
Ternary PdNiO nanocrystals-ornamented porous CeO2/onion-like carbon for electrooxidation of carbon monoxide: unveiling the effect of supports and electrolytes†
Adewale K.
Ipadeola
 ab,
Aderemi B.
Haruna
c,
Aboubakr M.
Abdullah
*a,
Rashid S.
Al-Hajri
d,
Roman
Viter
e,
Kenneth I.
Ozoemena
*c and
Kamel
Eid
*b
ab,
Aderemi B.
Haruna
c,
Aboubakr M.
Abdullah
*a,
Rashid S.
Al-Hajri
d,
Roman
Viter
e,
Kenneth I.
Ozoemena
*c and
Kamel
Eid
*b
aCenter for advanced materials, Qatar University, Doha 2713, Qatar. E-mail: bakr@qu.edu.qa
bGas Processing Center (GPC), College of Engineering, Qatar University, Doha 2713, Qatar. E-mail: kamel.eid@qu.edu.qa
cMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, PO Wits, Johannesburg 2050, South Africa. E-mail: Kenneth.ozoemena@wits.ac.za
dPetroleum and Chemical Engineering Department, Sultan Qaboos University, Muscat, Oman
eInstitute of Atomic Physics and Spectroscopy, University of Latvia, 19 Raina Blvd., LV-1586 Riga, Latvia
First published on 5th April 2023
Abstract
Porous ternary Pd-based catalysts are highly promising for various electrocatalytic applications, due to their low Pd mass, high surface area, accessible active sites, and tunable electronic structure; however, their activity for CO oxidation (COoxid) in different electrolytes is yet to be reported. Herein, ternary PdNiO nanocrystals supported on porous CeO2/onion-like carbon nanostructures (PdNiO–CeO2/OLC) were prepared by the sol–gel and impregnation approaches for electrochemical COoxid in different electrolytes at room temperature. Notably, porous CeO2/OLC acts as a support and nanoreactor for supporting the growth of PdNiO without the need for reducing agents or surfactants. The as-obtained PdNiO–CeO2/OLC had a porous sponge-like structure composed of ultra-small PdNiO nanocrystals (8 ± 1 nm) distributed on porous flower-like CeO2 and OLC, which possessed unique merits of multifunctional structure, clean surface, low mass of Pd (10 wt%), porosity (0.30 cm3 g−1), and high surface area (155.66 m2 g−1). The COoxid activity of PdNiO–CeO2/OLC was higher than those of PdNiO/OLC, PdNiO–CeO2, and commercial Pd/C catalyst, owing to the electronic interaction of PdNiO with CeO2/OLC support, which eases CO adsorption/activation alongside activation/dissociation of H2O to generate active oxygenated species (i.e., OH) needed for accelerating COoxid kinetics. The COoxid activity of PdNiO–CeO2/OLC in acidic electrolyte (HClO4) was better than those in alkaline (KOH) and neutral (NaHCO3) electrolytes. This study may open new doorways for understanding the effect of electrolytes and supports on the COoxid activity of porous ternary Pd-based catalysts.
Introduction
Electrochemical energy conversion utilizing different fuels, like water1–3 and small-molecule organic compounds,1,4,5 has emerged as one of the most promising renewable systems to replace the use of fossil fuels.1,2 However, carbon monoxide (CO) is a hindrance to the optimal operation of energy conversion systems because of its strong adsorption properties that devalue electrodes by blocking their active sites.6–9 Hence, studies have proved CO oxidation (COoxid) to be a prototypical reaction for heterogeneous catalytic processes with industrial and environmental applications,10–16 as it is converted to reusable fuels and suitable organic chemicals with efficient electrodes rather than deactivating them.17–20 Electrochemical COoxid is predominantly preferred to thermal COoxid, owing to its low energy/power consumption, high stability, and selectivity.21–24 Hence, electrochemical COoxid necessitates electrocatalysts that are tolerant to CO poisoning and convert it to useful products. Amongst the most sought-after electrocatalysts, palladium (Pd) is highly active and durable for COoxid electrocatalysis, due to the quick CO activation/adsorption and O2 dissociation energies at low overpotential.25–29 However, practical application is hindered because of the high cost, scarcity, and carbonaceous species poisoning of Pd.Efficient and durable Pd-based electrocatalysts are strategically prepared by adjusting their sizes, compositions, and morphologies supported on unique carbon platforms (i.e., onion-like carbon (OLC), carbon black (CB), carbon nanotubes, graphene etc.) to boost their electrical conductivity, charge mobility, and stability against aggregation.30–32 The impressive physicochemical merits of the OLC, including zero-dimensional concentric shell-like structures, nanosized diameter (<10 nm), high electrical conductivity and stability, low toxicity, and large surface area, are advantageous for improving Pd nanocatalysts' performance.33–35 Also, alloying Pd with earth-abundant and inexpensive transition metal oxides (M = CeO2, TiO2, Co2O3, Fe2O3, MnO2etc.) modifies its d-band centre close to the Fermi level that enable quick CO adsorption/activation on the active sites, besides the spillover effect that generates reactive oxygenated species (i.e., OH radicals) from H2O splitting,36–41 which are advantageously utilized for efficient electrocatalysis.25,42–46 For example, the addition of CeO2 to Pd/OLC (Pd–CeO2/OLC) significantly increased its hydrogen oxidation reaction (HOR) electrocatalytic activity by 13.2 and 15.0 times compared with Pd/OLC and Pd/CB, respectively, due the induced defects and modified electronic structure of Pd/OLC interfaced by CeO2.47 Hence, CeO2/OLC composite support enables great interfacial interaction with Pd for great electrocatalysis. The COoxid mechanism includes the adsorption of CO on Pd active sites and its removal by oxygen-containing species (i.e., OH). The spillover effect of co-metal oxides in the Pd-based nanocrystals promoted the generation of OH species for easy desorption of adsorbed CO, resulting in enhanced COoxid electrocatalysis.48–51 NiO/CeO2 showed high performance for electrooxidation of organic solvents (methanol, ethanol, phenol, etc.).52 The NiO/CeO2 composites due to higher concentration of surface sites promoted enhanced adsorption of CO and interaction with oxygen species.53 It was reported that Ni/Ce interface showed high electron coupling and supported adsorption of OH–, suitable for fast electrooxidation applications.54,55
Binary Pd-based nanocrystals are mostly fabricated for electrochemical COoxid. For example, Pd/Ni nanocrystals embedded on metal–organic framework-derived porous carbon nanosheets (Pd/Ni-MOF/PC) gave higher COoxid activity compared to Pd/C in acid, alkaline, and neutral conditions by factors of 5.0, 2.9, and 2.3, respectively, because of the large pores and abundant active sites of Pd/Ni–Nx in Pd/Ni-MOF/PC that enabled fast diffusion of reactant/intermediate species, CO activation/desorption and O2 dissociation at low overpotential.56 Similarly, the coordination of Pd and Co–Nx in Pd/ZIF-67/C significantly improved its COoxid electrocatalytic activity by 4.2- and 4.4-fold compared with Pd/C in HClO4 electrolyte, owing to the synergy between the catalytic merits of Pd (i.e., facile CO activation/adsorption and O2 dissociation at low potential) and physicochemical features of ZIF-67/C (i.e., porosity, rich electron density, defects, and electrical conductivity).22 Also, Rh nanoparticles were reported to effectively promote the COoxid catalysis of zeolite.57 As far as we are aware, there is no report on the use of ternary Pd-based nanocrystals for COoxid electrocatalysis, let alone using the OLC platform. Ternary Pd-based nanocrystals are expected to further improve the d-band centre, whereas OLC increases the electron/charge mobility that is maximally utilized for excellent COoxid. Although there have been great achievements in the synthesis of porous ternary Pd-based catalysts, their utilization in electrochemical COoxid is not yet reported. Also, the effect of electrolyte and support on the COoxid activity of porous ternary Pd-based electrocatalysts remains ambiguous.
This article reports the controlled fabrication of ternary PdNiO nanocrystals supported on porous CeO2/OLC nanostructures (PdNiO–CeO2/OLC). The synthetic approach entails the annealing of diamond to form OLC, then undergone sol–gel method with cerium salt to form flower-like CeO2/OLC nanostructure and then impregnation with Pd/Ni precursors to form porous sponge-like PdNiO–CeO2/OLC. The fabrication mechanism is attributed to the ability of CeO2/OLC to act as a reactor for promoting the reduction and growth of PdNiO in the absence of reducing agents or surfactants or organic solvents. The surface and bulk characterization techniques evidenced the good contact of porous PdNiO nanocrystals with 3D flower-like CeO2 and OLC nanostructures, which combine inimitable properties of the multifunctional structure, clean surface, low mass of Pd (10 wt%), porosity (0.30 cm3 g−1), and large surface area (155.66 m2 g−1). To investigate the effect of electrolytes and supports on the catalytic performance of PdNiO–CeO2/OLC, its electrochemical COoxid performance was tested and compared with that of PdNiO/OLC, PdNiO–CeO2, and commercial Pd/C in KOH, NaHCO3, and HClO4 electrolytes.
Experimental
Chemicals and materials
Cerium(II) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99%), nickel(II) chloride hexahydrate (NiCl2·6H2O, ≥99%), potassium palladium(II) chloride (K2PdCl4, ≥98%), potassium hydroxide pellets (KOH, ≥85%), anhydrous sodium bicarbonate (NaHCO3, 99.999%), perchloric acid (HClO4, 70%), and commercial Pd/C catalyst (Pd, 10 wt%) were purchased from Sigma-Aldrich Chemie GmbH (Munich, Germany).Preparation of PdNiO–CeO2/OLC, PdNiO/OLC, and PdNiO–CeO2 electrocatalysts
OLC was prepared by the direct annealing of high-purity nanodiamond powder in a muffle furnace at 1300 °C for 3 h in Ar as reported before.58 CeO2/OLC was prepared by mixing an aqueous solution of Ce(NO3)3 (1.50 g) and OLC (1.13 g) at pH 12 under magnetic stirring and heating at 80 °C for 1.5 h. The obtained black precipitate was centrifuged at 7000 rpm, washed with deionized water and dried at 80 °C for 1 h and then annealed at 250 °C for 2 h under air.Hierarchical porous PdNiO–CeO2/OLC nanostructure was synthesized by mixing aqueous solutions, containing K2PdCl4 (0.12 g), NiCl2 (0.15 g), NaOH (pH 12), and CeO2/OLC (0.30 g), under sonication at 25 °C for 30 min and then kept under reflux at 80 °C for 1 h. Finally, PdNiO–CeO2/OLC was purified via consecutive centrifugation at 7000 rpm for 20 min and washed severally with deionized water.
PdNiO/OLC and PdNiO/CeO2 were also prepared by the same method as that for PdNiO–CeO2/OLC, but in the absence of CeO2 or OLC supports, respectively.
Materials characterization
The as-formed nanocatalysts were characterized by a scanning electron microscope (SEM, S-4800, Hitachi, Tokyo, Japan) equipped with an energy dispersive X-ray analyzer (EDX) and a transmission electron microscope (TEM, TecnaiG220, FEI, Hillsboro, USA). X-ray photoelectron spectroscopy (XPS) was conducted with an Ultra DLD XPS (Kratos, Manchester, UK). X-ray diffraction (XRD) patterns were collected with an X'Pert-Pro MPD (PANalytical Co., Almelo, Netherlands). The N2 adsorption–desorption isotherm was conducted with a Micromeritics TriStar II 3000. Thermal gravimetric analysis was conducted with a PerkinElmer thermogravimetric analyzer (TGA 6000). Inductively coupled plasma optical emission spectrometry (ICP-OES) was conducted with an Agilent 5800 (USA).Electrochemical CO oxidation measurements
The electrochemical COoxid was conducted with a Gamry potentiostat (Reference 3000, Gamry Co., Warminster, PA, USA) using a three-electrode cell, consisting of Pt wire as a counter electrode, Ag/AgCl (KCl, 3 M) as reference electrode, and glassy carbon electrode (GCE) as a working electrode. The catalyst inks were prepared by dispersing each electrocatalyst (2 mg) in ethanol/H2O/Nafion (0.05 wt) (3/1/1 v/v/v ratio) and drop-casting onto the polished GCE and then left to dry in an oven under vacuum at 50 °C for 2 h before measurements. The mass loading of the Pd on the GCE was about 0.01 mgPd cm−2 as determined by ICP-OES. Cyclic voltammetry (CV), linear sweep voltammetry (LSV), electrochemical impedance spectroscopy (EIS), and chronoamperometry (CA) tests were carried out in aqueous solutions of HClO4, KOH, and NaHCO3 electrolytes at room temperature. The electrochemical active surface area (ECSA) of the nanocatalysts was determined by eqn (1):| ECSA = Q/(S × m) | (1) |
Results and discussion
Fig. 1a shows the fabrication process of PdNiO–CeO2/OLC involving the initial preparation of porous flower-like CeO2/OLC nanostructure via the sol–gel approach in the presence of the as-prepared OLC.58 Notably, adjusting the pH to 12 is the key to inducing the formation of a black precipitate; meanwhile, this method is green and allows the production of uniform flower-like CeO2 well dispersed over OLC. The as-formed CeO2/OLC was then impregnated with aqueous solutions of K2PdCl4 and NiCl2 at pH 12 under sonication and then refluxed at 80 °C for 1 h to form porous sponge-like PdNiO–CeO2/OLC nanostructure. CeO2/OLC somehow acts as a reactor for prompt reduction and growth of PdNiO nanocrystals without the need for reducing agents or surfactants, which is plausibly attributed to the negative reduction potential of CeO2/OLC relative to Pd and Ni ions. This is evidenced by the SEM images which reflect the formation of PdNiO–CeO2/OLC (Fig. 1b) and PdNiO/OLC (Fig. 1c) in 3D porous sponge-like nanostructure, whereas Pd/C possesses 3D spherical nanostructures (Fig. S1a†). The TEM images demonstrate that PdNiO–CeO2/OLC is composed of small PdNiO nanocrystals (2.54 nm) well anchored on flower-like CeO2 (20 nm) and OLC (30 nm) as indicated by the arrows in Fig. 1d, only porous sheets of PdNiO (4.34 nm) on OLC (30 nm) in PdNiO/OLC (Fig. 1e), and nanospheres of Pd (7.25 nm) in Pd/C (Fig. S1b and S2†). The good contact of PdNiO over CeO2/OLC is highly beneficial for its stabilization as well as electronic interaction during electrocatalytic CO oxidations, while porosity is important for maximizing atomic utilization and providing accessible active sites.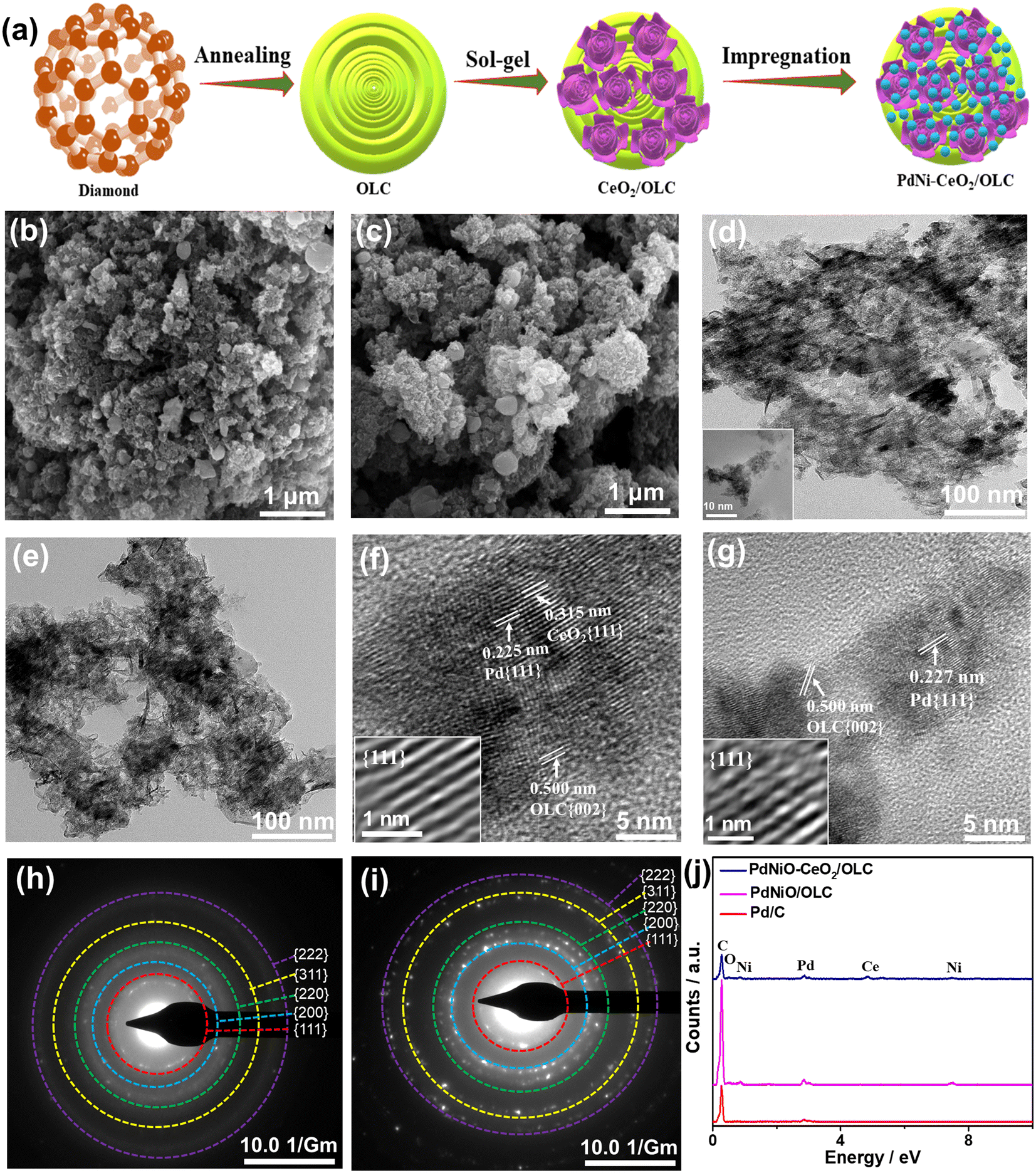 | ||
| Fig. 1 (a) Schematic preparation method. (b and c) SEM images, (d and e) TEM micrographs, (f and g) HRTEM images with Fourier-transform HRTEM, and (h and i) SAED patterns of PdNiO–CeO2/OLC and PdNiO/OLC, respectively, and (j) EDX analysis. | ||
The HRTEM micrographs of PdNiO–CeO2/OLC, PdNiO/OLC, and Pd/C show the lattice fringes with multiple crystalline defects (i.e., interfacial dislocation, intragranular dislocation, and stacking fault) as indicated by the lines in Fig. 1f and g and S1c.† Also, the lattice fringes are distributed in various directions, which implies the non-epitaxial growth of PdNiO, as usually observed in the formation of binary and ternary Pd-based catalysts. The defects may act as active catalytic sites during COoxid. The d-spacing of the resolved lattice fringes of PdNiO–CeO2/OLC is estimated to be 0.225 nm that is assigned to {111} facet of face-centred cubic (fcc) Pd, {111} index of CeO2 (0.315 nm), and {002} facet of OLC (0.500 nm) (Fig. 1f). Likewise, the resolved lattice fringes of PdNiO/OLC have a higher d-spacing of 0.227 nm for {111} facet of fcc Pd and 0.500 nm for {002} index of OLC, which are distributed in a different direction, but with fewer defects than PdNiO–CeO2/OLC (Fig. 1g). Pd/C has the highest d-spacing of 0.232 nm (Fig. S1c†). The selected area electron diffraction (SAED) patterns of PdNiO–CeO2/OLC and PdNiO/OLC reveal typical rings of {111}, {200}, {220}, {311}, and {222} facet of fcc Pd (Fig. 1h and i and S1d†). The EDX analysis reveals the existence of Pd/Ni/Ce/O/C in PdNiO–CeO2/OLC and Pd/Ni/C in PdNiO/OLC with atomic contents of 0.74/1.30/0.79/9.26/87.91% and 0.73/0.49/98.78%, respectively (Fig. 1j). The ICP-OES analysis reveals that the atomic contents are 9.79/11.47/29.37% for Pd/Ni/Ce in PdNiO–CeO2/OLC and 10.27/11.79% for Pd/Ni in PdNiO/OLC.
The XRD analyses of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C demonstrate the diffraction patterns assigned to {111}, {200}, {220}, {311}, and {222} facets of fcc Pd, besides {002} facet of graphitic carbon and two additional peaks assigned to {111} and {220} facets of NiO in PdNiO–CeO2/OLC and PdNiO/OLC (Fig. 2a). Notably, the diffraction peaks of PdNiO–CeO2/OLC are slightly shifted positively towards a higher 2θ angle, relative to those of PdNiO/OLC and Pd/C. This is due to the interaction of PdNiO with CeO2/OLC, which leads to the lattice contraction of Pd, which occurs via the integration of NiO and electronic interaction with CeO2/OLC. This is shown in the lower Pd crystallite sizes on the integration of NiO and NiO–CeO2, besides the lower crystallite sizes of PdNiO–CeO2/OLC (7.19 nm) than those of PdNiO/OLC (8.68 nm) and Pd/C (9.03 nm), except PdNiO–CeO2 (7.02 nm), as determined from the Scherrer equation.
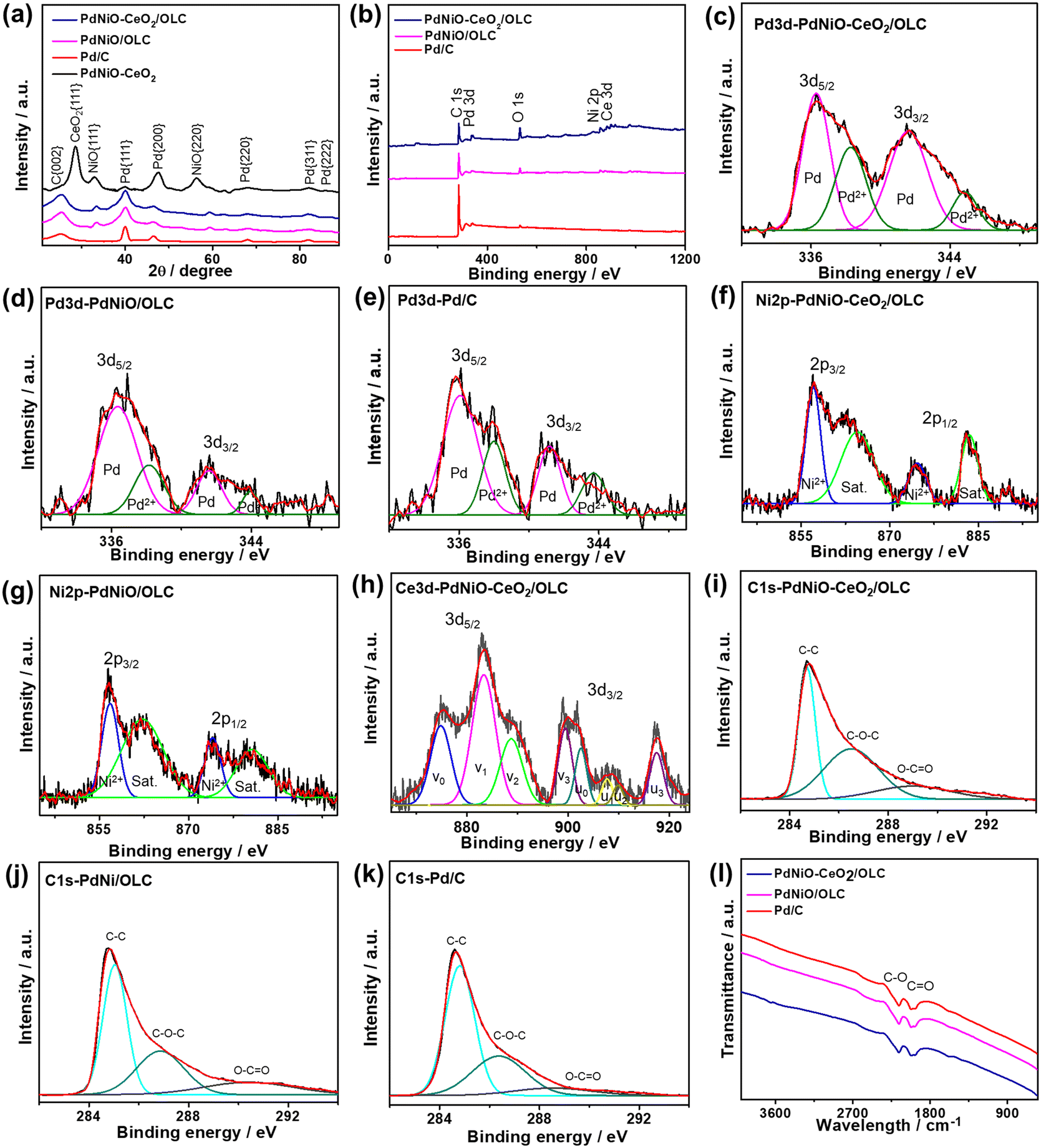 | ||
| Fig. 2 (a) XRD patterns, (b) XPS wide-survey scans, high-resolution XPS spectra of (c–e) Pd3d, (f and g) Ni2p, (h) Ce3d, (i–k) C1s, and (l) FTIR spectra of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C nanocrystals. | ||
The XPS full-survey scans display the valence states of Pd3d/Ni2p/Ce3d/O1s/C1s in PdNiO–CeO2/OLC, Pd3d/Ni2p/C1s in PdNiO/OLC, and Pd3d/C1s in Pd/C catalyst. The surface atomic contents of PdNiO–CeO2/OLC (Pd3d/Ni2p/Ce3d/O1s/C1s) are 10.61/9.85/11.21/15.87/52.46%, of PdNiO/OLC (Pd3d/Ni2p/C1s) are 9.90/6.03/84.07%, and of Pd/C (Pd3d/C1s) are 9.99/90.1% (Fig. 2b). The fitting of Pd3d spectra of PdNiO–CeO2/OLC, PdNiO/OLC, and Pd/C display Pd0(3d5/2 and 3d3/2) metallic phase as the major phase, besides the minor oxide phase of Pd2+(3d5/2 and 3d3/2)29 (Fig. 2c–e). The ratio of Pd0/Pd2+ is around 80% in both PdNiO–CeO2/OLC and PdNiO/OLC. However, the binding energies of Pd3d in PdNiO–CeO2/OLC are slightly shifted negatively relative to those in PdNiO/OLC and Pd/C, which serve as evidence for the increased d-band center of Pd in PdNiO–CeO2/OLC. This is plausibly owing to the interfacial interaction of PdNiO and its electronic interaction with CeO2 and OLC supports, i.e., increased screening of the Pd 3d nucleus by the Ce4+ 3d conduction band. Modulating the d-band center of Pd is one of the most efficient strategies to induce the adsorption and activation of reactants along with tolerating the adsorption of poisoning species during electrocatalytic oxidation reactions. The deconvolution of Ni2p spectra of PdNiO–CeO2/OLC and PdNiO/OLC reveals the major metal oxide phase of Ni2+(3d5/2 and 3d3/2) and their satellites (Fig. 2f and g); likewise, Ce3d spectra show the mixed oxide phases of Ce3+(3d5/2 and 3d3/2) representing v1 and u1, while those of Ce4+(3d5/2 and 3d3/2) are denoted as v0, v2, v3, u0, u2 and u3 in PdNiO–CeO2/OLC (Fig. 2h).
The C1s spectra are deconvoluted into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C as the main phase, besides C–O and CO phases in PdNiO–CeO2/OLC, PdNiO/OLC, and Pd/C (Fig. 2i–k).
C as the main phase, besides C–O and CO phases in PdNiO–CeO2/OLC, PdNiO/OLC, and Pd/C (Fig. 2i–k).
The FTIR analysis of PdNiO–CeO2/OLC and PdNiO/OLC displays stretching vibration bands at 1868–2087 cm−1 of CO vibration and at 2087–2334 cm−1 for C–O vibration (Fig. 2l). The bands of surface functional groups in PdNiO–CeO2/OLC are slightly broader than that in PdNiO/OLC, implying the better electronic interaction between PdNiO and CeO2/OLC.
The N2 adsorption–desorption isotherm and pore radius distribution of PdNiO–CeO2/OLC and PdNiO/OLC reveal the features close to those of a type IV curve with an H4 hysteresis loop, but with a larger area at 0.35 < P/P0 < 0.9 and two-step capillary condensation at P/P0 < 0.1 and P/P0 > 0.9 in PdNiO–CeO2/OLC, implying its multiple pores and higher surface area (Fig. S3†). Thereby, the surface area of PdNiO–CeO2/OLC (155.66 m2 g−1) is greater than that of PdNiO/OLC (119.70 m2 g−1) as calculated using the Brunauer–Emmett–Teller (BET) model from integrating the isotherm curves. Meanwhile, the pore volume of PdNiO–CeO2/OLC (0.30 cm3 g−1) is slightly higher than that of PdNiO/OLC (0.28 cm3 g−1), besides multiple pore size distribution ranging from 10 nm to 90 nm. The high surface area and multimodal porosity of PdNiO–CeO2/OLC are beneficial for providing accessible sites, enhancing reactant diffusion, and maximizing the utilization of active sites during electrocatalytic COoxid.
The electrocatalytic COoxid activities of PdNiO–CeO2/OLC, PdNiO/OLC, and PdNiO–CeO2 are benchmarked relative to Pd/C at wide pH ranges. The CV profiles of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C tested in N2-saturated HClO4 (0.1 M) present the standard voltammogram signatures for Pd-based catalysts, including double-layer hydrogen adsorption/desorption (Hads/des) and redox peaks of Pd (Fig. 3a). Notably, the onset potential (EOnset) of OH− adsorption on PdNiO–CeO2/OLC (0.78 V) is lower than those on PdNiO/OLC (0.80 V), PdNiO–CeO2 (0.80 V), and Pd/C (0.83 V), indicating the ease of H2O activation and subsequent OH− adsorption on PdNiO in PdNiO–CeO2/OLC, resulting from the electronic interaction with CeO2/OLC.
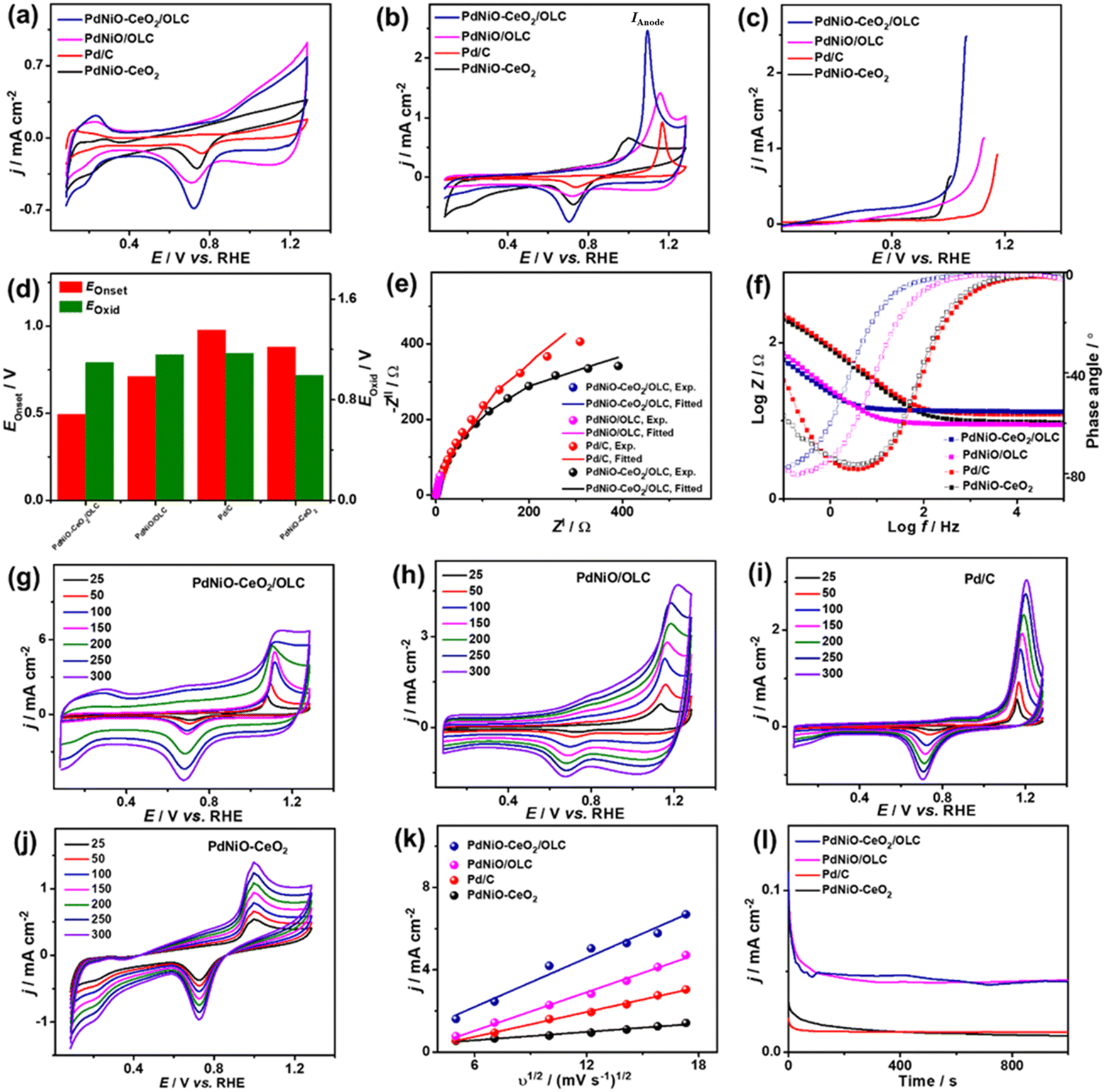 | ||
| Fig. 3 CV profiles at 50 mV s−1 in (a) N2-saturated HClO4 (0.1 M) and (b) CO-saturated HClO4 (0.1 M). (c) LSV at 50 mV s−1. (d) EOnset and EOxid. (e) Nyquist plots. (f) Bode plots. (g–j) Scan rate studies. (k) Randles–Sevcik plots (IAnodevs. ν1/2). (l) CA of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO, and PdC in CO-saturated HClO4 (0.1 M). | ||
Also, the Pd–O reduction peak of PdNiO–CeO2/OLC is slightly shifted negatively to a lower potential than those of PdNiO/OLC, PdNiO–CeO2, and Pd/C catalysts, indicating the ease of formation of Pd-oxygenated species, which is plausibly attributed to the O2-rich CeO2/OLC support that is highly desirable merit for enhanced COoxid performance.
The area of Hads/des for PdNiO–CeO2/OLC is larger than those for PdNiO/OLC, PdNiO–CeO2, and Pd/C, implying its higher surface area. The ECSA of PdNiO–CeO2/OLC (12.64 m2 g−1) is higher than PdNiO/OLC (11.40 m2 g−1), PdNiO–CeO2 (9.76 m2 g−1), and Pd/C (6.19 m2 g−1), implying its higher number of exposed active sites. The CV profiles of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C are evaluated in CO-saturated HClO4 (0.1 M), which reveal intense anodic current densities (IAnode) at higher potential assigned to oxidation of CO to CO2 and weak peak in the reverse direction at lower potential (Fig. 3b). PdNiO–CeO2/OLC has increased IAnode (2.50 mA cm−2), which is 1.74 times that of PdNiO/OLC (1.44 mA cm−2), 3.73 times that of PdNiO–CeO2 (0.67 mA cm−2), and 2.63 times that of Pd/C (0.95 mA cm−2), indicating the maximized utilization of active sites of PdNiO–CeO2/OLC. This is evident in the LSV curves, which imply the ability of PdNiO–CeO2/OLC to oxidize CO at a higher IAnode, but a lower applied potential compared with PdNiO/OLC, PdNiO–CeO2 and Pd/C (Fig. 3c). This implies the quick CO oxidation kinetics on PdNiO–CeO2/OLC as further seen in its lower CO oxidation EOnset/COoxid potential (Eoxid) (0.49 V/1.09 V) than PdNiO/OLC (0.71 V/1.16 V), PdNiO–CeO2 (0.88 V/1.00 V), PdNiO–CeO2 (0.80 V/0.99 V), and Pd/C (0.98 V/1.17 V) (Fig. 3d). This is owing to the synergism of PdNiO nanostructures, and their electronic interaction with CeO2/OLC which facilitates the formation of oxygenated species and accelerates the charge mobility needed for promoting CO oxidation at low potential.
This is proved by the EIS studies conducted to get more insights into the interfacial interaction between the nanocatalysts during the electrooxidation of CO. The Nyquist plots of the obtained PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C show deformed semi-circle curves in CO-saturated HClO4 (0.1 M) (Fig. 3e). The EIS data are fitted by a Voigt electrical equivalent circuit (EEC) model to determine the solution resistance (Rs), charge transfer resistance (Rct), and constant phase element (CPE) (Fig. S4a†). All the nanocatalysts have similar Rs values within the confinement of the error bars, implying the same ionic conductivity in the HClO4 electrolyte. However, PdNiO–CeO2/OLC has a lower Rct (36.22 Ω) than PdNiO/OLC (52.90 Ω) and Pd/C (278.11 Ω), revealing lower charge transfer resistance and consequent better reaction kinetics on PdNi–CeO2/OLC (Table 1).
| R s (Ω) | R ct (Ω) | CPE (μS s(1−a)) | n | |
|---|---|---|---|---|
| PdNiO–CeO2/OLC | 0.99 ± 0.07 | 36.22 ± 0.68 | 49.72 ± 2.31 | 0.80 |
| PdNiO/OLC | 1.02 ± 0.04 | 52.90 ± 0.52 | 43.05 ± 1.57 | 0.89 |
| Pd/C | 1.48 ± 0.75 | 278.11 ± 1.08 | 24.01 ± 0.22 | 0.90 |
| PdNiO–CeO2 | 1.28 ± 0.52 | 398.27 ± 3.32 | 18.44 ± 0.54 | 0.88 |
Following the power law of CPE impedance: ZCPE = 1/(Q(jω)n), with ideality factor n. The Pd-based nanocatalysts PdNiO–CeO2/OLC (49.72 μS s(1−a)) and PdNiO/OLC (43.05 μS s(1−a)) possess high CPE impedance relative to PdNi–CeO2 (18.44 μS s(1−a)) and Pd/C (24.01 μS s(1−a)), implying high charge transport on the roughness and porosity of multimetallic PdNi–CeO2/OLC. This is evident in the Bode plots (Fig. 3f), which proved the lower overall impedance in the low-frequency region of PdNiO–CeO2/OLC than PdNiO/OLC, and Pd/C. Also, the lower phase angle of PdNiO–CeO2/OLC (75.9°) than that of PdNiO/OLC (80.8°), PdNiO–CeO2 (82.7°), and Pd/C (83°) further proves superior diffusion of intermediates during CO oxidation on PdNiO–CeO2/OLC. The diffusion of intermediate species on the nanocatalysts during CO electrooxidation is corroborated by measuring the CV curves at different scan rates (υ, 25–300 mV s−1), which disclose that IAnode increases at high υ (Fig. 3g–j). The Randles–Sevcik plots express the linear relationship between IAnode and the square root of scan rate (υ1/2) that proves a diffusion-controlled process of CO on the tested nanocatalysts (Fig. 3k). Notably, PdNiO–CeO2/OLC exhibits a higher slope (0.40) than PdNiO/OLC (0.31), PdNiO–CeO2 (0.07), and Pd/C (0.20), evidencing the faster diffusion of CO intermediates on PdNiO–CeO2/OLC.
The stability of CO electrooxidation on the catalysts was investigated by CA and accelerated stability test (AST) in CO-saturated HClO4 (0.1 M) at 50 mV s−1. The CA results show gradual CO oxidation activity loss on all the nanocatalysts at steady-state conditions, but with a lower degradation, besides preserving a higher current density, on PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 3l). After 1000 stability cycles of CV curves in CO-saturated HClO4 (0.1 M), PdNiO–CeO2/OLC lost only 14.5% of its initial CO oxidation activity compared with PdNiO/OLC (27.8%), PdNiO–CeO2 (25.0%), and Pd/C (37.9%) (Fig. S4b–f†). This indicates the superior CO oxidation durability of PdNiO–CeO2/OLC as further proved by the TEM image obtained after AST, which showed the structural stability without any obvious morphological change (i.e., aggregation or attachment of PdNiO) (Fig. S4g†).
The COoxid electrocatalysis on the Pd-based nanocatalysts was investigated in KOH (0.1 M) medium. The CV curves measured in N2-saturated KOH (0.1 M) of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C display the voltammogram of Pd with a noticeable negative shift of the Pd–O reduction peak of PdNiO–CeO2/OLC relative to PdNiO/OLC, PdNiO–CeO2, and Pd/C. This implies the ease of generating oxygenated species on Pd. The ECSA of PdNiO–CeO2/OLC (53.09 m2 g−1) is greater than PdNiO/OLC (42.01 m2 g−1), PdNiO–CeO2 (31.76 m2 g−1), and Pd/C (25.76 m2 g−1) (Fig. 4a). The CV profiles of the PdNiO–CeO2/OLC, PdNiO/OLC, and Pd/C catalysts tested in CO-saturated KOH (0.1 M) show the CO oxidation features with a higher IAnode (2.486 mA cm−2) on PdNiO–CeO2/OLC than on PdNiO/OLC (1.46 mA cm−2), PdNiO–CeO2 (1.12 mA cm−2), and Pd/C (1.33 mA cm−2) (Fig. 4b). Also, the LSV results display the lower Eonset on PdNiO–CeO2/OLC (0.65 V) relative to PdNiO/OLC (0.68 V) and PdNiO–CeO2 (0.71 V) along with a higher generated IAnode than PdNiO–CeO2 and Pd/C under any applied voltage point on PdNiO–CeO2/OLC (Fig. 4c and d), indicating its faster CO electrooxidation kinetics. The mass (specific) activity of PdNiO–CeO2/OLC (245 mA mg−1 (4.61 mA cm−2)) are higher than those of PdNiO/OLC (146 mA mg−1 (3.58 mA cm−2)), PdNiO–CeO2 (82 mA mg−1 (1.93 mA cm−2)), and Pd/C (99 mA mg−1 (3.19 mA cm−2)) (Fig. 4d). This is based on an equivalent Pd mass, which implies the maximized utilization of Pd in PdNiO–CeO2/OLC, plausibly due to the porosity, electronic interaction, and higher concentration of surface sites.
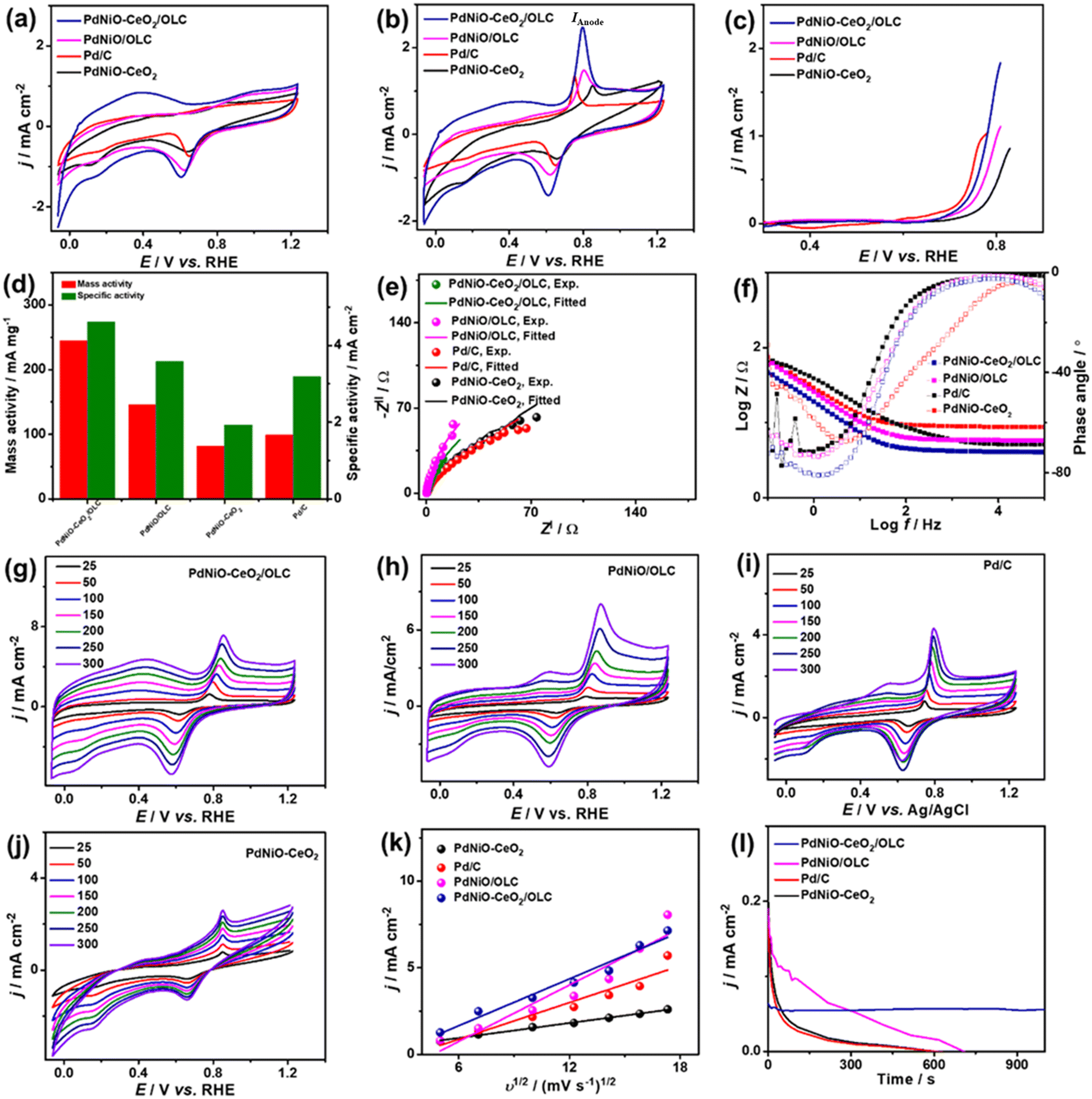 | ||
| Fig. 4 CV curves at 50 mV s−1 in (a) N2-saturated KOH (0.1 M) and (b) CO-saturated KOH (0.1 M). (c) LSV at 50 mV s−1. (d) Mass/specific activities. (e) Nyquist plots. (f) Bode plots. (g–j) Scan rate studies. (k) Randles–Sevcik plots (IAnodevs. υ1/2) and (l) CA of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C in CO-saturated KOH (0.1 M). | ||
The Nyquist plots show semi-circle curves but with a smaller diameter for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C in CO-saturated KOH (0.1 M), implying a better interfacial interaction between KOH electrolyte and PdNiO–CeO2/OLC. The fitting of EIS data demonstrates the lower Rs/Rct (0.98 Ω/45.52 Ω) for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 4e), indicating quicker COoxid kinetics and faster charge mobility on PdNiO–CeO2/OLC. This is validated by the Bode plots which depict the lower log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Z in the low-frequency region for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C, in addition to a lower phase angle (67°), indicating the fast diffusion of reactants on PdNiO–CeO2/OLC (Fig. 4f).
Z in the low-frequency region for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C, in addition to a lower phase angle (67°), indicating the fast diffusion of reactants on PdNiO–CeO2/OLC (Fig. 4f).
This is shown in the steady linear increase in IAnode of the tested catalysts with increasing υ from 25 to 300 mV s−1 and the linear relationship between IAnodevs. υ1/2 (Fig. 4g–j). This demonstrates the diffusion-controlled process for CO electrooxidation on PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C; however, the higher slope for PdNiO–CeO2/OLC (0.45) implies its faster COoxid kinetics (Fig. 4k). CA conducted in CO-saturated KOH (0.1 M) implies higher durability with lower degradation of IAnode for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 4l). Additionally, after 1000 CV cycles (AST), PdNiO–CeO2/OLC retains 94.2% of its initial IAnode compared to PdNiO/OLC (83.1%), PdNiO–CeO2 (86.8%), and Pd/C (73.4%) (Fig. S5†).
The CV curves measured in N2-saturated NaHCO3 (0.1 M) on PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C show the ideal voltammogram of Pd-based catalysts, but with an obvious higher peak area of PdO and negatively shifted peak on PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 5a). This implies a higher surface area and ease of generation of oxygenated species on PdNiO–CeO2/OLC and PdNiO/OLC. Thereby, the estimated ECSA are about 9.10, 6.78, 5.19, and 3.60 m2 g−1 for PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C, respectively. The CV curves measured in CO-saturated NaHCO3 (0.1 M) disclose the CO oxidation voltammogram, but with a greater IAnode on PdNiO–CeO2/OLC (1.23 mA cm−2) than PdNiO/OLC (0.69 mA cm−2), PdNiO–CeO2 (0.57 mA cm−2), and Pd/C (0.54 mA cm−2) by 1.76, 2.14, and 2.26 times correspondingly (Fig. 5b). The LSV test shows the ability of PdNiO–CeO2/OLC to oxidize CO and generate higher IAnode at a lower potential than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 5c), indicating faster CO electrooxidation kinetics on PdNiO–CeO2/OLC.
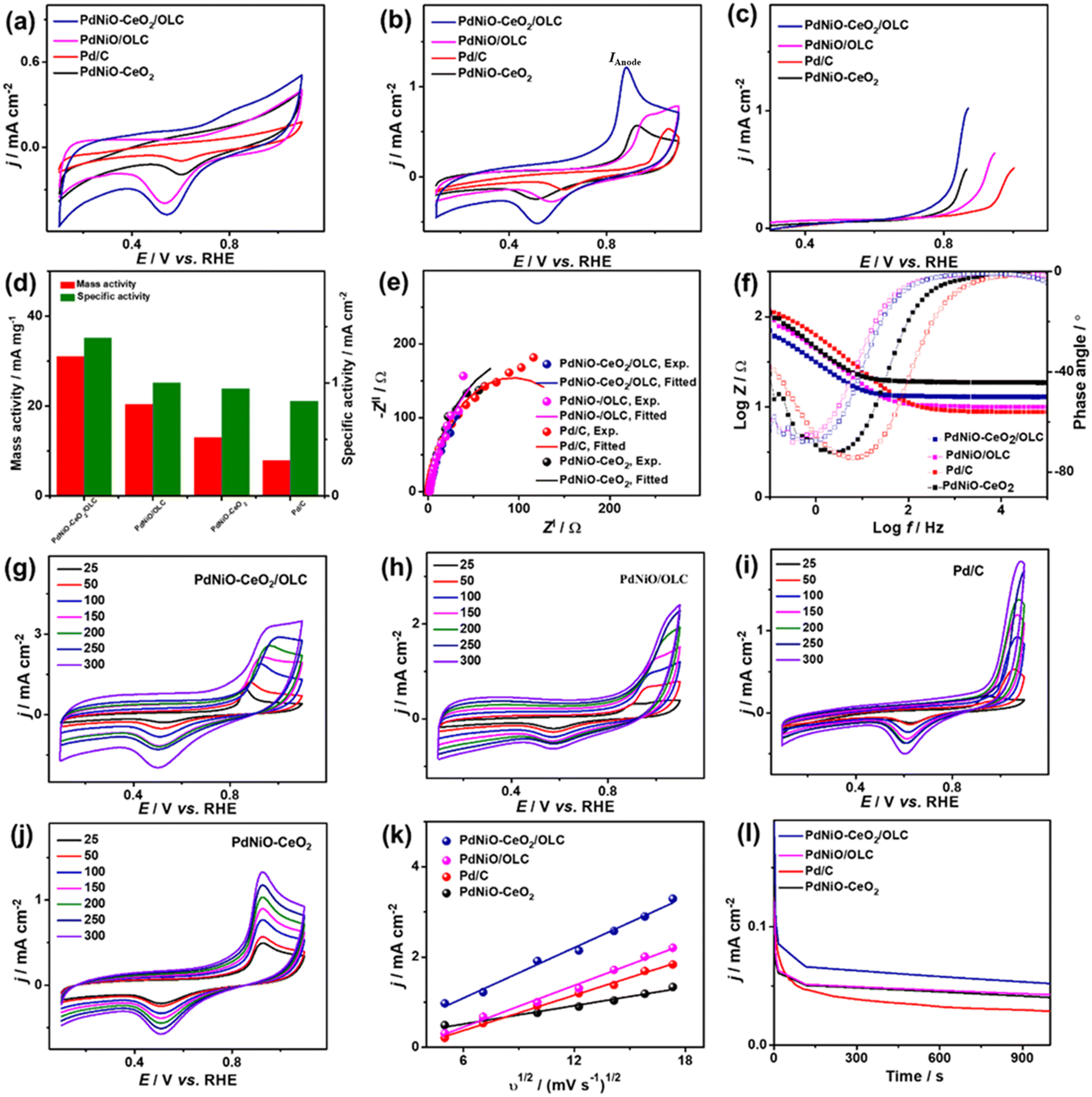 | ||
| Fig. 5 CV profiles at 50 mV s−1 in (a) N2-saturated NaHCO3 (0.1 M) and (b) CO-saturated NaHCO3 (0.1 M). (c) LSV at 50 mV s−1. (d) Mass/specific activities. (e) Nyquist plots. (f) Bode plots. (g–j) Scan rate studies. (k) Randles–Sevcik plots (IAnodevs. υ1/2) and (l) CA of PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and PdC in CO-saturated NaHCO3 (0.1 M). | ||
The mass (specific) activity of PdNiO–CeO2/OLC (31 mA mg−1 (1.40 mA cm−2)) are higher than those of PdNiO/OLC (20.34 mA mg−1 (1.00 mA cm−2)), PdNiO–CeO2 (12.98 mA mg−1 (0.95 mA cm−2)), and Pd/C (7.92 mA mg−1 (0.84 mA cm−2)) (Fig. 5d). The Nyquist plots obtained in CO-saturated NaHCO3 (0.1 M) show semi-circle curves, but with a lower diameter for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C, that could serve as evidence for the higher electrical conductivity. Fitting of EIS data using Voigt EEC displays the similar Rs of the tested catalysts but with smaller Rct for PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C, which indicates better electrolyte–electrode interaction and higher charge transfer on PdNiO–CeO2/OLC (Fig. 5e and Table S2†). This is also seen in the higher CPE impedance of PdNiO–CeO2/OLC (47.76 μS s(1−a)) than those of PdNiO/OLC (40.71 μS s(1−a)), PdNiO–CeO2 (25.78 μS s(1−a)), and Pd/C (37.74 μS s(1−a)) as further confirmed in the Bode plots, where PdNiO–CeO2/OLC exhibits low logZ in the low-frequency range alongside a lower phase angle (65.2°) relative to PdNiO/OLC (67.4°), PdNiO–CeO2 (71.8°), and Pd/C (74.7°) (Fig. 5f). This implies a better diffusion of reactants on the porous PdNiO–CeO2/OLC during COoxid.
The increase in IAnode with increasing υ from 25 to 250 mV s−1 on PdNiO–CeO2/OLC, PdNiO/OLC, PdNiO–CeO2, and Pd/C along with the linear relationship of IAnodevs. υ1/2 reveal a diffusion-controlled mechanism for the electrooxidation of CO (Fig. 5g–j). However, a greater slope for PdNiO–CeO2/OLC (0.19) than PdNiO/OLC (0.15), PdNiO–CeO2 (0.09), and Pd/C (0.13) (Fig. 5k) implies the improved diffusion of reactants and intermediate species on PdNiO–CeO2/OLC during COoxid. CA measured in CO-saturated NaHCO3 (0.1 M) for 1000 s displays the lower degradation of IAnode on PdNiO–CeO2/OLC than PdNiO/OLC, PdNiO–CeO2, and Pd/C (Fig. 5l). Meanwhile, after 1000 CO oxidation durability cycles, PdNiO–CeO2/OLC only lost 15.6% of its initial catalytic activity compared with PdNiO/OLC (27.6%), PdNiO–CeO2 (18.0%), and Pd/C (32.2%) (Fig. S6†). PdNiO–CeO2/OLC retained 84% of its initial ECSA relative to PdNiO/OLC (72%), PdNiO–CeO2 (82%), and Pd/C (68%) in addition to the absence of any voltammogram features related to degradation of PdNiO.
The CO electrooxidation results for PdNiO–CeO2/OLC, PdNiO/OLC, and PdNiO–CeO2 are compared with previous findings in the literature (Table S3†). The CO oxidation on PdNiO–CeO2/OLC is proposed to follow the Langmuir–Hinshelwood mechanism, presented in eqn (2)–(4):59
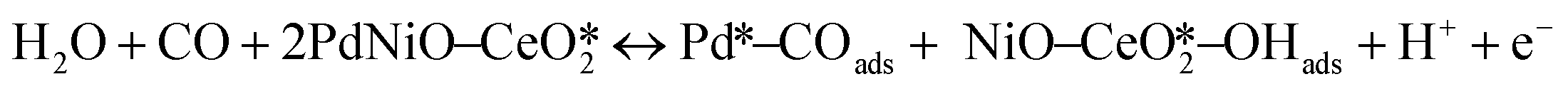 | (2) |
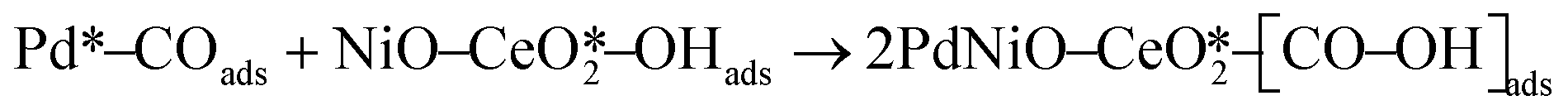 | (3) |
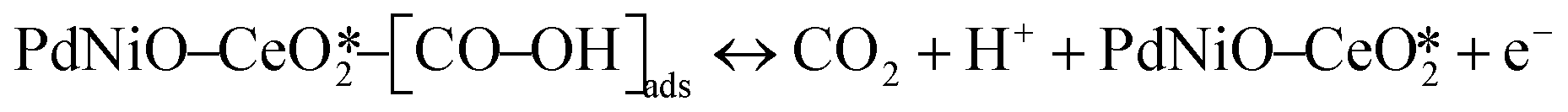 | (4) |
Conclusion
This article presents the controlled synthesis of ternary PdNiO nanocrystals supported on porous CeO2/onion-like carbon (PdNiO–CeO2/OLC) nanostructures based on the sol–gel, annealing, and impregnation approaches. Under typical conditions, flower-like CeO2/OLC supported the growth of PdNiO nanocrystals without the need for reducing agents or surfactants. This leads to the formation of porous sponge-like PdNiO–CeO2/OLC, coupled with the unique merits of multifunctional structures, low mass of Pd (10 wt%), pore volume (0.30 m3 g−1), and great BET surface area (155.66 m2 g−1). The effects of supports and electrolytes on the COoxid activity and durability of PdNiO–CeO2/OLC were investigated. The electrocatalytic COoxid activity of PdNiO–CeO2/OLC was superior to those of PdNiO/OLC by at least 1.66 times, PdNiO–CeO2 by at least 1.88 times, and commercial Pd/C by at least 2.12 times in all electrolytes, besides great durability after 1000 cycles. This is owing to the electronic interaction of PdNiO with the CeO2/OLC support, which eases CO adsorption/activation, alongside activation/dissociation of H2O to generate active oxygenated species (i.e., OH) needed for accelerating COoxid kinetics. Also, the COoxid of PdNiO–CeO2/OLC in different electrolytes followed the order of HClO4 > KOH > NaHCO3. This study may allow the synthesis of other ternary Pd-based electrocatalysts for CO oxidation along with an understanding of the effect of supports and electrolytes.Conflicts of interest
We declare no conflicts of interest.Acknowledgements
This work was supported by Qatar University High Impact Internal Grant (QUHI-CAM-22/23-550), Qatar National Research Fund (NPRP13S-0117-200095), and NRF/DSI/Wits SARChI Chair in Materials Electrochemistry and Energy Technologies (MEET) (UID No. 132739). The statements made herein are solely the responsibility of the authors.References
- K. Eid, Y. H. Ahmad, S. Y. AlQaradawi and N. K. Allam, Catal. Sci. Technol., 2017, 7, 2819–2827 RSC.
- K. Eid, K. A. Soliman, D. Abdulmalik, D. Mitoraj, M. H. Sleim, M. O. Liedke, H. A. El-Sayed, A. S. AlJaber, I. Y. Al-Qaradawi and O. M. Reyes, Catal. Sci. Technol., 2020, 10, 801–809 RSC.
- M. A. Ahsan, T. He, K. Eid, A. M. Abdullah, M. F. Sanad, A. Aldalbahi, B. Alvarado-Tenorio, A. Du, A. R. Puente Santiago and J. C. Noveron, ACS Appl. Mater. Interfaces, 2022, 14, 3919–3929 CrossRef CAS PubMed.
- F. Wu, K. Eid, A. M. Abdullah, W. Niu, C. Wang, Y. Lan, A. A. Elzatahry and G. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 31309–31318 CrossRef CAS PubMed.
- Q. Lu, J. Li, K. Eid, X. Gu, Z. Wan, W. Li, R. S. Al-Hajri and A. M. Abdullah, J. Electroanal. Chem., 2022, 916, 116361 CrossRef CAS.
- S. Sharma, M. Groves, J. Fennell, N. Soin, S. Horswell and C. Malardier-Jugroot, Chem. Mater., 2014, 26, 6142–6151 CrossRef CAS.
- A. K. Ipadeola, R. Barik, S. C. Ray and K. I. Ozoemena, Electrocatalysis, 2019, 10, 366–380 CrossRef CAS.
- K. I. Ozoemena, S. Musa, R. Modise, A. K. Ipadeola, L. Gaolatlhe, S. Peteni and G. Kabongo, Curr. Opin. Electrochem., 2018, 10, 82–87 CrossRef CAS.
- K. Eid, Y. H. Ahmad, H. Yu, Y. Li, X. Li, S. Y. AlQaradawi, H. Wang and L. Wang, Nanoscale, 2017, 9, 18881–18889 RSC.
- H. J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- R. M. Al Soubaihi, K. M. Saoud, M. T. Z. Myint, M. A. Göthelid and J. Dutta, Catalysts, 2021, 11, 131 CrossRef CAS.
- J. Lin, B. Qiao, L. Li, H. Guan, C. Ruan, A. Wang, W. Zhang, X. Wang and T. Zhang, J. Catal., 2014, 319, 142–149 CrossRef CAS.
- A. K. Ipadeola, M. Chitt, A. Abdelgawad, K. Eid and A. M. Abdullah, Int. J. Hydrogen Energy, 2023 DOI:10.1016/j.ijhydene.2023.01.208 , (in press).
- K. Eid, A. Gamal and A. M. Abdullah, Green Chem., 2023, 25, 1276–1310 RSC.
- Y. Zhang, Y. Cai, Y. Guo, H. Wang, L. Wang, Y. Lou, Y. Guo, G. Lu and Y. Wang, Catal. Sci. Technol., 2014, 4, 3973–3980 RSC.
- C. Hudy, J. Gryboś, K. Steenbakkers, K. Góra-Marek, F. Zasada and Z. Sojka, Catal. Sci. Technol., 2022, 12, 5723–5741 RSC.
- B. Singh, V. Sharma, R. P. Gaikwad, P. Fornasiero, R. Zboril and M. B. Gawande, Small, 2021, 17, 2006473 CrossRef CAS PubMed.
- R. G. dos Santos and A. C. Alencar, Int. J. Hydrogen Energy, 2020, 45, 18114–18132 CrossRef CAS.
- Q. Lu, K. Eid, W. Li, A. M. Abdullah, G. Xu and R. S. Varma, Green Chem., 2021, 23, 5394–5428 RSC.
- K. Eid, M. H. Sliem, M. Al-Ejji, A. M. Abdullah, M. Harfouche and R. S. Varma, ACS Appl. Mater. Interfaces, 2022, 14, 40749–40760 CrossRef CAS PubMed.
- K. Eid, M. H. Sliem and A. M. Abdullah, Nanoscale, 2019, 11, 11755–11764 RSC.
- A. K. Ipadeola, K. Eid, A. M. Abdullah and K. I. Ozoemena, Langmuir, 2022, 38, 11109–11120 CrossRef CAS PubMed.
- P. Van der Wal, N. De Rooij and M. Koudelka-Hep, Sens. Actuators, B, 1996, 35, 119–123 CrossRef CAS.
- A. Abdelgawad, B. Salah, K. Eid, A. M. Abdullah, R. S. Al-Hajri, M. Al-Abri, M. K. Hassan, L. A. Al-Sulaiti, D. Ahmadaliev and K. I. Ozoemena, Int. J. Mol. Sci., 2022, 23, 15034 CrossRef CAS PubMed.
- A. K. Ipadeola, P. V. Mwonga and K. I. Ozoemena, Electrochim. Acta, 2021, 390, 138860 CrossRef CAS.
- E. Madrid, C. Harabajiu, R. S. Hill, K. Black, L. Torrente-Murciano, A. J. Dickinson, P. J. Fletcher, K. I. Ozoemena, A. K. Ipadeola and E. Oguzie, ChemElectroChem, 2021, 8, 378–385 CrossRef CAS.
- A. K. Ipadeola, P. V. Mwonga, S. C. Ray, R. R. Maphanga and K. I. Ozoemena, Electroanalysis, 2020, 32, 3060–3074 CrossRef CAS.
- Y. Zhou, Z. Wang and C. Liu, Catal. Sci. Technol., 2015, 5, 69–81 RSC.
- B. Salah, K. Eid, A. M. Abdelgwad, Y. Ibrahim, A. M. Abdullah, M. K. Hassan and K. I. Ozoemena, Electroanalysis, 2022, 34, 677–683 CrossRef CAS.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- M. G. Hosseini, F. Hosseinzadeh, P. Zardari and M. Darbandi, Int. J. Hydrogen Energy, 2021, 46, 28513–28526 CrossRef CAS.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- J. K. McDonough and Y. Gogotsi, Electrochem. Soc. Interface, 2013, 22, 61 CrossRef CAS.
- N. Keller, N. I. Maksimova, V. V. Roddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlögl, Angew. Chem., Int. Ed., 2002, 41, 1885–1888 CrossRef CAS PubMed.
- I. Choi, Int. J. Hydrogen Energy, 2019, 44, 26589–26596 CrossRef CAS.
- J. M. Jaksic, N. V. Krstajic, L. M. Vracar, S. G. Neophytides, D. Labou, P. Falaras and M. M. Jaksic, Electrochim. Acta, 2007, 53, 349–361 CrossRef CAS.
- Y. Jin, G. Sun, F. Xiong, L. Ding and W. Huang, Eur. J. Chem., 2015, 21, 4252–4256 CrossRef CAS PubMed.
- K. Polychronopoulou, C. N. Costa and A. M. Efstathiou, Catal. Today, 2006, 112, 89–93 CrossRef CAS.
- S. Zeng, Y. Wang, S. Ding, J. J. Sattler, E. Borodina, L. Zhang, B. M. Weckhuysen and H. Su, J. Power Sources, 2014, 256, 301–311 CrossRef CAS.
- C.-J. Jia, M. Schwickardi, C. Weidenthaler, W. Schmidt, S. Korhonen, B. M. Weckhuysen and F. Schüth, J. Am. Chem. Soc., 2011, 133, 11279–11288 CrossRef CAS PubMed.
- D. Gu, J.-C. Tseng, C. Weidenthaler, H.-J. Bongard, B. Spliethoff, W. Schmidt, F. Soulimani, B. M. Weckhuysen and F. Schüth, J. Am. Chem. Soc., 2016, 138, 9572–9580 CrossRef CAS PubMed.
- A. K. Ipadeola, K. Eid, A. K. Lebechi, A. M. Abdullah and K. I. Ozoemena, Electrochem. Commun., 2022, 140, 107330 CrossRef CAS.
- A. K. Ipadeola, A. K. Lebechi, L. Gaolatlhe, A. B. Haruna, M. Chitt, K. Eid, A. M. Abdullah and K. I. Ozoemena, Electrochem. Commun., 2022, 136, 107207 CrossRef CAS.
- K. Eid, H. Wang, V. Malgras, Z. A. Alothman, Y. Yamauchi and L. Wang, J. Phys. Chem. C, 2015, 119, 19947–19953 CrossRef CAS.
- A. K. Lebechi, A. K. Ipadeola, K. Eid, A. M. Abdullah and K. I. Ozoemena, Nanoscale, 2022, 14, 10717–10737 RSC.
- A. K. Ipadeola, P. V. Mwonga, S. C. Ray, R. R. Maphanga and K. I. Ozoemena, ChemElectroChem, 2020, 7, 4562–4571 CrossRef CAS.
- J. J. Ogada, A. K. Ipadeola, P. V. Mwonga, A. B. Haruna, F. Nichols, S. Chen, H. A. Miller, M. V. Pagliaro, F. Vizza, J. R. Varcoe, D. M. Meira, D. M. Wamwangi and K. I. Ozoemena, ACS Catal., 2022, 12, 7014–7029 CrossRef CAS.
- J. M. Jaksic, D. Labou, G. D. Papakonstantinou, A. Siokou and M. M. Jaksic, J. Phys. Chem. C, 2010, 114, 18298–18312 CrossRef CAS.
- H. Na, H. Choi, J.-W. Oh, Y. S. Jung and Y. S. Cho, ACS Appl. Mater. Interfaces, 2019, 11, 25179–25185 CrossRef CAS PubMed.
- Y. Martynova, S. Shaikhutdinov and H. J. Freund, ChemCatChem, 2013, 5, 2162–2166 CrossRef CAS.
- L. P. Herrera, L. F. d. L. e Freitas, J. Hong, A. S. Hoffman, S. R. Bare, E. Nikolla and J. W. Medlin, Catal. Sci. Technol., 2022, 12, 1476–1486 RSC.
- C. Xu, Z. Tian and P. Shen, Electrochim. Acta, 2008, 53, 2610–2618 CrossRef CAS.
- W. Zou, C. Ge, M. Lu, S. Wu, Y. Wang, J. Sun, Y. Pu, C. Tang, F. Gao and L. Dong, RSC Adv., 2015, 5, 98335–98343 RSC.
- F. Yang, X. Zhang, L. Zhou, S. Lin, X. Cao, J. Jiang and X. Lu, Chem. Eng. J., 2022, 432, 134255 CrossRef CAS.
- A. S. Ismail, I. Garcia-Torregrosa, J. C. Vollenbroek, L. Folkertsma, J. G. Bomer, T. Haarman, M. Ghiasi, M. Schellhorn, M. Nachtegaal and M. Odijk, ACS Catal., 2021, 11, 12324–12335 CrossRef CAS.
- A. K. Ipadeola, K. Eid, A. M. Abdullah, R. S. Al-Hajri and K. I. Ozoemena, Nanoscale Adv., 2022, 4, 5044–5055 RSC.
- M. Lepage, T. Visser, F. Soulimani, A. M. Beale, A. Iglesias-Juez, A. M. van der Eerden and B. M. Weckhuysen, J. Phys. Chem. C, 2008, 112, 9394–9404 CrossRef CAS.
- M. Hantel, V. Presser, J. McDonough, G. Feng, P. T. Cummings, Y. Gogotsi and R. Kötz, J. Electrochem. Soc., 2012, 159, A1897 CrossRef CAS.
- B. N. Grgur, N. M. Marković, C. A. Lucas and P. N. Ross Jr, J. Serb. Chem. Soc., 2001, 66, 785–797 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00253e |
| This journal is © The Royal Society of Chemistry 2023 |