DOI:
10.1039/D3CC01517C
(Highlight)
Chem. Commun., 2023,
59, 7847-7862
Transition metal-catalysis in interrupted borrowing hydrogen strategy
Received
28th March 2023
, Accepted 23rd May 2023
First published on 24th May 2023
Abstract
In recent times, the transition metal-catalyzed borrowing hydrogen (BH) and interrupted borrowing hydrogen (IBH) strategies have attracted much attention and represent atom- and step-economic processes to access diverse building blocks via various C–C and C–heteroatom bond-forming reactions. The advantages of these approaches include (i) use of feedstock chemicals, (ii) high atom economy, (iii) no pre-activation of the substrates, and (iv) producing water as the only by-product. In this context, several synthetic strategies have been developed in this regime for the past few decades. To the best of our knowledge, no review article describes the important concepts of interrupted borrowing hydrogen (IBH) reaction. This review article highlights the recent advances in the IBH strategy and its application in sustainable chemical synthesis, particularly C–C bond formation using methanol as a C1 source, synthesis of 3,3′-bisindolylmethanes (3,3′-BIMs), α-branched ketones/diketones, and regioselective alkylation of N-heterocycles.
1. Introduction and brief historical perspective
The construction of diverse molecular frameworks via the carbon–heteroatom and carbon–carbon bond-forming reactions is fundamentally important in synthetic organic chemistry.1 Over the decades, several classical methods have been employed to access C–C and C–heteroatom bond formations. Recently, there has been a continuous challenging endeavor in the development of sustainable, efficient, and selective protocols under benign conditions by utilizing abundant feedstock chemicals.
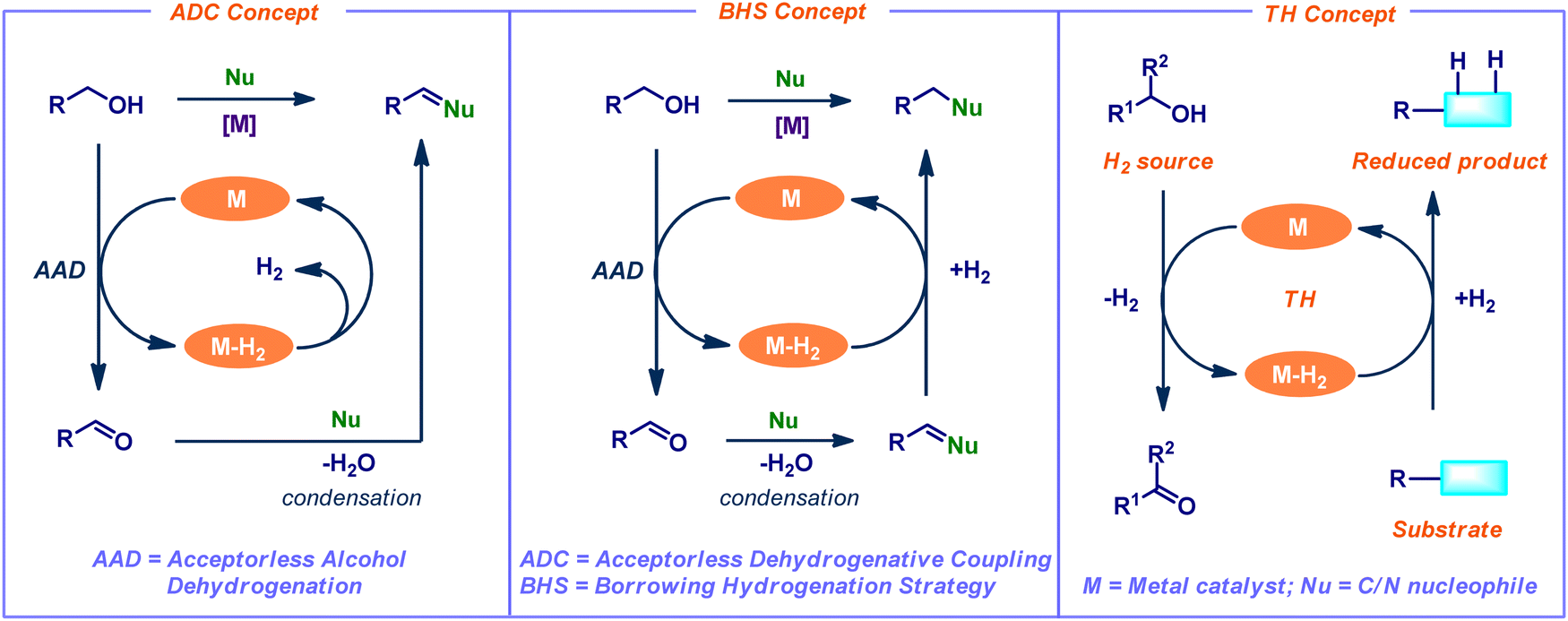
Dehydrogenation of alcohols is a significant process in organic chemistry, and its applications open a wide range of synthetically important transformations. In particular, the catalytic acceptorless alcohol dehydrogenation (AAD) strategy has attracted much interest in synthetic chemistry and chemical biology due to its advantageous properties, such as excellent step- and atom efficiency.2 This AAD method broadens the synthetic application of C–H, C–C, and C–heteroatom bond-forming reactions in current research by using straightforward and widely accessible feedstock molecules as the starting ingredients. In addition, the sole by-products are water and hydrogen gas. The application of AAD reactions is classified into three main concepts such as acceptorless dehydrogenative coupling (ADC), borrowing hydrogenation strategy (BHS), and transfer hydrogenation (TH) (Fig. 1). In the ADC process, dehydrogenation leads to condensation, which releases water and hydrogen gas while forming unsaturated molecules. In contrast, BHS consumes the generated hydrogen gas from the initial dehydrogenation step and forms saturated compounds as a final product. Typically, the BHS concept relies on the use of transition-metal catalysts. The concept involves borrowing hydrogen from one of the starting materials (mainly alcohols) as the hydrogen donor. Subsequently, the addition of hydrogen to the in situ generated an unsaturated functional group (hydrogen acceptor) of cross-coupled intermediate. Finally, it leads to the desired N- or C-alkylated products.3 Overall, this process is redox neutral and generates water as the only by-product (Fig. 1). Notably, conventional alkylation reactions involve enolate formation followed by an alkylation reaction under harsh conditions (e.g., lithium amide bases associated with halogenoalkane electrophiles). Hence, the BHS reaction is considered as an eco-friendly, efficient alternative to classical alkylation reactions. On the other hand, the TH concept involves the transfer of dihydrogen from one molecule to another. Thus, it liberates the dehydrogenated precursor as a co-product (Fig. 1).
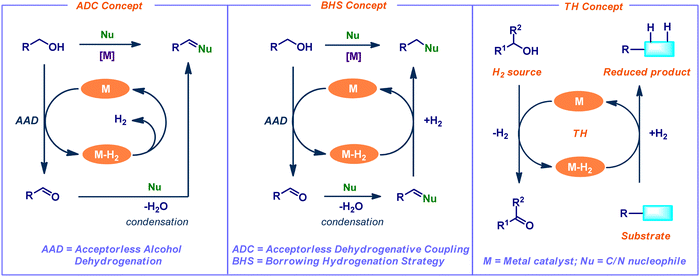 |
| | Fig. 1 Acceptorless alcohol dehydrogenation (AAD) and its applications in organic synthesis. | |
Transition metal-catalyzed C–X (X = carbon or heteroatom) bond-forming reactions via the borrowing hydrogen strategy (BHS) is one of the most promising atom-economical and eco-benign methods and has received much attention in recent years.4 The use of BHS catalysis in organic synthesis has facilitated the ability to specifically alkylate functional groups like amines, ketones, and active methylene compounds with alcohol.5 Conventionally, the ADC/BHS reactions were performed with precious metals,6–8 including Ir,9–16 Pd,11,12,16 Os,17–19 Rh,19 and Ru.11,12,19 Interestingly, Earth-abundant transition-metal catalysts were also developed to perform these reactions in recent years. For example, 3d-transition metals like Cu,19 Co,17,20,21,23,24 Mn,22–24 Ni,26 and Fe17,23–25 succeeded in catalyzing the ADC/BHS reactions.26 Significantly, employing methanol as a starting material for the alkylation reaction via BHS represents a highly challenging and thermodynamically uphill process. This is due to the initial dehydrogenation step of methanol, which is a highly energy-intensive process and requires more energy (ΔH = +84.0 kJ mol−1) than its higher analog alcohols, such as ethanol (ΔHEtOH = +68.0 kJ mol−1).27,28 In 2014, Beller and coworkers reported a soluble ruthenium-based catalytic system for selective methylation of 2-aryl ethanols using methanol as a C1 feedstock.29 However, very few reports describe the use of methanol and higher aliphatic alcohols, aliphatic alcohols, and other alcohols such as benzyl alcohol as feedstock alternatives to the corresponding aldehydes.30,31 Recently, the Earth-abundant first-row transition metals have been utilized for a simple and efficient catalytic β-C(sp3)-methylation of various primary alcohols using methanol as a sustainable alkylating agent.25,32 Indeed, there is a plethora of examples that deal with C–C and C–N bond-forming reactions in the field of borrowing hydrogen catalysis. On the other hand, the interrupted borrowing hydrogen (IBH) strategy reactions are less explored and rarely reported under transition metal catalysis.
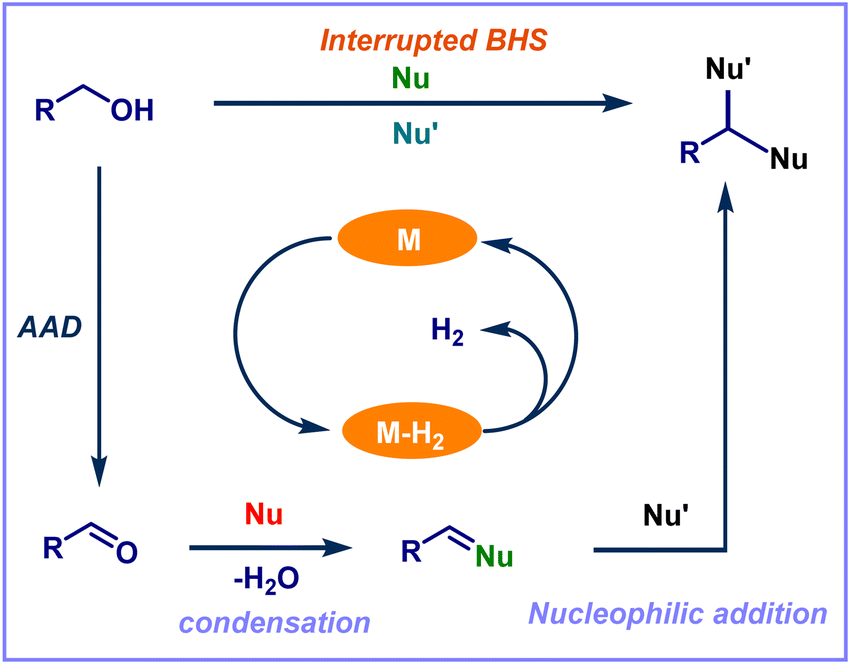
The research group of Donohoe and coworkers developed the first example of the IBH strategy. They described the process that stops unsaturated intermediate reduction and allows for the in situ conjugate addition of various nucleophiles, leading to diversified products named IBH strategy.31,33 In this scenario, along with an alkylation reaction, other functional group incorporation also occurs in the presence of various nucleophiles. Notably, there are limited precedents for intercepting metal hydrides by stopping/interrupting hydrogen borrowing using molecular oxygen or a reactive alkene (Fig. 2). These techniques, however, only enable the creation of unsaturated derivatives.34–37 Most of the transition metal-catalyzed IBH reactions were reported using Ru, Ir, and Co as the catalysts. The variety of transition metal catalysts accessible for this method and the many transformations carried out in this area are highlighted in the present review article. In addition, we also focused on several of the most important and recent advancements in the IBH strategy. This review article conveys a clear explanation and communication of the diversity of IBH strategy chemistry to the scientific audience, providing chances for further research. Moreover, the reactions based on the IBH strategy have never been discussed previously in any review articles.
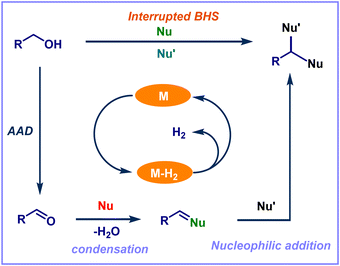 |
| | Fig. 2 Schematic representation of interrupted borrowing hydrogen (IBH) strategy. | |
2. Transition-metal-catalyzed interrupted borrowing hydrogen strategy
2.1. Iridium catalysis
The 3,3′-bisindolylmethane derivatives (3,3′-BIMs) are essential synthetic intermediates for the construction of many biologically active compounds and structural units of many natural and pharmaceutical products. The Friedel–Crafts reaction of indoles using aldehydes catalyzed by Lewis acids is a well-known method for the preparation of 3,3′-BIMs (Scheme 1).38
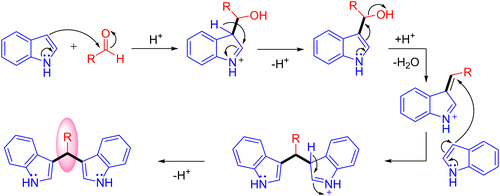 |
| | Scheme 1 Synthesis of 3,3′-BIMs by the Friedel–Crafts reaction of indoles with aldehydes. | |
However, the problem associated with this method is the use of highly toxic aldehydes that leads to severe environmental issues.51 Later, the transition-metal-catalyzed coupling of indoles with alcohols is an alternate method developed by various research groups.38–47 In 2007, Grigg and coworkers reported that the [{Cp*IrCl2}2]-catalyzed (i.e. Cat.1) coupling of indoles with aliphatic alcohols (except methanol) to form C3-alkylated indoles (only minor bisindolylmethanes were formed as by-products).39 Later, research groups, including Williams and Beller, reported the preparation of N1-alkylated indoles using the Shvo's catalyst (ruthenium complex).40
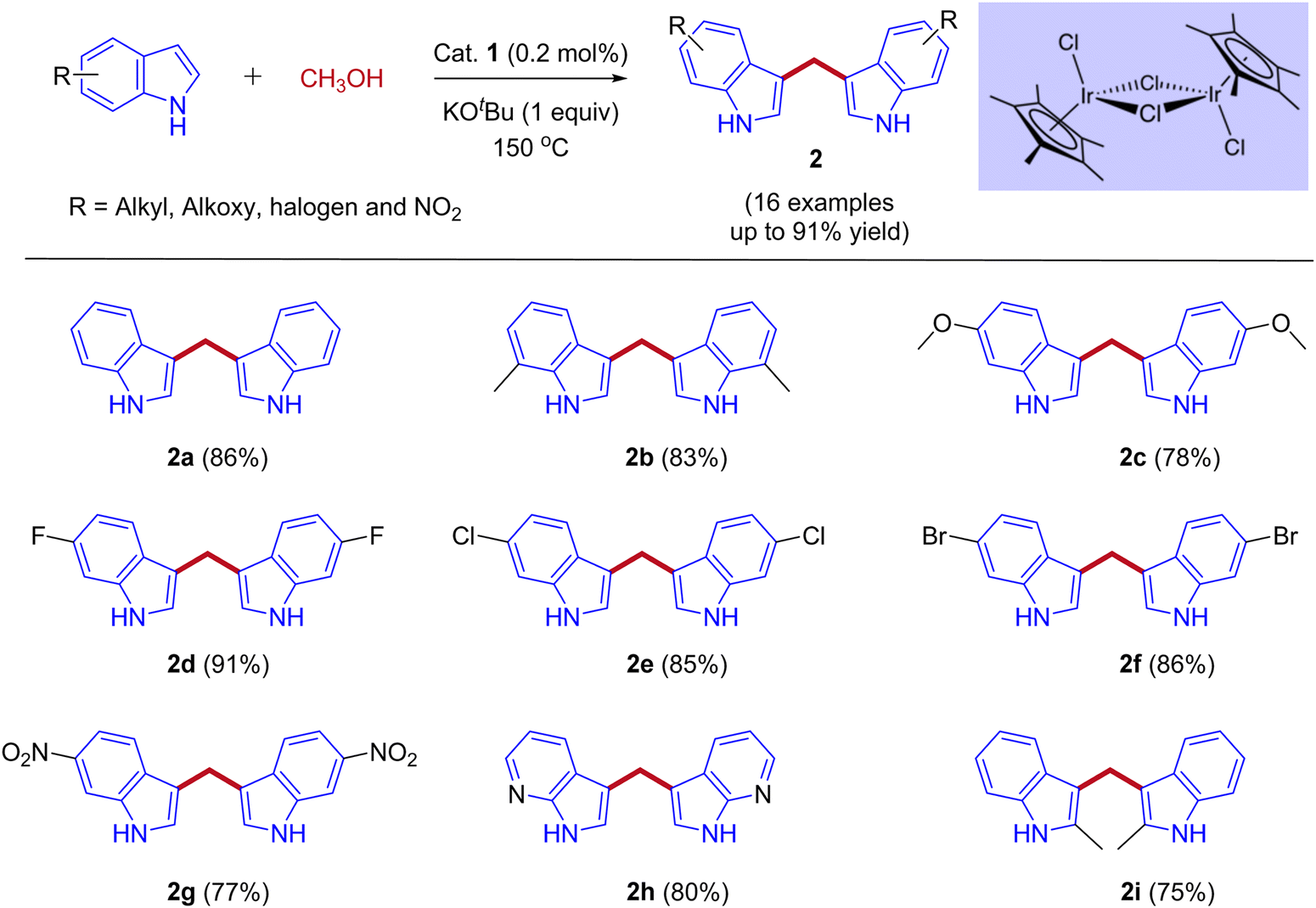
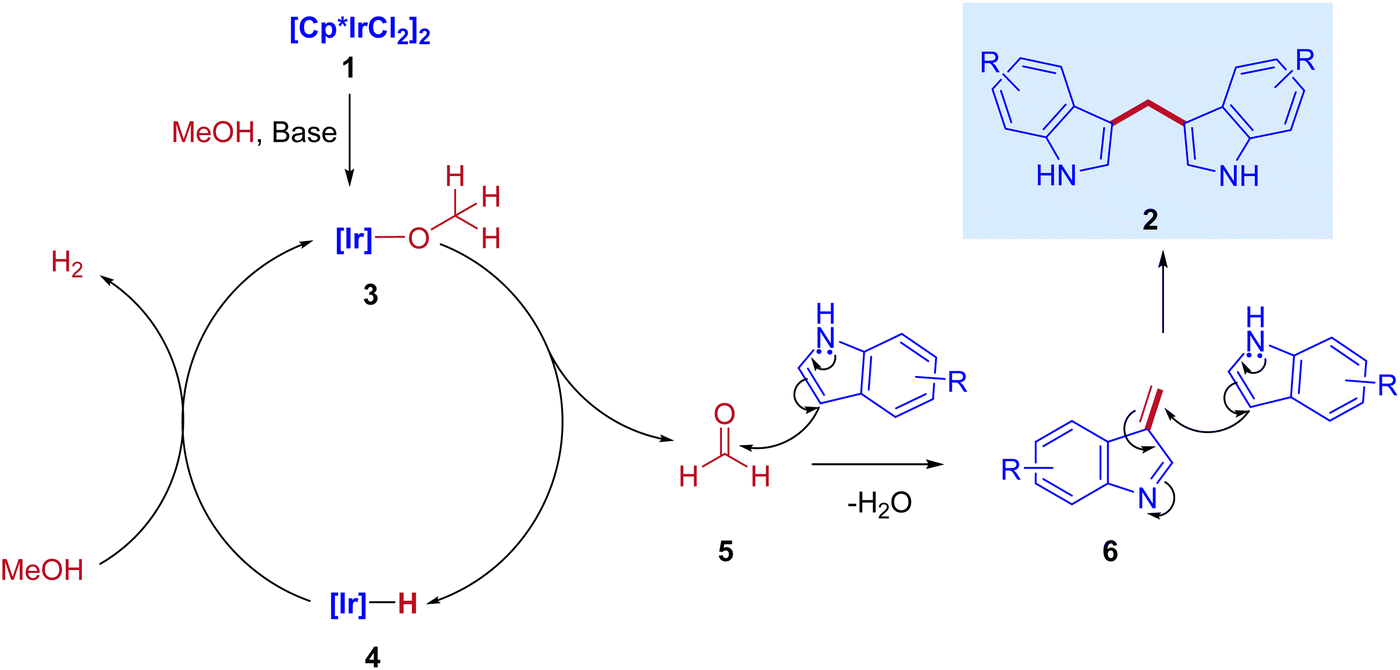
In 2013, Feng Li and coworkers reported the first example of the direct coupling of indoles with methanol to form 3,3′-bisindolylmethanes (3,3′- BIMs). The reaction was catalyzed by the commercially available [{Cp*IrCl2}2] as a catalyst via IBH strategy (Scheme 2).30
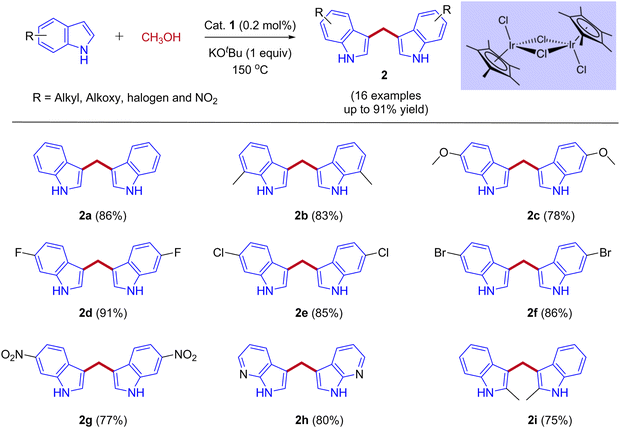 |
| | Scheme 2 Iridium-catalyzed coupling of indoles with methanol for the synthesis of 3,3′- BIMs. | |
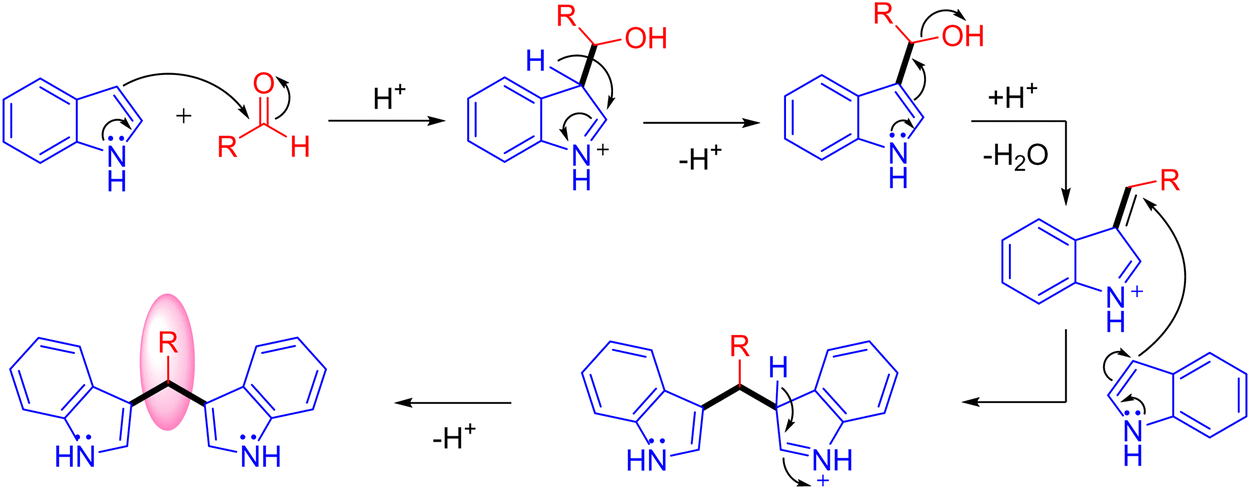
Optimization studies (Table 1) show that the reaction operates well with KOtBu as a base, and substrate scope was not affected by the electronic properties of substituents. During the reaction, the iridium catalyst (1) acts as a Lewis acid and a hydrogen acceptor. In the initial step of the reaction, the presence of a base that activates the methanol and affords iridium methoxide species 3 and subsequent β-hydride elimination of iridium methoxide complex to form iridium hydride species 4 and eliminates the reactive intermediate formaldehyde 5. Then, the coupling reaction between indole and formaldehyde forms an enamine intermediate 6. Finally, the enamine intermediate 6 further undergoes Michael addition in the presence of Lewis acid (Iridium catalyst which accepts a lone pair of electrons from the nitrogen of enamine intermediate)38 leading to the formation of 3,3′- BIMs 2 (Scheme 3). The key step of this reaction is the extremely low existence of metal hydride species under given reaction conditions which interrupted the borrowing hydrogen mechanism and promoted the Michael addition step. Thus, the reaction between the enamine intermediate and indole gives the corresponding 3,3′- BIMs exclusively in the presence of Lewis acid (Iridium catalyst).
Table 1 Optimization of reaction conditions for direct coupling of indoles with methanol to form 3,3′-bisindolylmethanes (3,3′- BIMs)
| Entry |
Catalyst |
Base |
Yield % (Isolated) |
| Reaction conditions: Indole (2 mmol), methanol (1 mL), catalyst (0.2 mol%), base (1 equiv.) heated at 150 °C for 12 h. |
| 1 |
[{Cp*IrCl2}2] |
KOH |
48 |
| 2 |
[{Cp*IrCl2}2] |
KOtBu |
86 |
| 3 |
No catalyst |
KOtBu |
0 |
| 4 |
[{Cp*RhCl2}2] |
KOtBu |
6 |
| 5 |
[{Ru(p-cymene)Cl2}2] |
KOtBu |
45 |
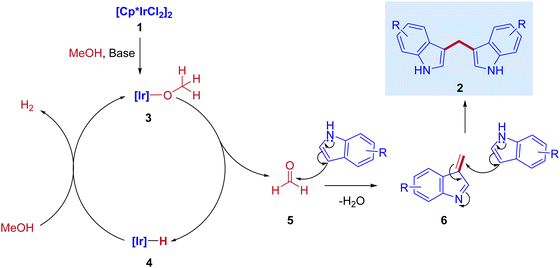 |
| | Scheme 3 Plausible mechanism for the direct synthesis of 3,3′- BIMs via coupling of indoles with methanol. | |
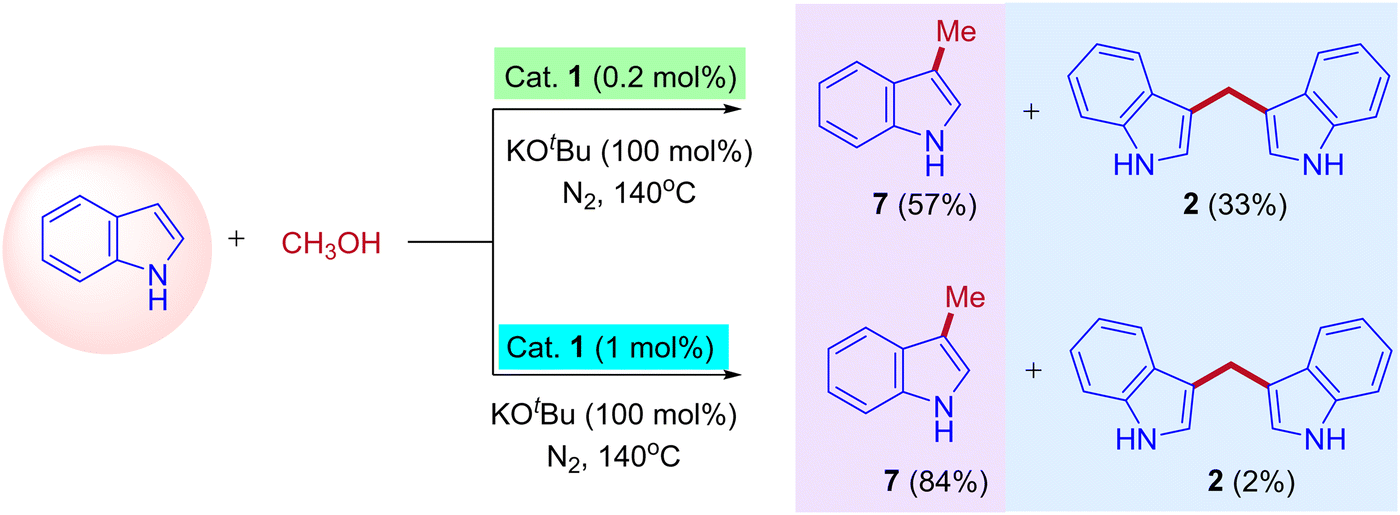
The research groups of Chun Cai supported the proposed mechanism.41 They observed the formation of expected C3-methylated indole product 7 (57% yield) and bisindolylmethane 2 as a side product (33% yield). The reaction was catalyzed by a low amount of iridium catalyst such as ([{Cp*IrCl2}2], 0.2 mol%). The formation of bisindolylmethane is due to the interruption in the borrowing hydrogen cycle. However, increasing the iridium catalyst loading ([{Cp*IrCl2}2], 1 mol%) leads to the exclusive formation of methylated indole product 7 (84% yield). This is due to the suppression of the Michael addition between enamine intermediate and indole under the high existence of iridium hydride species and favored the borrowing hydrogen cycle without any interruption (Scheme 4). Similarly, Ronald Grigg and coworkers also identified the bisindolylmethane as a side product due to the Michael addition between enamine intermediate and indole in the presence of [IrCp*Cl2]2 as a catalyst.39
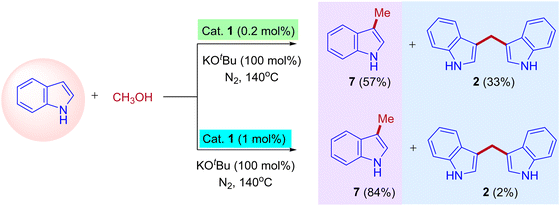 |
| | Scheme 4 Coupling of indoles with methanol leads to C3-methylated indole and 3,3′- BIMs under iridium catalysis. | |
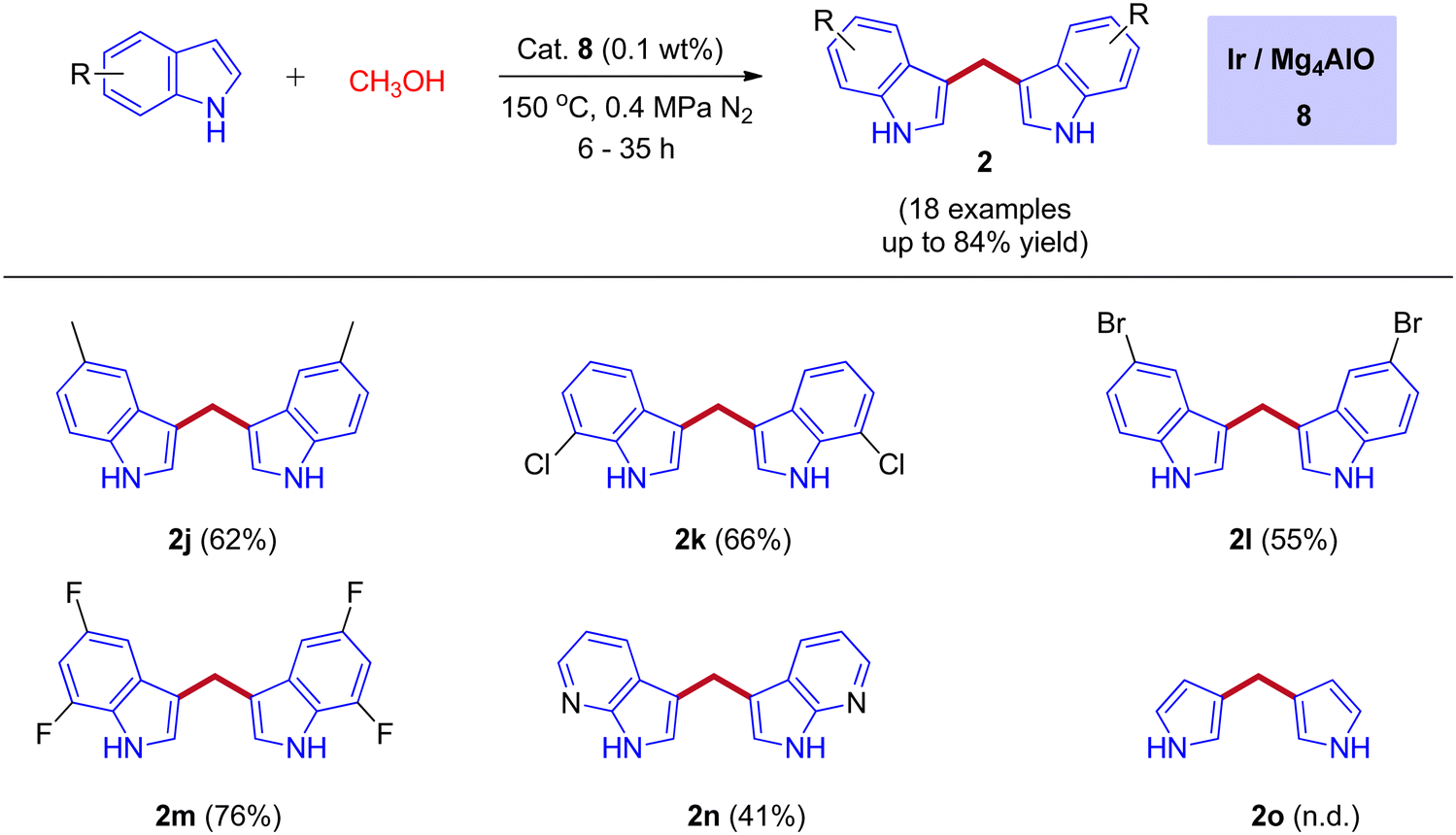
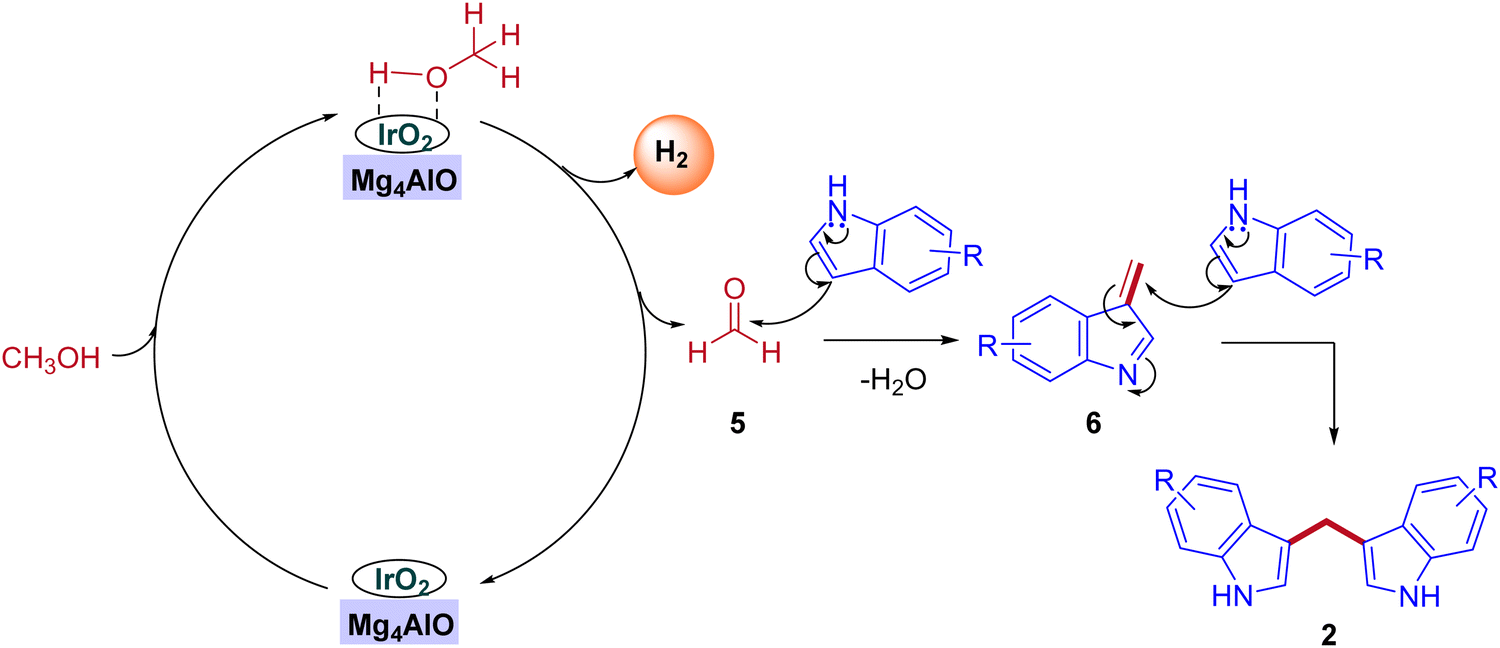
In 2019, the first heterogeneous additive free hydrotalcite-derived Mg–Al oxides supported iridium (Ir/MgxAlO) catalyst for the synthesis of BIMs via the activation of methanol was reported by Teck-Peng Loh and coworkers (Scheme 5).42 Simple coprecipitation method used to prepare the mixed oxides of Mg–Al. 0.1 wt% of iridium content was incorporated onto the surface of the support by an incipient-wetness impregnation method. The size of the metal particle plays a crucial role in product selectivity (Table 2). The cooperation between acidic-basic sites on the Mg4AlO and oxidized iridium species incorporation on the same surface suppresses the other possible products to get desired BIMs. Deuterium labeling studies and control experiments demonstrated that the reaction mechanism was consistent with the dehydrogenative condensation mechanism for the synthesis of BIMs, as depicted in Scheme 6. When simple methanol was replaced with deuterated methanol, the expected product was observed with a lower yield when compared with undeuterated methanol. In the absence of the catalyst, there is no product formation. Interestingly, 0.1 wt% Ir on the surface of Mg4AlO helps in the dehydrogenation step of methanol. The acidic-basic sites of Mg4AlO contributed condensation reaction between formaldehyde and indole.
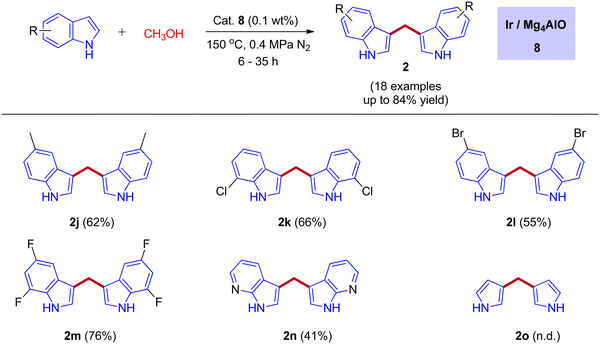 |
| | Scheme 5 Heterogeneous supported iridium catalyst for the coupling of indoles with methanol. | |
Table 2 Dehydrogenative coupling of indole and methanol with different catalysts
| Entry |
Catalyst |
Metal size (nm)a |
Yield % (Isolated) |
| Reaction conditions: Indole (1 mmol), catalyst (1 mol% metal based on 1a), methanol (5.0 mL), 0.4 MPa N2, 4 h; n.r. = no reaction. Metal sizes were evaluated by TEM. |
| 1 |
1 wt% Pt/Mg4AlO |
2.2 |
9 |
| 2 |
1 wt% Ru/Mg4AlO |
4.3 |
11 |
| 3 |
1 wt% Ir/Mg4AlO |
0.8 |
20 |
| 4 |
1 wt% Ir/TiO2 |
1.3 |
7 |
| 5 |
1 wt% Ir/MgO |
1.6 |
5 |
| 6 |
0.1 wt% Ir/Mg4AlO |
0.5 |
84 |
| 7 |
Mg4AlO |
— |
n.r. |
| 8 |
No catalyst |
— |
n.r. |
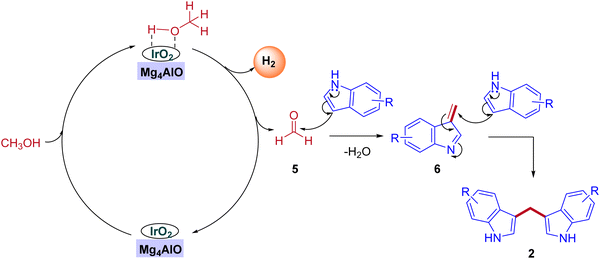 |
| | Scheme 6 Plausible reaction mechanism for the coupling of indoles with methanol under heterogeneous iridium catalysis. | |
The alternate way to conventional enolate production (or) alkylation reactions, widely established by numerous research groups, is the alkylation of ketone enolates produced by hydrogen-borrowing processes catalyzed by transition metals. However, these procedures proved ineffective for forming branching compounds from ketones from hindered alcohols. Donohoe group31,33 revealed the reaction that inhibits enone reduction and permits the in situ insertion of a nucleophile followed by conjugate addition to generate a variety of complex compounds. As a result, methanol was used to integrate the methylene functional group between the two nucleophilic units. Metal hydride interruption is also possible by stopping or interrupting hydrogen borrowing using molecular oxygen or a reactive alkene. However, these methods only enable the creation of unsaturated derivatives.34–37 The adjustment in the reactivity of hydrogen borrowing and hydrogen returning events is well balanced by using [{Ir(cod)Cl}2] complex in addition to various phosphine ligands and optimizing the reaction conditions.
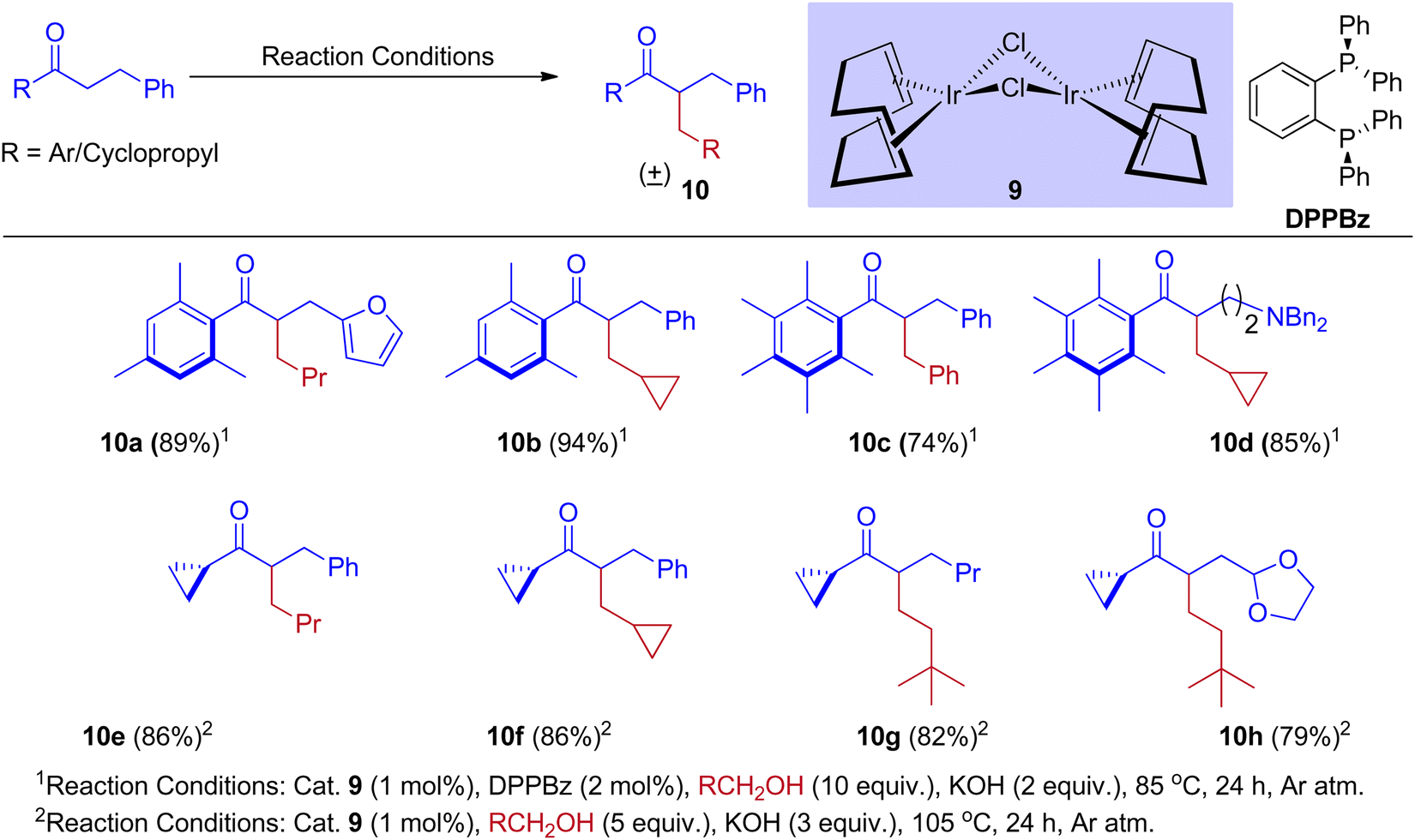
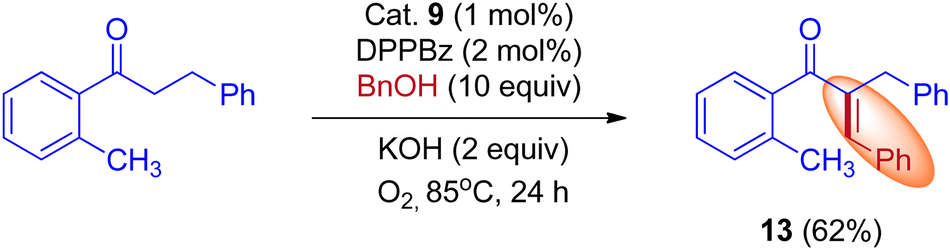
Furthermore, Donohoe and coworkers confirmed that the ligand plays a crucial role in deciding product formation. Gratifyingly, phosphine ligands that were sterically hindered are in combination with (cataCXium A – Di(1-adamantyl)-n-butylphosphine, DPPBz) molecular oxygen interrupts the cycle of hydrogen borrowing and stops enone reduction, due to this addition of a nucleophile to the in situ formed unsaturated intermediates will give more branched carbonyls (Scheme 7 and Scheme 8).31,33
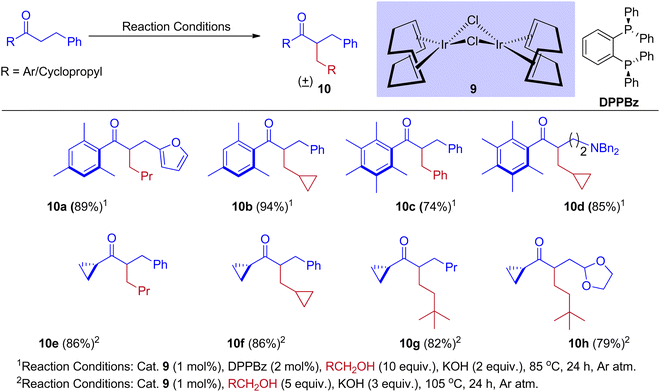 |
| | Scheme 7 Iridium-catalyzed BH reactions of ketones and alcohols. | |
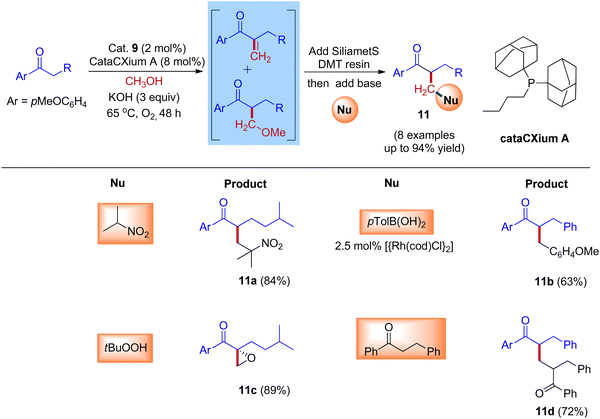 |
| | Scheme 8 Iridium-catalyzed IBH reactions of ketones and methanol in the presence of sterically hindered phosphine ligand (cataCXium A) and O2. | |
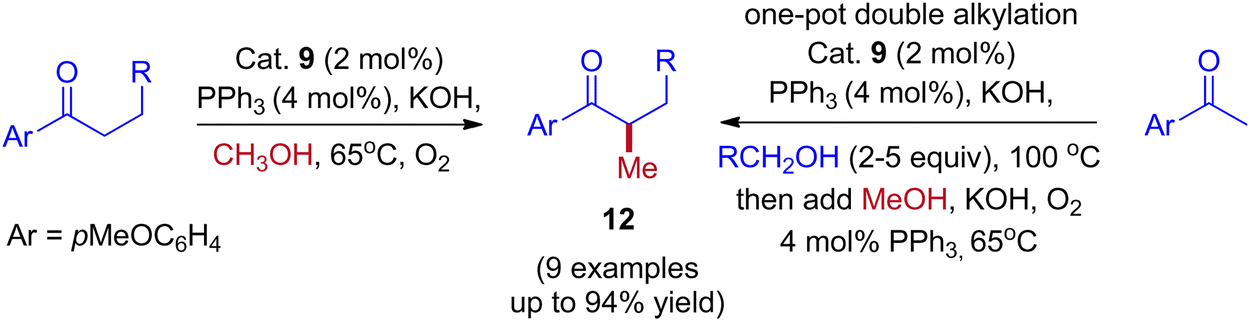
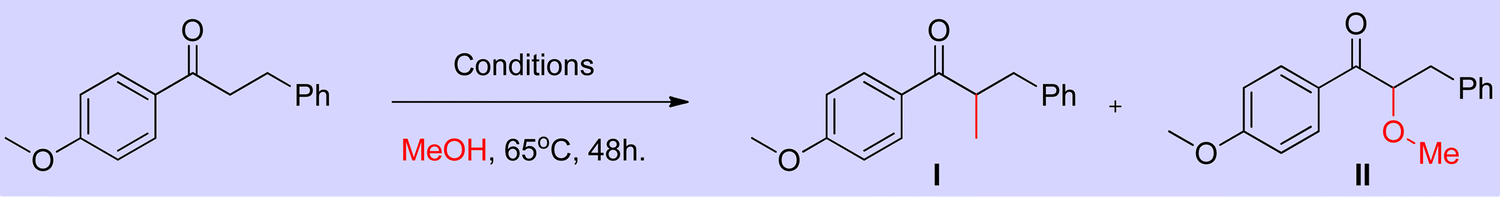
Employing simple and less hindered phosphine ligands such as PPh3 leads to the methylated product of ketone by involving borrowing hydrogen cycle without any interruption (Scheme 9). These results demonstrated that both BH (Scheme 7) and IBH (Scheme 8) strategies take place depending on the ligand system, and the reaction was performed in the presence of low catalyst loadings of iridium (I). In the hydrogen-borrowing cycle, enone reduction can be effectively interrupted by the coexistence of a hindered phosphine and an oxygen environment (Table 3, entry 4). The catalyst recycling process involves the oxidation of metal hydrides by O2. On the other hand, Feng Li and coworkers reported that the amount of catalyst loading plays a crucial role in deciding product formation in the reaction.30
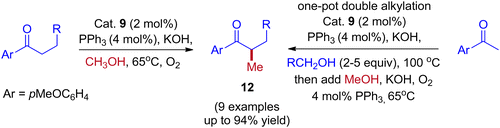 |
| | Scheme 9 Iridium-catalyzed methylation and double alkylation of aryl ketones with methanol. | |
Table 3 Ligand and atmosphere effect on yield of the product
In 2015, Donohoe and his group reported that the borrowing hydrogen reaction enabled α-branched ketones synthesis from alcohols by iridium catalyst.31 Employing bulkier phosphine ligands, for example, DPPBz (1,2-Bis(diphenylphosphino)benzene) under an oxygen atmosphere interrupted the borrowing hydrogen cycle and gave the olefinated product instead of α-branched ketone exclusively. This is due to the reactivity of O2 towards metal hydride in preference for enone (Scheme 10).
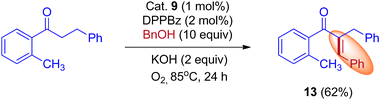 |
| | Scheme 10 Control experiment to prove that the O2 can interrupt the BH cycle. | |
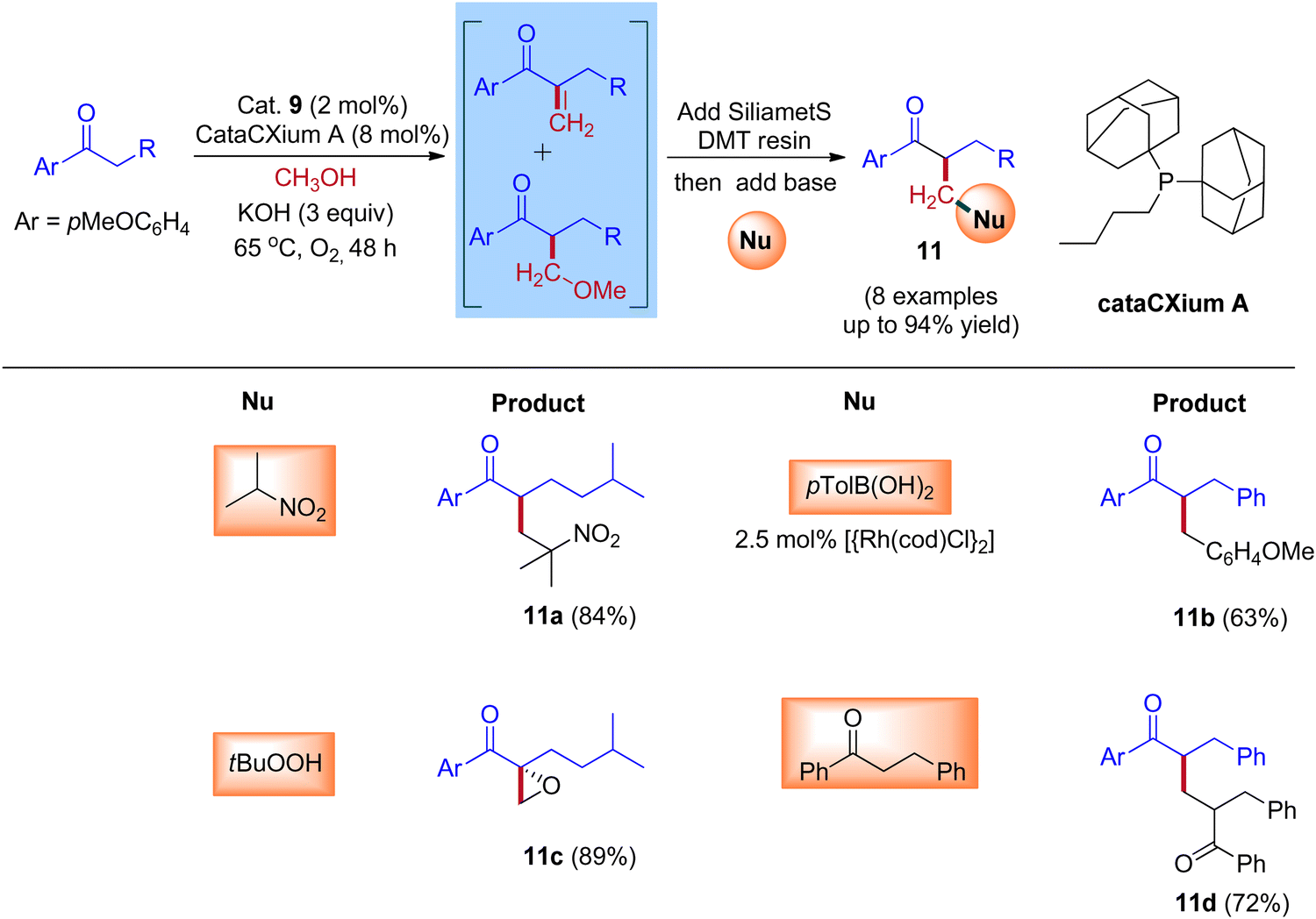
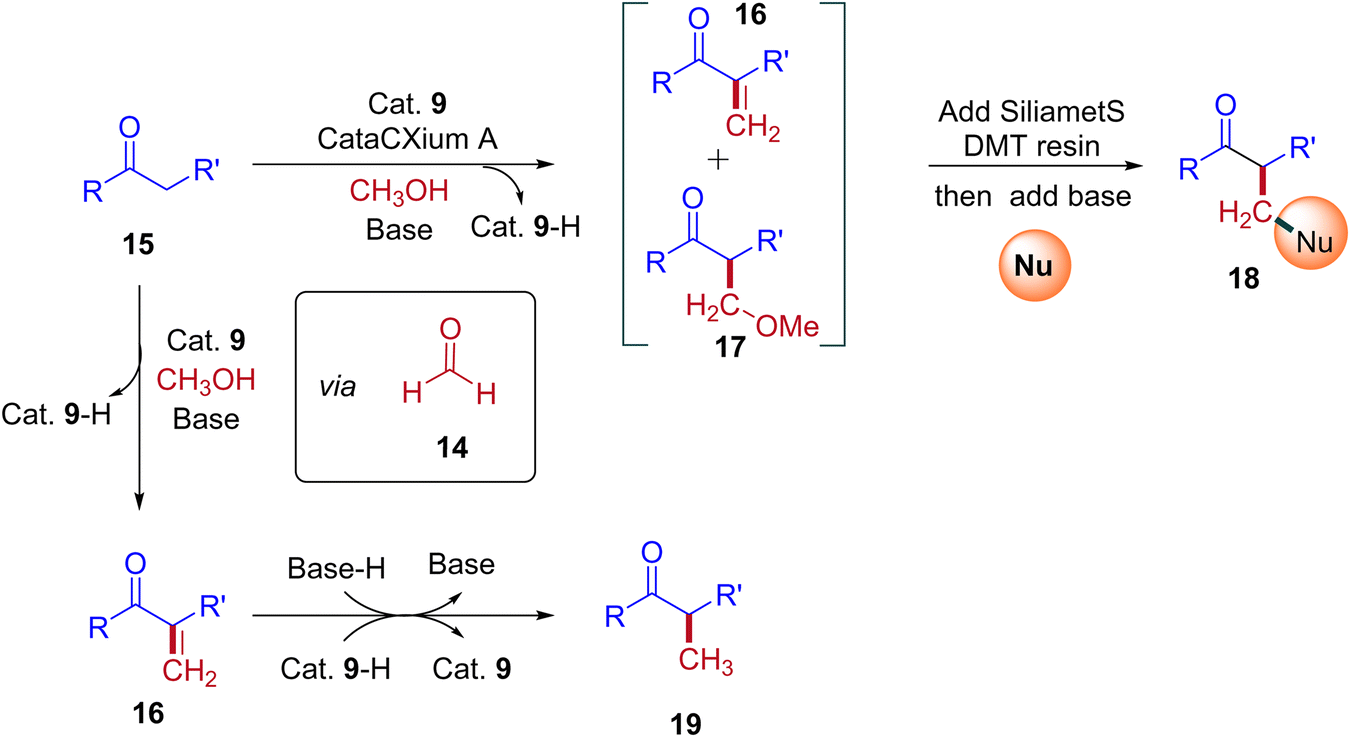
A plausible reaction mechanism for the formation of BH and IBH products is explained in Scheme 11. The research group of Donohoe believed that in the absence of sterically hindered phosphine ligand (cataCXium A) the reaction follows BH pathway and gives alkylated products (Scheme 7, Scheme 9 and Scheme 11). In this BH reaction pathway, in the presence of catalyst 9 the starting material alcohol is converted into its corresponding carbonyl compound (14) and reacts with 15 to give enone 16. The in situ generated enone further hydrogenated by 9-H (in the presence of a base) to give the expected BH product 19. On the other hand, in the presence of sterically hindered phosphine ligand (cataCXium A) along with the enone intermediate 16, a methoxy addition product 17 is also formed. This may be explained that in combination of hindered phosphine and an oxygen atmosphere with a low loading of catalyst 9 the BH cycle is interrupted, see Scheme 10 and Table 3 (entries 4 and 5). Indeed, it was observed that oxygen atmosphere and ligand play very important role in the formation of methoxy addition product. Notably, the formed intermediates 16 and 17 further reacts with nucleophile (NuH) to give the IBH product 18 (more branched carbonyl compound in Schemes 8 and 11). In case of IBH mechanism, closing the catalytic loop from the metal hydride is unclear. Although, there is no experimental evidence, it is postulated that oxidation of metal hydride under O2 atm is responsible for the regeneration of the active metal catalyst. It was observed that for accessing better yield of the IBH product, a metal scavenging resin treatment is recommended to the crude reaction mixture before the addition of base and the external nucleophile.
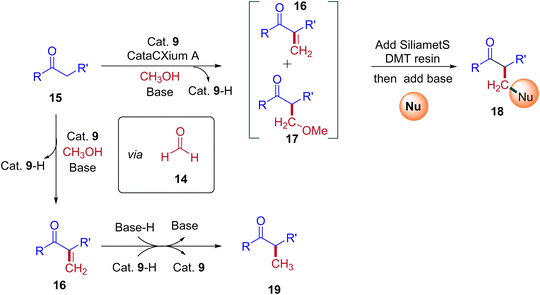 |
| | Scheme 11 A plausible reaction mechanism for the formation of IBH and BH products. | |
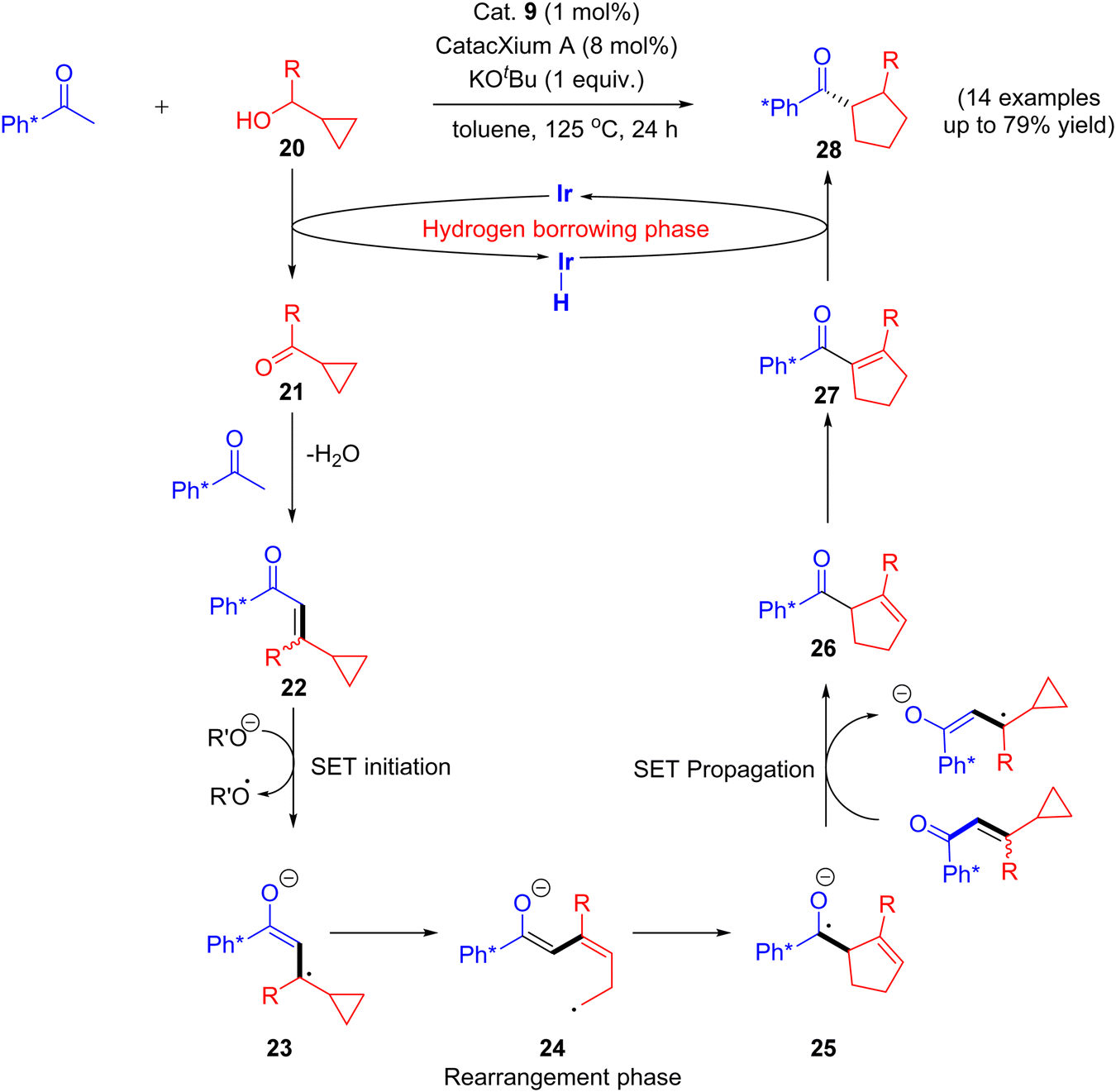
Recently in 2020, an attractive approach that describes the combination of both rearrangement and hydrogen borrowing cascade reaction catalyzed by [Ir(cod)Cl]2 complex was reported by Donohoe and coworkers. The reaction of aryl methyl ketone and cyclopropyl alcohols 20 under iridium-catalyzed hydrogen borrowing reaction to give substituted stereo-defined cyclopentanes 28via a vinyl cyclopropane 22 rearrangement. They identified that the delay of the enone 22 reductions would allow further rearrangement of enone intermediate and thus minimize the formation of directly alkylated products.
The reaction mechanism involves the initial vinyl cyclopropane ring expansion that leads to the cyclopentene carbonyl moiety via single electron transfer (SET) initiation (20–21) followed by rearrangement (23–25) and SET propagation (25–26) pathway. Finally, the selective hydrogenation of cyclopentene carbonyl moiety 27 gives substituted stereo-defined cyclopentane 28 as a final product.43 This stereo-defined cyclopentane product formation is due to the delay in enone reduction process that is caused by rearrangement of eneone intermediate (Scheme 12).
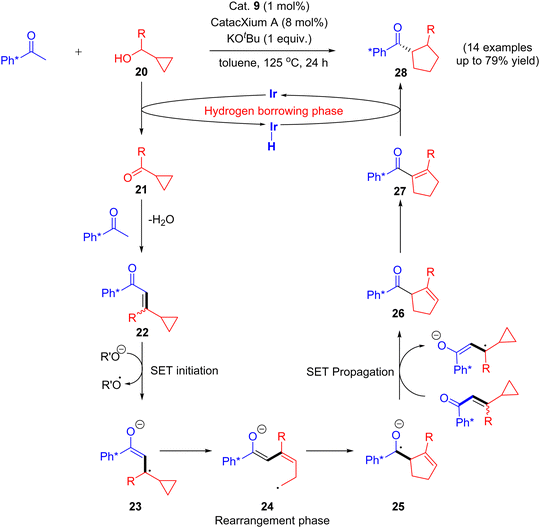 |
| | Scheme 12 Iridium-catalyzed hydrogen borrowing cascade reaction via a vinyl cyclopropane ring expansion. | |
2.2. Ruthenium catalysis
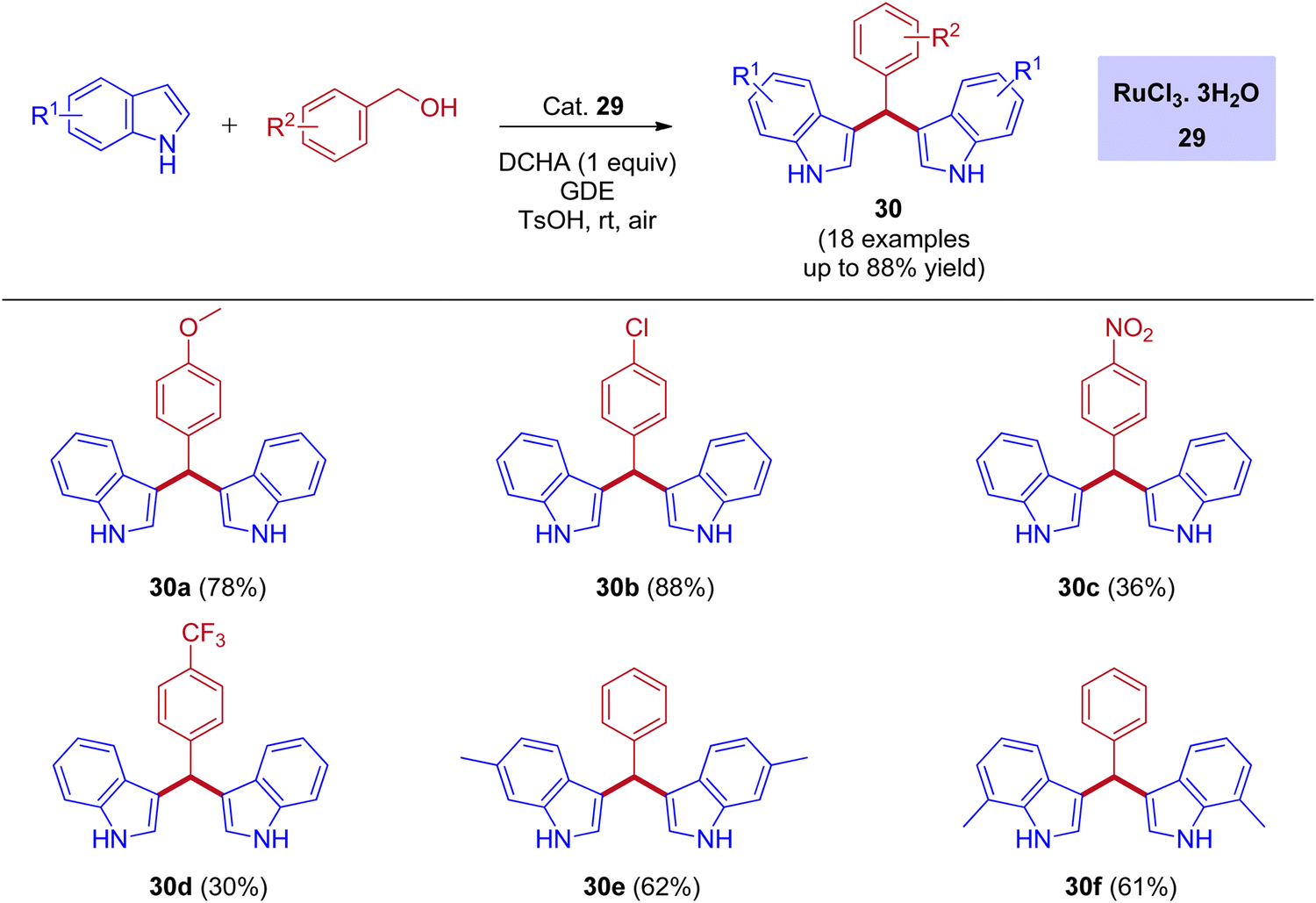
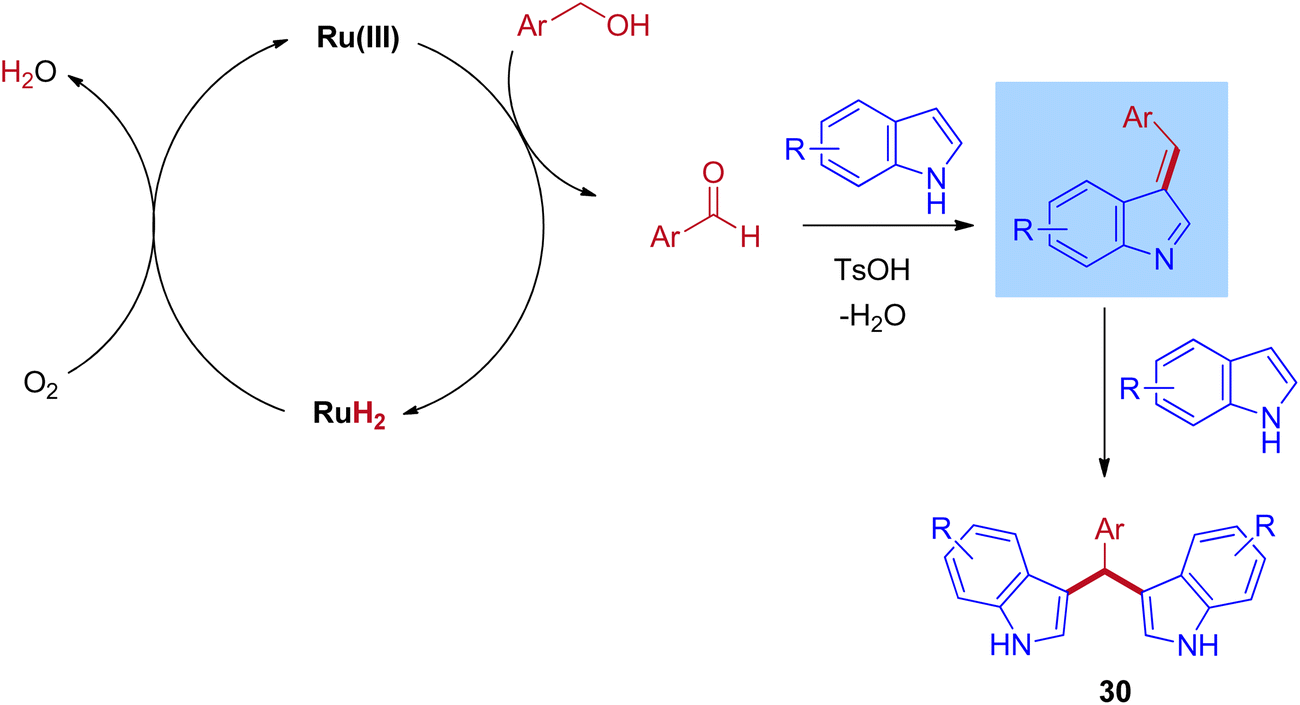
One of the most powerful tools in organic chemistry is transition metal catalysis, especially the ruthenium-based catalytic systems paid much attention due to its high efficiency and excellent chemoselectivity. Significantly, the ruthenium catalysis has also contributed to the area of BIMs synthesis with a few reports under the IBH methodology. In 2012, the first report for the bis(indolyl)methanes (BIMs) synthesis was disclosed by Liangxian Liu and workers using commercially available RuCl3·3H2O. This catalytic system is amazingly effective for the one-pot reactions of benzyl alcohols with indoles to give a wide range of BIMs. Indeed, moderate to excellent yields were achieved under an air atmosphere at room temperature. However, the intermolecular competitive experiment reveals that the reaction preferred primary alcohols over secondary alcohols of diols for the corresponding BIMs synthesis (Scheme 13).44
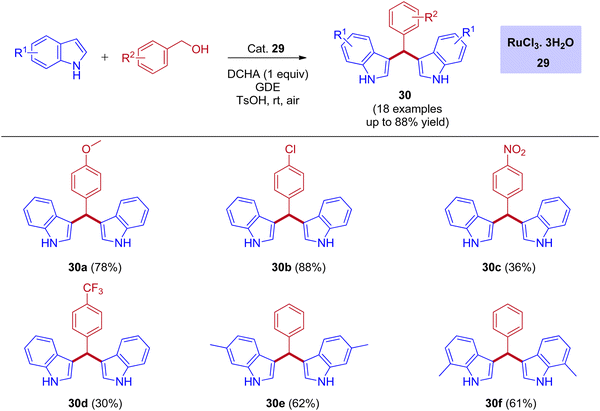 |
| | Scheme 13 Ru-catalyzed synthesis of bis(indolyl)methanes using benzyl alcohols and indoles via IBH strategy. | |
The plausible reaction mechanism for the ruthenium-catalyzed BIMs involves the initial dehydrogenation step of benzyl alcohol to give corresponding aldehyde and ruthenium hydride species. Then, the formed ruthenium hydride species react with oxygen in the atmospheric oxygen, regenerate the catalyst, and eliminate the water molecules. This is the key step in which the BH mechanism interrupts by the external oxygen source. After that, the formed benzaldehyde is activated by TsOH and undergoes an electrophilic substitution reaction with indole at C–3 positions to give mono(indolyl)methanes. Next, the elimination of water leads to an enamine intermediate. Further, the enamine intermediate interacts with another indole molecule to give the desired BIMs product, shown in Scheme 14. Advantageously, this reaction operated at room temperature under an air atmosphere and was catalyzed by a commercially available RuCl3·3H2O catalyst.
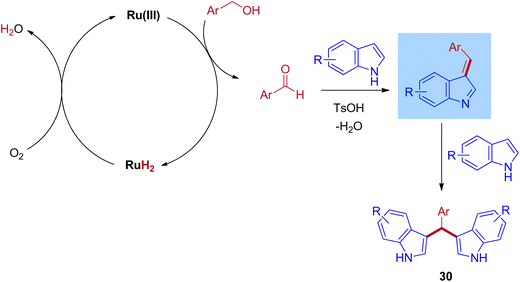 |
| | Scheme 14 Mechanism of Ru-catalyzed synthesis of bis(indolyl)methanes using benzyl alcohols and indoles via IBH strategy. | |
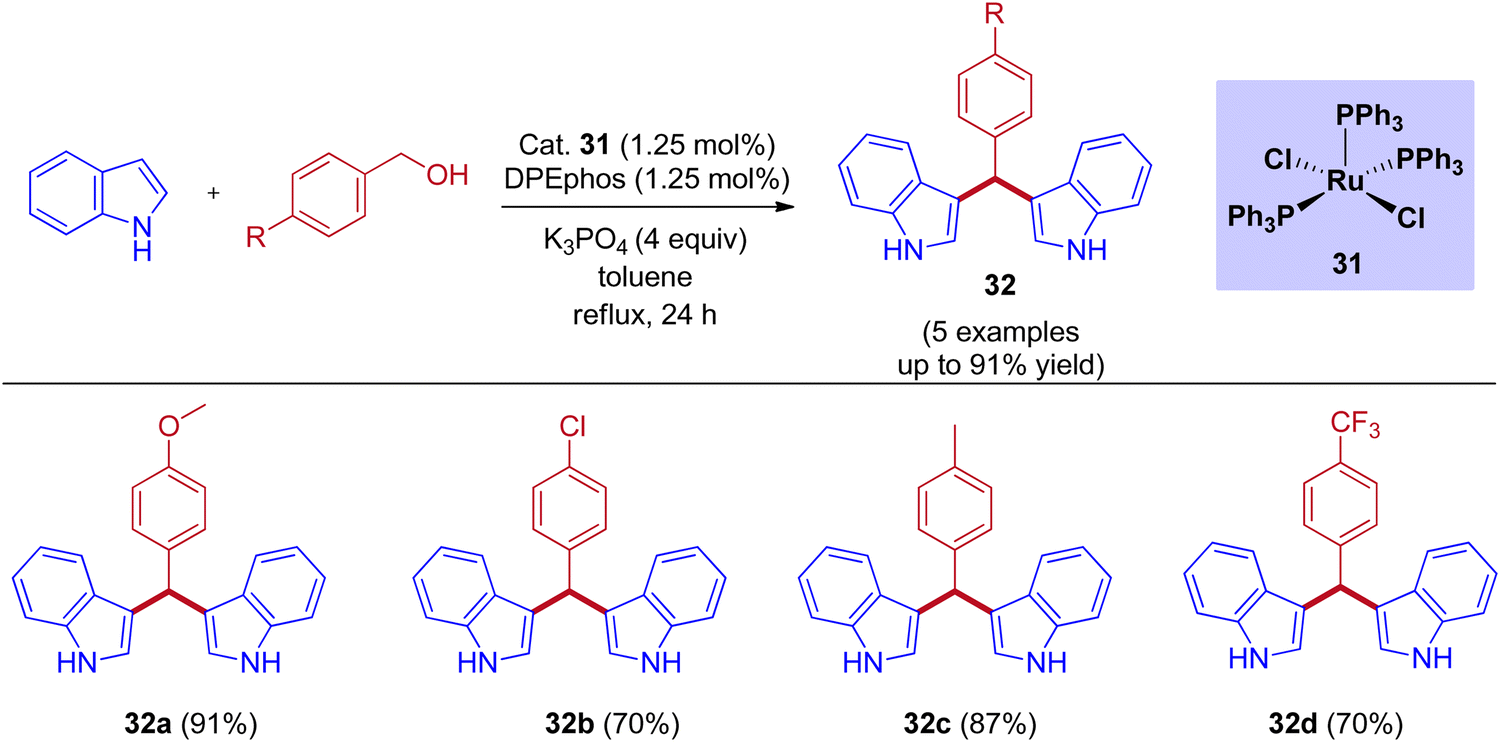
Tetsuo Ohta group reported that the RuCl2(PPh3)3-catalyzed bis(3-indolyl)phenylmethane preparation by taking DPEphos as a ligand under reflux conditions (Scheme 15).45 Examining various benzyl alcohols produced bis(3-indolyl)phenylmethane in up to 90% yield. When compared to electron-withdrawing substituents, excellent yields were observed for electron-releasing substituents in the para position of the benzyl alcohol.
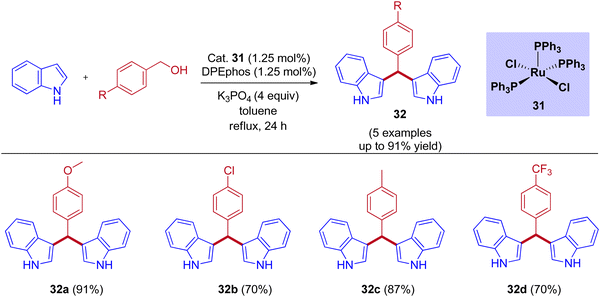 |
| | Scheme 15 Ruthenium-catalyzed synthesis of bis(3-indolyl)phenylmethanes. | |
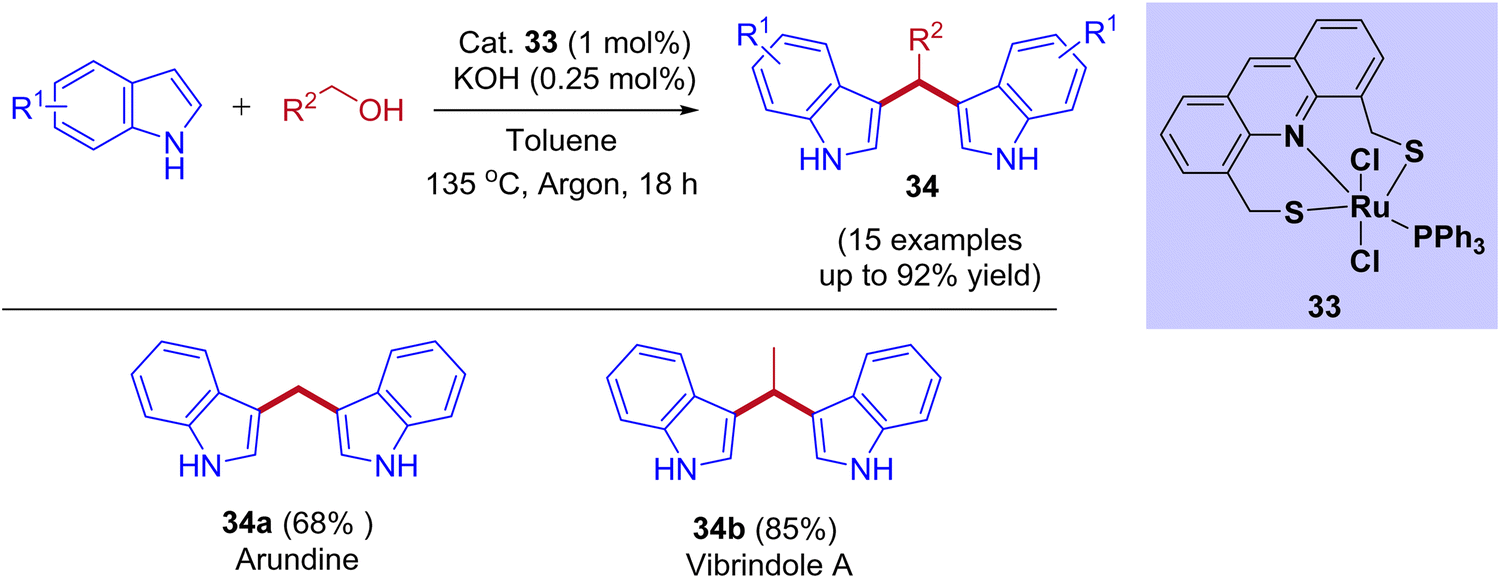
Recently, the ruthenium pincer complex-catalyzed BIMs synthesis from indoles and alcohols was reported by Srimani and coworkers (Scheme 16).46 Starting from lower-order alcohol such as methanol to higher-order alcohols such as octanol and benzyl alcohol reacted well with indoles. The reaction afforded excellent yields of corresponding BIMs derivatives using acridine-derived Ru-SNS pincer complex as a catalyst under reflux conditions. This BIMs synthesis was achieved by reducing 50% of the base loading compared to C3-alkylated products under standard reaction conditions. This result revealed that the interruption of the BH cycle happened by lowering the base amount (Table 4). Moreover, the present protocol was successfully applied to pharmaceutically important alkaloid derivatives such as arundine, vibrindole A, and tryptamine.
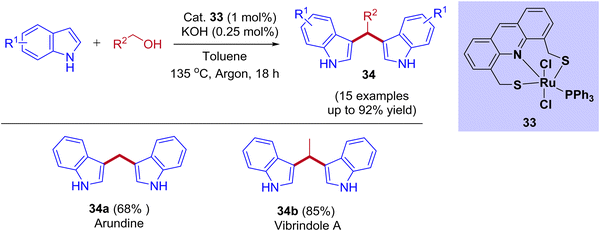 |
| | Scheme 16 Ru-SNS pincer-catalyzed synthesis of bis(3-indolyl)phenylmethane derivatives. | |
Table 4 Optimization of the reaction condition for the synthesis of bisindolylmethane
|

|
| Entrya |
Base (mmol) |
Indole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Alcohol :Alcohol |
Yield % |
|
Reaction conditions: Indole (1 mmol), 1-octanol (5 mmol), Cat. 36 (0.01 mmol, 1 mol%), pressure tube, 135 °C, 18 h.
Toluene as a solvent (1 mL).
Indole (1 mmol), alcohol (0.5 mmol), catalyst 1 (0.005 mmol, 1 mol%), KOH (0.25 mmol), toluene (1 mL), 135 °C, 18 h.
|
| 1 |
KOH (1) |
1:1 |
65 |
| 2 |
KOH (1) |
1:3 |
60 |
| 3 |
KOH (1) |
1:4 |
45 |
| 4 |
KOH (1) |
1:5 |
43 |
| 5 |
KOH (0.5) |
1:5 |
0 |
| 6 |
KOH (0.25) |
1:5 |
25 |
| 7b |
KOH (0.5) |
1:1 |
60 |
| 8c |
KOH (0.25) |
1:0.5 |
75 |
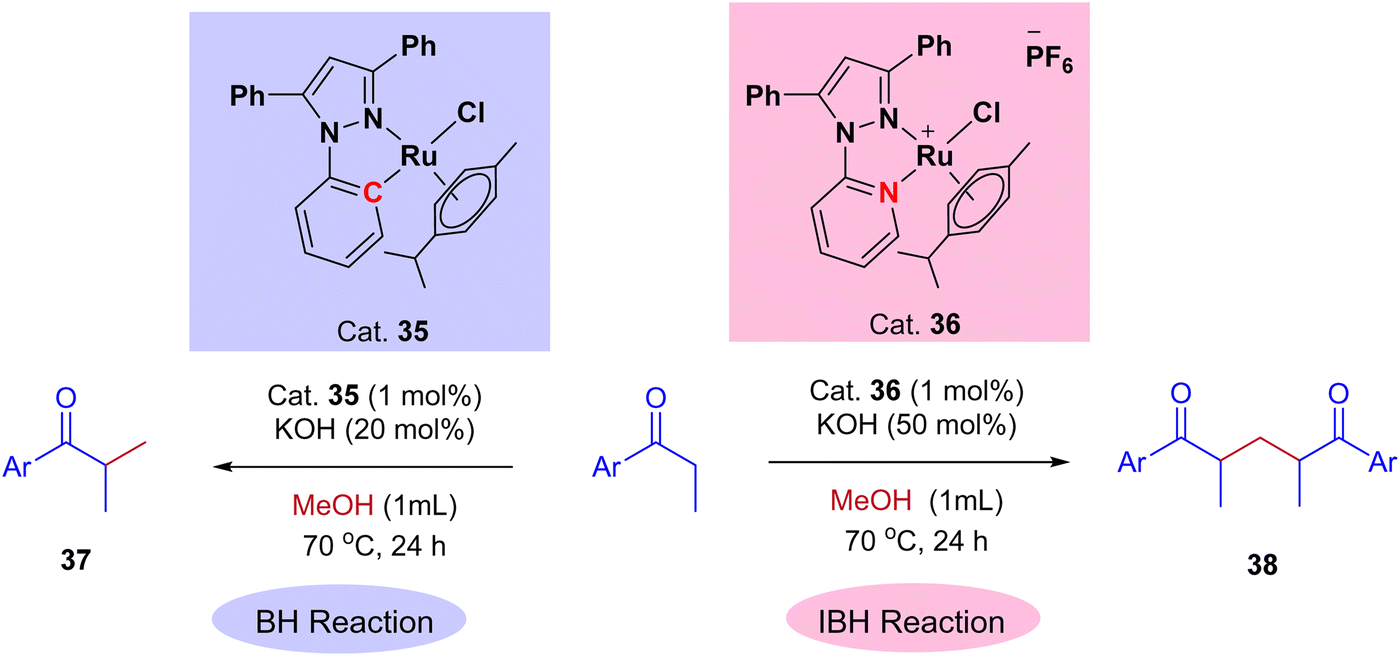
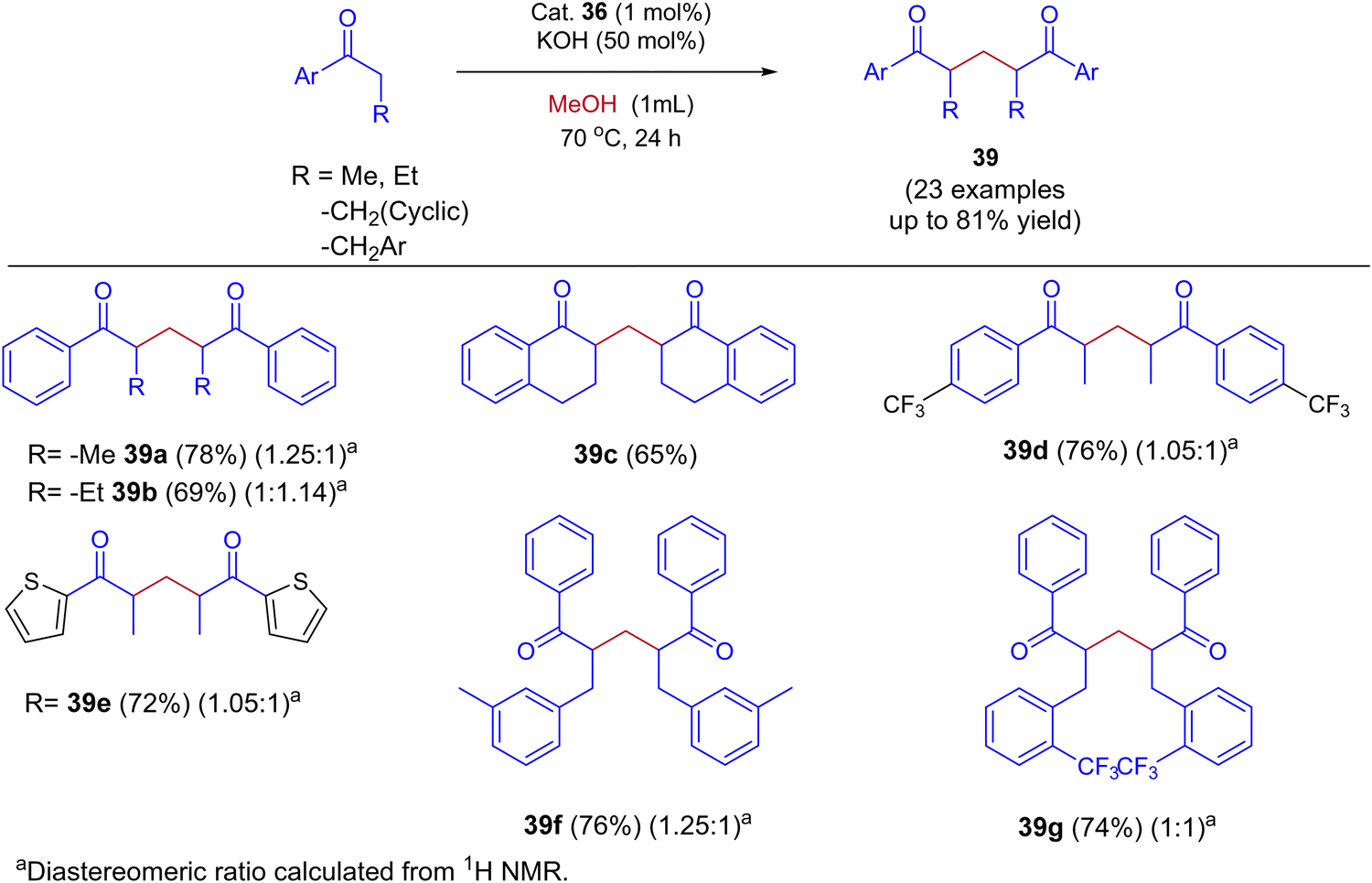
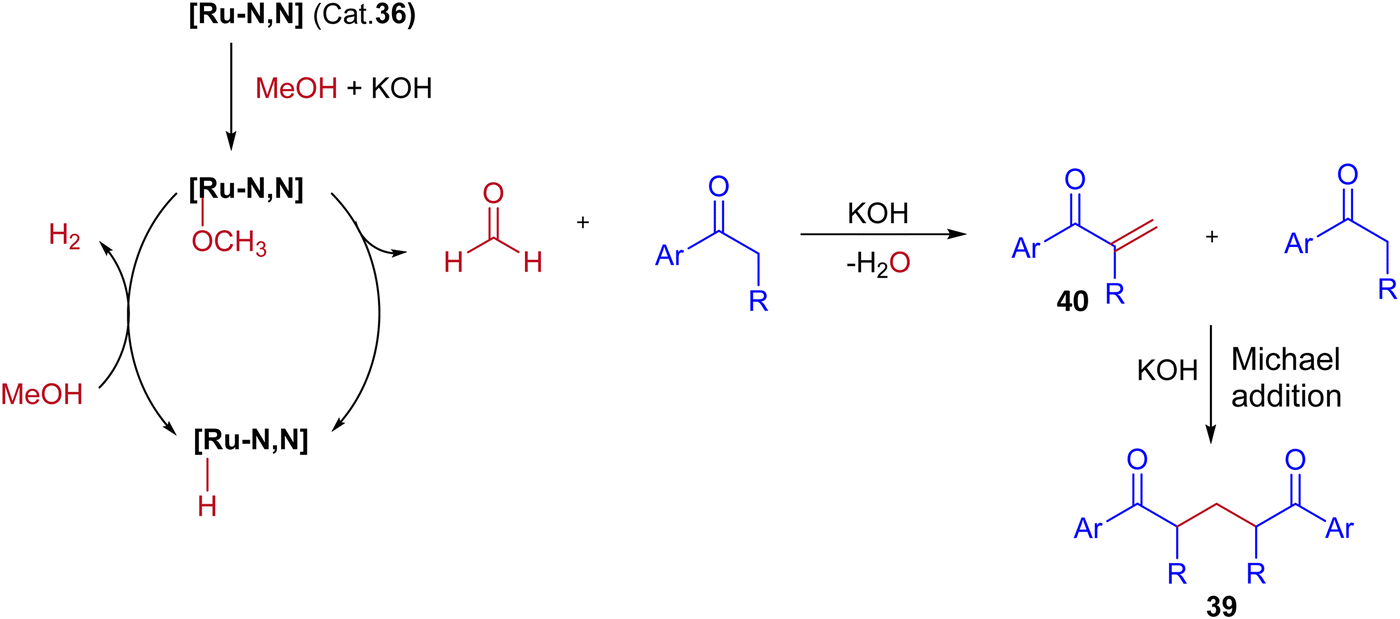
Very recently, Biswal et al., reported phosphine-free ligand-controlled ruthenium-catalyzed IBH functionalization of ketones for the formation of C–C bond using methanol as a C1 Source.47 In this report by changing the ligand system authors achieved both BH and IBH products as shown in Scheme 17. Optimization studies reveals that at low loading of base and Ru–N,C catalyst 35 gives the BH product (α-methyl propiophenone 37), wherein Ru–N,N catalyst 36 at high loading of base gives IBH product (1,5-diketones 38).
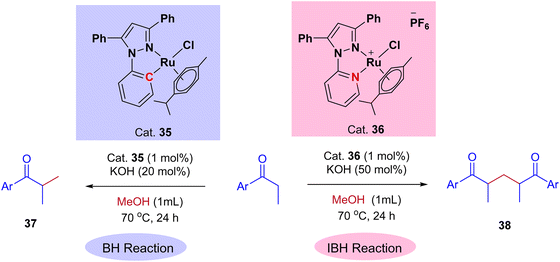 |
| | Scheme 17 Ru-catalyzed ligand controlled IBH and BH reaction of α-alkyl ketones for the formation of C–C bonds using methanol as a C1 source. | |
In this Ru-catalysed IBH reaction, both electron-releasing and -withdrawing substituents reacted smoothly to give the desired products in excellent yields. Importantly, alicyclic and heteroaryl ketones gave the expected diastereomeric products in good yields (Scheme 18). Control experiments and deuterium labeling experiments suggest that the in situ formed formaldehyde from methanol reacts with ketone to give enone 40. Intermediate 40 further reacts with another ketone molecule and forms 1,5-diketone 39via Michael addition (Scheme 19). The electronic effect of cationic complex 36 that contain pyridine moiety plays crucial role in irreversible hydrogenation reaction to liberate hydrogen gas.
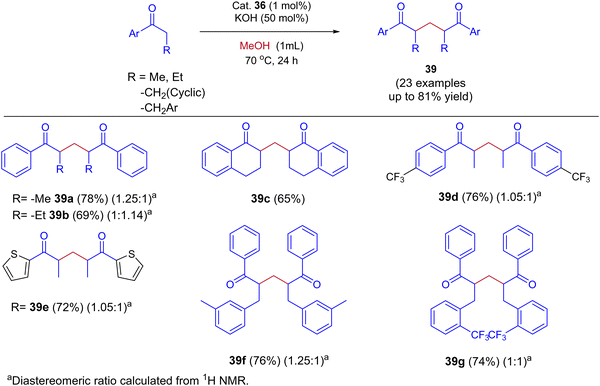 |
| | Scheme 18 Ru-catalyzed synthesis of 1,5-diketones by using methanol via the IBH strategy. | |
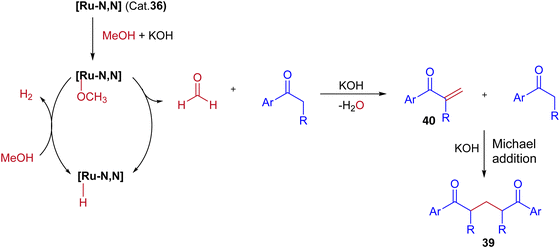 |
| | Scheme 19 A plausible reaction pathway for Ru-catalyzed synthesis of 1,5-diketones via IBH strategy. | |
2.3. Cobalt catalysis
Most of the BH and IBH reactions were reported with Ir and Ru-based catalysts. These noble metal catalyst systems have potential drawbacks, such as being expensive, having low earth abundance, difficulties in preparing complexes, and high air and moisture sensitivity. Thus, the replacement of noble metal catalysts by less expensive and highly efficient earth abundance non-noble metal catalysts system finds potential applications in the scientific community. As a result, in recent years, manganese, cobalt, iron, and nickel-based catalysts have been designed to replace noble metals. In this context, Chunshan Li and coworkers reported that the cobalt-catalyzed O2-IBH strategy synthesizes α,β-unsaturated esters from esters and alcohols (Scheme 20).48 They have demonstrated that the PN5P ligand stabilized cobalt complexes were highly active in catalyzing the dehydrogenation of alcohol into aldehyde and showed excellent activity in the IBH reaction of esters with alcohol. These complexes are very stable toward air and moisture, and the preparation follows a simple synthetic procedure (using PN5P ligands and CoCl2). Indeed, moderate to good yields were obtained for a wide range of substrates with high functional group tolerance, including alcohols, acetates, propionates, and diols. Mechanistic investigations illustrate that the activation of cobalt complex was done by KOtBu via double deprotonation of PN5P ligand and the removal of dichloro ligands, followed by alcohol activation and dehydrogenation. The oxygen atmosphere in the catalytic reaction is crucial for interrupting the hydrogen borrowing pathway to avoid the further hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in t-butyl cinnamate (Scheme 20).
C bonds in t-butyl cinnamate (Scheme 20).
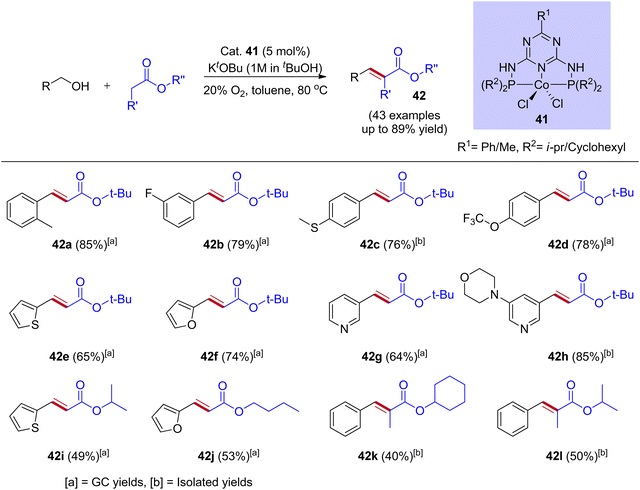 |
| | Scheme 20 Cobalt-catalyzed synthesis of α,β-unsaturated esters via mild O2-interrupted hydrogen borrowing strategy. | |
A wide variety of organic molecules, like heterocyclic and polyfunctional compounds, are prepared using diketones as building blocks. Due to their pharmacological and biological activities, 1,5-diketones gained a lot of attraction. In long-established methods, 1,5-diketones are prepared by the Michael addition and condensation between aldehydes and aryl methyl ketones. The cobalt (II) porphyrin-mediated IBH for the synthesis of 1,5-diketones from a variety of ketones using methanol as a C1 feedstock was reported by the research group of Chandrasekhar and Venkatasubbaiah (Scheme 21).49 Experimental mechanistic studies and deuterium labeling experiments revealed that the reaction mechanism proceeds through the IBH mechanism involving protonated cobalt(II)porphyrin methoxide complex as an intermediate followed by Michael addition of enolate ion and alpha methylenated ketone. This intermediate further generates formaldehyde along with metal hydride.
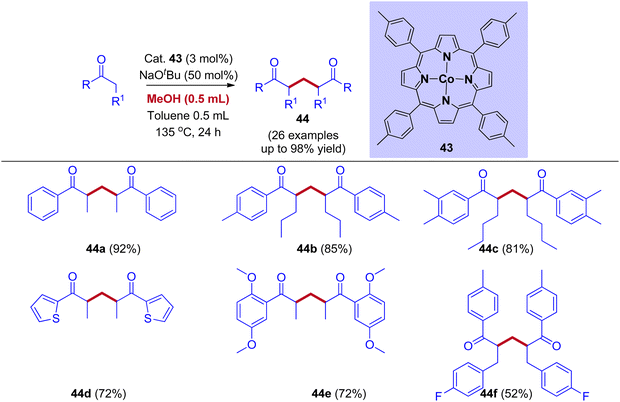 |
| | Scheme 21 Cobalt (II)porphyrin mediated selective synthesis of 1,5-diketones via IBH strategy. | |
2.4. Manganese catalysis
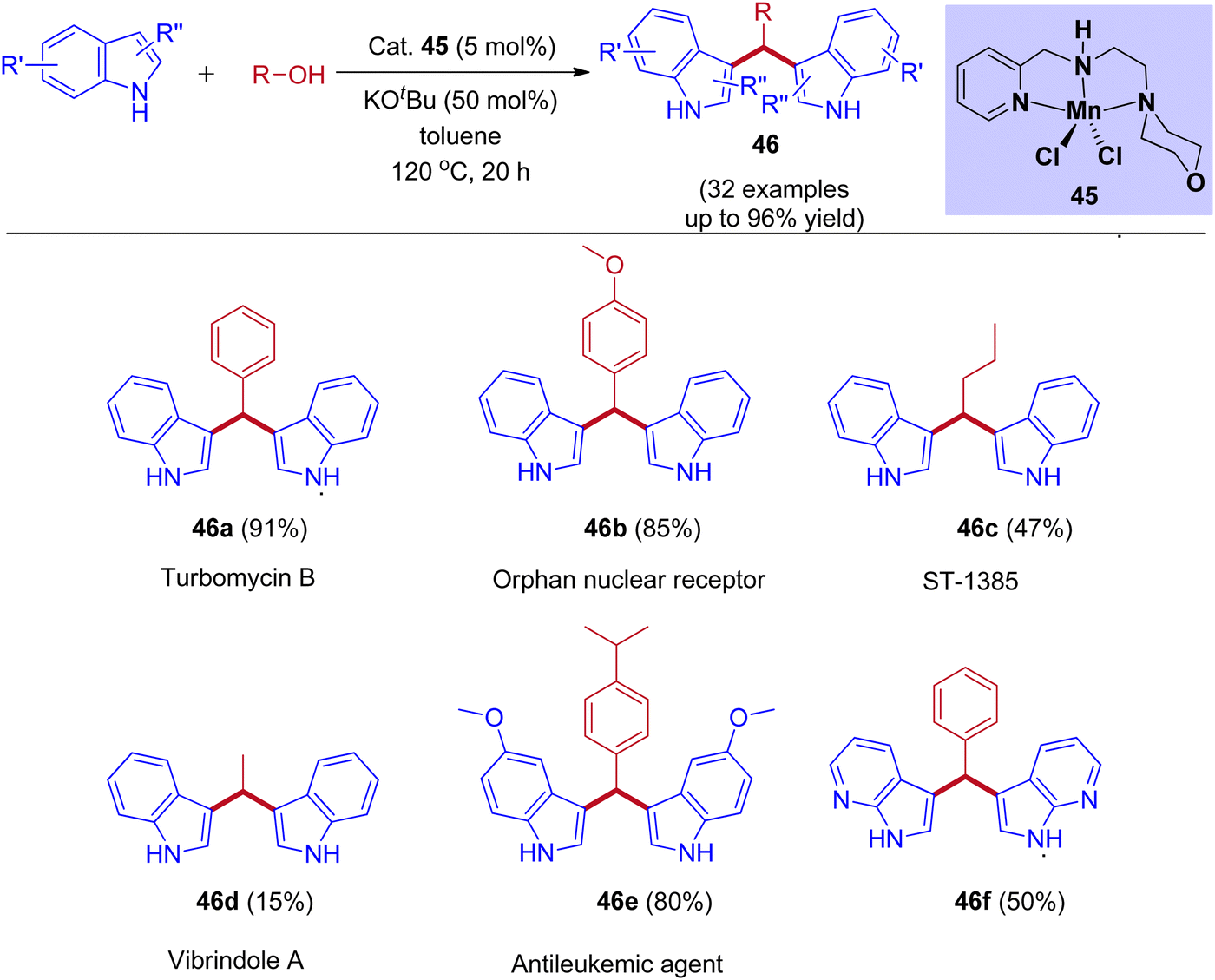
Recently, our group reported Mn(II) pincer complex-catalyzed synthesis of bis(indolyl)methane derivatives (Scheme 22), and this process is a phosphine ligand-free as well as an air-stable method.50 A wide range of alcohols, including aliphatic and aromatic alcohols, afforded the corresponding symmetrical and unsymmetrical BIMs derivatives in excellent yields. Besides, biologically important compounds such as Vibrindole A, Turbomycin B alkaloid, Antileukemic, and anticancer agents were also prepared by our protocol.
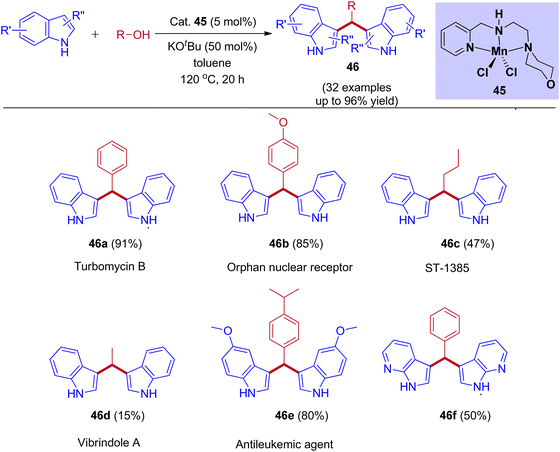 |
| | Scheme 22 Manganese-catalyzed coupling of alcohols with indoles to form BIMs derivatives via IBH strategy. | |
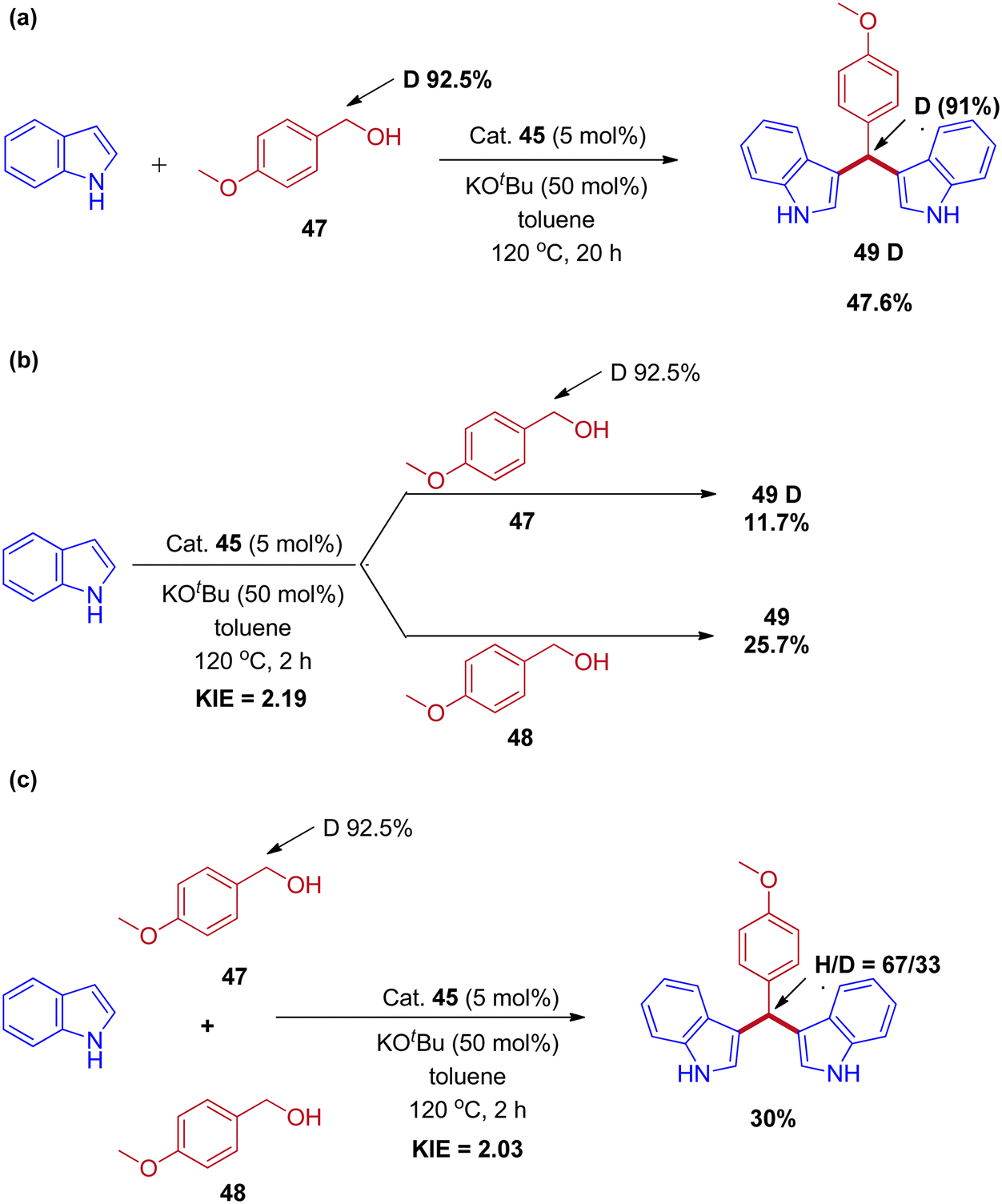
Controlled experiments and mechanistic studies were performed to establish the reaction mechanism. The reaction of deuterium-labeled alcohol (47) with indole was revealed under 91% deuterium incorporation in the product by 1H-NMR analysis (Scheme 23a). Next, parallel and competitive experiments of alcohol 48 and 47 with indole gave the kinetic isotope effect (KIE) (Schemes 23b and 23c). The KIE (KH/KD) value obtained in parallel and competitive experiments values 2.19 and 2.03 are in close agreement with each other and provide evidence that the dehydrogenation of the alcohol took place at or before the rate-determining step and revealed that the reaction proceeds via IBH strategy (Scheme 24).
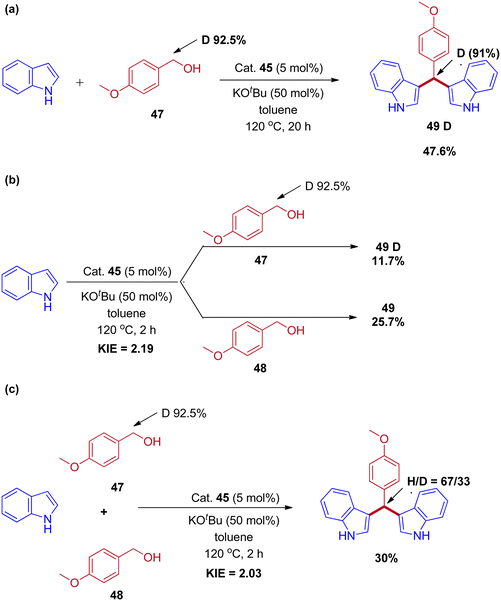 |
| | Scheme 23 Mechanistic experiments for Mn(II) pincer complex-catalyzed synthesis of bis(indolyl)methane derivatives. | |
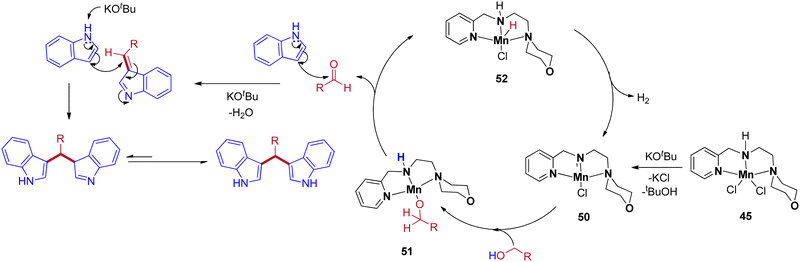 |
| | Scheme 24 Plausible reaction mechanism for Mn(II)-complex catalyzed synthesis of BIMs. | |
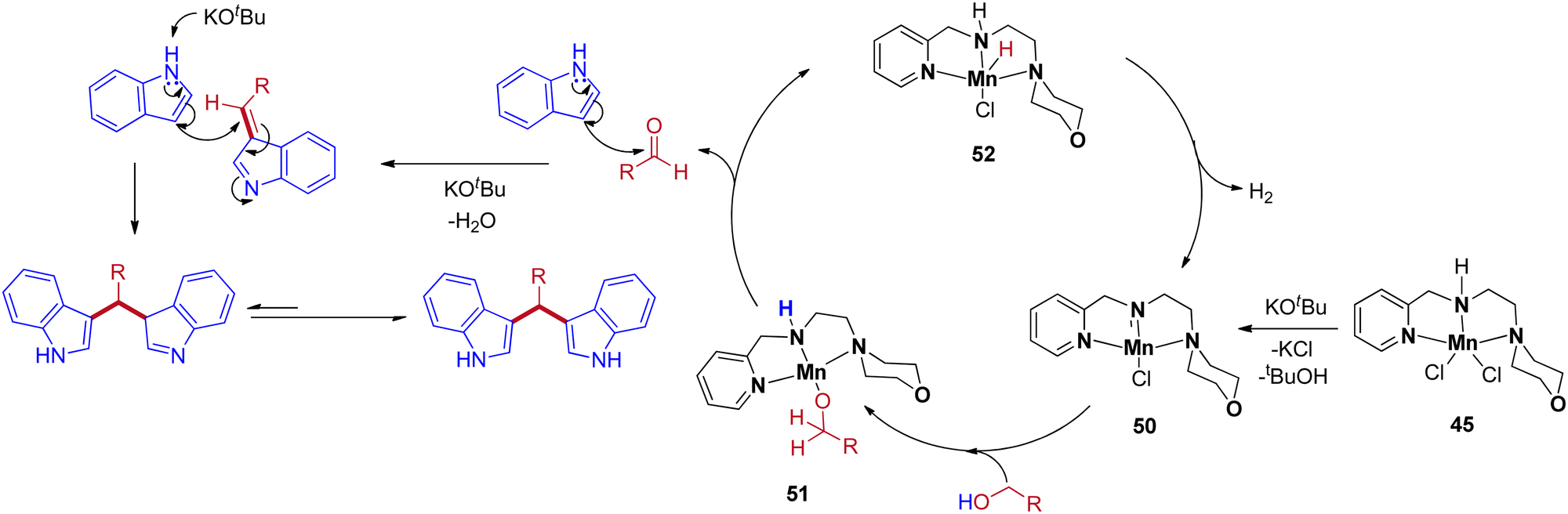
The precatalyst 45 produces the reactive intermediate 50 in the presence of a base, and further alcohol activated via metal–ligand cooperation (MLC)51 leads to the 51. The β-hydride elimination of intermediate 51 gives the corresponding carbonyl by the formation of intermediate 52. The reaction cycle continues with the liberation of H2 from intermediate 52 to give 50. Next, the in situ generated carbonyl gives BIM product by base-catalyzed condensation reaction with indole followed by Michael-type nucleophilic addition of another indole molecule (Scheme 24).
3. Conclusions
In this review article, we summarized recent efforts on IBH strategy to form a new C–C bond using abundant starting materials such as methanol and other alcohols as C1 feedstocks under transition-metal catalysis. Mainly, the reaction utilizes Ir, Ru, Co, and Mn-based complexes as catalysts for the synthesis of 3,3′-Bisindolylmethanes (3,3′-BIMs), alpha-branched ketones, diketones, and regioselective alkylation reaction via IBH strategy. Importantly, this methodology provided simple, mild, efficient, and eco-friendly methods for synthesizing various products. In addition, the metal catalysts and reaction conditions play a crucial role in such transformation, and the reaction mechanism was clearly explained. For all these reactions, the initial key step involves the dehydrogenation of alcohol to aldehyde, followed by the formation of metal hydride species. Consequently, the coupling reaction between the aldehyde and indole/methyl ketone forms enamine/enone intermediates. Then, the interruption of the borrowing hydrogen pathway was done by nucleophilic attack under the reaction conditions leading to a new functionalization or modification of the final product. The various parameters, including transition metal catalyst, reaction atmosphere, and ligand systems, play a significant role in this interruption of borrowing hydrogen cycle to give their corresponding desired products. On the other hand, developing new and efficient reaction systems (homogeneous and heterogeneous) and expanding reaction models including an enantioselective IBH strategy is highly desirable, especially with more abundant 3d-transition metal catalysts. The reported application of the IBH strategy is limited; however, it has been highly successful to date. Moreover, this concept can potentially broaden its synthetic scope shortly.
Author contributions
Conceptualization and outlook: E. Balaraman; methodology: M. Nallagangula and M. Subaramanian; writing – original draft preparation: M. Nallagangula, M. Subaramanian, and Rohit Kumar; writing – review & editing: E. Balaraman.
Conflicts of interest
The author declares that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this review article.
Acknowledgements
This work is supported by CSIR (Project No: 01/(3030)/21/EMR-II). EB acknowledges funding from Swarnajayanti Fellowship (SERB/F/5892/2020-2021). MN thanks SERB-NPDF for the fellowship (PDF/2022/000826).
References
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- A. Nandakumar, S. P. Midya, V. G. Landge and E. Balaraman, Angew. Chem., Int. Ed., 2015, 54, 11022–11034 CrossRef CAS PubMed.
- E. Balaraman, A. Nandakumar, G. Jaiswal and M. K. Sahoo, Catal. Sci. Technol., 2017, 7, 3177–3195 RSC.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- R. Yamaguchi, K.-I. Fujita and M. Zhu, Heterocycles, 2010, 81, 1093–1140 CrossRef CAS.
-
J. F. Bower and M. J. Krische, in Formation of C–C Bonds via Iridium-Catalyzed Hydrogenation and Transfer Hydrogenation, ed. P. G. Andersson, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 107–138 Search PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- G. Chidambaram and M. David, Science, 2013, 341, 1229712 CrossRef PubMed.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- R. H. Crabtree, Organometallics, 2011, 30, 17–19 CrossRef CAS.
- A. C. Marr, Catal. Sci. Technol., 2012, 2, 279–287 RSC.
- S. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef CAS.
- F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef CAS PubMed.
- A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed.
- J. F. Bower, E. Skucas, R. L. Patman and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 15134–15135 CrossRef CAS PubMed.
- Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC.
- L. Piccirilli, D. Lobo Justo Pinheiro and M. Nielsen, Catalysts, 2020, 10 Search PubMed.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed.
- F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC.
- M. Subaramanian, G. Sivakumar and E. Balaraman, Org. Biomol. Chem., 2021, 19, 4213–4227 RSC.
- M. Qian, M. A. Liauw and G. Emig, Appl. Catal., A, 2003, 238, 211–222 CrossRef CAS.
- W.-H. Lin and H.-F. Chang, Catal. Today, 2004, 97, 181–188 CrossRef CAS.
- Y. Li, H. Li, H. Junge and M. Beller, Chem. Commun., 2014, 50, 14991–14994 RSC.
- C. Sun, X. Zou and F. Li, Chem. – Eur. J., 2013, 19, 14030–14033 CrossRef CAS PubMed.
- J. R. Frost, C. B. Cheong, W. M. Akhtar, D. F. J. Caputo, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2015, 137, 15664–15667 CrossRef CAS PubMed.
- K. Polidano, J. M. J. Williams and L. C. Morrill, ACS Catal., 2019, 9, 8575–8580 CrossRef CAS PubMed.
- D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642–1645 CrossRef CAS PubMed.
- M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., Int. Ed., 2005, 44, 6913–6915 CrossRef CAS PubMed.
- M. I. Hall, S. J. Pridmore and J. M. J. Williams, Adv. Synth. Catal., 2008, 350, 1975–1978 CrossRef CAS.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Chem. Commun., 2008, 624–625 RSC.
- E. Y. Lee, Y. Kim, J. S. Lee and J. Park, Eur. J. Org. Chem., 2009, 2943–2946 CrossRef CAS.
- M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed.
- S. Whitney, R. Grigg, A. Derrick and A. Keep, Org. Lett., 2007, 9, 3299–3302 CrossRef CAS PubMed.
- S. Bähn, S. Imm, K. Mevius, L. Neubert, A. Tillack, J. M. J. Williams and M. Beller, Chem. – Eur. J., 2010, 16, 3590–3593 CrossRef PubMed.
- S.-J. Chen, G.-P. Lu and C. Cai, RSC Adv., 2015, 5, 70329–70332 RSC.
- W. Qiang, X. Liu and T.-P. Loh, ACS Sustainable Chem. Eng., 2019, 7, 8429–8439 CrossRef CAS.
- S. Wübbolt, C. B. Cheong, J. R. Frost, K. E. Christensen and T. J. Donohoe, Angew. Chem., Int. Ed., 2020, 59, 11339–11344 CrossRef PubMed.
- S. Zhang, W. Fan, H. Qu, C. Xiao, N. Wang, L. Shu, Q. Hu and L. Liu, Curr. Org. Chem., 2012, 16, 942–948 CrossRef CAS.
- A. E. Putra, K. Takigawa, H. Tanaka, Y. Ito, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6344–6354 CrossRef CAS.
- N. Biswas, R. Sharma and D. Srimani, Adv. Synth. Catal., 2020, 362, 2902–2910 CrossRef CAS.
- P. Biswal, M. Siva Subramani, S. Samser, V. Chandrasekhar and K. Venkatasubbaiah, J. Org. Chem., 2023, 88, 5135–5146 CrossRef PubMed.
- G. Wang, Z. Li, C. Li and S. Zhang, J. Catal., 2018, 368, 228–236 CrossRef CAS.
- P. Biswal, S. Samser, P. Nayak, V. Chandrasekhar and K. Venkatasubbaiah, J. Org. Chem., 2021, 86, 6744–6754 CrossRef CAS PubMed.
- V. Yadav, E. Balaraman and S. B. Mhaske, Adv. Synth. Catal., 2021, 363, 4430–4439 CrossRef CAS.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Murugan
Subaramanian
,
Rohit
Kumar
and
Ekambaram
Balaraman
,
Murugan
Subaramanian
,
Rohit
Kumar
and
Ekambaram
Balaraman