
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The selective oxidation of methane to methanol using in situ generated H2O2 over palladium-based bimetallic catalysts†
James H.
Carter‡
*a,
Richard J.
Lewis‡
 *a,
Nikolas
Demetriou
a,
Christopher
Williams
a,
Thomas E.
Davies
a,
Tian
Qin
b,
Nicholas F.
Dummer
a,
David J.
Morgan
ac,
David J.
Willock
d,
Xi
Liu
b,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
*a,
Nikolas
Demetriou
a,
Christopher
Williams
a,
Thomas E.
Davies
a,
Tian
Qin
b,
Nicholas F.
Dummer
a,
David J.
Morgan
ac,
David J.
Willock
d,
Xi
Liu
b,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
aMax Planck–Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: CarterJ5@cardiff.ac.uk; LewisR27@cardiff.ac.uk; Hutch@Cardiff.ac.uk
bSchool of Chemistry and Chemical, In situ Centre for Physical Sciences, Shanghai Jiao Tong University, 200240 Shanghai, P. R. China
cHarwellXPS, Research Complex at Harwell (RCaH), Didcot, OX11 OFA, UK
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 13th June 2023
Abstract
The selective oxidation of methane to methanol, using H2O2 generated in situ from H2 and O2 has been investigated using a series of TS-1 supported bimetallic palladium-based catalysts. The alloying of Pd with Au exhibited improved performance compared to monometallic Pd analogues, with the optimal AuPd catalyst stable over multiple uses. Complementary studies into catalytic performance towards the direct synthesis and subsequent degradation of H2O2 indicated that catalysts that offered moderate activity toward H2O2 synthesis and degradation were the most active for CH4 oxidation, balancing the high activity of the Pd-only formulation, with the greater selectivity of the Au-only analogue. In particular, the ability of Au to promote the release of oxygen-based radical species from catalytic surfaces is considered to be crucial in achieving improved reactivity, compared to monometallic Pd analogues. The alloying of Pd with more abundant secondary metals was also explored with the NiPd/TS-1 catalyst exhibiting comparable activity to the AuPd analogue. However, unlike over AuPd/TS-1, where methanol is the primary product, the production of formic acid was found to be favoured by the NiPd/TS-1 catalyst.
Introduction
The selective oxidation of methane to methanol, an important platform chemical with an estimated annual global demand of approximately 100 bn litres,1 is considered a grand challenge for catalytic chemistry, due to the large energy barrier for initial C–H bond activation and the need to prevent over oxidation to CO2. Methane is a thermally-stable molecule with four equivalent strong C–H bonds and its partial oxidation products are considerably more reactive than methane itself, making kinetic control of the partial oxidation reaction of paramount importance. Currently, methanol is formed indirectly in an energy-intensive two-step process: methane is initially converted into synthesis gas (CO + H2) via steam (or dry) reforming before methanol synthesis is performed.2 These steps require high temperature and pressure, adding to the capital costs of producing methanol and rendering the process efficient only on large scales. The conversion of methane to a value-added product via an alternative route could increase the range of applications of oxidative methane processing including dealing with low-level methane production at landfill sites, and anaerobic digesters while also reducing natural gas flaring at oil wells. A recent IEA report estimated flaring emissions globally to be approximately 250 Mt.3 Alternative dispositions for methane could result in the lowering of GHG emissions worldwide. Therefore, there is an urgent need to develop technologies to convert methane into useful chemicals.Several notable advances have been made in developing catalysts for the direct conversion of methane to methanol. Flytzani-Stephanopoulos and co-workers recently reported that mononuclear rhodium species supported on ZSM-5 can catalyse the conversion of methane into various oxygenates, including methanol, formic acid and acetic acid using O2, CO and H2O at 150 °C.4 Additionally, van Bokhoven and co-workers have demonstrated that methane could be anaerobically converted into methanol using H2O as an oxidant over Cu-exchanged mordenite.5 We have also reported that Au nanoparticles immobilised onto ZSM-5 are able to oxidise methane to methanol, formic acid and C2 oxygenates, with minimal formation of CO2, at relatively low temperatures and in a closed cycle.6
The use of H2O2 as an oxidant has yielded several breakthroughs in methane oxidation. In particular allowing for the reaction to be carried out at much lower temperatures (30–80 °C), than aerobic routes, and as such has been an area of considerable academic interest.7–12 However, the practical limits of using H2O2 as an oxidant to make methanol from methane are numerous. The technical and economic challenges associated with H2O2 manufacture via current industrial routes (dominated by the Anthraquinone Oxidation Process), in addition to concerns associated with safe transport and storage of the oxidant, would likely preclude the application of pre-formed H2O2 on an industrial scale. Furthermore, the cost of H2O2 is typically greater than that of methanol. Alternatively, the selective partial oxidation of methane via the in situ production of H2O2 from molecular H2 and O2 offers an attractive alternative and could reduce costs associated with the oxidant. Bimetallic AuPd catalysts, known to offer high activity towards H2O2 production,13 have been widely studied for a range of oxidative transformations via in situ production of the oxidant, including for methane valorisation.14–19 Recently, in an attempt to overcome reagent diffusion limitations and improve oxidant utilisation Jin et al. investigated the modification of the external surface of a AuPd@ZSM-5 catalyst, with a hydrophobic organosilane layer, which was found to both promote the localised concentration of reagents near active sites and confine the synthesised H2O2 near the AuPd nanoparticles for subsequent methane activation.20 In contrast to the reaction mechanism proposed for CuFe–ZSM-5 materials21 (another class of materials widely studied for methane oxidation when used in conjunction with H2O2), AuPd catalysed methane oxidation using H2O2 has been shown to proceed via the activation of the methane C–H bond through a hydrogen abstraction pathway, which is mediated by reactive oxygen species (ROS, ˙OOH, ˙OH and ˙O2−), which are generated from H2O2 over AuPd surfaces, and the resulting formation of a methyl radical (˙CH3). The termination of the methyl radical species with transient ROS is crucial to the formation of methanol, methyl hydroperoxide, formic acid and carbon dioxide, thus it is possible to draw a direct correlation between the rate of ROS formation and methane oxidation.14 The key reaction steps within the in situ H2O2 mediated activation of methane are reported in eqn (1)–(5) below and we wish to highlight the comprehensive study by Serra-Maia et al. for an in-depth discussion of the mechanism and kinetics of H2O2 driven methane oxidation over AuPd catalysts.22
| H2 + O2 → H2O2, ROS(˙OOH, ˙OH and ˙O2−) | (1) |
| H2O2 → ROS | (2) |
| ROS + CH4 → ˙CH3 | (3) |
| ˙CH3 + H2O2, ROS → CH3OOH → CO2 | (4) |
| ˙CH3 + ˙OH → CH3OH → CO2 | (5) |
Despite key advances in the area of methane oxidation using an in situ generated oxidant, there are still many opportunities to expand the number of known catalysts for this reaction and explore how different supports and metal compositions affect the catalysis. In this work, we investigate the role of the support and supported metal, as well as the mechanistic relevance of direct hydrogen peroxide synthesis and degradation to methane valourisation.
Experimental
Catalyst preparation
All catalysts were prepared by the wet co-impregnation of metal salts onto the support (TS-1, TiO2 (P25) or ZSM-5). The procedure to produce 2 g of 0.33 wt%Au–0.33 wt%Pd/TS-1, with an analogous methodology utilised for all catalyst formulations, is outlined as follows: PdCl2 (1.1 mL, [Pd] = 6 mg mL−1, Sigma Aldrich) and HAuCl4·3H2O solution (0.5388 mL, [Au] = 12.25 mg mL−1, Strem Chemicals) were placed in a 50 mL round bottom flask, with total volume fixed to 16 mL using H2O (HPLC grade, Fischer Scientific). The resulting mixture was heated to 65 °C in a thermostatically controlled oil bath with stirring (600 RPM). Upon reaching 65 °C, TS-1 (1.987 g, HighChem) was added over the course of 5 min. The resulting slurry was then heated to 85 °C for 16 h to allow for complete evaporation of water. The resulting solid material was ground prior to calcination (static air, 400 °C, 3 h, 10 °C min−1). Chloride-based precursors were utilised in each case (H2PtCl4, MnCl2.4H2O, CuCl2 and NiCl2·6H2O) using the same preparation as above and adjusting the amount of salt required.Catalyst testing
Methane oxidation via in situ H2O2 synthesis
The selective oxidation of methane was carried out in a 50 mL stainless steel Parr autoclave reactor equipped with a PTFE liner. The autoclave liner was charged with catalyst (0.028 g) and solvent (H2O, 10.0 mL, HPLC grade Fisher Scientific) and the autoclave was sealed. After purging with N2 (100 psi), N2, H2, CH4 and O2 were sequentially introduced to the reactor to achieve a composition of 21.55% N2, 0.86% H2, 75.86% CH4 and 1.72% O2 and a total pressure of 30.5 bar. Gaseous reagents were not continuously supplied. The autoclave was then heated to 50 °C before being stirred at 1500 RPM. After 0.5 h, stirring ceased and the reactor was cooled to 10 °C using ice water to minimise the loss of volatile products. The composition of the gas phase was quantified by gas bag analysis of the headspace using a Varian GC (SIL5CB column: 50 m, 0.33 mm internal diameter) equipped with a methaniser and flame ionisation detector. The liquid products were quantified using 1H NMR on a Bruker 500 MHz spectrometer. The reaction sample (0.7 mL) and D2O (0.1 mL, Merck) were added to the NMR sample tube with a calibrated insert (1% TMS in CDCl3) and spectra were integrated against this standard using predetermined calibrations.TOF calculation was determined based on actual metal loading, with this achieved through the ICP-MS analysis of fresh samples (using a combination of microwave-assisted aqua-regia digestion), and the analysis of post-reaction solutions.
Catalyst reusability in the oxidation of methane via in situ H2O2 synthesis
In order to determine catalyst reusability, a similar procedure to that outlined above for the oxidation of methane was followed using 0.075 g of catalyst. Following the initial test, the catalyst was recovered by filtration and dried (30 °C, 16 h, under vacuum). From the recovered sample, 0.028 g was used to conduct a standard methane oxidation reaction.Direct synthesis of H2O2 from H2 and O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 mL. To test each catalyst for H2O2 synthesis, the autoclave was charged with the catalyst (0.01 g) and solvent (5.6 g MeOH and 2.9 g H2O, both HPLC grade, Fischer Scientific). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5%H2/CO2 to a pressure of 420 psi, followed by the addition of 25% O2/CO2 (160 psi). Gaseous reagents were not continuously supplied. The temperature was then decreased to 2 °C followed by stirring (1200 RPM) of the reaction mixture for 0.5 h. The above reaction parameters represent the optimum conditions we have previously used for the synthesis of H2O2. H2O2 productivity was determined by titrating aliquots (approximately 0.5 g) of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.Degradation of H2O2
Catalytic activity towards H2O2 degradation was determined in a manner similar to the direct synthesis activity of a catalyst. The autoclave was charged with MeOH (5.6 g, HPLC grade, Fischer Scientific), H2O2 (50 wt% 0.69 g, Merck) H2O (2.21 g, HPLC grade, Fischer Scientific) and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution 2 aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The autoclave was purged with 5%H2/CO2 (100 psi) before being pressurised with 5%H2/CO2 (420 psi) and cooled to 2 °C. Upon reaching 2 °C the reaction mixture was stirred at 1200 RPM for 0.5 h. After the reaction was complete the catalyst was removed from the reaction solvents and as previously two aliquots (approximately 0.05 g) were titrated against an acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.Catalyst characterisation
X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were floated from the spectrometer by pressing onto Scotch-665 double-sided adhesive tape. Data was collected using monochromatic AlKα radiation (140 W, 10 mA × 14 kV) at pass energies of 160 eV for survey spectra, and 40 eV for the high-resolution scans with step sizes of 1 eV and 0.1 eV respectively. The system was operated in the Hybrid mode, using a combination of magnetic immersion and electrostatic lenses and acquired over an area of approximately 300 × 700 μm2. A magnetically confined charge compensation system was used to minimize charging of the sample surface, and all spectra were taken with a 90° take-off angle. The charge compensation system typically overcompensates the positive charge by approximately 3 eV, therefore all binding energies were referenced to the C (1s) binding energy of adventitious carbon contamination was taken to be 284.8 eV. Data were analysed using CasaXPS (v2.3.25)31 after subtraction of a Shirley background and using modified Wagner sensitivity factors as supplied by the manufacturer.Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dispersion in ethanol by sonication and deposited on 300 mesh copper grids coated with holey carbon film. Energy dispersive X-ray analysis (EDX) was performed using an Oxford Instruments X-MaxN 80 detector and the data were analysed using the Aztec software.
Aberration-corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected Hitachi HF5000 S/TEM, operating at 200 kV. The instrument was equipped with bright field (BF) and high angle annular dark field (HAADF) detectors for high spatial resolution STEM imaging experiments. This microscope was also equipped with a secondary electron detector and dual Oxford Instruments XEDS detectors (2 × 100 mm2) having a total collection angle of 2.02 sr. Additional aberration-corrected scanning transmission electron microscopy was performed using a ThermoFisher ThemisZ S/TEM, operating at 300 keV. The instrument was equipped with high angle annular dark field (HAADF) and a segmented DF4 detector for high spatial resolution STEM-HAADF and STEM-iDPC imaging experiments. The installed Super-X detector has a total area of 120 mm2 and 0.7 sr solid angle.
To allow for quantification of total metal loading catalytic samples were digested via microwave-assisted aqua-regia digestion of the as-prepared (dried only) catalyst samples, using a Milestone Connect Ethos UP microwave with an SK15 sample rotor. Samples were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent. Actual metal loadings of key catalytic samples are provided in Table S1.†
Total metal leaching from key catalysts was quantified via inductively coupled plasma mass spectrometry (ICP-MS). All samples were diluted by a factor of 10 using HPLC grade H2O (1%HNO3 and 0.5%HCl matrix). All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent.
Results and discussion
Selective oxidation of methane to methanol using H2 and O2
Initially, a series of supported AuPd catalysts were prepared by the wet co-impregnation of aqueous solutions of HAuCl4 and PdCl2 using a selection of catalyst supports. A 5 wt%AuPd/TiO2 catalyst was identified as the benchmark formulation as it was previously investigated by Ab Rahim et al. who reported that the catalyst produced 1.6 μmoles of oxygenates, under identical reaction conditions to that used in this work.14 AuPd nanoparticles supported on ZSM-5 and TS-1 supports were also prepared: ZSM-5 was selected due to its strong performance in methane to methanol catalysis while TS-1, as discussed above, has been widely reported to facilitate a range of selective oxidations using H2O2, both preformed and generated in situ. In particular, a 5 wt%AuPdPt/TS-1 catalyst was recently shown to be highly active for H2O2 synthesis.32 In this case, the introduction of Pt resulted in the electronic modification of Pd species, compared to the bimetallic AuPd parent material, with a resulting improvement in catalytic activity. The comparison of methane oxidation and H2O2 synthesis is a key aspect of the current investigation, hence the inclusion of the 5 wt%AuPdPt/TS-1 catalyst in the initial set of screening experiments. While each support could be considered a strong candidate, the porosity, reducibility, acid–base properties and specific surface area vary significantly across the set of catalysts. The catalytic results are shown in Table 1 below, which includes data from the previous investigation by Ab Rahim et al.14| Entry | Catalyst | Products (μmoles) | CH3OH selectivity (%) | Oxygenate selectivity (%) | TOFa (h−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CH3OH | CH3OOH | HCOOH | CO2 | Total products | Total oxygenates | |||||
Methane oxidation reaction conditions: catalyst (0.028 g), H2O (10.0 ml), 435 psi total pressure (0.86% H2, 1.72% O2, 75.86% CH4, 21.65% N2), 0.5 h, 50 °C, 1500 rpm.a With the exception of entry 2 turnover frequency (TOF) is calculated using the total moles of product and based on on actual metal loading as determined by ICP-MS analysis of digested catalyst samples.b 2.4 wt%Au–2.4 wt%Pd–0.2 wt%Pt/TS-1. Note: with the exception of entry 6 the metal loading of all catalysts is 5 wt% and for all formulations Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio is 1:1 (wt/wt). :Pd ratio is 1:1 (wt/wt). |
||||||||||
| 1 | Blank | 0.06 | 0.00 | 0.00 | 0.45 | 0.51 | 0.06 | 11 | 11 | — |
| 2 | AuPd/TiO2 (from ref. 12) | 1.31 | 0.29 | 0 | 0.32 | 1.92 | 1.60 | 68 | 83 | 0.38 |
| 3 | AuPd/TiO2 | 1.40 | 0.18 | 0.82 | 1.24 | 3.64 | 2.40 | 39 | 66 | 0.73 |
| 4 | AuPd/ZSM-5 | 0.38 | 0.07 | 1.54 | 0.75 | 2.74 | 1.99 | 14 | 73 | 0.54 |
| 5 | AuPdPt/TS-1b | 1.15 | 0.48 | 0.88 | 0.98 | 3.49 | 2.51 | 33 | 72 | 0.52 |
| 6 | 0.66 wt%AuPd/TS-1 | 0.48 | 0.00 | 0.11 | 0.52 | 1.11 | 0.59 | 43 | 53 | 1.68 |
As shown in Entry 1 of Table 1, in the absence of a catalyst very low levels of liquid products were observed. It is unlikely that the reaction would proceed non-catalytically and the concentration of methanol observed (0.06 μmoles) is on the limit of reliable detection and quantification. Furthermore, the observed CO2 in the case of the blank experiment is likely adventitious, resulting from the incomplete purging of the reaction solution and the off-line nature of our gaseous analysis. Although it is clear that CO2 may be formed through the over-oxidation of methane activation products, such as methanol, methyl hydroperoxide and formic acid. Entries 2 and 3 in Table 1 compare the previous work with our current investigations. These data are generally in good agreement, indicated by similar levels of methanol and methyl hydroperoxide production. However, an increased production of formic acid and CO2 was observed in the current work, and this is reflected in the different TOF values measured (0.73 compared to 0.38 in the previous study). The difference in performance may be related to the variation in the batch-on-batch synthesis of wet co-impregnation catalysts. Interestingly, the 5 wt%AuPd/ZSM-5 catalyst (Table 1, Entry 4) exhibited higher selectivity to formic acid than methanol, producing 1.99 μmoles of total oxygenates. The 5 wt%AuPdPt/TS-1 catalyst exhibited similar activity to that of 5 wt%AuPd/TiO2 (2.40 and 2.51 mol of total oxygenates produced over the TiO2 and TS-1 supported materials respectively), although the TS-1 based catalyst exhibited slightly lower CO2 production. Table 1 Entry 6 shows the catalytic activity of a 0.66 wt%AuPd/TS-1 catalyst. The objective of preparing this catalyst was to minimise the formation of large nanoparticles. Williams et al. have previously reported that 0.13 wt%AuPd/TiO2 catalysts were intrinsically more active for the selective oxidation of methane to methanol using preformed H2O2 compared to catalysts prepared with higher metal loadings.9 Specifically, lower metal loadings facilitated decreased H2O2 decomposition, which in turn increased the lifetime of reactive oxygen species in the reactor, leading to higher oxygenate production. Additionally, in the direct synthesis of H2O2, it has been shown that low-loaded AuPd catalysts are also highly active.33 The TOF of the 0.66 wt%AuPd/TS-1 catalyst was the highest of all the catalyst formulations screened, with a value of 1.68, approximately double that of the 5 wt%AuPd/TiO2 formulation, although the oxygenate selectivity was slightly reduced (53 and 66% for the 0.66 wt%AuPd/TS-1 and 5 wt%AuPd/TiO2 catalysts respectively). It is interesting to contrast the performance of the ZSM-5 and TS-1 supported materials (Table 1, Entries 4, 5 and 6). The AuPd/ZSM-5 catalyst mostly produced formic acid, while the TS-1 catalysts both favoured methanol and methyl hydroperoxide formation. The propensity of ZSM-5 to preferentially form formic acid over methanol was also reported by Hammond et al.34 The origin of overoxidation was suggested to be the surface decomposition of methyl hydroperoxide to methanol, which liberates ˙OH radicals that subsequently react with methanol to produce formaldehyde and formic acid.22 Both supports have a microporous MFI structure, but only ZSM-5 possesses strong Brønsted and Lewis acid sites. Methoxy groups may adsorb more strongly on ZSM-5 than TS-1 (or TiO2), increasing the surface lifetime of the intermediate and promoting overoxidation. TS-1, therefore, appears to be a more appropriate support for liquid-phase selective methane oxidation in the current work.
Based on the above data, a second series of low-loaded AuPd/TS-1 catalysts (total metal loading of 0.66 wt%) were prepared, via wet co-impregnation, with various Au:Pd ratios chosen in order to study the effect of nanoparticle composition on catalyst activity. Although the inclusion of Pt in the 5 wt%AuPd/TS-1 catalyst has been previously reported to be beneficial,25 the presence of a third metal introduces complexity to the catalyst structure. Therefore, in the present study, the Au:Pd ratio was investigated without the addition of Pt. The reactivity of the 0.66 wt%AuPd/TS-1 catalyst series is presented in Table 2.
:Pd ratio on the activity of 0.66 wt%AuPd/TS-1 catalysts
| Entry | Catalyst | Products (μmoles) | CH3OH selectivity (%) | Oxygenate selectivity (%) | TOFa (h−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CH3OH | CH3OOH | HCOOH | CO2 | Total products | Total oxygenates | |||||
| Methane oxidation reaction conditions: catalyst (0.028 g), H2O (10.0 g), 435 psi total pressure (0.86% H2, 1.72% O2, 75.86% CH4, 21.65% N2), 0.5 h, 50 °C, 1500 rpm.a Turnover frequency (TOF) calculated using the total moles of product and based on actual metal loading as determined by ICP-MS analysis of digested catalyst samples. | ||||||||||
| 1 | 0.66%Au | 0.17 | 0.00 | 0.09 | 0.49 | 0.75 | 0.26 | 22 | 34 | 1.61 |
| 2 | 0.55%Au–0.11%Pd | 0.22 | 0.00 | 0.04 | 0.46 | 0.72 | 0.26 | 30 | 36 | 1.37 |
| 3 | 0.44%Au–0.22%Pd | 0.21 | 0.00 | 0.39 | 0.37 | 0.96 | 0.59 | 22 | 62 | 1.63 |
| 4 | 0.33%Au–0.33%Pd | 0.48 | 0.00 | 0.11 | 0.52 | 1.11 | 0.59 | 43 | 53 | 1.68 |
| 5 | 0.11%Au–0.55%Pd | 0.40 | 0.00 | 0.07 | 0.27 | 0.74 | 0.47 | 54 | 63 | 0.92 |
| 6 | 0.66%Pd | 0.09 | 0.02 | 0.30 | 0.31 | 0.72 | 0.41 | 15 | 57 | 0.84 |
The rate of oxygenate formation varied across the series of catalysts. However, the greatest concentration was observed over the Pd-rich formulations, compared to the Au-rich analogues. In previous studies of selective methane oxidation over supported catalysts, methanol was the major oxygenate observed, which is generally the case in this work, with the exception of 0.66 wt%Pd/TS-1 and 0.44 wt%Au–0.22 wt%Pd/TS-1 catalysts. In the case of these catalysts, formic acid was the major oxygenate produced and CO2 production was lower compared to the rest of the catalytic series.
To further investigate catalyst performance and with a focus on the 0.33 wt%Au–0.33 wt%Pd/TS-1 formulation, catalyst activity was determined over multiple uses in the methane oxidation reaction (Table 3), with the extent of methanol and total oxygenate formation remaining consistent at ca. 0.6 μmol over each of the three reactions. Notably, we have previously reported the high stability of comparable materials, prepared by an analogous wet co-impregnation procedure, during application in both the direct synthesis of H2O235 and in situ oxidative valorisation of chemical feedstocks, under reaction conditions considered far more conducive towards metal leaching than those utilised for the in situ oxidation of methane.36 In keeping with these earlier studies, no leaching of Au was detected over the three reactions, although a minor loss of Pd was observed (Table S2†). Notably, the leaching of Pd was only measurable over the first two uses of the catalyst, with no further leaching upon use in the third reaction. However, further studies over extended reaction times are required in order to determine catalyst stability when utilised for methane activation, particularly given the formation of low concentrations of chelating agents such as formic acid.37
| Use | Products (μmol) | CH3OH selectivity (%) | Oxygenate selectivity (%) | TOFa (h−1) | |||||
|---|---|---|---|---|---|---|---|---|---|
| CH3OH | CH3OOH | HCOOH | CO2 | Total products | Total oxygenates | ||||
| Methane oxidation reaction conditions: catalyst (0.028 g), H2O (10.0 g), 435 psi total pressure (0.86% H2, 1.72% O2, 75.86% CH4, 21.65% N2), 0.5 h, 50 °C, 1500 rpm.a Turnover frequency (TOF) calculated using the total moles of product and based on actual metal loading as determined by ICP-MS analysis of digested catalyst samples and analysis of post-reaction solutions. | |||||||||
| 1 | 0.48 | 0.00 | 0.11 | 0.52 | 1.11 | 0.59 | 43.6 | 53 | 1.66 |
| 2 | 0.44 | 0.04 | 0.17 | 1.12 | 1.77 | 0.65 | 25.0 | 37 | 2.65 |
| 3 | 0.41 | 0.13 | 0.12 | 0.59 | 1.25 | 0.66 | 32.7 | 53 | 1.87 |
The catalyst testing results are promising and indicate that low-loaded Pd-based bimetallic catalysts can catalyse the selective oxidation of methane to methanol using H2 and O2. To rationalise the activity trends of the AuPd/TS-1 catalysts, and to gain a deeper understanding of the reaction mechanism, the samples were tested for H2O2 direct synthesis and degradation and characterised by XPS and TEM. H2O2 synthesis and degradation experiments (Fig. 1) were carried out under reaction conditions previously optimised to enhance H2O2 stability namely; sub-ambient temperature, a methanol co-solvent and a CO2 gaseous diluent, all of which have been shown to inhibit H2O2 degradation pathways.30
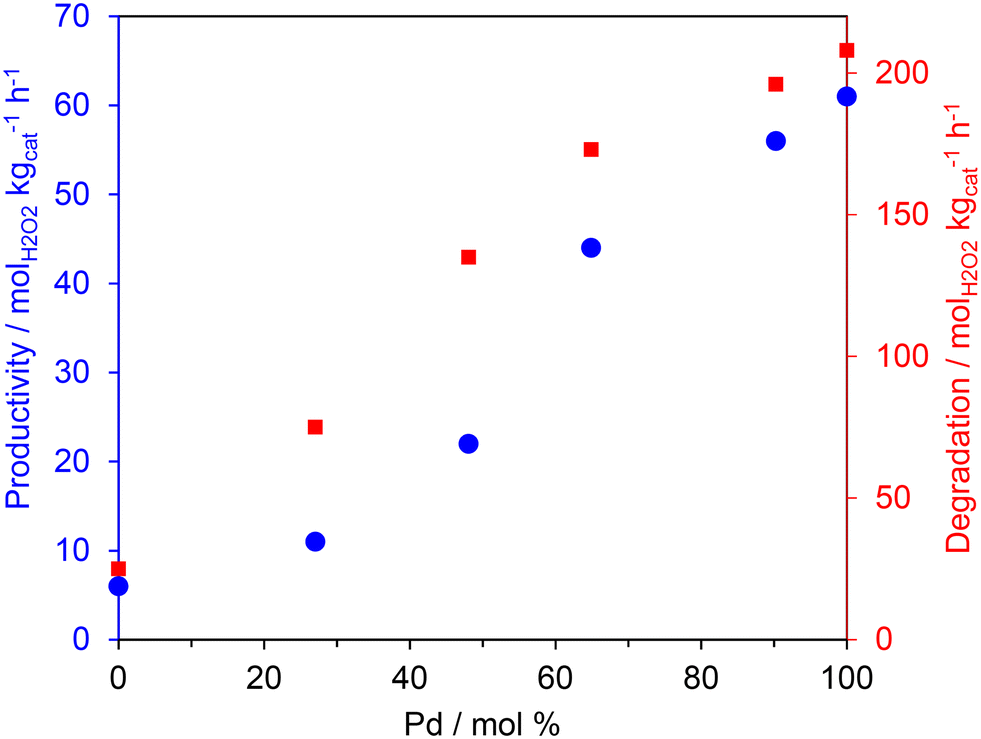 | ||
| Fig. 1 The effect of Pd: Au ratio on the catalytic activity of 0.66%PdAu/TS-1 catalysts towards the direct synthesis (●) and subsequent degradation (■) of H2O2. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g), H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. | ||
In keeping with earlier studies into AuPd nanoalloys immobilised on SiO238 and TS-132 supports, catalytic activity towards both the direct synthesis and subsequent degradation of H2O2 was found to correlate well with total Pd content. Indeed, the monometallic Pd catalyst offered the greatest activity towards both the direct synthesis of H2O2 (66 molH2O2 kgcat−1 h−1) as well as its subsequent degradation (209 molH2O2 kgcat−1 h−1) outperforming both the Au-only and bimetallic formulations. Such observations have been attributed to the poor mixing of the Au and Pd metallic components when immobilised onto the titanosilicate support.36 Catalytic performance towards H2O2 direct synthesis was not found to follow the same trend as that observed for methane oxidation (Table 2), with the bimetallic formulations achieving both higher oxygenate selectivity and TOFs than those observed over the monometallic Pd catalyst. When considered alongside previous investigations that report the crucial role of Au in promoting the release of ROS from catalyst surfaces,39,40 these observations indicate that the rate of methane oxidation is not simply a function of H2O2 production. There is likely an important contribution from the highly reactive radical species that are generated as intermediates during the formation of H2O2. As such it is possible that the observed improved activity of the bimetallic catalysts is related to the release of ROS, which are known to be crucial in the formation of methyl radicals, via H-abstraction, and subsequent formation of oxygenates.22
Fig. 2 shows the correlation between oxygenate formation and catalyst composition in addition to catalytic activity towards H2O2 synthesis and degradation. The observed trends approximate a volcano plot, where the highest rate of oxygenate production is associated with catalysts that offer moderate rates of H2O2 synthesis and subsequent degradation, that is those materials that consist of approximately equal weight loadings of Au and Pd. This relationship may also indicate the contribution of ROS in addition to H2O2 towards methane oxidation but also the importance of catalytic selectivity towards H2O2 synthesis and the need to inhibit competitive H2O2 degradation pathways.
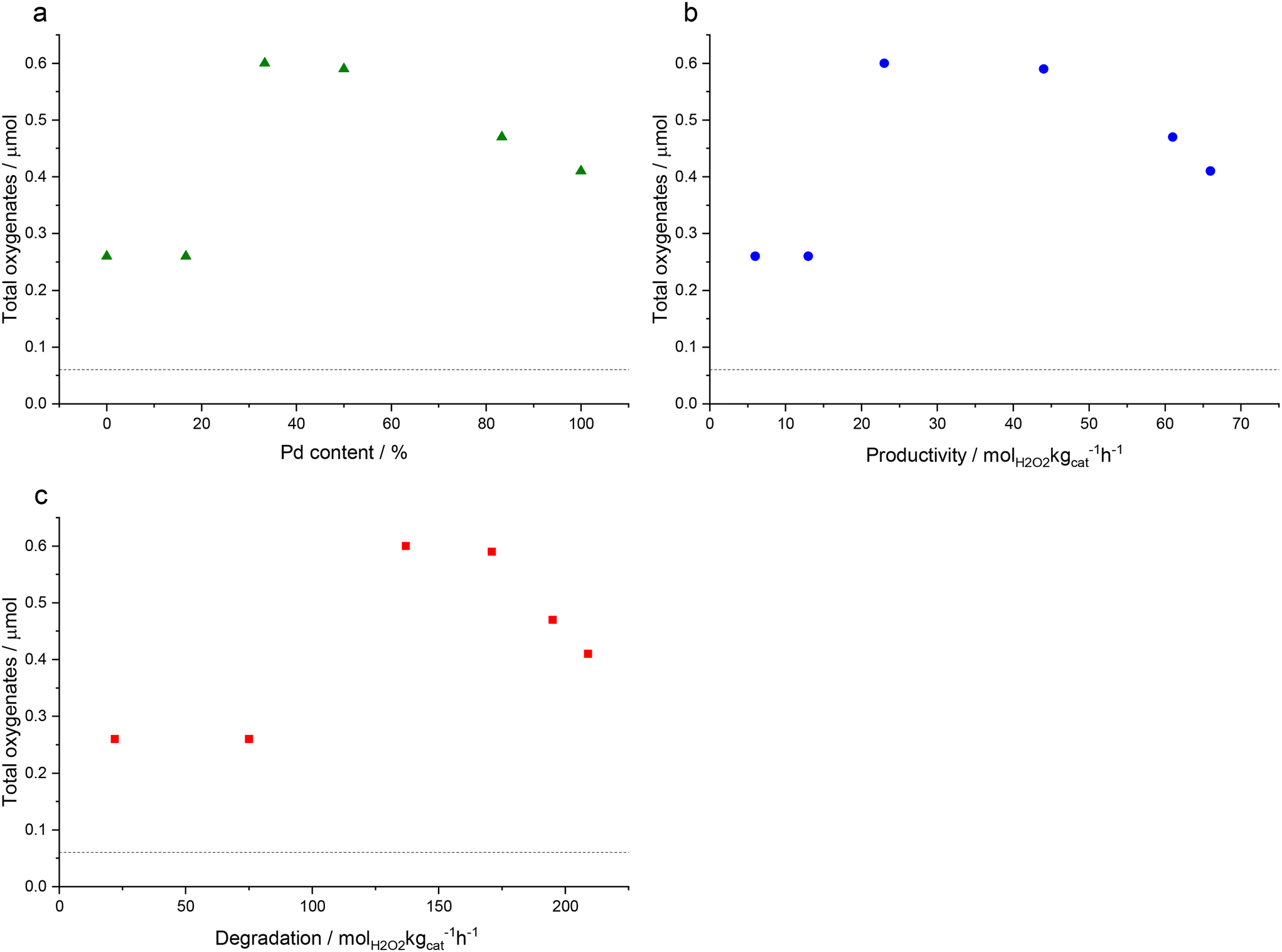 | ||
| Fig. 2 The relationship between total oxygenates formation during the oxidation of methane using H2 and O2 and (a) Pd content, (b) H2O2 synthesis activity and (c) H2O2 degradation rates. Methane oxidation reaction conditions: catalyst (0.028 g), H2O (10.0 g), 435 psi total pressure (0.86% H2, 1.72% O2, 75.86% CH4, 21.65% N2), 0.5 h, 50 °C, 1500 rpm. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g), H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. Note: the activity of the blank reaction is indicated by the dashed line. | ||
The differences in observed catalysis across the catalytic series could be associated with a number of factors, namely the particle size and the electronic properties of the nanoparticles (which will be dictated by nanoalloy composition among other factors). To investigate these properties, XPS and TEM analyses were carried out. The selectivity of Pd-based catalysts during H2O2 synthesis has been widely reported to be influenced by Pd oxidation state.41–43 Analysis of the series of supported 0.66 wt%AuPd/TS-1 catalysts via XPS (Fig. S1†) was challenging due to the low loadings of metal. In the case of the 0.66 wt%Pd/TS-1 catalyst, Pd was present almost entirely in the Pd2+ oxidation state, which was expected as the catalysts were exposed to an oxidative heat treatment. However, the introduction of Au resulted in a slight shift in Pd speciation towards Pd0. Similar shifts in the Au 4f binding energy were also observed, whereby the Au-only catalyst exhibited a binding energy of ∼84.0 eV and this shifted downward after Pd addition. Such observations are in keeping with previous studies into AuPd catalysts44 and suggest the formation of PdAu alloys; with such species widely reported to offer improved reactivity over monometallic analogues, for a range of reactions.45–47 It should be noted that in all cases, due to the low metal loading, the signal-noise ratio was relatively high, which prohibited the meaningful deconvolution of the spectra.
Numerous previous publications have also identified the role of nanoparticle size on catalyst efficiency. The effect of particle size on selectivity towards H2O2 was notably highlighted by Kim et al., who found that smaller Pd particles, which contain high proportions of defect sites, were less selective towards H2O2 and promoted the degradation of the oxidant to H2O.48 Williams et al. identified similar trends for the valorisation of methane to methanol when using preformed H2O2 over supported AuPd catalysts.9 Particle size distributions of the TS-1 supported catalysts were calculated using TEM analysis (Table 4, with corresponding electron micrographs reported in Fig. S2†), which revealed no clear trend: the 0.33 wt%Au–0.33 wt%Pd/TS-1 and 0.44 wt%Au–0.22 wt%Pd/TS-1 catalysts exhibited mean particle sizes of 23 and 17 nm, respectively, while all of the other samples were measured to be similar (7–11 nm). The particle size distributions, shown in Fig. S2,† indicate that most particles counted were <20 nm, but a minority of large particles (50–100 nm) were also detected, such observations are typical of the wet impregnation route to catalyst synthesis, particularly for AuPd catalysts, with a bimodal distribution of large Au-rich and small Pd-rich species widely reported.49 It should be noted that a positive correlation between Pd content and H2O2 synthesis rates was also observed under conditions optimal for H2O2 production. This, combined with the known ability of Au to promote ROS desorption from catalytic surfaces,40 as discussed above, means we are unable to definitively assign the underlying cause for the improved catalysis observed over the bimetallic formulations. However, such observations are particularly intriguing.
:Pd ratio as derived by TEM analysis
| Catalyst | Particle size/nm (standard deviation) |
|---|---|
| Catalysts exposed to an oxidative heat treatment (static air, 400 °C, 3 h, 10 °C min−1). 150 or more particles were counted for 0.66 wt%Pd/TS-1, 0.44 wt%Au–0.22 wt%Pd/TS-1, 0.33 wt%Au–0.33 wt%Pd/TS-1 and 0.11 wt%Au–0.55 wt%Pd/TS-1 catalysts. In the case of 0.66 wt%Au/TS-1 and 0.55 wt%Au–0.11 wt%Pd/TS-1 catalysts, imaging particles was more challenging and counting >50 particles was not possible. | |
| 0.66 wt%Au/TS-1 | 10.6 (4.5) |
| 0.55 wt%Au–0.11 wt%Pd/TS-1 | 22.6 (26.6) |
| 0.44 wt%Au–0.22 wt%Pd/TS-1 | 17.4 (18.4) |
| 0.33 wt%Au–0.33 wt%Pd/TS-1 | 9.3 (10.1) |
| 0.11 wt%Au–0.55 wt%Pd/TS-1 | 8.9 (10.8) |
| 0.66 wt%Pd/TS-1 | 7.2 (3.53) |
With a clear bi-modal particle size distribution evident from our TEM analysis, and with a focus on the 0.33 wt%Au–0.33 wt%Pd/TS-1 catalyst we subsequently conducted detailed HAADF-STEM and XEDS analysis, in order to determine the extent of alloy formation. These studies have confirmed the presence of both Pd-only nanoparticles (Fig. S3†) and AuPd nanoalloys (Fig. S4†), where the latter exist over a large particle size range, whereas no large (>20 nm) monometallic Pd species were detected. Notably, we have also confirmed that a considerable population of the AuPd alloyed particles adopt a Au-core Pd-shell morphology, which may be expected given the exposure to an oxidative heat treatment, and the propensity for Pd to undergo oxidation (Fig. 3).50
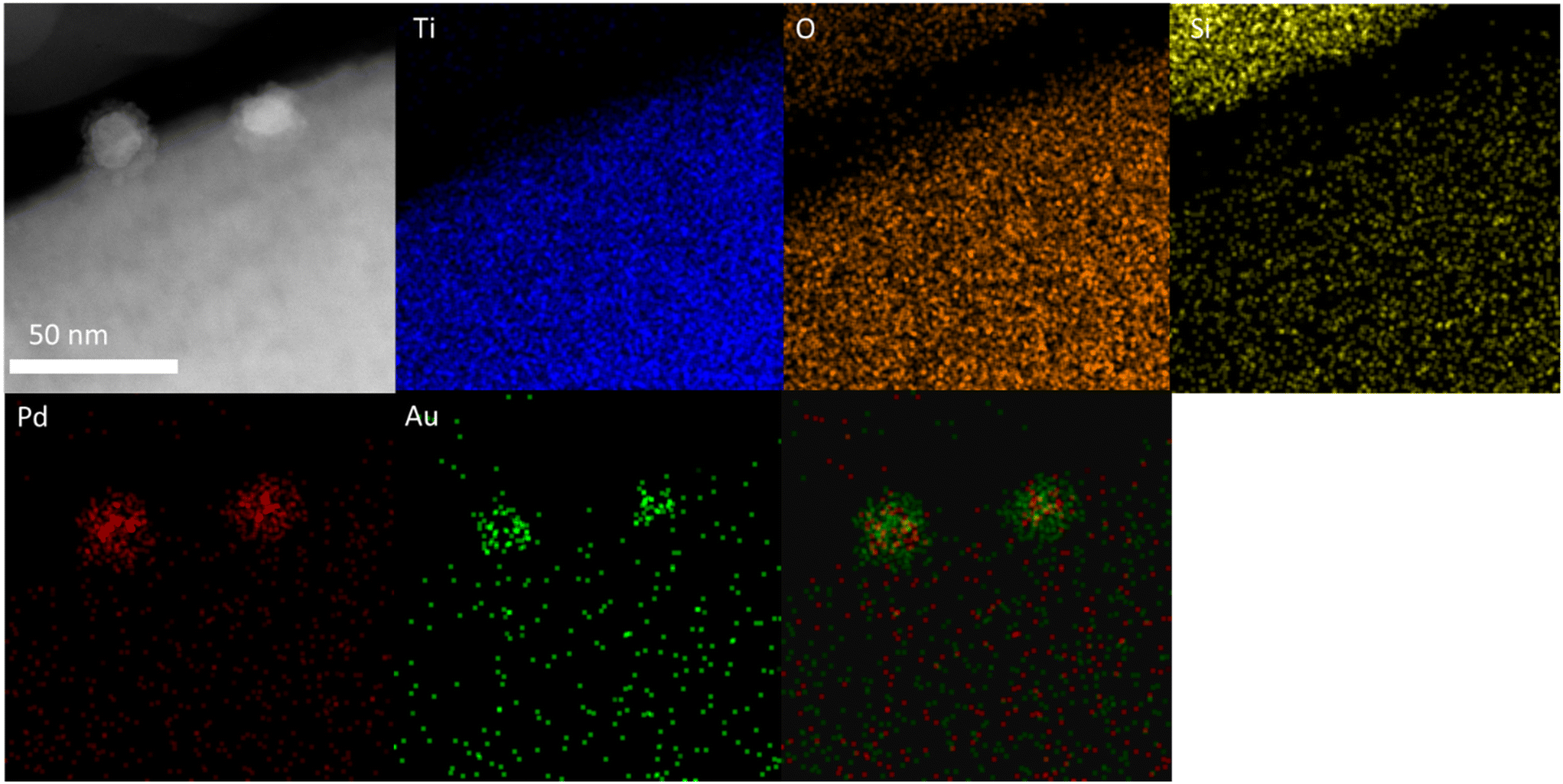 | ||
| Fig. 3 Microstructural analysis of the 0.33 wt%Au–0.33 wt%Pd/TS-1 catalyst including HAADF-STEM image and X-EDS mapping of the highlighted area showing the presence of AuPd nanoalloys with a Au-core Pd-shell morphology, Au (green), Pd (red), Ti (blue), and Si (yellow). Further analysis is reported in Fig. S3 and S4.† | ||
Although the performance of the 0.66 wt%AuPd/TS-1 catalysts toward the selective oxidation of methane to methanol with an oxidant produced in situ is promising, from an economic/green chemistry perspective minimising precious metal content would be desirable. The alloying of Pd with a range of base metals has been reported to offer improved catalytic performance compared to Pd-only analogues for a range of selective oxidation reactions,51,52 including methane oxidation53 and H2O2 direct synthesis.54–57 Such improvements have often been attributed to the modification of the Pd oxidation state and the disruption of contiguous Pd ensembles.58 As such, we subsequently evaluated the performance of a range of bimetallic MPd/TS-1 (total metal loading = 0.66 wt%, 1:1 weight ratio, where M = Mn, Cu or Ni) catalysts for the oxidation of methane using H2 and O2 (Table 5). Oxygenates were observed in each reaction. However, in the case of CuPd/TS-1 and MnPd/TS-1 catalysts, total oxygenate formation was approximately half of that observed over the AuPd analogue and less than that observed over the Pd-only catalyst. Such observations can be rationalised when considering the high activity of Mn towards H2O2 decomposition59 and the previous reports that hydroperoxyl (OOH*) and H2O2 formation over Cu-containing supported catalysts is kinetically unfavourable.60 Indeed, we have previously demonstrated that the introduction of Cu at high loadings into AuPd or Pd-only catalysts can significantly inhibit catalyst activity towards H2O2 synthesis and a range of H2O2 catalysed selective oxidative transformations, including the valorisation of methane.61,62 Although more recently the promotive effect that results from the introduction of dopant concentrations of Cu into AuPd nanoalloys has been identified, particularly for the direct synthesis of H2O2.63,64
| Catalyst | Product amount (μmol) | CH3OH selectivity (%) | Oxygenates selectivity (%) | TOFa (h−1) | |||||
|---|---|---|---|---|---|---|---|---|---|
| CH3OH | CH3OOH | HCOOH | CO2 | Total products | Total oxygenates | ||||
| Methane oxidation reaction conditions: catalyst (0.028 g), H2O (10.0 g), 435 psi total pressure (0.86% H2, 1.72% O2, 75.86% CH4, 21.65% N2), 0.5 h, 50 °C, 1500 rpm.a Turnover frequency (TOF) calculated using the total moles of product and based on actual metal loading as determined by ICP-MS analysis of digested catalyst samples. Note: in the case of the Pd-only catalyst total Pd loading is 0.66%. | |||||||||
| Pd | 0.09 | 0.02 | 0.30 | 0.31 | 0.72 | 0.41 | 15 | 57 | 0.84 |
| AuPd | 0.48 | 0.00 | 0.11 | 0.52 | 1.11 | 0.59 | 43 | 53 | 1.68 |
| CuPd | 0.32 | 0.01 | 0.00 | 0.20 | 0.54 | 0.33 | 60 | 62 | 0.47 |
| NiPd | 0.14 | 0.00 | 0.46 | 0.63 | 1.23 | 0.60 | 11 | 49 | 1.05 |
| MnPd | 0.24 | 0.00 | 0.00 | 0.44 | 0.68 | 0.24 | 35 | 35 | 0.56 |
Notably, the Ni-Pd/TS-1 catalyst was able to achieve a near-identical concentration of oxygenates (0.60 μmol) to that achieved over the AuPd analogue (0.59 μmol), although CO2 production was somewhat higher over the NiPd formulation. Interestingly, formic acid was the major product in this case rather than methanol or methyl hydroperoxide. It is noteworthy that 0.41 μmol of total oxygenates were observed over the monometallic 0.66 wt%Pd/TS-1 catalyst confirming that, in a similar manner to Au, the alloying of Pd with Ni is beneficial to the catalytic performance.
Conclusions
We have shown that the oxidation of methane to methanol using in situ synthesised H2O2 is catalysed by a range of supported Pd-based catalysts, with the introduction of Au into Pd, in particular, promoting catalytic performance. Evaluation of catalyst formulation, while maintaining total metal loading at 0.66 wt% revealed that the most active catalysts were those where the Au:Pd ratio was close to 1. Comparison of CH4 oxidation rates with H2O2 synthesis rates under idealised conditions indicated that the most active catalysts for CH4 oxidation were moderately active for H2O2 synthesis and degradation, with the enhanced performance of the AuPd alloyed catalysts considered to result from the ability of Au to both modify Pd oxidation states and promote the release of highly reactive oxygen-based radicals (ROS), which are formed as intermediates during H2O2 formation. Reusability tests confirmed that the optimal AuPd catalyst was stable for multiple uses, with minimal leaching of metal species detected, however further study over extended reaction times is still required. We have also demonstrated that Au can be substituted for more economically attractive metals. In particular, the NiPd/TS-1 catalyst exhibited similar oxygenate production to that observed over the AuPd analogue, although formic acid was the dominant product. These studies represent a promising basis for further exploration for developing catalysts for the selective oxidation of methane and other chemical feedstocks.
Author contributions
J. H. C., R. J. L., N. F. D., S. H. T. and G. J. H., contributed to the design of the study; J. H. C., R. J. L. and N. D. conducted experiments and data analysis. J. H. C., R. J. L., C. W., N. F. D., D. J. W., X. L., S. H. T. and G. J. H. provided technical advice and result interpretation. R. J. L., D. J. M., T. E. D., T. Q. and X. L. conducted catalyst characterization and corresponding data processing. J. H. C. and R. J. L., wrote the manuscript and the ESI† material, all authors commented on and amended both documents. All authors discussed and contributed to the work.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to thank ExxonMobil for providing technical advice and support. In particular, we would like to thank Sara Yacob, Pedro Serna and Randall J. Meyer for useful discussions. XPS data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under contract no. PR16195. The authors would like to thank the CCI-Electron Microscopy Facility which has been part-funded by the European Regional Development Fund through the Welsh Government and The Wolfson Foundation. Funding: J. H. C. and C. W. acknowledge ExxonMobil for funding. X. L. acknowledges financial support from National Key R&D Program of China (2021YFA1500300 and 2022YFA1500146) and National Natural Science Foundation of China (22072090 and 22272106). J. H. C., R. J. L., N. F. D., S. H. T. and G. J. H. also gratefully acknowledge Cardiff University and the Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support.References
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2022, 123(9), 6359–6411 CrossRef PubMed.
- IEA, Flaring Emissions, Paris, 2021, https://www.iea.org/reports/flaring-emissions, accessed 10.12. 2022.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- G. Qi, T. E. Davies, A. Nasrallah, M. A. Sainna, A. G. R. Howe, R. J. Lewis, M. Quesne, C. R. A. Catlow, D. J. Willock, Q. He, D. Bethell, M. J. Howard, B. A. Murrer, B. Harrison, C. J. Kiely, X. Zhao, F. Deng, J. Xu and G. J. Hutchings, Nat. Catal., 2022, 5, 45–54 CrossRef CAS.
- S. J. Freakley, N. Dimitratos, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Acc. Chem. Res., 2021, 54, 2614–2623 CrossRef CAS PubMed.
- R. J. Lewis, A. Bara-Estaun, N. Agarwal, S. J. Freakley, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2019, 149, 3066–3075 CrossRef CAS.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- S. Al-Shihri, C. J. Richard and D. Chadwick, ChemCatChem, 2017, 9, 1276–1283 CrossRef CAS.
- T. Moteki, N. Tominaga and M. Ogura, Appl. Catal., B, 2022, 300, 120742 CrossRef CAS.
- C. Wang, Y. Sun, L. Wang, W. Feng, Y. Miao, M. Yu, Y. Wang, X. Gao, Q. Zhao, Z. Ding, Z. Feng, S. Yu, J. Yang, Y. Hu and J. Wu, Appl. Catal., B, 2023, 329, 122549 CrossRef CAS.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS PubMed.
- Y. Xu, D. Wu, P. Deng, J. Li, J. Luo, Q. Chen, W. Huang, C. M. Shim, C. Jia, Z. Liu, Y. Shen and X. Tian, Appl. Catal., B, 2022, 308, 121223 CrossRef CAS.
- B. Wu, T. Lin, M. Huang, S. Li, J. Li, X. Yu, R. Yang, F. Sun, Z. Jiang, Y. Sun and L. Zhong, Angew. Chem., 2022, 61, e202204116 CAS.
- A. Delparish, S. Kanungo, J. van der Schaaf and M. F. Neira d'Angelo, Catal. Sci. Technol., 2019, 9, 5142–5149 RSC.
- E. D. Park, Y.-S. Hwang, C. W. Lee and J. S. Lee, Appl. Catal., B, 2003, 247, 269–281 CrossRef CAS.
- E. D. Park, Y. S. Hwang and J. S. Lee, Catal. Commun., 2001, 2, 187–190 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, ACS Catal., 2018, 8, 7961–7972 CrossRef PubMed.
- R. Serra-Maia, F. M. Michel, T. A. Douglas, Y. Kang and E. A. Stach, ACS Catal., 2021, 11, 2837–2845 CrossRef CAS.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2013, 52, 1168–1178 CrossRef CAS.
- J. Le Bars, J. Dakka and R. A. Sheldon, Appl. Catal., A, 1996, 136, 69–80 CrossRef CAS.
- C. P. Gordon, H. Engler, A. S. Tragl, M. Plodinec, T. Lunkenbein, A. Berkessel, J. H. Teles, A. Parvulescu and C. Copéret, Nature, 2020, 586, 708–713 CrossRef CAS PubMed.
- I. Moreno, N. F. Dummer, J. K. Edwards, M. Alhumaimess, M. Sankar, R. Sanz, P. Pizarro, D. P. Serrano and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 2425 RSC.
- K. Mori, Y. Miura, S. Shironita and H. Yamashita, Langmuir, 2009, 25, 11180–11187 CrossRef CAS PubMed.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Commun., 2007, 8, 247–250 CrossRef CAS.
- A. Prieto, M. Palomino, U. Díaz and A. Corma, Appl. Catal., A, 2016, 523, 73–84 CrossRef CAS.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci., 2021, 5, 100112 CrossRef.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- J. Brehm, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catal. Lett., 2022, 152, 254–262 CrossRef CAS.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Eur. J. Chem., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- J. Edwards, B. Solsona, P. Landon, A. Carley, A. Herzing, C. Kiely and G. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- R. Underhill, R. J. Lewis, S. J. Freakley, M. Douthwaite, P. J. Miedziak, J. K. Edwards, O. Akdim and G. J. Hutchings, Johnson Matthey Technol. Rev., 2018, 62, 417 CrossRef CAS.
- J. K. Edwards, S. F. Parker, J. Pritchard, M. Piccinini, S. J. Freakley, Q. He, A. F. Carley, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 812–818 RSC.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- L. Ouyang, P. Tian, G. Da, X. Xu, C. Ao, T. Chen, R. Si, J. Xu and Y. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- A. G. Gaikwad, S. D. Sansare and V. R. Choudhary, J. Mol. Catal. A: Chem., 2002, 181, 143–149 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163 CrossRef.
- L. Prati, A. Villa, F. Porta, D. Wang and D. Su, Catal. Today, 2007, 122, 386–390 CrossRef CAS.
- H. Duan, Y. Zeng, X. Yao, P. Xing, J. Liu and Y. Zhao, Chem. Mater., 2017, 29, 3671–3677 CrossRef CAS.
- S. Kim, D. Lee, K. Lee and E. A. Cho, Catal. Lett., 2014, 144, 905–911 CrossRef CAS.
- G. J. Hutchings and C. J. Kiely, Acc. Chem. Res., 2013, 46, 1759–1772 CrossRef CAS PubMed.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225–239 RSC.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. J. A. Slater, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2021, 151, 2762–2774 CrossRef CAS.
- A. Santos, R. J. Lewis, D. J. Morgan, T. E. Davies, E. Hampton, P. Gaskin and G. J. Hutchings, Catal. Sci. Technol., 2022, 12, 2943–2953 RSC.
- J. Kang, P. Puthiaraj, W. Ahn and E. D. Park, Appl. Catal., A, 2020, 602, 117711 CrossRef CAS.
- D. A. Crole, R. Underhill, J. K. Edwards, G. Shaw, S. J. Freakley, G. J. Hutchings and R. J. Lewis, Philos. Trans. R. Soc., A, 2020, 378, 20200062 CrossRef CAS PubMed.
- H. Xu, D. Cheng and Y. Gao, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS.
- T. Richards, R. J. Lewis, D. J. Morgan and G. J. Hutchings, Catal. Lett., 2023, 153, 931 CrossRef CAS.
- S. Wang, R. J. Lewis, D. E. Doronkin, D. J. Morgan, J. Grunwaldt, G. J. Hutchings and S. Behrens, Catal. Sci. Technol., 2020, 10, 1925–1932 RSC.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009–8020 RSC.
- L. Micoli, G. Bagnasco, M. Turco, M. Trifuoggi, A. Russo Sorge, E. Fanelli, P. Pernice and A. Aronne, Appl. Catal., B, 2013, 140–141, 516–522 CrossRef CAS.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. C, 2007, 111, 7384–7395 CrossRef CAS.
- F. Alotaibi, S. Al-Mayman, M. Alotaibi, J. K. Edwards, R. J. Lewis, R. Alotaibi and G. J. Hutchings, Catal. Lett., 2019, 149, 998–1006 CrossRef CAS.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins, J. A. Lopez-Sanchez, S. H. Taylor and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 3410–3418 RSC.
- A. Barnes, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catal. Sci. Technol., 2022, 12, 1986–1995 RSC.
- A. Barnes, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catalysts, 2022, 12, 1396 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00116d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |