
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High spatial resolution ambient tip-enhanced (multipolar) Raman scattering
Patrick Z.
El-Khoury

Physical Sciences Division, Pacific Northwest National Laboratory, P.O. Box 999, Richland, WA 99352, USA. E-mail: patrick.elkhoury@pnnl.gov
First published on 24th February 2023
Abstract
This article summarizes lessons learnt from ambient tip-enhanced Raman (TER) mapping of molecules interacting with plasmonic nanostructures. It is shown that numerous physical and chemical phenomena contribute to high-resolution TER spectral images. As a result, selectively tracking interfacial chemical transformations via TERS is more challenging than currently appreciated.
 Patrick Z. El-Khoury | Patrick El-Khoury received a BSc in chemistry from the American University of Beirut (2006), a PhD in photochemical sciences from Bowling Green State University (2010), and postdoctoral training at the University of California, Irvine (2012). Soon after, he joined Pacific Northwest National Laboratory (PNNL) as a Linus Pauling Distinguished Postdoctoral Research fellow (2013–2016). Patrick is currently a senior research scientist in the chemical physics and analysis group at PNNL. His work is focused on understanding molecules, low-dimensional (quantum) materials, plasmonic metals, and their interactions as gauged through multimodal (non)linear spectral nano-imaging. |
Raman spectroscopy is a powerful chemical identification technique that is widely used by physicists, chemists, and biologists alike. When performed in an optical microscope, Raman micro-spectroscopy is routinely used to record diffraction-limited hyperspectral chemical images of analytes as simple as small molecules and as complex as cellular communities. Despite its rich information content that stems far beyond the realm of mere chemical fingerprinting and spectral imaging, it is well-known that Raman scattering is a relatively inefficient process. This is why a large number of early works employed high laser powers that may compromise sample integrity and/or used long integration times (often hours) to obtain adequate signal-to-noise ratios, even from bulk samples. Advances in lasers, detectors, and optics somewhat improved the detection limits, but it wasn’t until the first observations of surface-enhanced Raman scattering (SERS1–3) that the ultimate detection limit (of a single/a few molecules) became possible. A large number of papers and review articles have been dedicated to SERS,3,4 and several aspects of the interplay between (individual) molecules and plasmonic metals are now well-appreciated.5 This is in part why SERS, much like conventional Raman scattering, is now widely adopted across different fields.5,6
Tip-enhanced Raman scattering (TERS7) is similar to SERS in that the interaction between a plasmonic nanostructure, now a plasmonic scanning probe, is used to enhance and localize the incident and scattered electromagnetic fields.8–13 The latter is possible when silver or gold probes are irradiated with visible light (linearly) polarized along their long axes. It is also possible in other geometries using other polarization modes of light.13 When the scanning probe is that of an atomic force microscope, enhanced Raman signals are recorded when the tip is in the immediate vicinity of the sample, often in direct contact. Moving the sample relative to the tip then allows recording spatially resolved tip-enhanced spectra. Signal enhancement aside, the spatial resolution in TERS is dictated by the spatial confinement of the nano-optical field, which in the case of an atomically terminated tip reaches the sub-nanometer domain.14 Moving away from the realm of low temperature measurements towards ambient/in situ TERS decreases the attainable spatial resolution, empirically to 1-few nanometers.12,15 This has as much to do with (thermal) noise and instrument instabilities as it has to do with the mobility of atoms at plasmonic junctions under ambient laboratory conditions.16
Even though the signal magnitudes in both SERS and TERS predominantly arise from the resonant interaction between light and plasmonic nanostructures, the recorded signals themselves are molecular in origin. Dynamic molecular properties therefore govern plasmon-enhanced Raman spectra and images. Molecular (re)orientation dynamics, plasmon-induced redox and chemical events, and modified optical selection rules are among the physical and chemical processes that come to mind in this context. These phenomena together comprise the topic of this article. Emphasis is otherwise put on high spatial resolution ambient TERS measurements that address ultra-small molecular ensembles interacting with plasmonic metal nanostructures, and inevitably, with the plasmonic probe.
Orientation-dependent Raman scattering
Traditionally, Raman spectroscopy and microscopy measurements address ensembles of randomly oriented molecules, even though most lasers used to perform these measurements are polarized. This is why most Raman simulation software output Raman scattering cross-sections (Sm) as17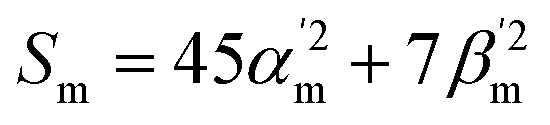 | (1) |
In the above equation, primes are used to denote derivatives with respect to the mth vibrational eigenstate and 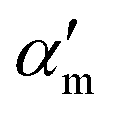 and
and 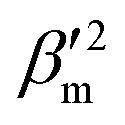 are the isotropic and anisotropic polarizabilities. Note that the factors (45 and 7) are derived assuming orthogonal excitation/collection and otherwise ensemble averaging to account for randomly oriented molecules, e.g., in a liquid.
are the isotropic and anisotropic polarizabilities. Note that the factors (45 and 7) are derived assuming orthogonal excitation/collection and otherwise ensemble averaging to account for randomly oriented molecules, e.g., in a liquid.
In ultrasensitive/single molecule TERS and in TERS measurements that address oriented molecular ensembles (e.g., a self-assembled monolayer), the above treatment is inadequate. Rather, the scattering tensor that governs TERS scattering is re-written as18,19
 | (2) |
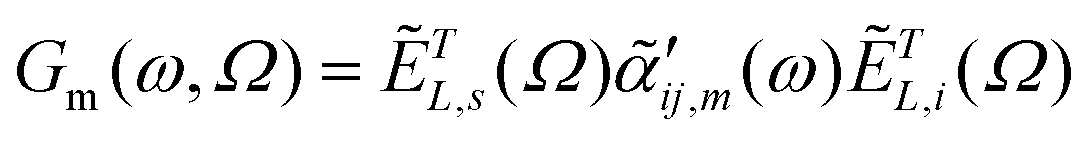 | (3) |
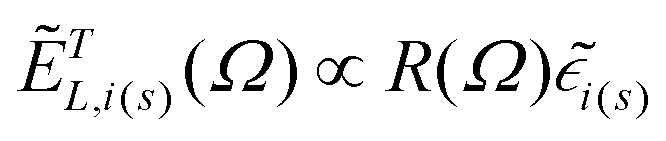 | (4) |
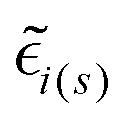 are the local optical fields along the incident (i) and scattered (s) directions,
are the local optical fields along the incident (i) and scattered (s) directions, 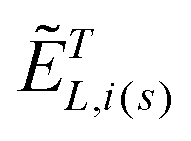 are the same local fields but now projected along a common (laboratory) reference frame, Ω represents the Euler angles that determine molecular orientation relative to the local fields, R is a rotation matrix, and
are the same local fields but now projected along a common (laboratory) reference frame, Ω represents the Euler angles that determine molecular orientation relative to the local fields, R is a rotation matrix, and 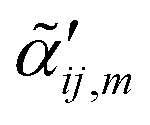 (i,j = x,y,z) are individual components of the polarizability tensor. Eqn (3) admits that the experimentally observed ultrasensitive TERS relative intensities of the (Raman active) states depend on molecular orientation relative to the enhanced local optical fields. In the TERS geometry the latter is typically along the long axis of the probe, which is orthogonal to the sample plane.
(i,j = x,y,z) are individual components of the polarizability tensor. Eqn (3) admits that the experimentally observed ultrasensitive TERS relative intensities of the (Raman active) states depend on molecular orientation relative to the enhanced local optical fields. In the TERS geometry the latter is typically along the long axis of the probe, which is orthogonal to the sample plane.
In Fig. 1, a familiar molecular reporter (thiophenol, TP) is used to show how the removal of orientational averaging affects the observables in ultrasensitive/single molecule TERS measurements. The ensemble averaged response computed for an isolated TP molecule is plotted in Fig. 1A. In this plot, the relative intensities of the observable vibrations were obtained following eqn (1). The calculated spectrum agrees reasonably well with its experimental ensemble averaged analogue, as discussed elsewhere.20 Removal of orientational averaging following eqn (3) yields the spectra stacked in Fig. 1B for rotation around the Y-axis (defined in the inset of Fig. 1B). Here, the incident and scattered fields are along Z to account for enhanced local fields along the long axis of a plasmonic probe. Several aspects of the orientation-dependent response are noteworthy. Evidently, distinct relative intensities of the modes are observed for different orientations of the molecule relative to the surface. This is immediately noticeable for the 990/1585 cm−1 modes. In fact, the 1585 cm−1 that predominates the ensemble averaged spectrum (see Fig. 1A) is dim/barely noticeable at small rotation angles (0–20°) towards the bottom of panel B. Interestingly, the 1472 cm−1 peak that is only weakly active in the ensemble measurement is brighter than its 1585 cm−1 analogue for the flatter TP geometries. The situation is reversed when the long axis of the molecule is (nearly) orthogonal to the surface in the upper spectra (70–90°), wherein the 1585 cm−1 mode has the strongest scattering activity in the spectrum. These observations emphasize that the disappearance/stronger contributions of certain bands to the overall spectrum may arise from modified optical selection rules, as opposed to chemical transformations, molecular diffusion out of the probing volume, or resonance (vibronic) effects. In fact, molecular (re)orientation is the most mundane effect that should be considered first in comparing TERS to conventional (micro)Raman spectra. On a final note, it is important to stress that even though some conformations may have weaker optical signatures/absolute scattering cross-sections, ample enhancement in the TERS geometry render even the faintest of signatures detectable. After all, single molecules are routinely detected via TERS.
 | ||
| Fig. 1 (A) Ensemble averaged Raman spectrum of an isolated TP molecule computed at the B3LYP/6-311++G** level of theory. (B) Molecular orientation-dependent Raman scattering from TP, simulated following eqn (5) in the main text with incident and scattered fields along Z. The spectra were obtained by rotating the molecule around Y (see the lower right inset of panel B, and the color coded numbers correspond to rotation angles in degrees. The inset shows a schematic representation of the molecule atop the surface in the laboratory frame. All spectra were scaled with a single factor of 0.975. The orientation-dependent spectra were otherwise individually normalized. | ||
Spatially-resolved 3D molecular GPS
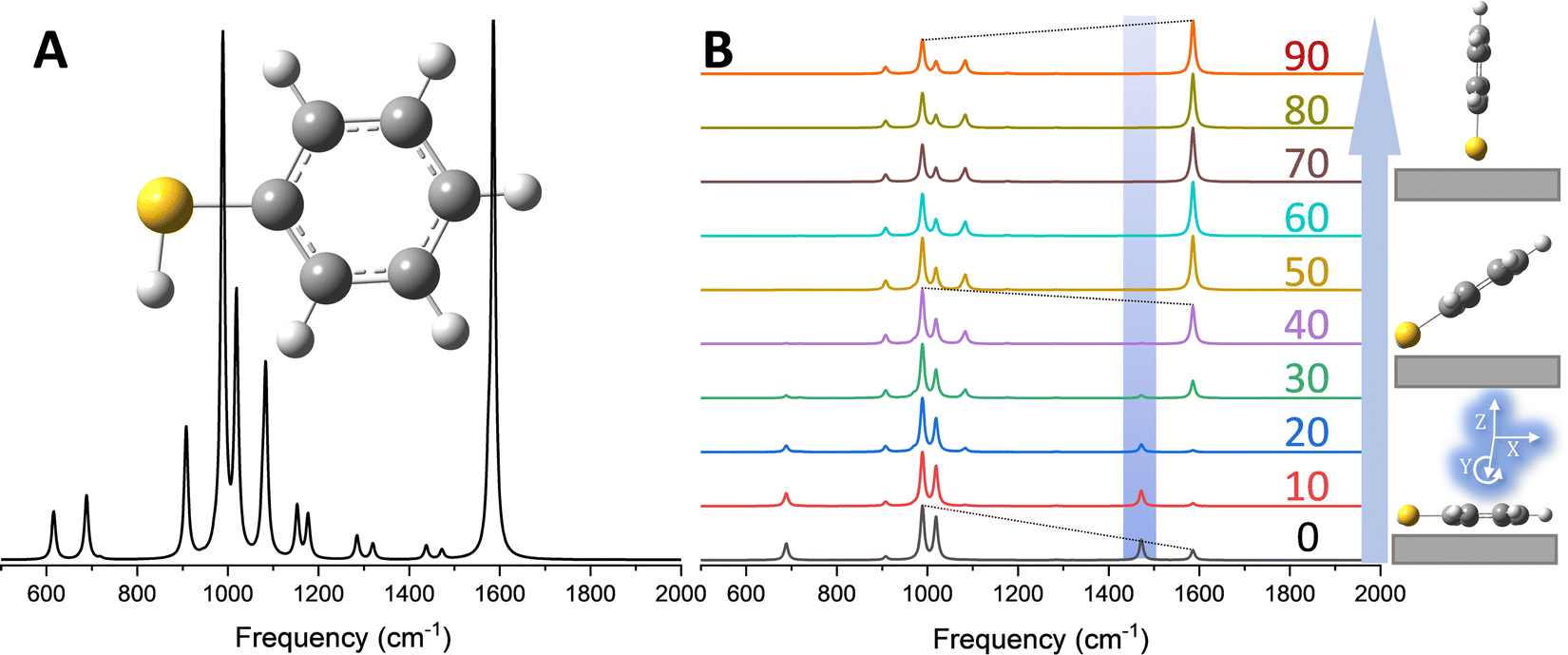
The analysis exemplified in Fig. 1 can be used to extract the 3D orientation of surface-bound species with ultra-high spatial resolution.18 In Fig. 2, the concept is illustrated through TERS mapping of a silver nanocube coated with 4-mercaptobenzonitrile (MBN). As well-established in several prior reports,12,21 TERS maps of chemically functionalized silver nanocubes trace their edges and reveal regions of highly localized (1–2 nm) and enhanced Raman scattering. Spectral nano-imaging near the edge of the cube results in the image shown in Fig. 2B (here traced at ∼1589 cm−1). Spatial averaging of the local optical response atop the cube results in a distinct scattering pattern, which is plotted in Fig. 2C (grey trace). Using eqn (3), the best match to the recorded spectrum was obtained and used to infer the preferred molecular orientation. The nearly flat geometry of MBN on the silver surface is schematically illustrated in the inset. As noted in prior work, the same approach may also be used to image the vector components of local electromagnetic fields.19 This naturally requires knowledge of molecular orientation, which can be simulated or even directly measured using complimentary scanning probe and nonlinear optical approaches.22 More generally, and beyond the realm of ultrasensitive spectral nano-imaging, TERS maps of plasmonic metal nanoparticles and nanostructures track the magnitudes,12 resonances,23 and (non)local character24,25 of the local optical field.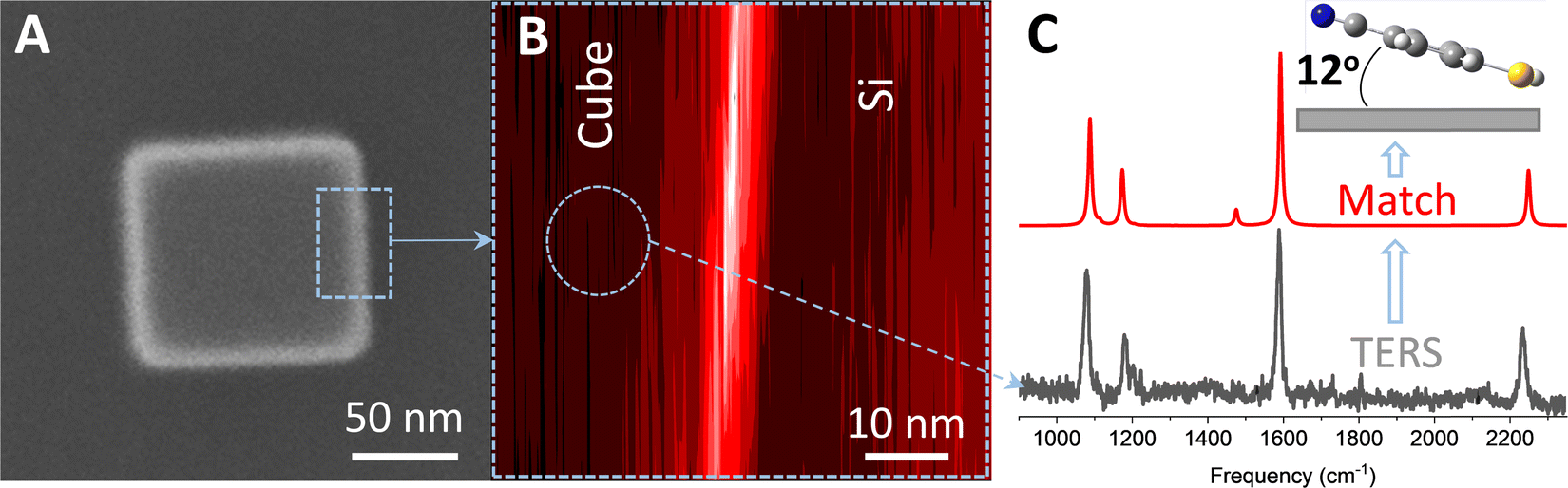 | ||
| Fig. 2 (A) Helium ion image of a 100 nm silver nanocube coated with MBN. The dashed rectangular area highlighted in this panel is imaged using TERS in (B). Spatial averaging of the optical response in the circular area highlighted in B results in the spectrum plotted in the lower part of C (grey trace). The best match to the experimental spectrum, obtained using eqn (3) in the text, is shown in panel C (red trace). The predominant molecular orientation that leads to the observed relative intensity pattern is schematically illustrate in the inset of (C). The same laboratory frame used in Fig. 1 is also used here. See ref. 18 for more details. | ||
Signatures of multipolar Raman scattering in TERS
The dipole approximation does not always hold in high spatial resolution TERS measurements.26–28 Besides empirical evidence, i.e., the direct observations and assignments of multipolar Raman scattering using density functional theory calculations, theoretical and experimental analyses both reveal that large spatiotemporal optical field gradients are operative in the plasmon-enhanced Raman scattering.29–31 Once again, using TP as a model system, we plot three spectra in Fig. 3 that may be used to inspect contributions from the generalized molecular polarizabilities32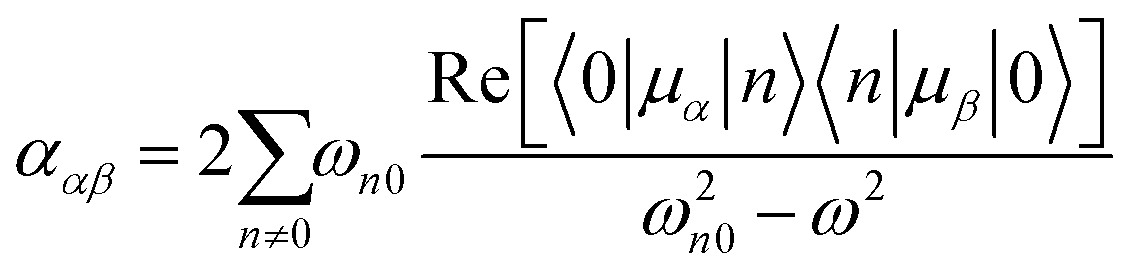 | (5) |
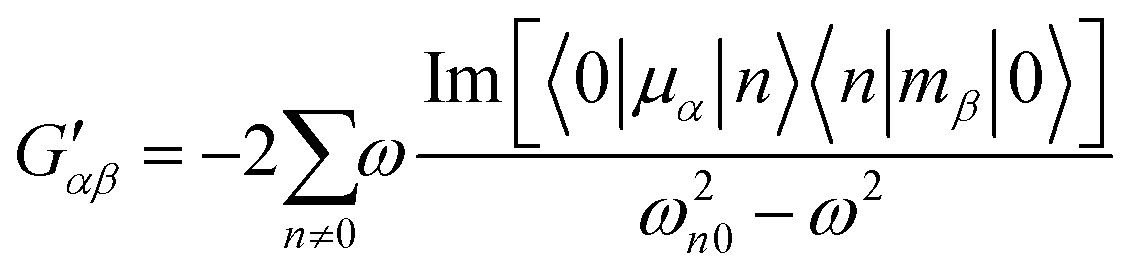 | (6) |
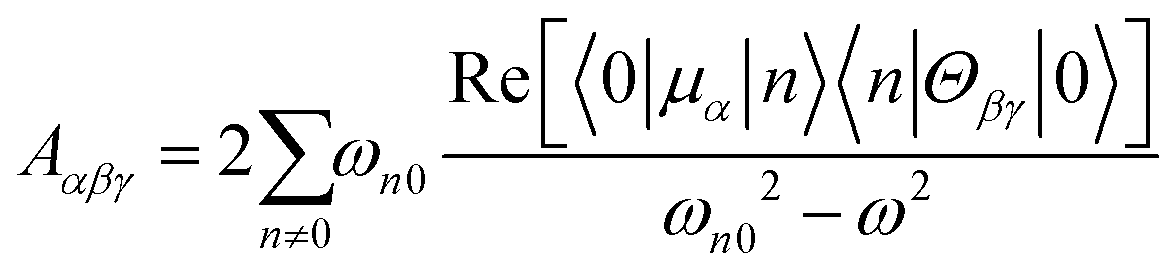 | (7) |
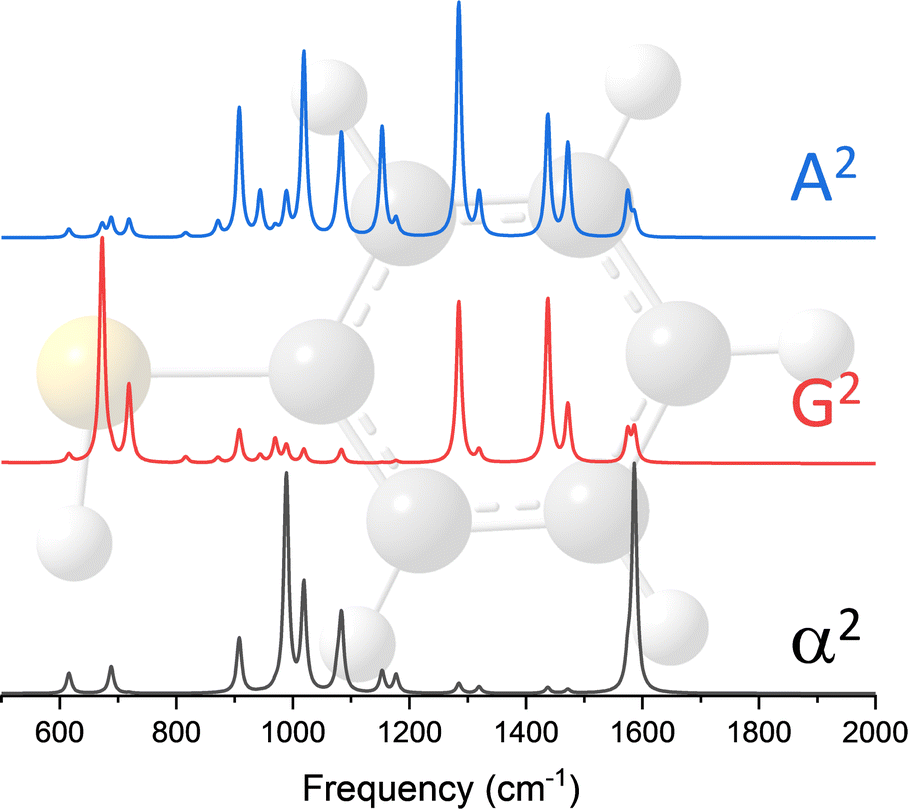 | ||
| Fig. 3 Power spectra of the dipole–dipole (α2), electric dipole-magnetic dipole (G2), and electric dipole-electric quadrupole (A2) polarizabilities of thiophenol. These spectra were computed at the B3LYP/6-311++G** level of theory. The frequency axes were scaled using a single factor of 0.975 and the intensities were all individually normalized for comparison. | ||
When the incident light source is not resonant with an excited electronic state of molecular or mixed molecular-metallic character, observations of Raman inactive modes in TERS may be understood on the basis of the above analysis. Naturally, the observation of spectral patterns – that for the case of TP are separately shown in Fig. 3 for each of the polarizabilities considered in our present analysis – as opposed to a single/a few peaks would be needed to make the case that tip-enhanced multipolar Raman scattering is operative. This is challenging, particularly in the realm of ultrasensitive TERS measurements, where tensorial Raman scattering is also operative. Nonetheless, satisfying matches and convincing spectral assignments were made in recent papers, which we discussed below and use to make our case.
Case of MBN
Our first observation of tip-enhanced multipolar Raman scattering was documented throughout the course of TERS mapping of MBN-functionalized plasmonic silver nanocubes.26 Even though the majority of the recorded spectra could be recovered by only considering conventional electric dipole-electric dipole polarizabilities (see Fig. 2C), the brightest spectra within the hyperspectral image cube contained spectral patterns that closely resemble the G2 and A2 spectra that are shown in Fig. 4. The two spectra we select for the purpose of our first example are shown in the lower panel of Fig. 4. Both spectra contain the 3 familiar Raman-active modes (highlighted using red rectangles) that derive their intensities through α.20,26 The blue trace contains an additional dominant mode (highlighted using a blue rectangle) and otherwise closely resembles the G2 spectrum that is dominated by the same resonance. The black experimental trace is distinct, containing three peaks that all derive their intensities from A2. Overall, both spectra can be described by a linear combination of the α2 on one hand and either the G2 (blue trace) or A2 spectra (black trace) on the other. In this particular example, and besides the latter, spectral assignments were simplified by the fact that no vibrational resonances in the 1225–1500 cm−1 spectral region derive their intensities from α.20,26 This allowed us to make our initial proposition that higher order polarizabilities contribute to TERS spectral images. It is important to note that we analyze the brightest spectra contained in a hyperspectral TERS image cube because part of our goal is to understand spatially averaged measurements that are more abundant in literature. The latter may be lower resolution TERS maps recorded using a coarse TERS probe or even ensemble-averaged surface-enhanced Raman spectra. That said, signatures of multipolar scattering are also often observed throughout a given field of view and at sites featuring lower overall TERS signal magnitudes.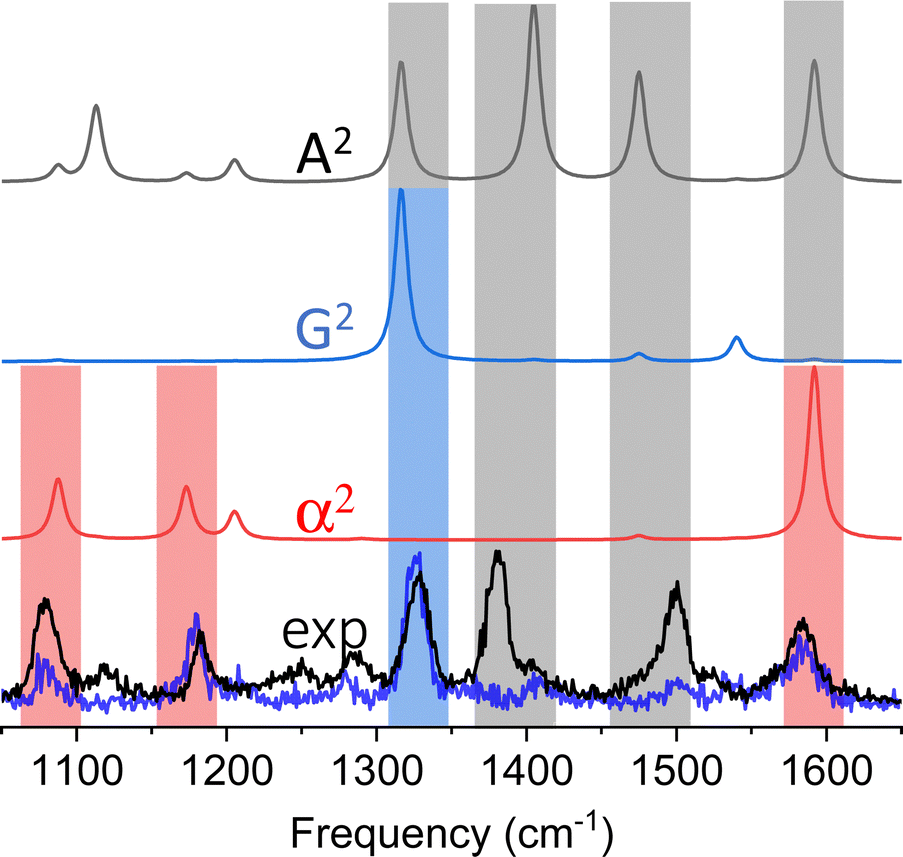 | ||
| Fig. 4 Experimental TERS spectra (lower two overlapped spectra) are compared to power spectra of the dipole–dipole (α2), electric dipole-magnetic dipole (G2), and electric dipole-electric quadrupole (A2) polarizabilities of thiophenol. The theoretical spectra are color-coded, and the rectangular bars that are overlapped with the experimental spectra designate modes that derived their intensities from α (red), G (blue), and A (black). In two cases, the peak intensities can derive their intensities from different contributions. These peaks are highlighted using vertically stacked rectangular bars. | ||
Case of 4-mercaptopyridine (MPY)
The above analysis is particularly important to consider in the context of TERS-based chemical reaction mapping.28 Namely, the fact that plasmons may be used to both induce chemical transformations and to enhance Raman scattering from ultrasmall volumes has motivated quests to utilize TERS to monitor interfacial chemical reactions in real space.11 Since the (dis)appearance of peaks in ultrasensitive (or equivalently ultrahigh spatial resolution) TERS may arise from a combination of tensorial and multipolar Raman scattering, caution needs to be exercised in associating spectral changes with the onset of interfacial chemical transformations. The latter is exemplified through the analysis presented in Fig. 5. In addition to the standard Raman-active vibrations of MPY that are contained within the areas highlighted using red rectangles in both panels of Fig. 5, spectral patterns that can only be recovered by considering G2 (panel A) and A2 (panel B) were observed.28 Taking both tensorial and multipolar Raman scattering into account results in theoretical spectra that closely match their experimental analogues. In this example, it is clear that the spectra shown, parts of which may be easily confused with the onset of plasmon-induced/enhanced chemistry, all arise from MPY.28 The numerous peaks that appear in concert in addition to the standard Raman-active lines arise from resonances that derived their intensities from the G2 and A2 terms.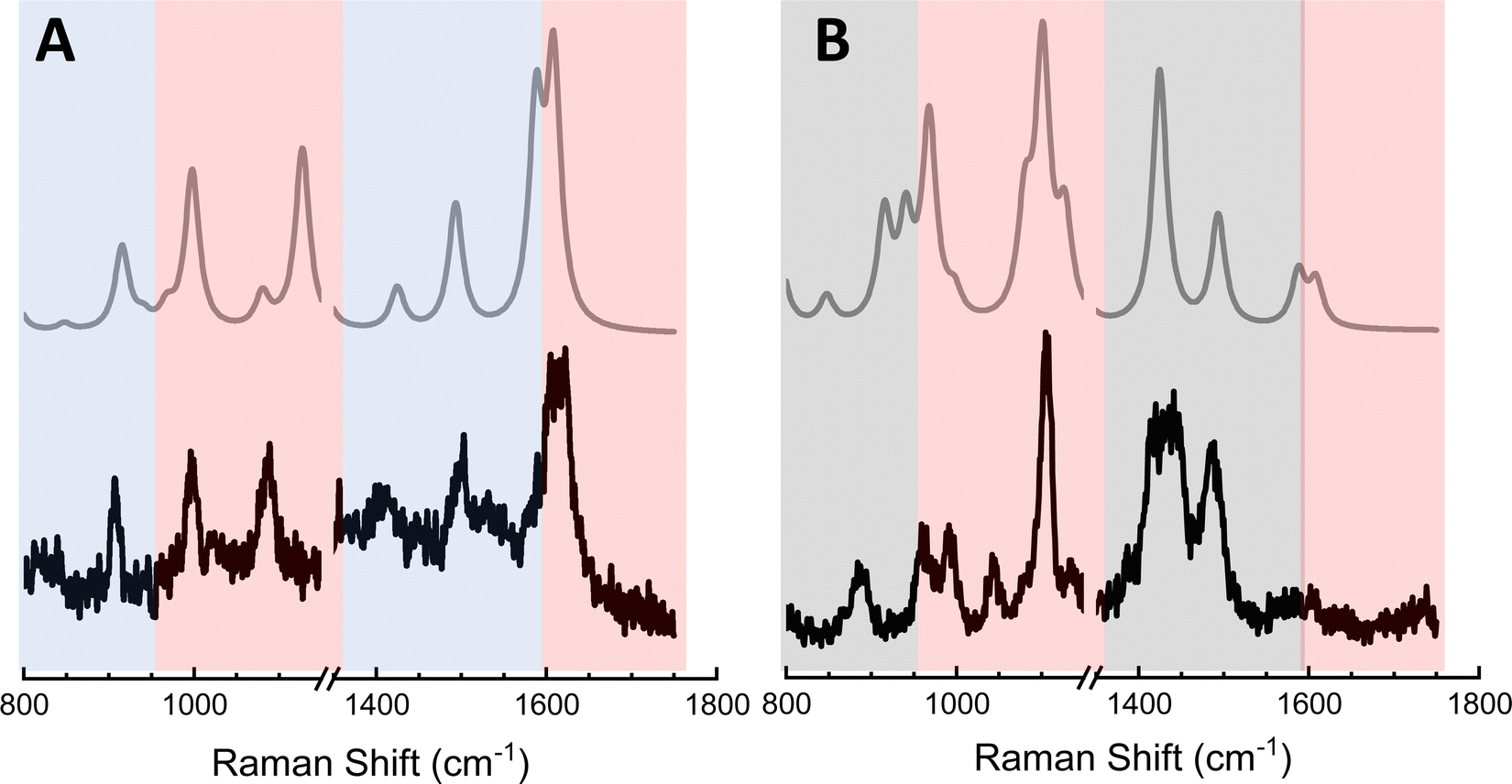 | ||
| Fig. 5 Experimental TERS spectra (lower traces in both panels) are compared to theoretical spectra of MPY that take both tensorial Raman scattering as well as multipolar Raman scattering into consideration. The difference between the two panels is that only modes that derive their intensities from G are considered in panel A, whereas only terms that derive their intensities from A are considered in panel B. The color-coded rectangular areas highlight modes that derive their intensities from α (red, both panels), G (blue, panel A), and A (black, panel B). | ||
Case of 4,4′-dimercaptostilbene (DMS)
The above example of MPY does not mean that chemical transformations do not take place in the TERS geometry. Besides well-documented prior work from several groups,11,33 we recently explored the isomerization of DMS at the edges of plasmonic silver nanocubes using TERS.27 We isolate and discuss this particular study because several observations therein are worth recounting for the purpose of our current discussion. Namely, we were able to assign a total of 4 distinct species in the TERS spectral images of chemically functionalized silver nanocubes. In addition to the trans and cis forms of DMS, we also observed the anions of both species,27 which is generally consistent with prior observations of anions under similar experimental conditions.25,34 All 4 species were marked through their dipolar and multipolar tip-enhanced Raman spectra,27 which we were able to match using variants of the methods that are described in the above sections. Our group also revisited the dimerization of p-nitrothiophenol (NTP) to dimercaptoazobenzene at gold surfaces. Our work was distinct from numerous prior studies in that our chemical reaction nano-imaging measurements were performed at the solid–liquid interface.35 The same measurements also showed that the magnitudes of plasmonic fields may be imaged at the solid–liquid interface using our technique of choice.Conclusions and outlook
The work summarized in this article unveils some of the exquisite complexities of TERS, the information content of which stems far beyond the realm of mere chemical imaging. Indeed, this technique may be used not only to observe and identify molecules, but also to track redox reactions and interfacial chemical transformations with nanometer precision. Ascertaining the latter requires careful analysis of Raman nano-images, particularly in high spatial resolution TERS under ambient laboratory conditions. A detailed analysis of TERS spectral images also makes it possible to image spatially varying magnitudes, vector components, resonances, gradients, among various aspects of the operative local optical fields. Overall, most these measurements are possible at solid–air and solid–liquid interfaces.Evidently, there are several exiting avenues to pursue from here both on the molecular and local optical field sides of the equation. Understanding how local resonances affect local chemistry is an obvious area that warrants immediate attention. This is true for local plasmon resonances that vary on the nm length scale. This is also true for molecular resonances that must also depend on local environments and spatially varying molecular conformations. The latter is particularly important in the quest to move far beyond the realm of thiolated aromatic systems that we have thus far used for fundamental TERS studies. Indeed, complex chemical and biological structures that don’t necessarily self-assemble or even adopt well-defined conformations or protonation/charge states throughout the sample area necessitate an even more detailed analysis of coupled molecular, vibrational, and electronic structures on plasmonic metals. Besides perceived difficulties in distinguishing between resonance effects and multipolar TERS under resonant excitation conditions, more exhaustive theoretical analyses that treat plasmonic metals, molecules, and their interactions on equal footing seem unavoidable at this stage of evolution in this field. Alternatively, multimodal (non)linear nano-optical measurements that may be used to dissect the TERS response seem promising and more tractable in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Condensed Phase and Interfacial Molecular Science program, FWP 16248. Some of the measurements that are described in this work were performed using equipment that was partially developed using funding from the Laboratory Directed Research and Development program at Pacific Northwest National Laboratory, through the Chemical Dynamics Initiative.References
- M. Fleischmann, P. J. Hendra and A. J. Mcquillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Vanduyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, Annu. Rev. Phys. Chem., 2012, 63, 65–87 CrossRef CAS PubMed.
- A. B. Zrimsek, N. H. Chiang, M. Mattei, S. Zaleski, M. O. McAnally, C. T. Chapman, A. I. Henry, G. C. Schatz and R. P. Van Duyne, Chem. Rev., 2017, 117, 7583–7613 CrossRef CAS PubMed.
- J. Langer, D. J. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguie, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. G. de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Ka, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhofer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlucker, L. L. Tay, K. G. Thomas, Z. Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzan, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- P. El-Khoury, N. Jiang and Z. Schultz, J. Phys. Chem. C, 2022, 126, 17471–17473 CrossRef CAS.
- R. M. Stockle, Y. D. Suh, V. Deckert and R. Zenobi, Chem. Phys. Lett., 2000, 318, 131–136 CrossRef CAS.
- B. Pettinger, P. Schambach, C. J. Villagomez and N. Scott, Annu. Rev. Phys. Chem., 2012, 63, 379–399 CrossRef CAS PubMed.
- S. Mahapatra, L. F. Li, J. F. Schultz and N. Jiang, J. Chem. Phys., 2020, 153 CrossRef CAS PubMed.
- P. Verma, Chem. Rev., 2017, 117, 6447–6466 CrossRef CAS PubMed.
- Z. D. Li and D. Kurouski, Acc. Chem. Res., 2021, 54, 2477–2487 CrossRef CAS PubMed.
- P. Z. El-Khoury, Acc. Chem. Res., 2021, 54, 4576–4583 CrossRef CAS PubMed.
- N. Kazemi-Zanjani, S. Verdraine and F. Lagugne-Labarthet, Opt. Express, 2013, 21, 25271–25276 CrossRef PubMed.
- J. Lee, K. T. Crampton, N. Tallarida and V. A. Apkarian, Nature, 2019, 568, 78–82 CrossRef CAS PubMed.
- C. F. Wang and P. Z. El-Khoury, J. Phys. Chem. Lett., 2021, 12, 10761–10765 CrossRef CAS PubMed.
- B. Yang, G. Chen, A. Ghafoor, Y. F. Zhang, Y. Zhang, Y. Zhang, Y. Luo, J. L. Yang, V. Sandoghdar, J. Aizpurua, Z. C. Dong and J. G. Hou, Nat. Photonics, 2020, 14, 693–699 CrossRef CAS.
- W. F. Murphy, W. Holzer and H. J. Bernstein, Appl. Spectrosc., 1969, 23, 211–218 CrossRef CAS.
- P. Z. El-Khoury and E. Apra, J. Phys. Chem. C, 2020, 124, 17211–17217 CrossRef CAS.
- A. Bhattarai, A. G. Joly, W. P. Hess and P. Z. El-Khoury, Nano Lett., 2017, 17, 7131–7137 CrossRef CAS PubMed.
- E. Apra, A. Bhattarai, E. Baxter, S. Y. Wang, G. E. Johnson, N. Govind and P. Z. El-Khoury, Appl. Spectrosc., 2020, 74, 1350–1357 CrossRef CAS PubMed.
- A. Bhattarai, I. V. Novikova and P. Z. El-Khoury, J. Phys. Chem. C, 2019, 123, 27765–27769 CrossRef CAS.
- Z. F. Cai, L. Q. Zheng, Y. Zhang and R. Zenobi, J. Am. Chem. Soc., 2021, 143, 12380–12386 CrossRef CAS PubMed.
- A. Bhattarai, B. T. O'Callahan, C. F. Wang, S. Y. Wang and P. Z. El-Khoury, J. Phys. Chem. Lett., 2020, 11, 2870–2874 CrossRef CAS PubMed.
- P. Z. El-Khoury, D. H. Hu, V. A. Apkarian and W. P. Hess, Nano Lett., 2013, 13, 1858–1861 CrossRef CAS PubMed.
- C. F. Wang, B. T. O’Callahan, D. Kurouski, A. Krayev, Z. D. Schultz and P. Z. El-Khoury, J. Phys. Chem. Lett., 2020, 11, 5890–5895 CrossRef CAS PubMed.
- C. F. Wang, Z. H. Cheng, B. T. O’Callahan, K. T. Crampton, M. R. Jones and P. Z. El-Khoury, J. Phys. Chem. Lett., 2020, 11, 2464–2469 CrossRef CAS PubMed.
- B. T. O'Callahan and P. Z. El-Khoury, J. Phys. Chem. Lett., 2022, 13, 3886–3889 CrossRef PubMed.
- A. B. C. Mantilla, C. F. Wang, Y. Gu, Z. D. Schultz and P. Z. El-Khoury, J. Am. Chem. Soc., 2022, 144, 20561–20565 CrossRef CAS PubMed.
- J. K. Sass, H. Neff, M. Moskovits and S. Holloway, J. Phys. Chem., 1981, 85, 621–623 CrossRef CAS.
- M. Moskovits and D. P. Dilella, J. Chem. Phys., 1982, 77, 1655–1660 CrossRef CAS.
- C. M. Aikens, L. R. Madison and G. C. Schatz, Nat. Photonics, 2013, 7, 508–510 CrossRef CAS.
- K. Ruud, T. Helgaker and P. Bour, J. Phys. Chem. A, 2002, 106, 7448–7455 CrossRef CAS.
- E. M. van Schrojenstein Lantman, T. Deckert-Gaudig, A. J. G. Mank, V. Deckert and B. M. Weckhuysen, Nat. Nanotechnol., 2012, 7, 583–586 CrossRef CAS PubMed.
- C. F. Wang, B. T. O'Callahan, D. Kurouski, A. Krayev and P. Z. El-Khoury, J. Phys. Chem. Lett., 2020, 11, 3809–3814 CrossRef CAS PubMed.
- A. Bhattarai and P. Z. El-Khoury, J. Phys. Chem. Lett., 2019, 10, 2817–2822 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |