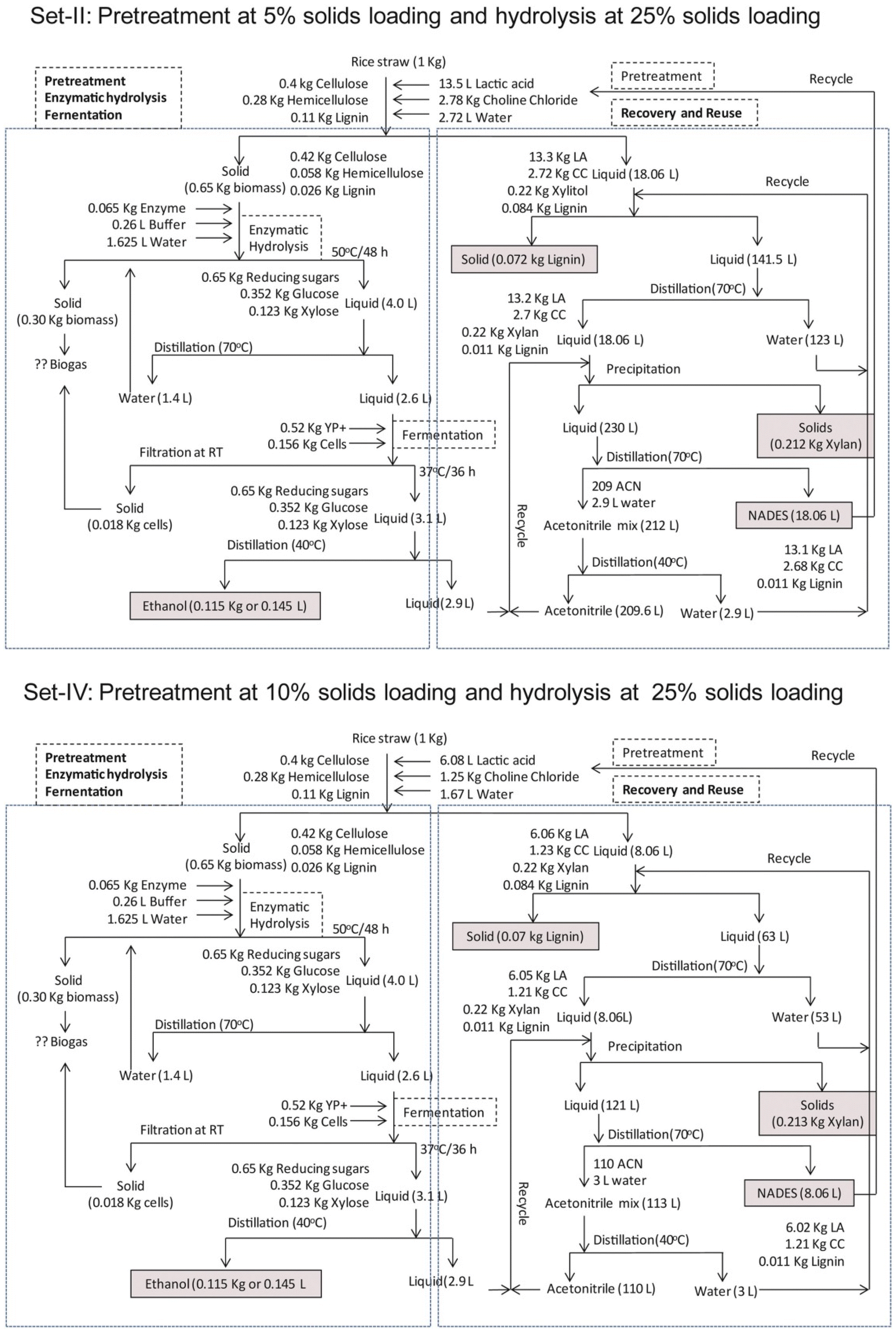 | ||
| Fig. 25 Pretreatments process flow steps of 5 wt% (set-II, up) and 10 wt% (set-IV, bottom) solid loading, adjusted from reference. Reproduced from ref. 356 with permission from Elsevier, copyright 2018. | ||
In the above techno-economic analyses, the potential issues related to DES recycling and diminishing performances over time are almost completely overlooked. This is despite the problems reported by Morais et al.357 concerning the esterification of cellulose, degradation of xylan, and polymerization of LA using ChCl:LA (1:10) as medium, and the drastic reduction in furfural production when the recycled DES is used, in the case of ChCl:EG (1:2) with 1 wt% of H2SO4.358 Therefore, further studies are needed to thoroughly unveil the actual cost contribution of DESs in biorefineries, beyond the simple cost of raw material for their initial preparation. In the case of ILs, a few more techno-economic analyses of different processes have been carried out,359 which span from biogas upgrading360 and CO2 capture,361 to production of fuel-grade tert-butyl alcohol362 and synthesis of platinum nanoparticles.363
Within the specific theme of the review, Blanch et al. highlighted already in 2011364 how reduction of ILs’ cost, reduction of ILs’ loading and increase of ILs’ recycling were key areas to be tackled for increasing the profitability of a lignocellulosic ethanol biorefinery based on IL pretreatment. Furthermore, the authors studied the effect of selling lignin-derived products to reduce the minimum selling price of the ethanol product. Overall, the IL-based processes share the same issues as the DES-based processes, with the additional concern of the IL price. The potential impact on profitability of the IL/biomass ratio, followed by the other criticalities mentioned above, was further corroborated by the analysis presented by Ferrari et al.365 for a biorefinery designed to transform sugarcane into ethanol. In this work, the biomass pretreatment process is carried out with a mixture of [H3N(CH2CH2OH)]OAc and of [H3N(CH2CH2OH)]OHex. Of note is also the recent comparative techno-economic analysis of Eucalyptus globulus wood pretreatment carried out either with [C2C1im]OAc or [Ch]OAc.366 In particular, the amount of water needed to remove residual IL from dissolved cellulose during the washing step, the effect on the subsequent enzymatic hydrolysis and the IL recycling step all indicate the possibility of lower operating costs for the imidazolium-containing IL. Despite these findings, additional features such as the potential environmental footprints and the overall toxicity should be considered in future developments.
The most informative case study is, however, represented by the Ionosolv process and its development over time. Indeed, its evolution exemplifies well the potential of ILs in biomass pretreatment in conjunction with the need to thoroughly study each step and the necessity to devise or consider even hybrid options when solving problems or increasing efficiency. The Ionosolv process proposed by Hallet et al. is summarised in Fig. 26 and is based on PILs. As mentioned previously, the cost of PILs is often comparable to and in some cases lower than conventional organic solvents.351,352 The first study on the Ionosolv process entailed the use of [N2220]HSO4 plus 20 wt% of water at 120 °C and a 1:10 solid loading.367 This setting allowed for the complete dissolution of hemicellulose and of 85 wt% of lignin in the fractionation process of the grass Miscanthus x giganteus, leaving behind a cellulose-rich pulp. Studies on the saccharification step and on the quality of the lignin recovered were also performed as well as an assessment of the recovery of the IL (ca. 98% for each cycle).367 The potential effect of inorganic salts brought in by the biomass on the IL quality was also investigated, an aspect often almost completely overlooked, and finally the presence of IL degradation products was ascertained. The preliminary techno-economic analysis identified the solid loading and water usage during lignin recovery as the critical aspects.
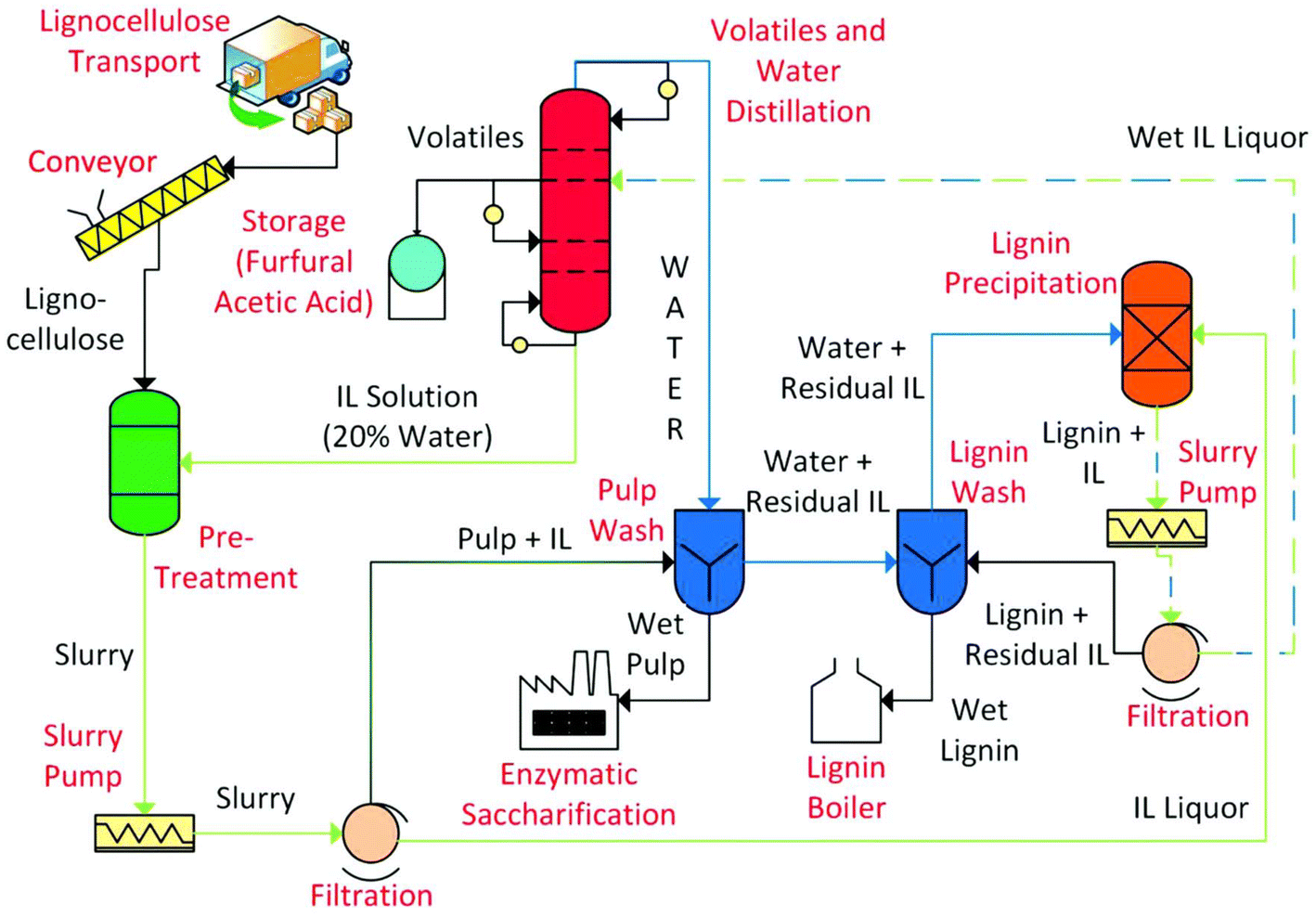 | ||
| Fig. 26 Process flow diagram for the Ionosolv process taken from ref. 367. Reproduced from ref. 367 with permission from The Royal Society of Chemistry, copyright 2017. | ||
Intensification and diversification strategies and an in-depth analysis were then carried out to further investigate pro and cons of the Ionosolv process. In particular, the effect of higher loading (20 wt% solid loading), time, temperature368 as well as particle size and scaling up from 10 mL to 1 L369 were assessed. It is worth stressing that the scaling up required optimization of the washing protocols and agitator design, with the latter not required at the bench-scale stage.
A different PIL, N,N-dimethylbutylammonium hydrogen sulfate, [N1140]HSO4, was also employed in the Ionosolv process and was proved to be effective. It actually outperformed [N2220]HSO4 in the pretreatment of recalcitrant softwood pine with a remarkable 1:2 solid loading at 170 °C during 30 minutes.370
As a further possible variation, the hybrid combined Ionosolv–Organosolv strategy, which entails the use of a PILs, [N2220]HSO4 or [N1140]HSO4, and selected organic solvents (ethanol, butanol or acetone), was also explored in the pretreatment of two representative feedstocks, Miscanthus and pine.371 Beneficial effects were found in terms of higher loading, up to 1:2 solid loading with 60 wt% IL and 40 wt% ethanol at 120 °C, and lower energy consumption for the IL regeneration step, especially at high organic solvent concentration. Both aspects have direct implications for the economic profitability of the process.
The Ionosolv example thus highlights the need for an in-depth investigation of multiple variables before claiming the potential profitability of a biomass pretreatment/biorefinery process on the basis of limited lab-scale results. This important reminder must be kept in mind when assessing both IL-based and DES-based processes.
6. Outlook and future perspective
The intention of this review is to highlight, without providing an exhaustive survey of the present literature, that there is no winner or loser between ILs and DESs, and none should even be sought. The possible cation–anion combinations for preparing ILs are almost limitless, and the number of new mixtures of compounds that yield DES systems is constantly increasing (hydrophobic DESs were disclosed in recent years too).372 It is thus logical that some systems will be better suited for a given application and others for a different one. It could even be possible that one among the mixtures of ILs (which could also give rise to DES systems in some cases), ILs with co-solvents/water, DESs with water or ILs-HBD mixtures will be the ideal matching medium for a given application, considering the whole life cycle of the process. Ultimately, since ILs and DESs are distinct classes of solvents, each one of them with distinct properties, they should be seen as options to choose from in the design and development of environmentally friendly processes, both as alternatives to volatile organic solvents rather than alternatives to each other.The dynamic research in the DES field in recent years should avoid repeating the same mistakes made initially in the ILs area, in which a deleterious tendency to generalise behaviours and properties observed for specific cases took place. This generated frustration and misplaced expectations of ILs over time. It seems nowadays that DES research area is following the same path by claiming their superiority over ILs. The urge to define DESs as a viable alternative to ILs probably arises from the need to overcome the initial misleading classification of DESs as a new family of ILs. The main aim of this review is to defend the DES field from future “scientific recriminations” by placing them in the correct place within the green media context. This should pave the way for an objective evaluation of their pro and cons, under specific situations.
From the same point of view stems also the need to revaluate eutectic solvents (ESs) and low melting mixtures (LMMs)373,374 as other alternative media to VOCs endowed with the same potential as DESs and ILs to achieve more sustainable processes and systems. Nowadays, the term “DES” has an undeniable appeal within the scientific community, and far too often new eutectic systems are just presented as DESs although their phase diagrams are not known. It is thus desirable that researchers working in the field will encourage a further understanding of the nature of DESs, through thermodynamic studies and the molecular and laboratorial scale, which will help to categorize the actual nature of the systems. Ultimately, it will always be impossible to decide a priori if a DES is more suited for a certain application than an IL, an ES or an LMM or even a bio-based solvent.
What is also clear from the above-discussed examples is that further investigation is needed both at the fundamental and the applied levels. In the former case, it is not a surprise that the ILs field is more mature given that, for instance, the first studies of polysaccharides dissolution date back to 1934,375 while for DESs, some features still need to be defined such as the amount of water tolerated by each system. On the other hand, process-oriented studies which involve, for instance, techno-economic evaluations, system recycling efforts and life cycle assessments376 are urgently needed for both ILs and DESs and would provide invaluable data for further conscious development.
Abbreviations
| [AC1im]Cl | 1-Allyl-3-methylimidazolium chloride |
| [BEMPH]NTf2 | 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorinium bis(trifluoromethane)sulfonimide |
| [C10C1IM]FeCl4 | 1-Decyl-3-methylimidazolium tetrachloroferrate |
| [C10C1IM]NTF2 | 1-Decyl-3-methylimidazolium bis(trifluoromethane)sulfonimide |
| [C10C1IM]PF6 | 1-Decyl-3-methylimidazolium hexafluorophosphate |
| [C10C1im]Phe | 1-Decyl-3-methylimidazolium phenolate |
| [C10DABCO]Br | 1-Decyl-1,4-diazabicyclooctane bromide |
| [C14C1im]Cl | 1-Tetradecyl-3-methylimidazolium chloride |
| [C14Qn]Br | 1-Tetradecylquinolinium bromide |
| [C16C1im]Cl | 1-Hexadecyl-3-methylimidazolium chloride |
| [C16C1im]NTf2 | 1-Hexadecyl-3-methylimidazolium bis(trifluoromethane)sulfonimide |
| [C1C1im]OAc | 1,3-Dimethylimidazolium acetate |
| [C1C4mor]Br | 4-Butyl-4-methylmorpholin-4-ium bromide |
| [C1C4mor]CF3SO3 | 4-Butyl-4-methylmorpholin-4-ium trifluoromethanesulfonimide |
| [C1C4mor]N(CN)2 | 4-Butyl-4-methylmorpholin-4-ium dicyanamide |
| [C1C4mor]NTf2 | 4-Butyl-4-methylmorpholin-4-ium bis(trifluoromethane)sulfonimide |
| [C2C1im]Br | 1-Ethyl-3-methylimidazolium bromide |
| [C2C1im]Cl | 1-Ethyl-3-methylimidazolium chloride |
| [C2C1im]DMP | 1-Ethyl-3-methylimidazolium dimethyl phosphate |
| [C2C1im]OAc | 1-Ethyl-3-methylimidazolium acetate |
| [C2C1im]Phe | 1-Ethyl-3-methylimidazolium phenolate |
| [C2C2im]OAc | 1,3-Diethylimidazolium acetate |
| [C2DABCO]Br | 1-Etyl-1,4-diazabicyclooctane bromide |
| [C4C1im]BF4 | 1-Butyl-3-methylimidazolium tetrafluoroborate |
| [C4C1im]Br | 1-Butyl-3-methylimidazolium bromide |
| [C4C1im]Cl | 1-Butyl-3-methylimidazolium chloride |
| [C4C1im]OAc | 1-Butyl-3-methylimidazolium acetate |
| [C4C1im]Phe | 1-Butyl-3-methylimidazolium phenolate |
| [C4DABCO]Br | 1-Butyl-1,4-diazabicyclooctane bromide |
| [C6C1im]OAc | 1-Hexyl-3-methylimidazolium acetate |
| [C6C1im]Phe | 1-Hexyl-3-methylimidazolium phenolate |
| [C6DABCO]Br | 1-Hexyl-1,4-diazabicyclooctane bromide |
| [C8C1im]Cl | 1-Octyl-3-methylimidazolium chloride |
| [C8C1im]OAc | 1-Octyl-3-methylimidazolium acetate |
| [C8C1im]Phe | 1-Octyl-3-methylimidazolium phenolate |
| [C8DABCO]Br | 1-Octyl-1,4-diazabicyclooctane bromide |
| [C9(C1im)2][NTf2]2 | 1,9-Bis(3-methylimidazolium-1-yl)nonane bis(trifluoromethane)sulfonimide |
| [Ch]Arg | Cholinium arginate |
| [Ch]Gl | Cholinium glycolate |
| [Ch]Lev | Cholinium levulinate |
| [Ch]Lys | Cholinium lysinate |
| [Ch]Mal | Cholinium malonate |
| [Ch]OAc | Cholinium acetate |
| [DBNH]OAc | 1,5-Diaza-bicyclo[4.3.0]non-5-enium |
| [DBNH]OPr | 1,5-Diazabicyclo[4.3.0]non-5-enium propionate |
| [DBUH]OAc | 1,5-Diaza-bicyclo[4.3.0]undec-5-enium acetate |
| [H2N(CH2CH2OH)2]OAc | Bis(2-hydroxyethyl)methylammonium acetate |
| [H2N(CH2CH2OH)2]OBut | Bis(2-hydroxyethyl)methylammonium butanoate |
| [H2N(CH2CH2OH)2]OEt | Bis(2-hydroxyethyl)methylammonium ethanoate |
| [H2N(CH2CH2OH)2]OiBut | Bis(2-hydroxyethyl)methylammonium isobutyrate |
| [H2N(CH2CH2OH)2]OPent | Bis(2-hydroxyethyl)methylammonium pentanoate |
| [H3N(CH2CH2OH)]OHex | 2-Hydroxyethylammonium hexanoate |
| [H3N(CH2CH2OH)]NO3 | 2-Hydroxyethylammonium nitrate |
| [H3N(CH2CH2OH)]OAc | 2-Hydroxyethylammonium acetate |
| [H3N(CH2CH2OH)]OBut | 2-Hydroxyethylammonium butanoate |
| [H3N(CH2CH2OH)]OF | 2-Hydroxyethylammonium formate |
| [H3N(CH2CH2OH)]OPr | 2-Hydroxyethylammonium propanoate |
| [HC1im]Cl | 1-Methylimidazolium chloride |
| [HC1im]HSO4 | 1-Methylimidazolium hydrogen sulfate |
| [HC1im]OAc | 1-Methylimidazolium acetate |
| [HC1im]OEt | 1-Methylimidazolium ethanoate |
| [HC1im]OF | 1-Methylimidazolium formate |
| [HC1im]OPr | 1-Methylimidazolium propanoate |
| [HC4im]HSO4 | 1-Butylimidazolium hydrogen sulfate |
| [HN(CH2CH2OH)3]MeSO3 | Tris(2-hydroxyethyl)methylammonium methanesulfonate |
| [HN(CH2CH2OH)3]OAc | Tris(2-hydroxyethyl)methylammonium acetate |
| [HN(CH2CH2OH)3]OBut | Tris(2-hydroxyethyl)methylammonium butanoate |
| [HOC2C1im]Cl | 1-Hydroxyethyl3-methylimidazolium chloride |
| [HOC2C1im]ClO4 | 1-Hydroxyethyl3-methylimidazolium perchlorate |
| [HOC2C1im]N(CN)2 | 1-Hydroxyethyl3-methylimidazolium dicyanamide |
| [HOC2C1im]NO3 | 1-Hydroxyethyl3-methylimidazolium nitrate |
| [HOC2C1im]NTf2 | 1-Hydroxyethyl3-methylimidazolium bis(trifluoromethane)sulfonimide |
| [HOC2C1im]PF6 | 1-Hydroxyethyl3-methylimidazolium hexafluorophosphate |
| [mDBN]DMP | 5-Methyl-1,5-diazabicyclo[4.3.0]non-5-enium dimethylphosphate |
| [mTBDH]NTf2 | 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-enium bis(trifluoromethane)sulfonimide |
| [mTBDH]OAc | 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-enium acetate |
| [N1140]HSO4 | N,N-Dimethyl-N-butylammonium hydrogen sulfate |
| [N2000]Gl | Ethylammonium glicolate |
| [N2000]NO3 | Ethylammonium nitrate |
| [N2000]OAc | Ethylammonium acetate |
| [N2000]OF | Ethylammonium formate |
| [N2200]OF | Diethylammonium formate |
| [N2220]HSO4 | Triethylammonium hydrogensulfate |
| [N2222]Cl | Tetraethylammonium chloride |
| [N3333]Cl | Tetrapropylammonium chloride |
| [N4000]OF | Butylammonium formate |
| [N4444]Br | Tetrabutylammonium bromide |
| [N4444]Cl | Tetrabutylammonium chloride |
| [N5000]OF | Pentylammonium formate |
| [N8880]TfO | Trioctylammonium triflate |
| [P4440]MsO | Tributylaphosphonium mesilate |
| [P4440]NO3 | Tributylaphosphonium nitrate |
| [P4440]NTf2 | Tributylphosphonium bis(trifluoromethane)sulfonimide |
| [P4440]TfO | Tributylphosphonium triflate |
| [P4444]Br | Tetrabutylphosphonium bromide |
| [P8880]TfO | Tetraoctylphosphonium |
| [Ph3NH]TfO | Triphenylammonium triflate |
| AILs | Aprotic Ionic Liquids |
| AL | Alkali lignin |
| BA | Boric acid |
| Bet | Betaine |
| BetHCl | Betaine hydrochloride |
| ChBr | Cholinium bromide |
| ChCl | Cholinium chloride |
| ChI | Cholinium iodide |
| ChOAc | Cholinium acetate |
| ClChCl | Chlorocholinium chloride |
| CNC | Cellulose nanocrystalline |
| CNF | Cellulose nanofiber |
| DA | Degrees of acetylation |
| DMAc | N,N-Dimethylacetamide |
| DMSO | Dimethyl sulfoxide |
| EG | Ethylene glycol |
| G | Guaiacyl lignin subunit |
| Gly | Glycerol |
| GVL | γ-Valerolactone |
| H | p-Hydroxyphenyl lignin subunit |
| H2SO4 | Sulfuric acid |
| HFIP | Hexafluoroisopropanol |
| Im | Imidazole |
| LA | Lactic acid |
| MA | Malic acid |
| NMMO·H2O | N-Methylmorpholine-N-oxide monohydrate |
| OA | Oxalic acid |
| OES | Organic electrolyte solutions |
| PA | Propionic acid |
| PEG-200 | Polyethylene glycol-200 |
| PILs | Protic Ionic Liquids |
| pTSA | p-Toluenesulfonic acid |
| S | Syringyl lignin subunit |
| U | Urea |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Afonso gratefully acknowledges the financial support of FCT/MCTES (Portugal) for PhD fellowship 2021.07949.BD. This work was financed by FCT/MCTES (Portugal) through the project PTDC/EAM-AMB/2023/2021, Centro de Química Estrutural (UIDB/00100/2020 and UIDP/00100/2020) and Institute of Molecular Sciences (LA/P/0056/2020).References
- F. Guthrie, The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 2010, vol. 17, pp. 462–482 Search PubMed.
- A. Diedrichs and J. Gmehling, Ind. Eng. Chem. Res., 2011, 50, 1757–1769 CrossRef CAS.
- H. Suzuki and H. Sunada, Chem. Pharm. Bull., 1997, 45, 1688–1693 CrossRef CAS PubMed.
- I. Pasquali, R. Bettini and F. Giordano, J. Therm. Anal. Calorim., 2007, 90, 903–907 CrossRef CAS.
- E. Palomo Del Barrio, R. Cadoret, J. Daranlot and F. Achchaq, Sol. Energy Mater. Sol. Cells, 2016, 155, 454–468 CrossRef CAS.
- H. Schmit, C. Rathgeber, P. Hennemann and S. Hiebler, J. Therm. Anal. Calorim., 2014, 117, 595–602 CrossRef CAS.
- D. Wei, S. Han and B. Wang, Fluid Phase Equilib., 2014, Complete, 84–88 CrossRef.
- A. E. Hoyt and S. J. Huang, J. Macromol. Sci., 2006, 32, 1931–1945, DOI:10.1080/10601329508009371.
- G. W. Smith, Mol. Cryst. Liq. Cryst., 2007, 30, 101–107, DOI:10.1080/15421407508082845.
- R. I. Nessim, Thermochim. Acta, 2000, 343, 1–6 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- M. H. Zainal-Abidin, M. Hayyan, A. Hayyan and N. S. Jayakumar, Anal. Chim. Acta, 2017, 979, 1–23 CrossRef CAS.
- C. Li, D. Li, S. Zou, Z. Li, J. Yin, A. Wang, Y. Cui, Z. Yao and Q. Zhao, Green Chem., 2013, 15, 2793–2799 RSC.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- C. Florindo, F. S. Oliveira, L. P. N. Rebelo, A. M. Fernandes and I. M. Marrucho, ACS Sustainable Chem. Eng., 2014, 2, 2416–2425 CrossRef CAS.
- N. Rodriguez Rodriguez, A. van den Bruinhorst, L. J. B. M. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- Y. H. Choi, J. van Spronsen, Y. Dai, M. Verberne, F. Hollmann, I. W. C. E. Arends, G. J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701–1705 CrossRef CAS PubMed.
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- D. O. Abranches, M. A. R. Martins, L. P. Silva, N. Schaeffer, S. P. Pinho and J. A. P. Coutinho, Chem. Commun., 2019, 55, 10253–10256 RSC.
- N. Schaeffer, D. O. Abranches, L. P. Silva, M. A. R. Martins, P. J. Carvalho, O. Russina, A. Triolo, L. Paccou, Y. Guinet, A. Hedoux and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2021, 9, 2203–2211 CrossRef CAS.
- C. Florindo, L. G. Celia-Silva, L. F. G. Martins, L. C. Branco and I. M. Marrucho, Chem. Commun., 2018, 54, 7527–7530 RSC.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 CrossRef CAS.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- D. H. Zaitsau, G. J. Kabo, A. A. Strechan, Y. U. Paulechka, A. Tschersich, S. P. Verevkin and A. Heintz, J. Phys. Chem. A, 2006, 110, 7303–7306 CrossRef CAS PubMed.
- O. Aschenbrenner, S. Supasitmongkol, M. Taylor and P. Styring, Green Chem., 2009, 11, 1217–1221 RSC.
- Q. Zhang, K. de Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- A. van den Bruinhorst, L. J. B. M. Kollau, M. Vis, M. M. R. M. Hendrix, J. Meuldijk, R. Tuinier and A. C. C. Esteves, J. Chem. Phys., 2021, 155, 014502 CrossRef CAS.
- N. López-Salas, J. M. Vicent-Luna, S. Imberti, E. Posada, M. J. Roldán, J. A. Anta, S. R. G. Balestra, R. M. Madero Castro, S. Calero, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, ACS Sustainable Chem. Eng., 2019, 7, 17565–17573 CrossRef.
- N. López-Salas, J. M. Vicent-Luna, E. Posada, S. Imberti, R. M. Madero-Castro, S. Calero, C. O. Ania, R. J. Jiménez-Rioboó, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, ACS Sustainable Chem. Eng., 2020, 8, 12120–12131 CrossRef.
- M. E. di Pietro, O. Hammond, A. van den Bruinhorst, A. Mannu, A. Padua, A. Mele and M. Costa Gomes, Phys. Chem. Chem. Phys., 2021, 23, 107–111 RSC.
- Y. Marcus, ACS Sustainable Chem. Eng., 2017, 5, 11780–11787 CrossRef CAS.
- H. Yu, Z. Xue, X. Lan, Q. Liu, R. Shi and T. Mu, Cellulose, 2020, 27, 6175–6188 CrossRef.
- D. Deng, C. Zhang, X. Deng and L. Gong, Energy Fuels, 2019, 34, 665–671 CrossRef.
- B. Kabane and G. G. Redhi, J. Mol. Liq., 2020, 311, 113216 CrossRef CAS.
- Z. L. Li, L. sen Zhou, Y. H. Wei, H. L. Peng and K. Huang, Ind. Eng. Chem. Res., 2020, 59, 13696–13705 CrossRef CAS.
- S. Wang, Z. Zhu, D. Hao, T. Su, C. Len, W. Ren and H. Lü, J. CO2 Util., 2020, 40, 101250 CrossRef CAS.
- W. J. Jiang, J. B. Zhang, Y. T. Zou, H. L. Peng and K. Huang, ACS Sustainable Chem. Eng., 2020, 8, 13408–13417 CrossRef CAS.
- K. Sheng, D. Li and Y. Kang, J. Mol. Liq., 2021, 337, 116432 CrossRef CAS.
- T. Zhao, J. Liang, Y. Zhang, Y. Wu and X. Hu, Chem. Commun., 2018, 54, 8964–8967 RSC.
- D. Yang, S. Zhang and D. E. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 9086–9091 CrossRef CAS.
- X. Yang, Y. Zhang, F. Liu, P. Chen, T. Zhao and Y. Wu, Sep. Purif. Technol., 2020, 250, 117273 CrossRef CAS.
- D. Yang, S. Zhang, D. E. Jiang and S. Dai, Phys. Chem. Chem. Phys., 2018, 20, 15168–15173 RSC.
- D. Yang, Y. Han, H. Qi, Y. Wang and S. Dai, ACS Sustainable Chem. Eng., 2017, 5, 6382–6386 CrossRef CAS.
- G. Long, C. Yang, X. Yang, T. Zhao and M. Xu, J. Mol. Liq., 2020, 302, 112538 CrossRef CAS.
- Z. L. Li, L. sen Zhou, Y. H. Wei, H. L. Peng and K. Huang, Ind. Eng. Chem. Res., 2020, 59, 13696–13705 CrossRef CAS.
- G. Long, C. Yang, X. Yang, T. Zhao, F. Liu and J. Cao, ACS Sustainable Chem. Eng., 2020, 8, 2608–2613 CrossRef CAS.
- W. J. Jiang, J. B. Zhang, Y. T. Zou, H. L. Peng and K. Huang, ACS Sustainable Chem. Eng., 2020, 8, 13408–13417 CrossRef CAS.
- H. Zhang, J. M. Vicent-Luna, S. Tao, S. Calero, R. J. Jiménez Riobóo, M. L. Ferrer, F. del Monte and M. C. Gutiérrez, ACS Sustainable Chem. Eng., 2022, 10, 1232–1245 CrossRef CAS.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS.
- A. P. Abbott, K. J. Edler and A. J. Page, J. Chem. Phys., 2021, 155, 150401 CrossRef CAS PubMed.
- J. C. Sternberg, J. Chem. Educ., 1957, 34, 442 CrossRef CAS.
- W. Chen, A. J. Haslam, A. Macey, U. v. Shah and C. Brechtelsbauer, J. Chem. Educ., 2016, 93, 920–926 CrossRef CAS.
- C. L. Yaws and M. A. Satyro, The Yaws Handbook of Vapor Pressure, 2015, pp. 1–314 Search PubMed.
- K. Xin, I. Roghair, F. Gallucci and M. van Sint Annaland, J. Mol. Liq., 2021, 325, 115227 CrossRef CAS.
- A. M. Bernhard, I. Czekaj, M. Elsener, A. Wokaun and O. Kröcher, J. Phys. Chem. A, 2011, 115, 2581–2589 CrossRef CAS.
- C. H. J. T. Dietz, J. T. Creemers, M. A. Meuleman, C. Held, G. Sadowski, M. van S. Annaland, F. Gallucci and M. C. Kroon, ACS Sustainable Chem. Eng., 2019, 7, 4047–4057 CrossRef CAS.
- K. Shahbaz, F. S. Mjalli, G. Vakili-Nezhaad, I. M. AlNashef, A. Asadov and M. M. Farid, J. Mol. Liq., 2016, 222, 61–66 CrossRef CAS.
- S. Ravula, N. E. Larm, M. A. Mottaleb, M. P. Heitz and G. A. Baker, ChemEngineering, 2019, 3, 42 CrossRef CAS.
- J. N. Canongia Lopes and P. N. Rebelo, Phys. Chem. Chem. Phys., 2010, 12, 1948–1952 RSC.
- X. Zhu, Y. Wang and H. Li, Phys. Chem. Chem. Phys., 2011, 13, 17445–17448 RSC.
- F. Lima, C. H. J. T. Dietz, A. J. D. Silvestre, L. C. Branco, J. C. Lopes, F. Gallucci, K. Shimizu, C. Held and I. M. Marrucho, J. Phys. Chem. B, 2020, 124, 10386–10397 CrossRef CAS PubMed.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Phys. Chem. Chem. Phys., 2018, 20, 8382–8402 RSC.
- W. Chen, Z. Xue, J. Wang, J. Jiang, X. Zhao and T. Mu, Acta Phys.-Chim. Sin., 2018, 34, 904–911 CAS.
- H. Ghaedi, M. Ayoub, S. Sufian, B. Lal and Y. Uemura, J. Mol. Liq., 2017, 242, 395–403 CrossRef CAS.
- H. Ghaedi, M. Ayoub, S. Sufian, S. M. Hailegiorgis, G. Murshid and S. N. Khan, J. Chem. Thermodyn., 2018, 116, 50–60 CrossRef CAS.
- M. F. Majid, H. F. Mohd Zaid, C. F. Kait, N. A. Ghani and K. Jumbri, J. Mol. Liq., 2019, 294, 111588 CrossRef CAS.
- Y. Chen and T. Mu, Encyclopedia of Ionic Liquids, 2020, pp. 1–13 Search PubMed.
- C. Florindo, A. J. S. McIntosh, T. Welton, L. C. Branco and I. M. Marrucho, Phys. Chem. Chem. Phys., 2017, 20, 206–213 RSC.
- E. Buncel and S. Rajagopal, Acc. Chem. Res., 2002, 23, 226–231 CrossRef.
- C. Reichardt, Chem. Rev., 2002, 94, 2319–2358 CrossRef.
- A. K. Dwamena and D. E. Raynie, J. Chem. Eng. Data, 2020, 65, 640–646 CrossRef CAS.
- D. Kundu, P. S. Rao and T. Banerjee, Ind. Eng. Chem. Res., 2020, 59, 11329–11339 CrossRef CAS.
- P. P. Madeira, H. Passos, J. Gomes, J. A. P. Coutinho and M. G. Freire, Phys. Chem. Chem. Phys., 2017, 19, 11011 RSC.
- A. R. R. Teles, E. v. Capela, R. S. Carmo, J. A. P. Coutinho, A. J. D. Silvestre and M. G. Freire, Fluid Phase Equilib., 2017, 448, 15–21 CrossRef CAS PubMed.
- H. Schneider, Y. Badrieh, Y. Migron and Y. Marcus, Z. Phys. Chem., 1992, 177, 143–156 CrossRef CAS.
- J. Sherwood, J. Granelli, C. R. McElroy and J. H. Clark, Molecules, 2019, 24, 2209 CrossRef CAS.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- K. Fumino, S. Reimann and R. Ludwig, Phys. Chem. Chem. Phys., 2014, 16, 21903–21929 RSC.
- D. Yalcin, C. J. Drummond and T. L. Greaves, Phys. Chem. Chem. Phys., 2019, 22, 114–128 RSC.
- S. K. Shukla, N. D. Khupse and A. Kumar, Phys. Chem. Chem. Phys., 2012, 14, 2754–2761 RSC.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 2002, 48, 2877–2887 CrossRef.
- C. Laurence, P. Nicolet, M. T. Dalati, J. L. M. Abboud and R. Notario, J. Phys. Chem., 2002, 98, 5807–5816 CrossRef.
- R. S. Moog, D. D. Kim, J. J. Oberle and S. G. Ostrowski, J. Phys. Chem. A, 2004, 108, 9294–9301 CrossRef CAS.
- A. Pandey and S. Pandey, J. Phys. Chem. B, 2014, 118, 14652–14661 CrossRef CAS PubMed.
- K. Arora, G. Singh, G. Singh and T. S. Kang, ACS Omega, 2018, 3, 13387–13398 CrossRef PubMed.
- K. L. E. Kaiser, Environ. Health Perspect., 1998, 106, 583 CAS.
- A. K. Halder and M. N. D. S. Cordeiro, ACS Sustainable Chem. Eng., 2019, 7, 10649–10660 CrossRef CAS.
- Microtox M500 ® Industry-leading toxicity detection, https://www.modernwater.com/assets/downloads/Brochures/MW_Factsheet_MicroM500_LOWRES.pdf, (accessed 4 December 2021).
- P. de Morais, F. Gonçalves, J. A. P. Coutinho and S. P. M. Ventura, ACS Sustainable Chem. Eng., 2015, 3, 3398–3404 CrossRef CAS.
- D. Lapeña, D. Errazquin, L. Lomba, C. Lafuente and B. Giner, Environ. Sci. Pollut. Res., 2021, 28, 8812–8821 CrossRef.
- I. P. E. Macário, F. Jesus, J. L. Pereira, S. P. M. Ventura, A. M. M. Gonçalves, J. A. P. Coutinho and F. J. M. Gonçalves, Chemosphere, 2018, 212, 890–897 CrossRef PubMed.
- I. P. E. Macário, S. P. M. Ventura, J. L. Pereira, A. M. M. Gonçalves, J. A. P. Coutinho and F. J. M. Gonçalves, Ecotoxicol. Environ. Saf., 2018, 165, 597–602 CrossRef PubMed.
- R. N. Das, T. E. Sintra, J. A. P. Coutinho, S. P. M. Ventura, K. Roy and P. L. A. Popelier, Toxicol. Res., 2016, 5, 1388–1399 CrossRef CAS PubMed.
- K. L. E. Kaiser and V. S. Palabrica, Water Qual. Res. J., 1991, 26, 361–431 CrossRef CAS.
- Y. Ilieva, L. Dimitrova, M. M. Zaharieva, M. Kaleva, P. Alov, I. Tsakovska, T. Pencheva, I. P. el Tibi, H. Najdenski and I. Pajeva, Toxics, 2021, 9, 92 CrossRef CAS PubMed.
- I. P. E. Macário, H. Oliveira, A. C. Menezes, S. P. M. Ventura, J. L. Pereira, A. M. M. Gonçalves, J. A. P. Coutinho and F. J. M. Gonçalves, Sci. Rep., 2019, 9, 1–9 CrossRef.
- R. Arunkumar, A. N. Abraham, R. Shukla, C. J. Drummond and T. L. Greaves, J. Mol. Liq., 2020, 314, 113602 CrossRef CAS.
- M. McLaughlin, M. A. Gilea, M. J. Earle, K. R. Seddon, B. F. Gilmore and S. A. Kelly, Chemosphere, 2021, 270, 129432 CrossRef CAS.
- M. Benlebna, M. Ruesgas-Ramón, B. Bonafos, G. Fouret, F. Casas, C. Coudray, E. Durand, M. Cruz Figueroa-Espinoza and C. Feillet-Coudray, J. Agric. Food Chem., 2018, 66, 6205–6212 CrossRef CAS PubMed.
- Y. Dai, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef CAS PubMed.
- J. Zhang, Q. Wang and Z. Cao, Chin. Phys. B, 2020, 29, 087804 CrossRef CAS.
- O. S. Hammond, D. T. Bowron, K. J. Edler, S. Hammond, K. J. Edler and D. T. Bowron, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. Ma, A. Laaksonen, C. Liu, X. Lu and X. Ji, Chem. Soc. Rev., 2018, 47, 8685–8720 RSC.
- Z. Fei, F. D. Bobbink, E. Pəunescu, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2015, 54, 10504–10512 CrossRef CAS PubMed.
- J. M. Vicent-Luna, D. Dubbeldam, P. Gómez-Álvarez and S. Calero, ChemPhysChem, 2016, 17, 380–386 CrossRef CAS PubMed.
- P. Makoś, E. Słupek and J. Gębicki, Microchem. J., 2020, 152, 104384 CrossRef.
- C. Florindo, L. C. Branco and I. M. Marrucho, Fluid Phase Equilib., 2017, 448, 135–142 CrossRef CAS.
- H. S. Salehi, O. A. Moultos and T. J. H. Vlugt, J. Phys. Chem. B, 2021, 125, 12303–12314 CrossRef CAS PubMed.
- L. P. Silva, L. Fernandez, J. H. F. Conceição, M. A. R. Martins, A. Sosa, J. Ortega, S. P. Pinho and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2018, 6, 10724–10734 CrossRef CAS.
- V. Zullo, A. Iuliano and L. Guazzelli, Molecules, 2021, 26, 2052 CrossRef CAS PubMed.
- C. L. Boldrini, N. Manfredi, F. M. Perna, V. Capriati and A. Abbotto, ChemElectroChem, 2020, 7, 1707–1712 CrossRef CAS.
- A. Brzęczek-Szafran, B. Gaida, A. Blacha-Grzechnik, K. Matuszek and A. Chrobok, Int. J. Mol. Sci., 2021, 22, 10426 CrossRef PubMed.
- A. Le Donne and E. Bodo, Biophys. Rev., 2021, 13, 147–160 CrossRef CAS.
- V. Sang Sefidi and P. Luis, Ind. Eng. Chem. Res., 2019, 58, 20181–20194 CrossRef CAS.
- P. B. Sánchez, B. González, J. Salgado, J. J. Parajó and Á. Domínguez, J. Chem. Thermodyn., 2019, 131, 517–523 CrossRef.
- A. Khan, R. Gusain, M. Sahai and O. P. Khatri, J. Mol. Liq., 2019, 293, 111444 CrossRef CAS.
- M. K. Ali, R. M. Moshikur, R. Wakabayashi, Y. Tahara, M. Moniruzzaman, N. Kamiya and M. Goto, J. Colloid Interface Sci., 2019, 551, 72–80 CrossRef CAS PubMed.
- C. Florindo, L. Romero, I. Rintoul, L. C. Branco and I. M. Marrucho, ACS Sustainable Chem. Eng., 2018, 6, 3888–3895 CrossRef CAS.
- C. Florindo, L. C. Branco and I. M. Marrucho, ChemSusChem, 2019, 12, 1549–1559 CrossRef CAS PubMed.
- Y. Wang, X. Meng, K. Jeong, S. Li, G. Leem, K. H. Kim, Y. Pu, A. J. Ragauskas and C. G. Yoo, ACS Sustainable Chem. Eng., 2020, 8, 12542–12553 CrossRef CAS.
- M. Nasrollahzadeh, M. Ghasemzadeh, H. Gharoubi and Z. Nezafat, J. Mol. Liq., 2021, 342, 117559 CrossRef CAS.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- Outlook for the Global Dissolving Pulp Market, https://www.risiinfo.com/product/outlook-for-the-global-dissolving-pulp-market/.
- World Water Day: the cost of cotton in water-challenged India, https://www.theguardian.com/sustainable-business/2015/mar/20/cost-cotton-water-challenged-india-world-water-day#:~:text=The global average water footprint,just8%2C000litresperkg.
- D. Neiva, L. Fernandes, S. Araújo, A. Lourenço, J. Gominho, R. Simões and H. Pereira, Ind. Crops Prod., 2015, 66, 89–95 CrossRef CAS.
- H. Sixta, Handbook of Paper and Board, Wiley, 2006 Search PubMed.
- P. J. Kleppe, Tappi, 1970, 53, 35–47 CAS.
- D. J. G. P. van Osch, L. J. B. M. Kollau, A. van den Bruinhorst, S. Asikainen, M. A. A. Rocha and M. C. Kroon, Phys. Chem. Chem. Phys., 2017, 19, 2636–2665 RSC.
- T. N. Kleinert, US74832068A, US3585104A, 1971.
- A. Johansson, O. Aaltonen and P. Ylinen, Biomass, 1987, 13, 45–65 CrossRef CAS.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- M. Ouellet, S. Datta, D. C. Dibble, P. R. Tamrakar, P. I. Benke, C. Li, S. Singh, K. L. Sale, P. D. Adams, J. D. Keasling, B. A. Simmons, B. M. Holmes and A. Mukhopadhyay, Green Chem., 2011, 13, 2743 RSC.
- A. J. Greer, J. Jacquemin and C. Hardacre, Molecules, 2020, 25, 5207 CrossRef CAS PubMed.
- R. Muazzam, A. M. Asim, M. Uroos, N. Muhammad and J. P. Hallett, RSC Adv., 2021, 11, 19095–19105 RSC.
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC.
- W.-C. Tu, L. Weigand, M. Hummel, H. Sixta, A. Brandt-Talbot and J. P. Hallett, Cellulose, 2020, 27, 4745–4761 CrossRef CAS.
- S. O. Anuchi, K. L. S. Campbell and J. P. Hallett, Sci. Rep., 2022, 12, 6108 CrossRef CAS PubMed.
- C. L. Chambon, T. Y. Mkhize, P. Reddy, A. Brandt-Talbot, N. Deenadayalu, P. S. Fennell and J. P. Hallett, Biotechnol. Biofuels, 2018, 11, 247 CrossRef CAS PubMed.
- C. L. Chambon, M. Chen, P. S. Fennell and J. P. Hallett, Front. Chem., 2019, 7, 246 CrossRef CAS PubMed.
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC.
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC.
- A. R. Abouelela, A. Al Ghatta, P. Verdía, M. Shan Koo, J. Lemus and J. P. Hallett, ACS Sustainable Chem. Eng., 2021, 9, 10524–10536 CrossRef CAS.
- L. Weigand, S. Mostame, A. Brandt-Talbot, T. Welton and J. P. Hallett, Faraday Discuss., 2017, 202, 331–349 RSC.
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161–5178 RSC.
- F. A. Ferrari, J. F. B. Pereira, G.-J. Witkamp and M. B. S. Forte, ACS Sustainable Chem. Eng., 2019, 7, 12779–12788 CrossRef CAS.
- F. A. Ferrari, G. P. Nogueira, T. T. Franco, M. O. S. Dias, C. K. N. Cavaliero, G. J. Witkamp, L. A. M. van der Wielen and M. B. S. Forte, Green Chem., 2021, 23, 9126–9139 RSC.
- X.-D. Hou, A.-L. Li, K.-P. Lin, Y.-Y. Wang, Z.-Y. Kuang and S.-L. Cao, Bioresour. Technol., 2018, 249, 261–267 CrossRef CAS PubMed.
- X. Liang, Y. Zhu, B. Qi, S. Li, J. Luo and Y. Wan, Bioresour. Technol., 2021, 336, 125312 CrossRef CAS PubMed.
- Z.-J. Huang, G.-J. Feng, K.-P. Lin, F.-L. Pu, Y.-M. Tan, W.-C. Tu, Y.-L. Han, X.-D. Hou, H.-M. Zhang and Y. Zhang, Ind. Crops Prod., 2020, 152, 112515 CrossRef CAS.
- W. Lin, S. Xing, Y. Jin, X. Lu, C. Huang and Q. Yong, Bioresour. Technol., 2020, 306, 123163 CrossRef CAS.
- E. S. Morais, A. M. Da Costa Lopes, M. G. Freire, C. S. R. Freire and A. J. D. Silvestre, ChemSusChem, 2021, 14, 686–698 CrossRef CAS PubMed.
- A. K. Kumar, S. Sharma, G. Dixit, E. Shah, A. Patel and G. Boczkaj, Biofuels, Bioprod. Biorefin., 2020, 14, 746–763 CrossRef CAS.
- K. H. Kim, T. Dutta, J. Sun, B. Simmons and S. Singh, Green Chem., 2018, 20, 809–815 RSC.
- S. L. Bradbury and W. B. Jakoby, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2373–2376 CrossRef CAS PubMed.
- R. Wahlström, J. Hiltunen, M. Pitaluga de Souza Nascente Sirkka, S. Vuoti and K. Kruus, RSC Adv., 2016, 6, 68100–68110 RSC.
- V. Sharma, P. Nargotra, S. Sharma and B. K. Bajaj, Renewable Energy, 2021, 163, 1910–1922 CrossRef CAS.
- B. Ai, W. Li, J. Woomer, M. Li, Y. Pu, Z. Sheng, L. Zheng, A. Adedeji, A. J. Ragauskas and J. Shi, Green Chem., 2020, 22, 6372–6383 RSC.
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef.
- D. Huang, R. Li, P. Xu, T. Li, R. Deng, S. Chen and Q. Zhang, Chem. Eng. J., 2020, 402, 126237 CrossRef CAS.
- D. Huang, R. Li, P. Xu, T. Li, R. Deng, S. Chen and Q. Zhang, Chem. Eng. J., 2020, 402, 126237 CrossRef CAS.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- T. Dutta, N. G. Isern, J. Sun, E. Wang, S. Hull, J. R. Cort, B. A. Simmons and S. Singh, ACS Sustainable Chem. Eng., 2017, 5, 10116–10127 CrossRef CAS.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- A. K. Kumar, S. Sharma, G. Dixit, E. Shah, A. Patel and G. Boczkaj, Biofuels, Bioprod. Biorefin., 2020, 14, 746–763 CrossRef CAS.
- L. Matsakas, V. Raghavendran, O. Yakimenko, G. Persson, E. Olsson, U. Rova, L. Olsson and P. Christakopoulos, Bioresour. Technol., 2019, 273, 521–528 CrossRef CAS PubMed.
- D. J. G. P. van Osch, L. J. B. M. Kollau, A. van den Bruinhorst, S. Asikainen, M. A. A. Rocha and M. C. Kroon, Phys. Chem. Chem. Phys., 2017, 19, 2636–2665 RSC.
- P. Varanasi, P. Singh, R. Arora, P. D. Adams, M. Auer, B. A. Simmons and S. Singh, Bioresour. Technol., 2012, 126, 156–161 CrossRef CAS PubMed.
- N. Sathitsuksanoh, K. M. Holtman, D. J. Yelle, T. Morgan, V. Stavila, J. Pelton, H. Blanch, B. A. Simmons and A. George, Green Chem., 2014, 16, 1236–1247 RSC.
- Y. Sun and B. Xue, Ind. Crops Prod., 2018, 123, 600–609 CrossRef CAS.
- Q.-P. Liu, X.-D. Hou, N. Li and M.-H. Zong, Green Chem., 2012, 14, 304–307 RSC.
- X.-D. Hou, Q.-P. Liu, T. J. Smith, N. Li and M.-H. Zong, PLoS One, 2013, 8, e59145 CrossRef CAS.
- Y.-X. An, M.-H. Zong, H. Wu and N. Li, Bioresour. Technol., 2015, 192, 165–171 CrossRef CAS.
- T. Dutta, G. Papa, E. Wang, J. Sun, N. G. Isern, J. R. Cort, B. A. Simmons and S. Singh, ACS Sustainable Chem. Eng., 2018, 6, 3079–3090 CrossRef CAS.
- Y.-X. An, N. Li, H. Wu, W.-Y. Lou and M.-H. Zong, ACS Sustainable Chem. Eng., 2015, 3, 2951–2958 CrossRef CAS.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- L. Weigand, S. Mostame, A. Brandt-Talbot, T. Welton and J. P. Hallett, Faraday Discuss., 2017, 202, 331–349 RSC.
- A. Ovejero-Pérez, V. Rigual, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Int. J. Biol. Macromol., 2022, 197, 131–140 CrossRef.
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161–5178 RSC.
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC.
- S. Hong, X.-J. Shen, Z. Xue, Z. Sun and T.-Q. Yuan, Green Chem., 2020, 22, 7219–7232 RSC.
- Z.-K. Wang, S. Hong, J. Wen, C.-Y. Ma, L. Tang, H. Jiang, J.-J. Chen, S. Li, X.-J. Shen and T.-Q. Yuan, ACS Sustainable Chem. Eng., 2020, 8, 1050–1057 CrossRef CAS.
- Q. Xia, Y. Liu, J. Meng, W. Cheng, W. Chen, S. Liu, Y. Liu, J. Li and H. Yu, Green Chem., 2018, 20, 2711–2721 RSC.
- Z. Guo, Q. Zhang, T. You, X. Zhang, F. Xu and Y. Wu, Green Chem., 2019, 21, 3099–3108 RSC.
- Q. Liu, X. Zhao, D. Yu, H. Yu, Y. Zhang, Z. Xue and T. Mu, Green Chem., 2019, 21, 5291–5297 RSC.
- C. Alvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133–5141 RSC.
- X. Yue, T. Suopajärvi, O. Mankinen, M. Mikola, A. Mikkelson, J. Ahola, S. Hiltunen, S. Komulainen, A. M. Kantola, V.-V. Telkki and H. Liimatainen, J. Agric. Food Chem., 2020, 68, 15074–15084 CrossRef CAS PubMed.
- X.-J. Shen, J.-L. Wen, Q.-Q. Mei, X. Chen, D. Sun, T.-Q. Yuan and R.-C. Sun, Green Chem., 2019, 21, 275–283 RSC.
- X.-J. Shen, T. Chen, H.-M. Wang, Q. Mei, F. Yue, S. Sun, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2020, 8, 2130–2137 CrossRef CAS.
- S. Hong, X.-J. Shen, B. Pang, Z. Xue, X.-F. Cao, J.-L. Wen, Z.-H. Sun, S. S. Lam, T.-Q. Yuan and R.-C. Sun, Green Chem., 2020, 22, 1851–1858 RSC.
- A. M. da Costa Lopes, J. R. B. Gomes, J. A. P. Coutinho and A. J. D. Silvestre, Green Chem., 2020, 22, 2474–2487 RSC.
- D. Smink, A. Juan, B. Schuur and S. R. A. Kersten, Ind. Eng. Chem. Res., 2019, 58, 16348–16357 CrossRef CAS.
- J. Hiltunen, L. Kuutti, S. Rovio, E. Puhakka, T. Virtanen, T. Ohra-Aho and S. Vuoti, Sci. Rep., 2016, 6, 32420 CrossRef PubMed.
- Y. Ci, F. Yu, C. Zhou, H. Mo, Z. Li, Y. Ma and L. Zang, Green Chem., 2020, 22, 8713–8720 RSC.
- Z. Chen, X. Bai, L. A. H. Zhang and C. Wan, ACS Sustainable Chem. Eng., 2020, 8, 9783–9793 CrossRef CAS.
- Z. Chen, W. D. Reznicek and C. Wan, Bioresour. Technol., 2018, 263, 40–48 CrossRef CAS PubMed.
- B. Ai, W. Li, J. Woomer, M. Li, Y. Pu, Z. Sheng, L. Zheng, A. Adedeji, A. J. Ragauskas and J. Shi, Green Chem., 2020, 22, 6372–6383 RSC.
- H. Ji and P. Lv, Green Chem., 2020, 22, 1378–1387 RSC.
- S. De, A. S. Burange and R. Luque, Green Chem., 2022, 24, 2267–2286 RSC.
- M. Sajid, U. Farooq, G. Bary, M. M. Azim and X. Zhao, Green Chem., 2021, 23, 9198–9238 RSC.
- Z. Zhou, D. Liu and X. Zhao, Renewable Sustainable Energy Rev., 2021, 146, 111169 CrossRef CAS.
- C. Zhang, R. Liu, J. Xiang, H. Kang, Z. Liu and Y. Huang, J. Phys. Chem. B, 2014, 118, 9507–9514 CrossRef CAS PubMed.
- S. Wang, A. Lu and L. Zhang, Prog. Polym. Sci., 2016, 53, 169–206 CrossRef CAS.
- J. Zhou and L. Zhang, Polym. J., 2000, 32, 866–870 CrossRef CAS.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- A.-L. Dupont, Polymer, 2003, 44, 4117–4126 CrossRef CAS.
- A. J. Hunt, A. S. Matharu, A. H. King and J. H. Clark, Green Chem., 2015, 17, 1949–1950 RSC.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- E. S. Morais, A. M. da C. Lopes, M. G. Freire, C. S. R. Freire, J. A. P. Coutinho and A. J. D. Silvestre, Molecules, 2020, 25, 3652 CrossRef CAS PubMed.
- F. Hermanutz, F. Gähr, E. Uerdingen, F. Meister and B. Kosan, Macromol. Symp., 2008, 262, 23–27 CrossRef CAS.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- A. Xu and F. Wang, Green Chem., 2020, 22, 7622–7664 RSC.
- K. M. Gupta and J. Jiang, Chem. Eng. Sci., 2015, 121, 180–189 CrossRef CAS.
- L. M. Haverhals, W. M. Reichert, H. C. De Long and P. C. Trulove, Macromol. Mater. Eng., 2010, 295, 425–430 CrossRef CAS.
- A. J. Greer, J. Jacquemin and C. Hardacre, Molecules, 2020, 25, 5207 CrossRef CAS PubMed.
- https://www.ditf.de/en/index/current/press-releases/detail/novel-hemp-based-cellulose-fibers-natural-material-from-environmentally-friendly-production.html .
- A. Michud, M. Tanttu, S. Asaadi, Y. Ma, E. Netti, P. Kääriainen, A. Persson, A. Berntsson, M. Hummel and H. Sixta, Text. Res. J., 2016, 86, 543–552 CrossRef CAS.
- Ioncell, https://ioncell.fi/.
- S. Elsayed, B. Viard, C. Guizani, S. Hellsten, J. Witos and H. Sixta, Ind. Eng. Chem. Res., 2020, 59, 20211–20220 CrossRef CAS.
- A. H. Tullo, The time is now for ionic liquids, https://cen.acs.org/materials/ionic-liquids/time-ionic-liquids/98/i5.
- G. A. Buchner, K. J. Stepputat, A. W. Zimmermann and R. Schomäcker, Ind. Eng. Chem. Res., 2019, 58, 6957–6969 CrossRef CAS.
- J. Zhang, L. Xu, J. Yu, J. Wu, X. Zhang, J. He and J. Zhang, Sci. China: Chem., 2016, 59, 1421–1429 CrossRef CAS.
- Y. Li, J. Wang, X. Liu and S. Zhang, Chem. Sci., 2018, 9, 4027–4043 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, ChemPhysChem, 2012, 13, 3126–3133 CrossRef CAS PubMed.
- B. Lu, A. Xu and J. Wang, Green Chem., 2014, 16, 1326–1335 RSC.
- A. Xu and F. Wang, Green Chem., 2020, 22, 7622–7664 RSC.
- L. K. J. Hauru, M. Hummel, A. W. T. King, I. Kilpeläinen and H. Sixta, Biomacromolecules, 2012, 13, 2896–2905 CrossRef CAS PubMed.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- Y.-B. Huang, P.-P. Xin, J.-X. Li, Y.-Y. Shao, C.-B. Huang and H. Pan, ACS Sustainable Chem. Eng., 2016, 4, 2286–2294 CrossRef CAS.
- A. J. Holding, A. Parviainen, I. Kilpeläinen, A. Soto, A. W. T. King and H. Rodríguez, RSC Adv., 2017, 7, 17451–17461 RSC.
- A. Mezzetta, S. Becherini, C. Pretti, G. Monni, V. Casu, C. Chiappe and L. Guazzelli, New J. Chem., 2019, 43, 13010–13019 RSC.
- A. J. Holding, M. Heikkilä, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2014, 7, 1422–1434 CrossRef CAS PubMed.
- S. Elsayed, S. Hellsten, C. Guizani, J. Witos, M. Rissanen, A. H. Rantamäki, P. Varis, S. K. Wiedmer and H. Sixta, ACS Sustainable Chem. Eng., 2020, 8, 14217–14227 CrossRef CAS.
- I. Schlapp-Hackl, J. Witos, K. Ojha, P. Uusi-Kyyny, V. Alopaeus and H. Sixta, Ind. Eng. Chem. Res., 2022, 61, 259–268 CrossRef CAS.
- F. M. S. Ribeiro, C. F. R. A. C. Lima, A. M. S. Silva and L. M. N. B. F. Santos, ChemPhysChem, 2018, 19, 2364–2369 CrossRef CAS PubMed.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 CrossRef CAS.
- A. Parviainen, R. Wahlström, U. Liimatainen, T. Liitiä, S. Rovio, J. K. J. Helminen, U. Hyväkkö, A. W. T. King, A. Suurnäkki and I. Kilpeläinen, RSC Adv., 2015, 5, 69728–69737 RSC.
- M. S. Miran, H. Kinoshita, T. Yasuda, Md. A. B. H. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178 RSC.
- G. Sharma, K. Takahashi and K. Kuroda, Carbohydr. Polym., 2021, 267, 118171 CrossRef CAS PubMed.
- H. Ren, C. Chen, S. Guo, D. Zhao and Q. Wang, BioResources, 2016, 11, 8457–8469 CAS.
- H. Fu, X. Wang, H. Sang, Y. Hou, X. Chen and X. Feng, J. Mol. Liq., 2020, 299, 112140 CrossRef CAS.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149 RSC.
- H. Ren, C. Chen, Q. Wang, D. Zhao and S. Guo, BioResources, 2016, 11, 5435–5451 CAS.
- Y.-L. Chen, X. Zhang, T.-T. You and F. Xu, Cellulose, 2019, 26, 205–213 CrossRef CAS.
- J. A. Sirviö, M. Visanko and H. Liimatainen, Biomacromolecules, 2016, 17, 3025–3032 CrossRef PubMed.
- Y. Liu, B. Guo, Q. Xia, J. Meng, W. Chen, S. Liu, Q. Wang, Y. Liu, J. Li and H. Yu, ACS Sustainable Chem. Eng., 2017, 5, 7623–7631 CrossRef CAS.
- X.-Q. Wu, P.-D. Liu, Q. Liu, S.-Y. Xu, Y.-C. Zhang, W.-R. Xu and G.-D. Liu, RSC Adv., 2021, 11, 14071–14078 RSC.
- H. Zhang, J. Lang, P. Lan, H. Yang, J. Lu and Z. Wang, Materials, 2020, 13, 278 CrossRef CAS PubMed.
- E. Zhou and H. Liu, Asian – J. Chem., 2014, 26, 3626–3630 CrossRef CAS.
- T.-M. Tenhunen, M. Hakalahti, J. Kouko, A. Salminen, T. Härkäsalmi, J. Pere, A. Harlin and T. Hänninen, BioResources, 2016, 11, 2492–2503 CrossRef CAS.
- V. Klar, H. Orelma, H. Rautkoski, P. Kuosmanen and A. Harlin, ACS Omega, 2018, 3, 10918–10926 CrossRef CAS PubMed.
- H. V. D. Nguyen, R. De Vries and S. D. Stoyanov, ACS Sustainable Chem. Eng., 2020, 8, 14166–14178 CrossRef CAS.
- H. Ren, R. Gong, M. Li, Y. Liu, H. Zhu, C. Wang and E. Duan, J. Mol. Liq., 2020, 312, 113282 CrossRef CAS.
- F. Liu, Z. Xue, X. Zhao, H. Mou, J. He and T. Mu, Chem. Commun., 2018, 54, 6140–6143 RSC.
- Z. Yang, T.-A. Asoh and H. Uyama, Polym. Degrad. Stab., 2019, 160, 126–135 CrossRef CAS.
- J. A. Sirviö, J. Ukkola and H. Liimatainen, Cellulose, 2019, 26, 2303–2316 CrossRef.
- X. Yang, H. Xie, H. Du, X. Zhang, Z. Zou, Y. Zou, W. Liu, H. Lan, X. Zhang and C. Si, ACS Sustainable Chem. Eng., 2019, 7, 7200–7208 CrossRef CAS.
- J. Jiang, N. C. Carrillo-Enríquez, H. Oguzlu, X. Han, R. Bi, M. Song, J. N. Saddler, R.-C. Sun and F. Jiang, ACS Sustainable Chem. Eng., 2020, 8, 7182–7191 CrossRef CAS.
- Z. Ling, J. V. Edwards, Z. Guo, N. T. Prevost, S. Nam, Q. Wu, A. D. French and F. Xu, Cellulose, 2019, 26, 861–876 CrossRef CAS.
- L. Douard, J. Bras, T. Encinas and M. N. Belgacem, Carbohydr. Polym., 2021, 252, 117136 CrossRef CAS PubMed.
- W. Yu, C. Wang, Y. Yi, H. Wang, Y. Yang, L. Zeng and Z. Tan, Cellulose, 2021, 28, 175–188 CrossRef CAS.
- J. A. Sirviö, K. Hyypiö, S. Asaadi, K. Junka and H. Liimatainen, Green Chem., 2020, 22, 1763–1775 RSC.
- W. Yu, C. Wang, Y. Yi, H. Wang, L. Zeng, M. Li, Y. Yang and Z. Tan, ACS Omega, 2020, 5, 5580–5588 CrossRef CAS PubMed.
- S. Wang, X. Peng, L. Zhong, S. Jing, X. Cao, F. Lu and R. Sun, Carbohydr. Polym., 2015, 117, 133–139 CrossRef CAS PubMed.
- F. M. Gírio, C. Fonseca, F. Carvalheiro, L. C. Duarte, S. Marques and R. Bogel-Łukasik, Bioresour. Technol., 2010, 101, 4775–4800 CrossRef PubMed.
- X. Liu, Q. Lin, Y. Yan, F. Peng, R. Sun and J. Ren, Curr. Med. Chem., 2019, 26, 2430–2455 CrossRef CAS PubMed.
- K. S. Mikkonen, K. Parikka, A. Ghafar and M. Tenkanen, Trends Food Sci. Technol., 2013, 34, 124–136 CrossRef CAS.
- S. K. Bhaduri, I. N. Ghosh and N. L. Deb Sarkar, Ind. Crops Prod., 1995, 4, 79–84 CrossRef CAS.
- A. Escalante, A. Gonçalves, A. Bodin, A. Stepan, C. Sandström, G. Toriz and P. Gatenholm, Carbohydr. Polym., 2012, 87, 2381–2387 CrossRef CAS.
- P. Oinonen, D. Areskogh and G. Henriksson, Carbohydr. Polym., 2013, 95, 690–696 CrossRef CAS PubMed.
- J. Berglund, D. Mikkelsen, B. M. Flanagan, S. Dhital, S. Gaunitz, G. Henriksson, M. E. Lindström, G. E. Yakubov, M. J. Gidley and F. Vilaplana, Nat. Commun., 2020, 11, 4692 CrossRef CAS PubMed.
- F. Delbecq, Y. Wang, A. Muralidhara, K. El Ouardi, G. Marlair and C. Len, Front. Chem., 2018, 6, 146 CrossRef PubMed.
- W. Geng, R. A. Venditti, J. J. Pawlak, T. De Assis, R. W. Gonzalez, R. B. Phillips and H. Chang, Biofuels, Bioprod. Biorefin., 2020, 14, 225–241 CrossRef CAS.
- A. M. da Costa Lopes, K. G. João, D. F. Rubik, E. Bogel-Łukasik, L. C. Duarte, J. Andreaus and R. Bogel-Łukasik, Bioresour. Technol., 2013, 142, 198–208 CrossRef CAS PubMed.
- C. Froschauer, M. Hummel, M. Iakovlev, A. Roselli, H. Schottenberger and H. Sixta, Biomacromolecules, 2013, 14, 1741–1750 CrossRef CAS PubMed.
- A. Roselli, M. Hummel, J. Vartiainen, K. Nieminen and H. Sixta, Carbohydr. Polym., 2017, 168, 121–128 CrossRef CAS PubMed.
- P. Wollboldt, M. Strach, A. Russler, S. Jankova and H. Sixta, Holzforschung, 2017, 71, 611–618 CAS.
- A. Roselli, M. Hummel, A. Monshizadeh, T. Maloney and H. Sixta, Cellulose, 2014, 21, 3655–3666 CrossRef CAS.
- B. Yang, X. Qin, C. Duan, Z. He and Y. Ni, Cellulose, 2021, 28, 1311–1320 CrossRef CAS.
- S. Shokri, S. Hedjazi, H. Q. Lê, A. Abdulkhani and H. Sixta, Carbohydr. Polym., 2022, 288, 119364 CrossRef CAS PubMed.
- F. Cheng, H. Wang, G. Chatel, G. Gurau and R. D. Rogers, Bioresour. Technol., 2014, 164, 394–401 CrossRef CAS PubMed.
- H.-Y. Li, X. Chen, Y.-J. Li, X.-F. Cao, S.-N. Sun and R.-C. Sun, Sep. Purif. Technol., 2018, 191, 364–369 CrossRef CAS.
- N. Sun, R. Parthasarathi, A. M. Socha, J. Shi, S. Zhang, V. Stavila, K. L. Sale, B. A. Simmons and S. Singh, Green Chem., 2014, 16, 2546–2557 RSC.
- G. Colombo Dugoni, A. Mezzetta, L. Guazzelli, C. Chiappe, M. Ferro and A. Mele, Green Chem., 2020, 22, 8680–8691 RSC.
- H. Yu, Z. Xue, X. Lan, Q. Liu, R. Shi and T. Mu, Cellulose, 2020, 27, 6175–6188 CrossRef.
- L.-X. Zhang, H. Yu, H.-B. Yu, Z. Chen and L. Yang, Chin. Chem. Lett., 2014, 25, 1132–1136 CrossRef CAS.
- Q. Yu, A. Zhang, W. Wang, L. Chen, R. Bai, X. Zhuang, Q. Wang, Z. Wang and Z. Yuan, Bioresour. Technol., 2018, 247, 705–710 CrossRef CAS PubMed.
- X. Liu, W. Wei and S. Wu, Bioresour. Technol., 2019, 290, 121775 CrossRef CAS PubMed.
- E. S. Morais, P. V. Mendonça, J. F. J. Coelho, M. G. Freire, C. S. R. Freire, J. A. P. Coutinho and A. J. D. Silvestre, ChemSusChem, 2018, 11, 753–762 CrossRef CAS PubMed.
- R. A. Sequeira, D. Mondal and K. Prasad, Green Chem., 2021, 23, 8821–8847 RSC.
- M. Yadav, P. Goswami, K. Paritosh, M. Kumar, N. Pareek and V. Vivekanand, Bioresour. Bioprocess., 2019, 6, 8 CrossRef.
- Y. Zhong, J. Cai and L.-N. Zhang, Chin. J. Polym. Sci., 2020, 38, 1047–1060 CrossRef CAS.
- I. Latańska, P. Rosiak, P. Paul, W. Sujka and B. Kolesińska, Modulating the Physicochemical Properties of Chitin and Chitosan as a Method of Obtaining New Biological Properties of Biodegradable Materials, IntechOpen, 2021 Search PubMed.
- J. L. Shamshina, P. Berton and R. D. Rogers, ACS Sustainable Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- S. I. Ahmad, R. Ahmad, M. S. Khan, R. Kant, S. Shahid, L. Gautam, G. M. Hasan and Md. I. Hassan, Int. J. Biol. Macromol., 2020, 164, 526–539 CrossRef CAS PubMed.
- Global Chitin Market Size By Derivative Type, By End-User, By Geographic Scope And Forecast, https://www.verifiedmarketresearch.com/product/chitin-market/.
- J. Dai, F. Li and X. Fu, ChemSusChem, 2020, 13, 6498–6508 CrossRef CAS PubMed.
- D. Zhou, D. Shen, W. Lu, T. Song, M. Wang, H. Feng, J. Shentu and Y. Long, Molecules, 2020, 25, 541 CrossRef CAS PubMed.
- J. C. Roy, F. Salaün, S. Giraud, A. Ferri, G. Chen and J. Guan, in Solubility of Polysaccharides, InTech, 2017 Search PubMed.
- D. G. Ramírez-Wong, M. Ramírez-Cardona, R. J. Sánchez-Leija, A. Rugerio, R. A. Mauricio-Sánchez, M. A. Hernández-Landaverde, A. Carranza, J. A. Pojman, A. M. Garay-Tapia, E. Prokhorov, J. D. Mota-Morales and G. Luna-Bárcenas, Green Chem., 2016, 18, 4303–4311 RSC.
- E. S. Morais, A. M. da C. Lopes, M. G. Freire, C. S. R. Freire, J. A. P. Coutinho and A. J. D. Silvestre, Molecules, 2020, 25, 3652 CrossRef CAS PubMed.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968 RSC.
- J. Kadokawa, RSC Adv., 2015, 5, 12736–12746 RSC.
- A. Takegawa, M. Murakami, Y. Kaneko and J. Kadokawa, Carbohydr. Polym., 2010, 79, 85–90 CrossRef CAS.
- R. A. Mantz, D. M. Fox, J. M. Green, P. A. Fylstra, H. C. De Long and P. C. Trulove, Z. Naturforsch., A: Phys. Sci., 2007, 62, 275–280 CrossRef CAS.
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC.
- J. Sun, J. Shi, N. V. S. N. M. Konda, D. Campos, D. Liu, S. Nemser, J. Shamshina, T. Dutta, P. Berton, G. Gurau, R. D. Rogers, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2017, 10, 154 CrossRef PubMed.
- G. Gurau, H. Wang, Y. Qiao, X. Lu, S. Zhang and R. D. Rogers, Pure Appl. Chem., 2012, 84, 745–754 CrossRef CAS.
- M. Shimo, M. Abe and H. Ohno, ACS Sustainable Chem. Eng., 2016, 4, 3722–3727 CrossRef CAS.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149 RSC.
- SAFETY DATA SHEET of Thiurea, https://www.sigmaaldrich.com/IT/en/product/sial/t8656?context=product.
- S. Idenoue, K. Yamamoto and J. Kadokawa, ChemEngineering, 2019, 3, 90 CrossRef CAS.
- T. Uto, S. Idenoue, K. Yamamoto and J. Kadokawa, Phys. Chem. Chem. Phys., 2018, 20, 20669–20677 RSC.
- J. Horinaka, Y. Urabayashi, T. Takigawa and M. Ohmae, J. Appl. Polym. Sci., 2013, 130, 2439–2443 CrossRef CAS.
- H. B. Wineinger, A. Kelly, J. L. Shamshina and R. D. Rogers, Green Chem., 2020, 22, 6001–6007 RSC.
- Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer), 2008, 49, 2321–2327 CrossRef CAS.
- C. Florindo, A. J. S. McIntosh, T. Welton, L. C. Branco and I. M. Marrucho, Phys. Chem. Chem. Phys., 2018, 20, 206–213 RSC.
- W. M. Reichert, A. E. Visser, R. P. Swatloski and R. D. Rogers. Presented at the 222nd ACS National Meeting, Chicago, IL, August 26–30, 2001, Abstract I&EC025, I&EC 052.
- O. Zavgorodnya, J. L. Shamshina, J. R. Bonner and R. D. Rogers, ACS Sustainable Chem. Eng., 2017, 5, 5512–5519 CrossRef CAS.
- P. S. Barber, C. S. Griggs, J. R. Bonner and R. D. Rogers, Green Chem., 2013, 15, 601 RSC.
- J. L. Shamshina, O. Zavgorodnya, J. R. Bonner, G. Gurau, T. Di Nardo and R. D. Rogers, ChemSusChem, 2017, 10, 106–111 CrossRef CAS PubMed.
- L. Deng and L.-M. Zhang, Colloids Surf., A, 2020, 586, 124220 CrossRef CAS.
- X. Shen, J. L. Shamshina, P. Berton, J. Bandomir, H. Wang, G. Gurau and R. D. Rogers, ACS Sustainable Chem. Eng., 2016, 4, 471–480 CrossRef CAS.
- C. A. King, J. L. Shamshina, O. Zavgorodnya, T. Cutfield, L. E. Block and R. D. Rogers, ACS Sustainable Chem. Eng., 2017, 5, 11660–11667 CrossRef CAS.
- H. Ma, B. S. Hsiao and B. Chu, Polymer, 2011, 52, 2594–2599 CrossRef CAS.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- T. Di Nardo, C. Hadad, A. Nguyen Van Nhien and A. Moores, Green Chem., 2019, 21, 3276–3285 RSC.
- H. Hirayama, J. Yoshida, K. Yamamoto and J. Kadokawa, Carbohydr. Polym., 2018, 200, 567–571 CrossRef CAS PubMed.
- E. Husson, C. Hadad, G. Huet, S. Laclef, D. Lesur, V. Lambertyn, A. Jamali, S. Gottis, C. Sarazin and A. Nguyen Van Nhien, Green Chem., 2017, 19, 4122–4131 RSC.
- Q. Ma, X. Gao, X. Bi, Q. Han, L. Tu, Y. Yang, Y. Shen and M. Wang, Carbohydr. Polym., 2020, 230, 115605 CrossRef CAS PubMed.
- C. Mukesh, D. Mondal, M. Sharma and K. Prasad, Carbohydr. Polym., 2014, 103, 466–471 CrossRef CAS PubMed.
- S.-L. Cao, W.-M. Gu, W.-D. Ou-Yang, D.-C. Chen, B.-Y. Yang, L.-H. Lai, Y.-D. Wu, Y.-J. Liu, J. Zhu, W.-J. Chen, Z.-Q. Gai, X.-D. Hou, Y.-Z. Ma and Y.-X. An, Carbohydr. Polym., 2019, 213, 304–310 CrossRef CAS PubMed.
- Y. Yuan, S. Hong, H. Lian, K. Zhang and H. Liimatainen, Carbohydr. Polym., 2020, 236, 116095 CrossRef CAS PubMed.
- N. L. Mai, K. Ahn and Y.-M. Koo, Process Biochem., 2014, 49, 872–881 CrossRef CAS.
- J. Zhou, H. Sui, Z. Jia, Z. Yang, L. He and X. Li, RSC Adv., 2018, 8, 32832–32864 RSC.
- A. Isci and M. Kaltschmitt, Biomass Convers. Biorefin., 2022, 12, 197–226 CrossRef.
- K. Haerens, S. Van Deuren, E. Matthijs and B. Van der Bruggen, Green Chem., 2010, 12, 2182 RSC.
- X. Liang, Y. Fu and J. Chang, Bioresour. Technol., 2017, 245, 760–767 CrossRef CAS PubMed.
- X. Liang, Y. Fu and J. Chang, Sep. Purif. Technol., 2019, 210, 409–416 CrossRef CAS.
- W. Vereycken, S. Riaño, T. Van Gerven and K. Binnemans, ACS Sustainable Chem. Eng., 2022, 10, 946–955 CrossRef CAS PubMed.
- H. Baaqel, J. P. Hallett, G. Guillén-Gosálbez and B. Chachuat, ACS Sustainable Chem. Eng., 2022, 10, 323–331 CrossRef CAS.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- H. Baaqel, I. Díaz, V. Tulus, B. Chachuat, G. Guillén-Gosálbez and J. P. Hallett, Green Chem., 2020, 22, 3132–3140 RSC.
- A. K. Kumar, S. Sharma, G. Dixit, E. Shah, A. Patel and G. Boczkaj, Biofuels, Bioprod. Biorefin., 2020, 14, 746–763 CrossRef CAS.
- G. Zang, A. Shah and C. Wan, Biofuels, Bioprod. Biorefin., 2020, 14, 326–343 CrossRef CAS.
- J. Peng, H. Xu, W. Wang, Y. Kong, Z. Su and B. Li, Ind. Crops Prod., 2021, 172, 114036 CrossRef CAS.
- A. K. Kumar, S. Sharma, E. Shah and A. Patel, J. Mol. Liq., 2018, 260, 313–322 CrossRef CAS.
- E. S. Morais, A. M. Da Costa Lopes, M. G. Freire, C. S. R. Freire and A. J. D. Silvestre, ChemSusChem, 2021, 14, 686–698 CrossRef CAS PubMed.
- Z. Chen, X. Bai, A. Lusi and C. Wan, ACS Sustainable Chem. Eng., 2018, 6, 12205–12216 CrossRef CAS.
- L. R. Karadaghi, N. Malmstadt, K. M. Van Allsburg and R. L. Brutchey, ACS Sustainable Chem. Eng., 2021, 9, 246–253 CrossRef CAS.
- J. Haider, M. Abdul Qyyum, A. Riaz, A. Naquash, B. Kazmi, M. Yasin, A.-S. Nizami, M. Byun, M. Lee and H. Lim, Fuel, 2022, 314, 123064 CrossRef CAS.
- Y. Xie, J. Björkmalm, C. Ma, K. Willquist, J. Yngvesson, O. Wallberg and X. Ji, Appl. Energy, 2018, 227, 742–750 CrossRef CAS.
- V. Aniya, D. De, A. Singh and B. Satyavathi, Energy, 2018, 144, 1013–1025 CrossRef CAS.
- L. R. Karadaghi, N. Malmstadt, K. M. Van Allsburg and R. L. Brutchey, ACS Sustainable Chem. Eng., 2021, 9, 246–253 CrossRef CAS.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels, Bioprod. Biorefin., 2011, 5, 562–569 CrossRef CAS.
- F. A. Ferrari, G. P. Nogueira, T. T. Franco, M. O. S. Dias, C. K. N. Cavaliero, G. J. Witkamp, L. A. M. van der Wielen and M. B. S. Forte, Green Chem., 2021, 23, 9126–9139 RSC.
- A. Ovejero-Pérez, M. Ayuso, V. Rigual, J. C. Domínguez, J. García, M. V. Alonso, M. Oliet and F. Rodriguez, ACS Sustainable Chem. Eng., 2021, 9, 8467–8476 CrossRef.
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC.
- F. J. V. Gschwend, F. Malaret, S. Shinde, A. Brandt-Talbot and J. P. Hallett, Green Chem., 2018, 20, 3486–3498 RSC.
- C. L. Chambon, P. Verdía, P. S. Fennell and J. P. Hallett, Sci. Rep., 2021, 11, 15383 CrossRef CAS PubMed.
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC.
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161–5178 RSC.
- C. Florindo, F. Lima, L. C. Branco and I. M. Marrucho, ACS Sustainable Chem. Eng., 2019, 7, 14739–14746 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- C. Ruß and B. König, Green Chem., 2012, 14, 2969–2982 RSC.
- G. Charles, Cellulose Solution. US1943176A, 1934.
- Q. Zaib, M. J. Eckelman, Y. Yang and D. Kyung, Green Chem., 2022, 24, 7924–7930 RSC.
Footnote |
| † Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2023 |