
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired complexes confined in well-defined capsules: getting closer to metalloenzyme functionalities
Donglin
Diao
,
A. Jalila
Simaan
 ,
Alexandre
Martinez
and
Cédric
Colomban
*
,
Alexandre
Martinez
and
Cédric
Colomban
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: cedric.colomban@univ-amu.fr
First published on 14th March 2023
Abstract
Reproducing the key features offered by metalloprotein binding cavities is an attractive approach to overcome the main bottlenecks of current open artificial models (in terms of stability, efficiency and selectivity). In this context, this featured article brings together selected examples of recent developments in the field of confined bioinspired complexes with an emphasis on the emerging hemicryptophane caged ligands. In particular, we focused on (1) the strategies allowing the insulation and protection of complexes sharing similarities with metalloprotein active sites, (2) the confinement-induced improvement of catalytic efficiencies and selectivities and (3) very recent efforts that have been made toward the development of bioinspired complexes equipped with weakly binding artificial cavities.
 Donglin Diao | Donglin Diao (middle-right) completed his PhD degree in bioinspired complexes confined in hemicryptophane cages, in 2022, under the guidance of C. Colomban, A. Martinez and A. J. Simaan. Ariane Jalila Simaan (left) obtained her PhD degree in bioinorganic chemistry with J.-J. Girerd and F. Banse, before joining the group of P. Hildebrandt as a postdoc. She is now the CNRS research director at Aix-Marseille University with interest in bioinspired catalysis and enzymatic studies. Alexandre Martinez (middle-left) obtained his PhD degree in asymmetric catalysis with B. Meunier. He then works on supramolecular chemistry with J. Lacour (as Postdoc) and J.-P. Dutasta (as lecturer). He is now a full professor at the Ecole Centrale Marseille with interest in stereochemistry, catalysis, and supramolecular chemistry. Cédric Colomban (right) completed his PhD degree in bioinspired catalysis with A. Sorokin. He undertook postdocs on self-assembled cages with M. Costas and X. Ribas and covalent hosts with A. Martinez. Since 2020, he is a CNRS researcher at University Aix-Marseille, with interest in Bioinspired Confined Catalysis. |
Introduction
Metalloprotein catalysis is particularly inspiring for synthetic chemists because these enzymatic machineries accomplish a broad range of difficult chemical transformations, under mild conditions. Such biological catalysts combine an active site – usually based on bioavailable metal ions (Cu, Fe, Zn, and Mn) supported by natural ligands (O, S and N-donors) – with a three-dimensional enzymatic architecture. The latter provides additional interactions at the secondary (and further) coordination sphere level(s). Within metalloenzymes, remarkable structural tuning of the catalytic efficiency is observed thanks to these cavities (or channels) that guide and orient small substrates to the metal active site, promoting precise selectivity and product release. For instance, it has been experimentally1 and computationally2 evidenced that substrate positioning in SyrB2, WelO5 and BesD halogenases is critical to achieve highly chemo- and regio-selective C–H bond functionalization. This specific adjustment between the substrate and the active-site results from the substrate orientation, thanks to its stabilization by the protein environment (being sensitive to substrate's charge and polarity). These findings suggest that, within the same family of metalloenzymes, active sites and secondary structures might be substrate, enzyme and even reaction specific. The enzymatic pocket is therefore associated with several key structural features that strongly impact metalloprotein catalysis. This allows for the protection of the active site and the control of its nuclearity, stabilization of highly reactive intermediates, substrate positioning and product release.These identified features account for the high specificity, selectivity and efficiency observed with these remarkable catalysts.
Over the past few decades, there has been growing interest for bioinspired chemistry and, in particular, bioinspired catalysis. Since the pioneering work of Breslow3,4 and Kimura,5 metalloprotein mimics based on artificial complexes have attracted considerable attention. These minimalistic metal-based structures aim at reproducing the essential features of an enzyme active site, without recreating the whole protein scaffold. This approach has been widely reported as an efficient strategy to (i) reproduce metalloprotein spectroscopic signatures to help understand the structures and electronic properties of their active sites (structural models) and (ii) mirror enzyme reactivities to both elucidate reaction mechanisms and the nature of metastable intermediates (functional models). The classical bioinspired strategy has first focused on the development of complexes reproducing the first-coordination sphere of one (or more) metal ion(s). These open complexes usually lack stability and cannot fully reproduce the high catalytic efficiency and selectivity of enzymes. In order to demonstrate the core role of the enzymatic hydrophobic binding pocket, artificial complexes displaying second coordination spheres6–8 and/or confined within artificial cavities9 have then been targeted. To better reproduce metalloprotein catalysis, these supramolecular artificial catalysts must provide insulations of both the substrate and the active site from the bulk medium (recognition) and guarantee their preferential arrangement (orientation). The challenge is therefore to reproduce such key features, while keeping a minimalistic structure allowing for straightforward synthesis, high solubility, ease of spectroscopic characterization and recyclability.
Artificial catalysts performing reactions identical to those promoted by enzymes have been extensively reported over the past few years. In several cases, nanomaterials that are unrelated to the principles of biochemistry or enzymatic catalysis have been described as nanozymes. But it has been recently emphasized by the catalytic community that such inappropriate analogies have led to the overuse of the “bioinspired”, “enzyme mimic” and “nanozyme” terms.10,11 To deserve this label, bioinspired models should share some structural characteristics and/or similar mechanisms (active species, substrates, products, and kinetics) with the related enzymes.
In this context, this featured article details recent advances on metal complexes resembling a metalloprotein active site and being confined in a well-defined molecular cage structure. It is designed to reflect the “state-of-the-art” strategies that have been lately employed to (i) confine metalloprotein structural models, (ii) protect reactive active sites and (iii) reach superior catalytic behaviours by the confinement effect. In particular, we summarize our efforts to use hemicryptophane-based complexes as catalytically active confined models with improved efficiency and selectivity. We finally outline our recent progression toward the development of complexes equipped with chiral and functionalized artificial cavities, aiming at getting one step closer to the metalloprotein key structural features. Supramolecular catalysts based on non-biological metals, artificial metalloenzymes or nanomaterial frameworks (MOF) are beyond the scope of this work which focus on discrete caged artificial models.
Discussion
Several strategies could be used to confine artificial complexes to three-dimensional architectures. For instance, complexes could be incorporated in a biological streptavidin scaffold12 or in micelles,13 confined within a self-assembled supramolecular cage by means of host–guest chemistry, or covalently grafted to an organic cage-like unit. In this last category, the metal complex could be situated inside (endo-functionalization) or above the cage structure. For example Costas, Lledó and coworkers reported, in 2018, bio-inspired FeII and MnII complexes based on the bis(pyridyl)dipyrrolidine (pdp) ligand, attached to a resorcinarene cavitand FeII/MnII(1-pdp)(OTf)2.14 This self-folding deep cavitand provides a well-defined hydrophobic cavity in very close proximity to the metal center (Fig. 1). The structure did not allow for a complete endohedral functionalization of the complex, which is bound to the cavitand by a single acetal bridge. In contrast to its parent complex FeII(pdp)(OTf)2, the supramolecular complex displays an iron center in a low spin-state, indicating that the presence of the cavitand modulates the metal electronic properties. Interestingly, the resulting supramolecular complex could activate H2O2 to perform C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond oxidations without suffering from product inhibition. However, no improved selectivities were observed compared to the parent “cavitand-free” complex. On this basis, rigid catalyst–host linkages and/or endohedral functionalization of the active site might be the key to allow selectivity enhancement via substrate recognition at a near-by hydrophobic cavity.
C bond oxidations without suffering from product inhibition. However, no improved selectivities were observed compared to the parent “cavitand-free” complex. On this basis, rigid catalyst–host linkages and/or endohedral functionalization of the active site might be the key to allow selectivity enhancement via substrate recognition at a near-by hydrophobic cavity.
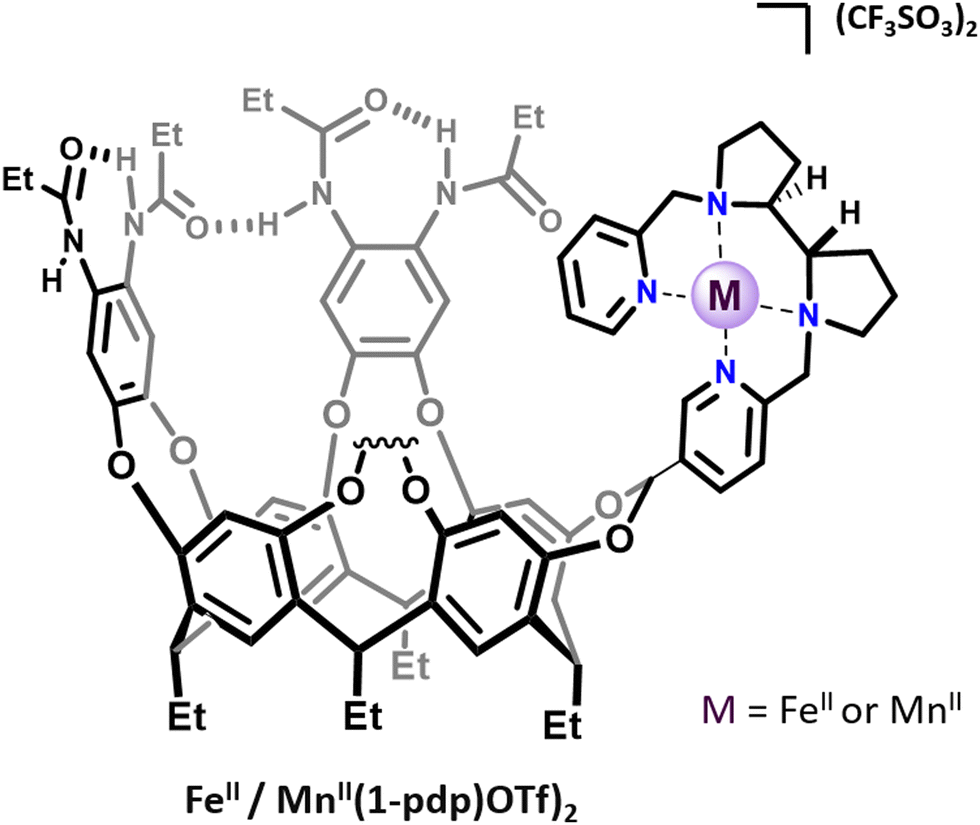 | ||
| Fig. 1 Representation of cavitand-tailored bioinspired complexes FeII(1-pdp)(OTf)2 and MnII(1-pdp)(OTf)2. | ||
The two following sections will focus on the selected examples of bioinspired complexes confined in the interior of either covalent or self-assembled cages.
Imidazole-based complexes confined in covalent cages
The imidazole ligand is particularly interesting as it reproduces the histidine–copper coordination found in many mono-oxygenase enzymes (such as particulate methane monooxygenase (pMMO), lytic polysaccharide monooxygenase (LPMO), and tyrosinase). Examples of ZnII complexes based on cyclodextrins15 or calix[6]arene hosts16 functionalized by imidazole coordinating units have been reported. However, in the case of these open ligands, the metal was not fully encapsulated in a well-defined cavity. It is not before 2019 that a tripodal azacyclophane cage 2, decorated by three imidazole ligands pointing toward its interior, was described by the Otte group (Fig. 2a).17 The design of this organic capsule allows for the coordination of CuI, which is situated in the middle of the cage's cavity. A rare T-shaped geometry was revealed by XRD analysis. Interestingly, this T-shaped CuI coordination mode, with typical Cu-imidazole distances (1.9–2.1 Å), reproduces well the copper-coordination geometry found in some enzymes like LPMOs and pMMO. The authors finally demonstrate that the confined complex CuI(2)(BF4) remains catalytically active. It performs the oxidation of benzyl alcohol substrates into aldehydes, in the presence of TEMPO, under air. However, the cage structure does not result in an improvement of the catalytic efficiency, compared to other open copper-based catalysts. Indeed, CuI(2)(BF4) and the bipyridine-based Cu(I) catalyst developed by Stahl18 display similar efficiency for the oxidation of 4-methoxybenzyl alcohol to 4-anisaldehyde in the presence of N-methylimidazole, with 37% and 39% yields, respectively.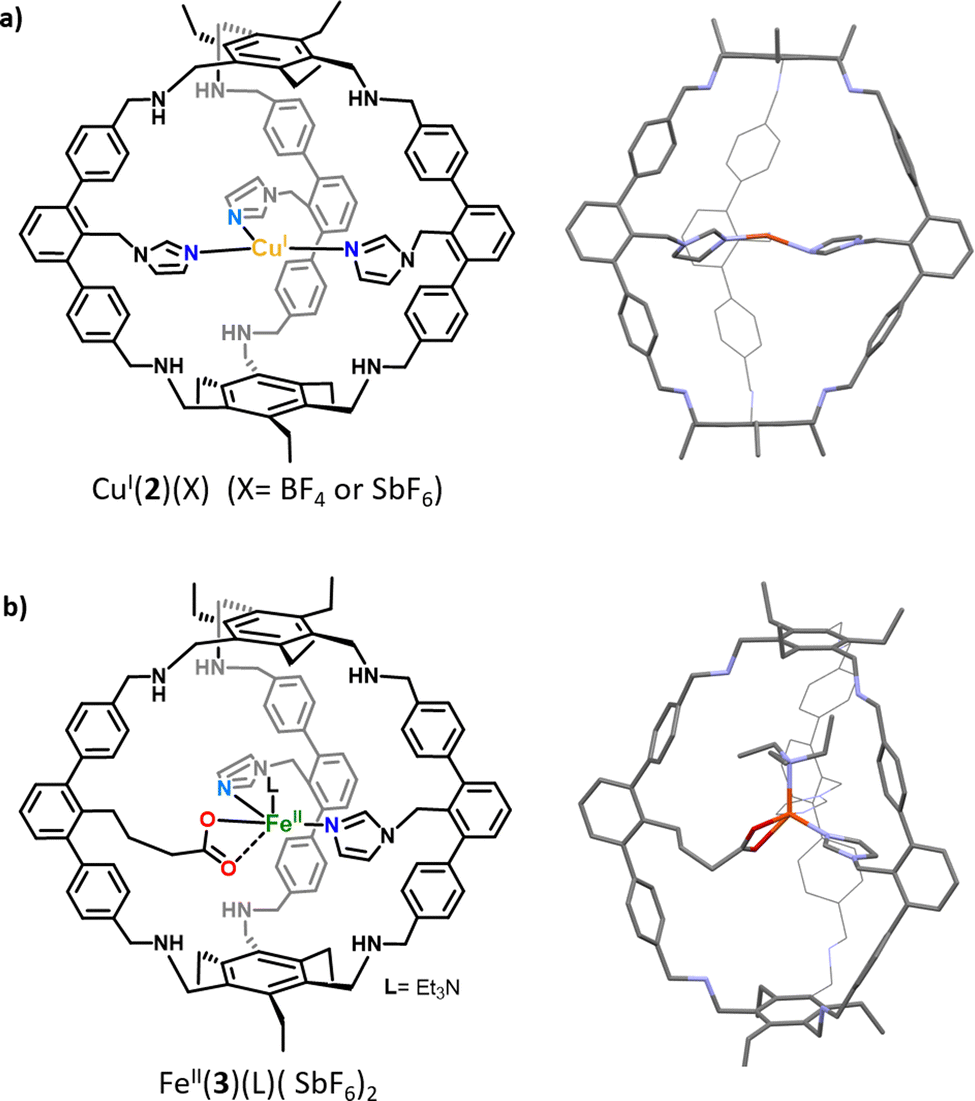 | ||
| Fig. 2 Schematic representations and XRD structures of (a) the confined copper complex CuI(2) and (b) heteroleptic caged iron complex FeII(3). | ||
The same group reported, in 2021, a derivative of 2 in which one of the imidazole arms is replaced by a carboxylate ligand, resulting in cage 3 (Fig. 2b).19 It should be noted that, compared to previously reported cage-ligands which display a high degree of symmetry, 3 provides an heteroleptic ligand environment. Discovering new strategies to decrease the level of symmetry of caged ligands is particularly important in the context of bioinspired chemistry.20 It indeed allows for introducing non-equivalent ligands around the metal core, as it is observed in several metalloproteins. For example, well-known α-ketoglutarate (αKG)-dependent iron oxygenases display a characteristic 2-histidine-1-carboxylate facial triad at their first coordination sphere. In this context, it is worth noting that the covalent cage-based strategy seems particularly appropriate for the construction of heteroleptic assembly. Indeed, in the case of the self-assembly technique, favoring the formation of a mixed-ligand cage (social sorting) over a homoleptic analogue (one kind of ligand, narcissistic self-sorting) is a more challenging task due to the formation of statistical mixtures of both cage complexes, without selectivity.21 The corresponding quasi heteroleptic zinc and iron complexes ZnII(3)(SbF6)2 and FeII(3)(SbF6)2 display an endohedral functionalization provided by the cage structure. This functionalization was unambiguously confirmed by NMR spectroscopy and XRD studies. Importantly, the solid-state structure of FeII(3)(NEt3)(SbF6)2 displays an iron coordination comprising an external molecule of triethylamine (Et3N, crystallisation co-solvent, Fig. 2b), the two imidazole moieties, and the carboxylate unit that binds in an asymmetric bidentate fashion. This carboxylate coordination, although being unusual for mononuclear artificial complexes, has been interestingly observed in some enzyme active sites (e. g. naphthalene dioxygenase NDO).22
In sharp contrast to most of the “open” carboxylate-based iron complexes, which lead to dimers or polymeric structures,23 the caged ligand 3 ensures the sole formation of the targeted monomeric Fe complex. These results highlight the benefits of confining complexes within a cage scaffold that offers protection against dimerization or polymerization reactions. This control of the active site nuclearity, via insulation of the metal core, is also a characteristic feature of metalloproteins. Finally, although FeII(3)(SbF6)2 did not react with dioxygen, it can bind the α-KG cofactor at a low temperature (−80 °C), resulting in an intermediate that reacts upon warming, in the presence of O2. These encouraging early findings are particularly interesting for further uses of FeII(3) as functional mimics of non-heme iron oxygenases. This represents a remarkable achievement since Fe(3) reproduces, for the first time, the 2-histidine-1carboxylate facial triad found in many enzymatic active sites, inside a well-defined cavity.
Very lately, this cage-ligand was also used for Cu coordination at the two-imidazole one-carboxylate ligand, without the formation of polymeric species.24 Both Cu(I) and Cu(II) complexes have been confined. Interestingly, the cage flexibility allows for the formation of Cu-complexes with different coordinating properties. The CuI(3) complex displays a trigonal planar geometry while its oxidized analogue reveals a CuII complex in a highly distorted octahedral geometry. The CuII-coordination comprises the two imidazoles, two external THF molecules and a bidentate carboxylate unit. Upon O2 exposure of CuI(3), oxidation of the cage benzamine backbone into benzimine was observed. Additionally, the EPR spectrum of the CuII(3)(PF6) complex, in the presence of water, displays a resemblance with the one of the subunits of a purified particulate methane monooxygenase enzyme (pMMO).25 According to (i) its two imidazole one carboxylate triad coordinating Cu(I/II) with two different modes, (ii) its oxidizing abilities under aerobic conditions and (iii) its spectroscopic resemblance to the enzyme active site, the CuI/II(3) complex could therefore be considered as a promising artificial model of the pMMO active site. Introducing the 2-imidazole-1-carboxylate coordination triad inside an organic cage therefore appears as a particularly encouraging approach that might lead to a new kind of bioinspired catalysts for highly challenging oxidation of strong C–H bonds via O2 activation.
Bioinspired complexes confined within self-assembled cages
The second main strategy to confine metal-complexes in an artificial cage architecture is based on supramolecular hosts obtained via the self-assembly technique. Compared to the covalent cages, this approach usually requires less synthetic steps and results in more straightforward preparations and purifications. Although remarkable examples of metal-based catalysts confined in self-assembled cages – for improvement of both their catalytic efficiency and selectivity – have been reported over the past decade,26–28 very few bioinspired complexes have been included in self-assembled capsules.A first example of the encapsulation of non-heme ZnII-, CuII- and FeIII- complexes displaying a bis(2-pyridylmethyl)amine (BPA) coordinating unit was reported in 2018 by Costas, Ribas and coworkers.29 The supramolecular host was a palladium-based self-assembled nanocage. It consists of two cofacial Zn–porphyrin units linked by four bridging bis-palladium macrocyclic walls, via Pd–carboxylate bonds. A BPA ligand, tailored with a bipyridine unit acting as an anchor binding both the Zn–porphyrin units of the cage, was designed. It was demonstrated that the bipyridine anchor successfully drives the encapsulation of the complexes, by host–guest interactions. The BPA unit remains available upon encapsulation, to further coordinate Zn, Cu or Fe biometals in the confined space (Fig. 3a).
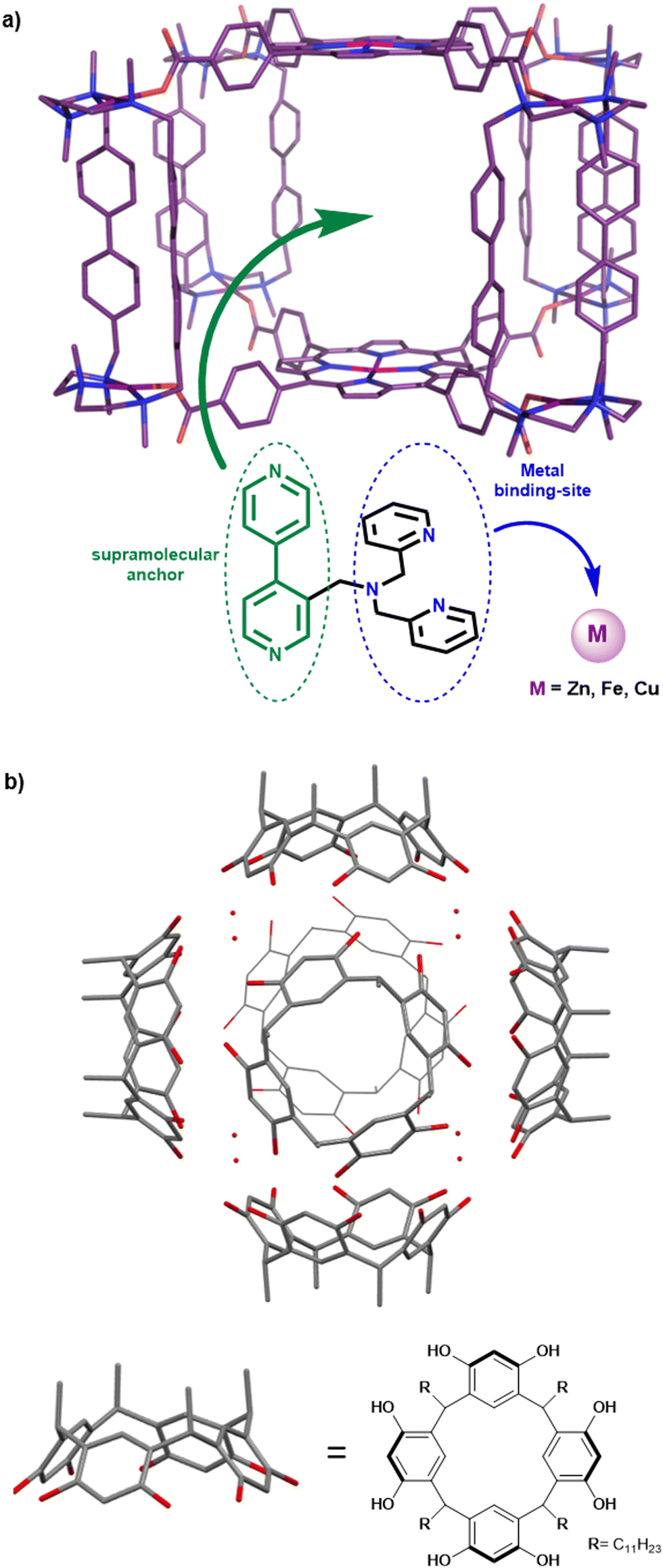 | ||
| Fig. 3 (a) Encapsulation of bioinspired Zn, Fe and Cu complexes within the cavity of a porphyrin-based supramolecular cage, via host–guest interactions. (b) Representation of the hexameric self-assembled resorcinarene capsule used for the encapsulation of Zn(II) and Cu(II) TPA-based complexes. | ||
Recently, a promising approach consisting of the encapsulation of bioinspired complexes within a purely organic self-assembled capsule was reported by Colasson and coworkers.30 This organic supramolecular host is a hexameric resorcinarene cage, previously described as a powerful nanovessel for supramolecular organocatalysis.31,32 It is composed of six resocin[4]arene units and eight water molecules, linked together by a network of sixty hydrogen bonds (Fig. 3b). One key advantage of this capsule is its straightforward preparation that does not require synthetically complex procedures.
Interestingly, in contrast to self-assembled metallacages, this cage is devoid of bridging metal nodes. As a result, it displays a sole metal center that corresponds to the caged bioinspired complexes, resulting in easier characterization. The authors demonstrated that Zn(II) and Cu(II) complexes based on the well-known tris(2-pyridylmethyl)amine (TPA) bioinspired ligand could be efficiently entrapped inside the resorcinarene-based cavity. Encapsulated complexes have been fully characterized by 1H-NMR analysis coupled with docking simulations. Importantly, hydrogen-bond donor groups present in the supramolecular assembly (eight water molecules) were found to interact with the azido ligand of an encapsulated CuII(TPA)(N3) complex. Studying confined azidocopper(II) adducts is particularly interesting because they are known as structural and electronic analogues of the end-on superoxo intermediate (CuIIO2˙−) found in several copper monooxygenases. Its stabilization, by the second coordination sphere offered by the cage, is therefore particularly encouraging toward further applications of such Cu(I) confined bioinspired complexes as O2 activating model catalysts.
Confined Cu(I) complexes as enhanced O2 activating catalysts
In biology, vital oxidation and oxygenation reactions are performed by O2-activating metalloenzymes. Copper- or iron-containing oxidase and oxygenase are indeed able to perform a challenging reaction (e. g. CH4 oxidation) by generating highly reactive metastable intermediates arising from the activation of O2. In this context, the enzyme cavity is essential to stabilize these active species and avoid undesired side reactions such as the thermodynamically favored formation of O-bridged dimers. A typical example of this key nuclearity control is the stabilization of CuII(O2˙−) intermediates by the enzyme cavity of Cu-based oxygenases and oxidases.33 Indeed, in the absence of steric protection, the unstable mononuclear cupric superoxide species is easily converted to peroxo-bridged dicopper(II) complexes. As a consequence, O2 activation in open artificial CuI(TREN) or CuI(TPA) models usually results in the formation of less reactive [(Cu(L))2(O2)] peroxo dimers (Fig. 4).34–36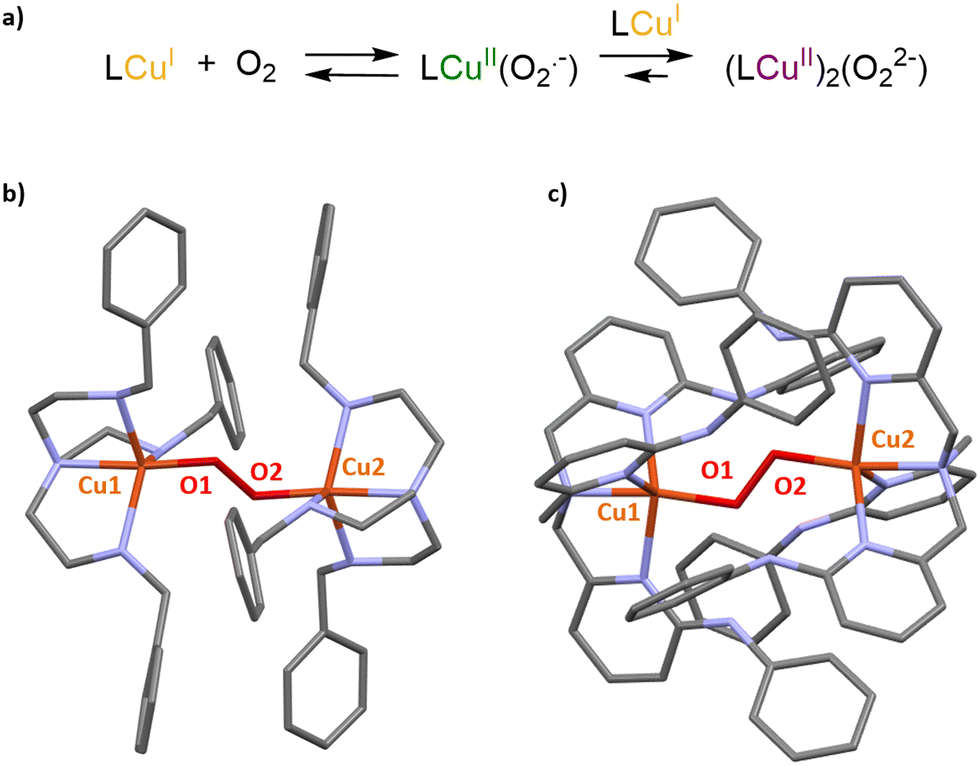 | ||
| Fig. 4 (a) Formation of primary and secondary O2 adducts arising from O2 activation in open Cu(I) complexes. XRD structures of [(Cu(L))2(O2)] peroxo dimers observed upon O2 activation in (b) CuI(TREN) and (c) CuI(TPA) derivatives. | ||
This undesired dimerization could be avoided by using (i) very low temperatures,37,38 (ii) open Cu(I) ligands with significant steric shields,39 and (iii) caged bioinspired Cu(I) complexes.
It is in this last category that Reinaud et al. have reported calix[6]arene-caged Cu(I) complexes for O2 activation studies.40
CuI(TREN) and CuI(TPA) units have been connected to the calix[6]arene cap, via methylene bridges, to form the caged complexes CuI(4)41 and CuI(5)42, respectively (Fig. 5).
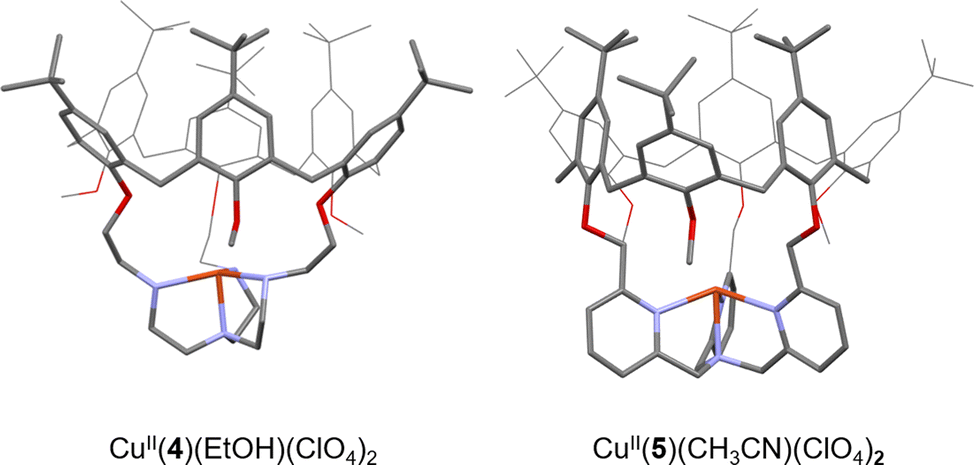 | ||
| Fig. 5 Views of the XRD structures of CuII complexes based on the 4 and 5 calix[6]arene-based ligands. Counter ions and bounded molecules of solvent (EtOH and CH3CN, respectively) have been omitted for clarity. | ||
Insulation of the Cu core inside the cage ligands 3 and 4 interestingly prevents the formation of thermodynamically favored peroxo dimers. Instead, it was demonstrated that CuI(4) and CuI(5) could activate O2 to perform intramolecular oxidation of a methylene –CH2– linker. This four-electron oxidation is mediated by the cupric superoxide intermediate CuII(4/5)(O2˙−), arising from O2 activation. This results in stable CuI complexes with a mono-oxidized ligand 4O or 5O displaying an ester moiety (Fig. 6). It should be noted that the TPA-based complex CuI(5) was less reactive than its TREN analogue. Indeed, CuI(5) solely activates O2 when used in the solid state. This behavior could be explained by the presence of an encapsulated solvent molecule within the cavity of 5, preventing the reaction of the CuI center with O2, in solution.
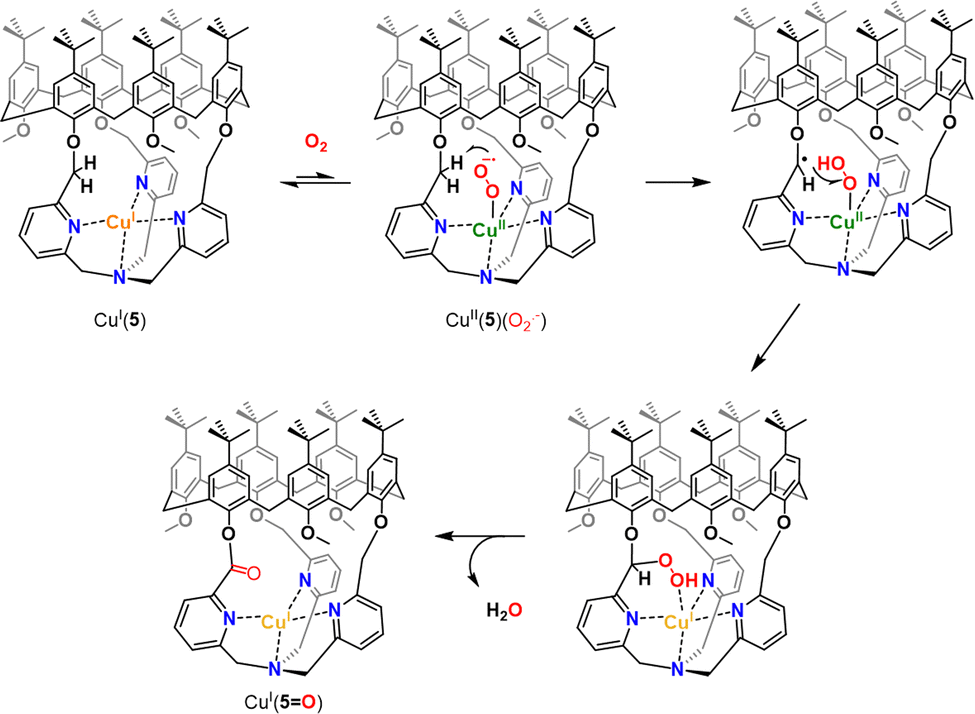 | ||
| Fig. 6 Proposed mechanism for intramolecular C–H bond oxidation upon O2 activation in CuI(5). A similar pathway is proposed for CuI(4). | ||
These results clearly indicate that isolating highly reactive intermediates, within well-defined capsules, could be an efficient strategy to reach other reactivity than the one observed in the bulk solution. In these cases, confining the biorelevant CuII(O2˙−) intermediate to the profit of an O2-mediated C–H oxidation reaction prevents the formation of peroxo dimers. However, this reactivity was limited to intramolecular oxidations. The development of caged O2 activating catalysts, able to perform C–H oxidation on external substrates, with several turnover numbers is therefore still needed.
It should be noted that controlling the active site nuclearity via cage-like ligands has also been used to achieve the activation of another important gas: dinitrogen (N2). Small tripodal cages of the cyclophane-type, displaying two or three identical ligands, have been used to respectively template the formation of bi-43 and trinuclear44 iron clusters. The caged-ligands do not only account for the formation of multinuclear complexes, but also impose specific steric constraints and N2-coordination modes that allow for unprecedented examples of the cleavage of the N2 triple bond in multimetallic complexes.
Catalytically active caged models with improved efficiencies
Hydrogenase-inspired catalysts represent an interesting class of artificial models that have been confined to artificial cages to improve their efficiency. The first attempt of confining small models of the [FeFe]-hydrogenase active site was reported in 2010. Two β-CD cyclodextrins, assembled via a sodium bridge, were used as the host in (Fe2S2)(β-CD)2Na. In this system, the typical Fe2S2 cluster was functionalized at its S-to-S linker by an aryl sulfonate unit. This anchoring group was used to further include the cluster in the bis-cyclodextrin hydrophobic channel (Fig. 7a.). However, the confined model resulted in a less efficient proton reduction, which was attributed to the architecture rigidity that hampers the essential rearrangement of the [Fe–Fe] core.45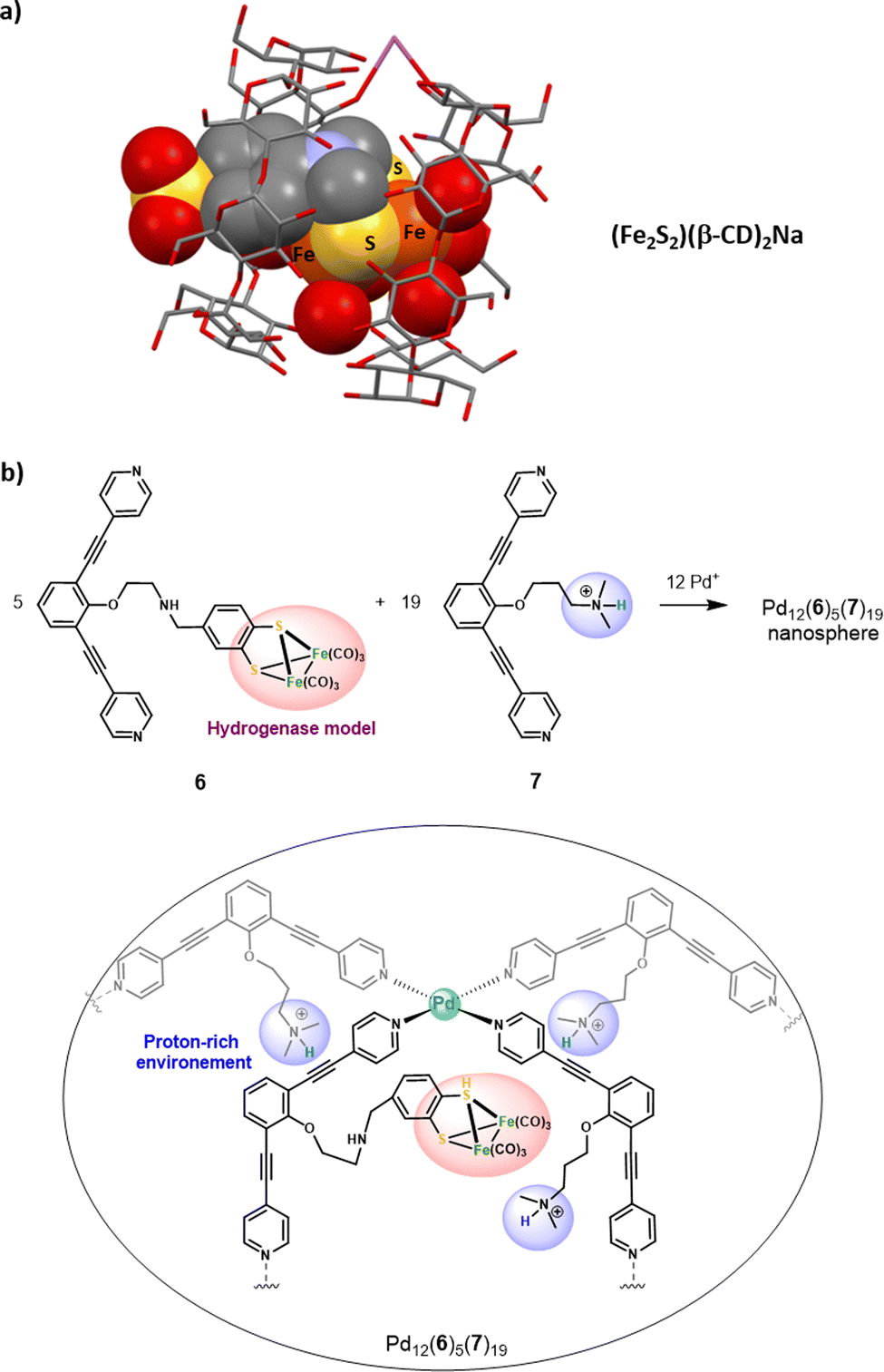 | ||
| Fig. 7 (a) Representation of the XRD structure of (Fe2S2)(β-CD)2Na. (b) Confinement of five hydrogenase mimicking clusters in a proton reach the Pd12(6)5(7)19 nanosphere (top), along with the schematic representation of one of the five catalytic centers found in the nanosphere (bottom). | ||
More recently, Reek and co-workers have extended their initial work on the encapsulation of single hydrogenase mimics in a metallacage aiming at accessing lower overoxidation potentials46 toward larger metal-based nanospheres that could incorporate multiple hydrogenase active sites.47 The M12L24 Fujita-type nanosphere Pd12(6)5(7)19 was used to confine five [Fe2S2] clusters in an ammonium-rich nano-environment. This large metallacage is based on two different bis(pyridyl) ditopic ligands bridged with Pd metal nodes, offering a very wide cavity (5 nm diameter). It arises from the combination of ligands 6 and 7, being respectively appended with a [Fe2S2] cluster and an ammonium unit (Fig. 7b). Inside the nanosphere cavity, the five diiron catalysts are therefore surrounded by a proton-rich environment provided by the nineteen ligand 7. Interestingly, this particular supramolecular environment results in enhanced electrocatalytic proton reduction performances. This positive confinement effect was attributed to the conjunction of two factors: (i) insulation of the catalyst that reduces the catalytic overpotential and (ii) preorganization of the proton substrate and intermediate stabilization, arising from the co-confinement of the ammonium salts. This remarkable enhancement of catalytic efficiency underlines the benefits of combining cage-like hosts with a functionalized second coordination sphere. On this basis, decorated cavities reproducing the key amino-acid residues found around the enzyme active site appear as a key feature for future progression in the field of bioinspired catalysis.
Among the different kinds of covalent cage-ligands, the hemicryptophane-type, which is developed in our team, has recently emerged as a useful scaffold for catalytically active bioinspired complexes. Hemicryptophanes are organic covalent cages built from a northern cyclotriveratrilène (CTV) unit and connected to another C3 symmetrical moiety by three linkers. Interestingly, we found that these hosts display a remarkable rigidity-flexibility balance allowing their use as supramolecular catalysts that usually do not suffer from inhibition by the product.48 Hemicryptophanes possess a hydrophobic cavity that has been used for the improvement of the efficiency of caged organocatalysts.49,50 In particular, these structures take advantage of (i) their flexibility allowing product ejection, (ii), their hydrophobic cavity for substrate recognition and (iii) their chiral CTV cap for building enantiopure hosts. On this basis, we reasoned that hemicryptophanes might be an interesting platform to confine bioinspired catalysts inside a hydrophobic cavity. To achieve this goal, tripodal bioinspired ligands have been incorporated at the southern part of the edifice and the corresponding complexes have been studied as confined catalysts.
Martinez and coworkers have investigated the enzyme-like kinetic behavior of the oxido-vanadium(V)-based catalyst confined in hemicryptophane displaying binaphthol linkers (OVv)(8) (Fig. 8).51 (OVv)(8) has been studied as a catalyst for sulfide to sulfoxide oxidation by the activation of the cumene hydroperoxide oxidant (CHP). The strong impact of its hydrophobic cavity was evidenced by the observation of a major improvement of the catalytic efficiency, with up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 turnovers. Indeed, a 33-fold faster reaction rate was observed for the caged catalyst, compared to its open analogue devoid of the CTV cap (OVv)(TKA). This confinement effect was explained by efficient hosting of the thioanisol substrate and realising of the oxidized product from the cavity. Remarkably, kinetic studies revealed that the oxidation reaction catalysed by (OVv)(8) obeys the Michaelis–Menten kinetic model enzymatic reactions. Furthermore, inhibition of the reaction was observed in the presence of the tetramethylammonium Me4N+ competitive guest which displays strong affinity for the cage cavity thanks to cation–π interactions with the CTV cap (Fig. 8). It should be noted that Michaelis–Menten kinetic behaviour and reaction competitive-inhibition are two typical features of enzymes that are very rarely observed with artificial catalysts. Clearly, equipping metal-based oxidation catalysts with the appropriate hydrophobic cavity is a strong asset to get one step closer to the catalytic behaviour of metalloproteins.
000 turnovers. Indeed, a 33-fold faster reaction rate was observed for the caged catalyst, compared to its open analogue devoid of the CTV cap (OVv)(TKA). This confinement effect was explained by efficient hosting of the thioanisol substrate and realising of the oxidized product from the cavity. Remarkably, kinetic studies revealed that the oxidation reaction catalysed by (OVv)(8) obeys the Michaelis–Menten kinetic model enzymatic reactions. Furthermore, inhibition of the reaction was observed in the presence of the tetramethylammonium Me4N+ competitive guest which displays strong affinity for the cage cavity thanks to cation–π interactions with the CTV cap (Fig. 8). It should be noted that Michaelis–Menten kinetic behaviour and reaction competitive-inhibition are two typical features of enzymes that are very rarely observed with artificial catalysts. Clearly, equipping metal-based oxidation catalysts with the appropriate hydrophobic cavity is a strong asset to get one step closer to the catalytic behaviour of metalloproteins.
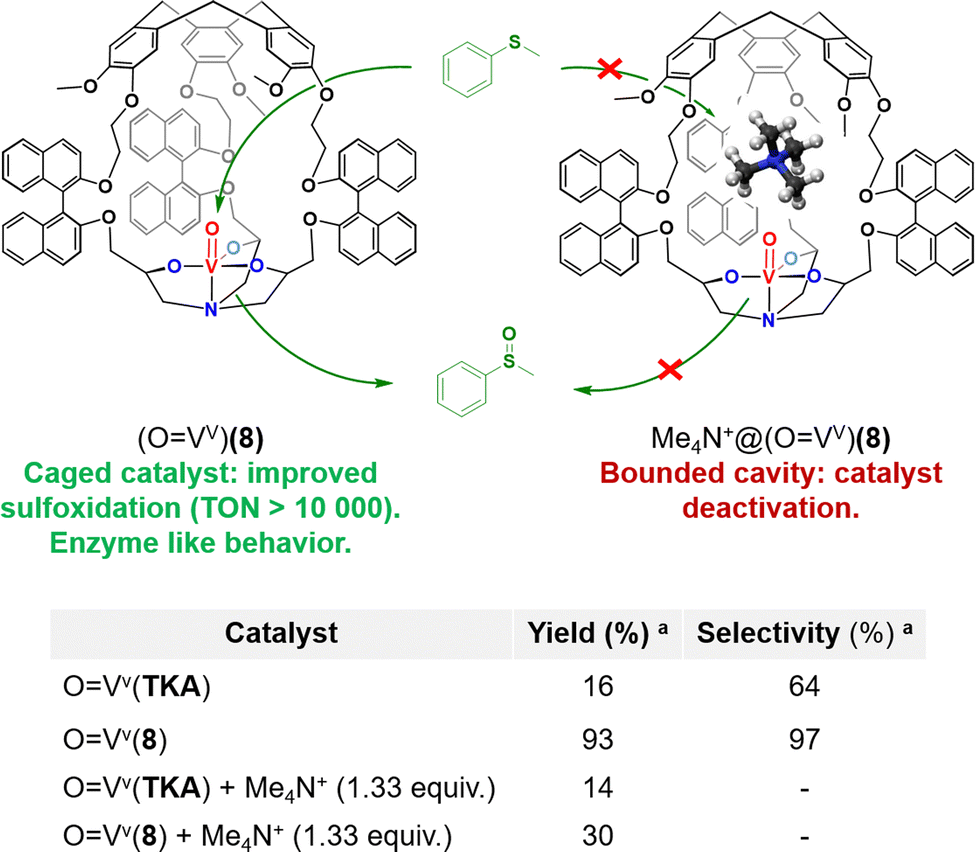 | ||
| Fig. 8 (Top) Representation of the efficient sulfoxidation observed in caged oxido-vanadium catalyst (OVv)(8) (left), and catalyst inhibition observed upon tetramethylammonium binding in the catalyst cavity (right). (Bottom) Comparison with its open analogue devoid of the CTV cap (OVv)(TKA).aOxidation of thioanisol in the presence of the catalyst (1.5 mol%) and CHP (1.0 equiv.) for 180 min in CH2Cl2 at 0 °C. | ||
Improved selectivity by the release of primary oxidation products
Enhancing the selectivity of artificial catalysts, by creating a constrained reaction site, is highly interesting. For example, it was recently demonstrated that equipping an iron porphyrin catalyst with a glycoluril-based cage structure hosting potassium ions52 results in a large selectivity improvement, in CO2 to CO reduction catalysis.53 This strategy is also particularly attractive to overcome one of the major issues of bioinspired oxidations: controlling the selectivity toward primary oxidation products. Indeed, classical open catalysts usually lack selectivity when applied to the oxidation of very strong C–H bonds. In particular, this is a major problem of the direct methane to methanol oxidation. The exceptional stability of CH4 bonds indeed requires harsh oxidative conditions that lead to overoxidation of the primary oxidation product (CH3OH) to undesired formic acid and even CO2 products.54,55 In this context, the bioinspired approach is highly interesting since, in nature, soluble (sMMO) and particulate (pMMO) methane monooxygenases selectively convert CH4 to CH3OH, by using confined iron and copper metal ions.These enzymes take advantages of their constrained hydrophobic environments that promote methanol release, preventing further overoxidation. By connecting a CTV cap with the tripodal pyridine-based ligand TPA, via phenyl linkers, we have prepared the caged bio-inspired Cu and Fe complexes CuII/FeII(Hm-Ph-TPA) (Fig. 9). These H2O2-activating catalysts have been further applied in C–H bond oxidation reactions with enhanced selectivity. Inspired by the superior efficiency of copper(II) hemicryptophane catalysts in the oxidation of cyclohexane C–H bonds (via H2O2 activation),56 we indeed reasoned that such caged catalysts could also favour methanol ejection from the cage cavity, preventing overoxidation. These complexes were supported onto silica and studied as heterogeneous H2O2-activating catalysts for CH4 to CH3OH oxidation, in water. We interestingly demonstrated that equipping the iron complex Fe(TPA), with the hemicryptophane cavity, indeed limits CH3OH overoxidation.57 The FeII(Hm-Ph-TPA) catalyst displayed a 4-fold increase of the yield in mono-oxidized products compared to its “open” analogue. Such a confinement effect was explained by the preferential binding of the hydrophobic CH4 and release of the more polar CH3OH to the bulk water solvent.
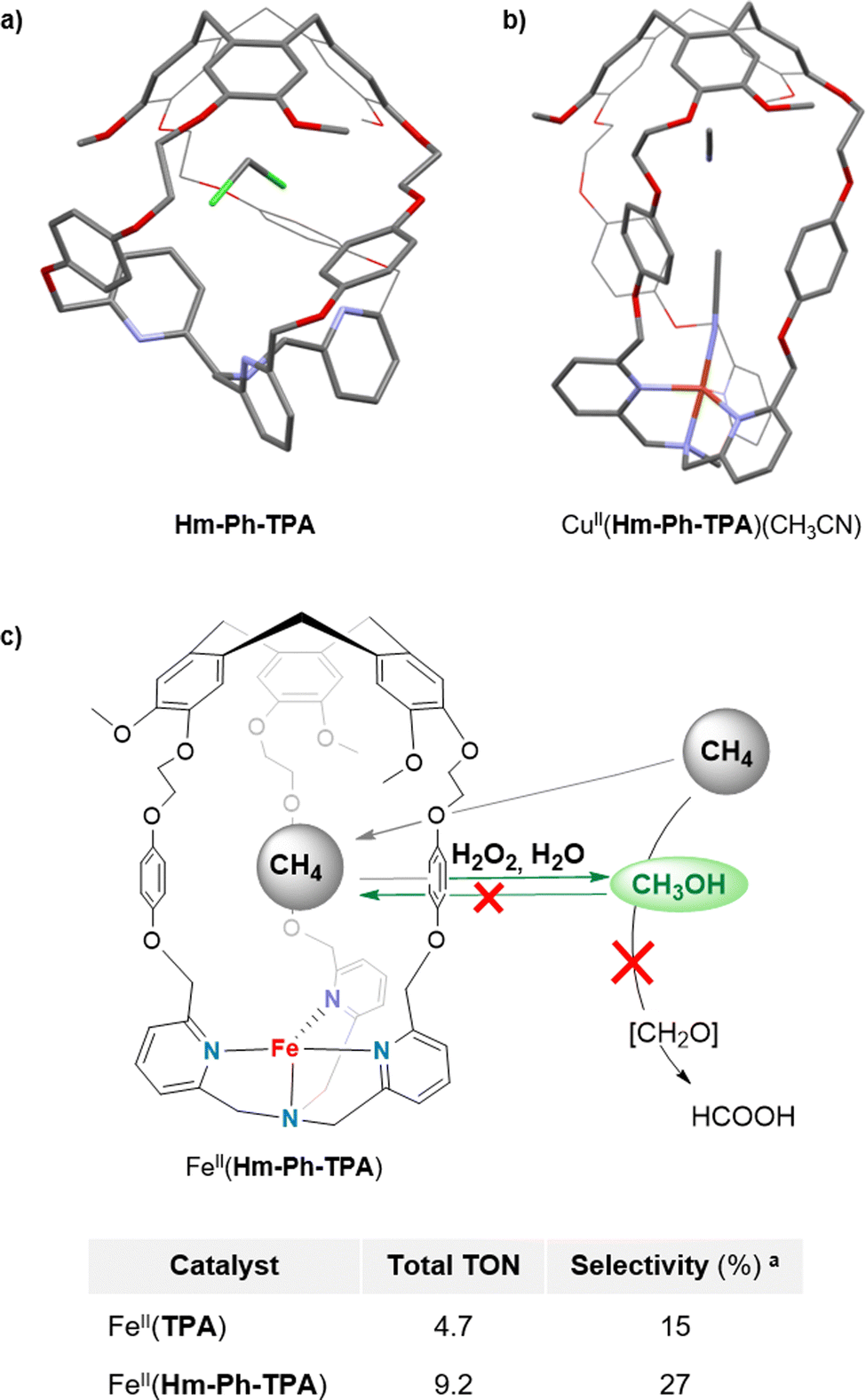 | ||
| Fig. 9 Representation of the XRD structures of (a) Hm-Ph-TPA and (b) CuII(Hm-Ph-TPA). (c) Concept of the selective methane to methanol oxidation via H2O2 activation in FeII(Hm-Ph-TPA), in water along with the comparison table between the open Fe(TPA) and the caged catalysts. a Selectivity toward primary oxidation products CH3OH and CH3OOH. | ||
Although these results reveal the benefits of the endohedral hydrophobic cavity in terms of selectivity, very low TON (<10) was observed. Therefore, pursuing these studies toward caged catalysts displaying more reactive active sites appears as an encouraging perspective to achieve both efficient and selective CH4 to CH3OH conversions.
Reaction in complex media
A key structural feature of metalloproteins is their ability to catalyse specific reactions within the complex in vivo media. For example, several copper-containing enzymes use CuI centers to activate O2, without suffering from the competition of external biological CuI–ligands such as biothiols (like gluthathione or cysteine). Interactions between these biothiols and the CuI center are indeed prevented by having the metal core deeply buried inside the metalloprotein scaffold. Artificial CuI-catalysts for in vivo chemistry have recently attracted considerable attention. Reported examples aimed at achieving (i) reactive oxygen species (ROS) production in CuI/CuII-cycling complexes for anticancer drug development58,59 or (ii) Cu-catalyzed azide-alkyne cycloaddition (CuAAC) bioconjugation reactions.60 However, the development of catalysts that remain active in the complex mixtures found in living media (containing strong CuI biological chelators) remains highly challenging. Indeed, deactivation of open Cu-catalysts via copper-coordination by external nucleophiles is the main explanation for their usual scarce in vivo efficiency. For example, CuI coordination by the gluthathione reduced (GSH) tripeptide is considered as the main event preventing CuAAC bioconjugation reactions in living cells.61On this basis, we wondered if building an organic hemicryptophane cage around the widely used CuI(TBTA) CuAAC catalysts (TBTA = tris(benzyltriaolemethyl)amine) could result in an efficient protection of the CuI core. We therefore reported, in 2021, the first covalent caging of the canonical TBTA ligand, using the hemicryptophane cage Hm-TBTA.62 To evaluate the benefits of the cage-shielded structure, the catalytic behaviours of the open CuI(TBTA) and caged CuI(Hm-TBTA) catalysts, in the presence of external bulky Cu–ligands, were compared (Fig. 10). Both catalysts were engaged in a typical CuAAC benchmark reaction. We first demonstrated that the flexibility of the caged ligand allows for product ejection from its cavity. Indeed, triazole products were not blocked within the cage and catalytic transformations were observed with 99 TON. This behaviour contrasts with other confined CuAAC CuI-catalysts based on the cucurbit[7]uril host, which displays a complete suppression of the catalytic behavior,63 and highlight the benefits of the hemicryptophane structure. Remarkably, the confined catalyst was tolerant to the presence of the notorious gluthathione reduce inhibitor.
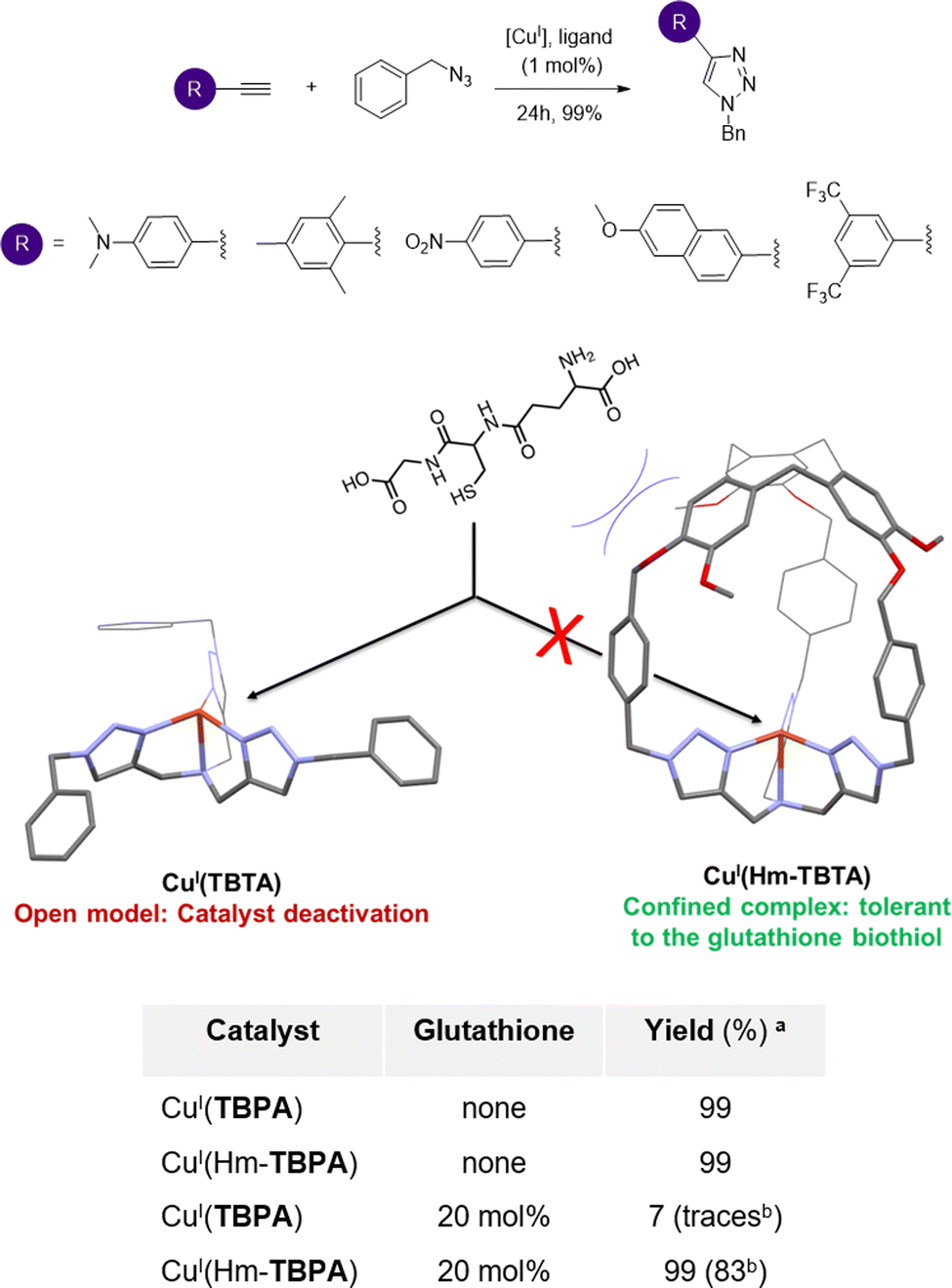 | ||
| Fig. 10 Typical CuAAC reactions catalysed by CuI(Hm-TBTA) with quantitative yields (top). The CuAAC reaction in the presence of the gluthathione biothiol: no catalyst inhibition was observed in the case of the caged complex, in contrast to the classical open CuAAC catalyst (bottom). a Catalyst (1 mol%), MeOH/CH2Cl2 1:10, 25 °C, and argon atmosphere.b Isolated yields. | ||
Indeed, the catalytic efficiency of CuI(Hm-TBTA) remained unchanged even in the presence of 20 equivalents of GSH. Under identical conditions, the catalytic efficiency of the canonical open complex CuI(TBTA) was totally suppressed. Moreover, it was previously reported that the addition of only 2 equivalents of GSH was sufficient to inhibit the catalytic activity of common tris(triazole)-based CuAAC catalysts.64 This behaviour also highlights the benefits of the hemicryptophane flexibility–rigidity balance, since Cu-catalysts encapsulated in azacryptand caged ligands were found to be too structurally flexible to efficiently prevent the GSH-induced deactivation of the confined catalysts.65
Clearly, confining the CuI active center inside a CTV-based organic cage offers an efficient protection of the active core against its deactivation by the abundant biological Cu(I) ligand, GSH. This strategy, inspired by Cu-containing metalloprotein pockets, is therefore encouraging for the future forging of artificial metal catalysts efficiently operating in complex biological media.
Toward chiral and functionalized artificial cavities
Stereoselective recognitions of natural substrates are particularly important events for several natural biochemical catalytic processes. In order to reproduce these selective bindings, the preparation of chiral artificial cages has recently attracted considerable attention.66 In this context, the hemicryptophane structure is particularly interesting due to the inherent chirality of the CTV cap, which displays a M or P configuration.67,68 Our team has developed remarkable preparation and/or purification techniques (e. g.chiral HPLC resolution of enantiomers), allowing for the efficient preparation of enantiopure hemicryptophane cages on the gram scale.69 These hosts could be considered as promising ligands for the preparation of both caged and enantiopure bioinspired complexes.70We therefore wondered: could the chirality of the northern CTV cap be transferred to a southern bioinspired ligand? To answer this question, we have designed hemicryptophane Hm-CH2-TPA, displaying a southern tris(2-pyridylmethyl)amine (TPA) ligand and a CTV unit, linked in close proximity by short methylene –CH2– spacers. We investigated if a helical arrangement of the TPA could be induced and controlled by the CTV cap, in this structurally contracted cage (Fig. 11). Indeed, controlling the interconversion of the TPA's pyridines could lead to a propeller-like arrangement of the ligand, which becomes a chiral coordinating unit. We were pleased to observe that such propagation of the CTV chirality could indeed be achieved in Hm-CH2-TPA. The CTV unit was found to control the ligand helicity, with the P-CTV imposing a right-handed propeller arrangement of the TPA's pyridines (while the M-CTV provides a left-handed arrangement).
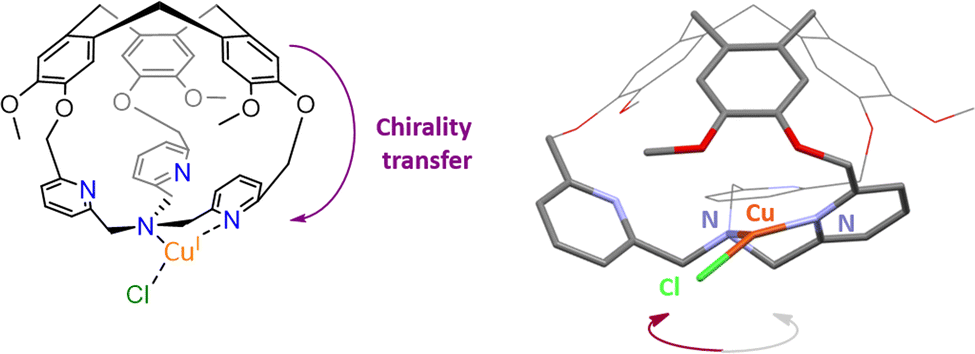 | ||
| Fig. 11 Schematic representation of the chirality transfer observed in M-CuI(Hm-CH2-TPA)(Cl) (left), along with view of its XRD structure (right). | ||
Importantly, this chirality transfer remains intact in the corresponding CuI complex M-CuI(Hm-CH2-TPA)(Cl). XRD, 1H-NMR spectroscopy, ECD analysis and DFT calculations confirm the formation of a highly unusual T-shaped TPA-CuI core with a controlled helicity.71,72 Connecting tripodal ligands with CTV-based hosts could be therefore used to reach both optically pure and caged bioinspired complexes that might find interesting applications in enantioselective catalysis.
Another major structural feature of metalloenzymes is their binding cavities that guide the reactivity and selectivity of reactions via (i) substrate positioning and (ii) stabilization of extremely reactive intermediates. Several non-covalent second coordination sphere interactions exist in their hydrophobic channels (electrostatic or hydrogen-bonding interactions, salt-bridges and long-ranges charge-effects). Such weak binding events enable high chemo-, regio- and stereoselectivity by finely positioning the substrate. This behaviour has been demonstrated for both heme73 and non-heme iron oxygenases.74 Particularly important H-bonding interactions are also found in O2 activating copper-containing metalloproteins. In these systems, H-bonding could account for the stabilization of highly reactive intermediates. The strong impact of bonding second coordination spheres has also been demonstrated with tailored open artificial catalysts.75 For instance, Karlin and Solomon reported efficient stabilization of end-on superoxo CuII(TPA)(O2˙−) intermediates by introducing the H-bonding group at the second coordination sphere of the canonical TPA ligand.76–78 Furthermore, highly precise and predictable C–H oxidation reactions, enabled by substrate positioning at crown-ether tailored Mn or Fe bioinspired catalysts, were reported by Costas, Olivo and Di Stephano.79–81 However, despite both progresses in the development of (i) caged bioinspired complexes and (ii) open models displaying the functional second coordination sphere, the preparation of bioinspired complexes combining a hydrophobic cavity with weak binding units has rarely been explored.82 For instance, the impact of the functionalized second coordination sphere on metal ion lability and host–guest binding was very lately evidenced using confined Zn(II) complexes displaying a calix[6]arene-based cavity decorated with phenol or quinone units.83 To better reproduce metalloprotein key structural features, bioinspired catalysts able to precisely orientate substrates inside an artificial cavity will represent a major breakthrough in the field.
Our first efforts toward the construction of such kind of caged complexes involved the building of a TPA-ligand surmounted with a triazole-decorated cavity. We reasoned that triazoles could represent an attractive building block since it is a convenient connecting unit to covalently link the north (CTV) and south (ligand) parts of a cage via a well-known “click” CuAAC reaction. Additionally, triazoles could be used as H-bond donor moieties. Ctriazole–H are indeed good H-bond donors that have been, for instance, incorporated in the most efficient chloride-binding covalent cage to date.84 We therefore design Hm-TriA-TPA, where the bioinspired TPA ligand is connected to the hemicryptophane's CTV by three triazole links (Fig. 12a). This cage was the first TPA-based ligand surmounted with a Ctriazole–H H-bonding cavity.85 The structure of the corresponding zinc complex ZnII(Hm-TriA-TPA) was elucidated by means of 1H-NMR spectroscopy and DFT studies. An endohedral functionalization of the ZnII(TPA) complex, with the three triazole units remaining available for further H-bonding events, was observed (Fig. 12b). The XRD structure of a cage complex was elucidated by replacing zinc with a copper(II) metal ion in CuII(Hm-TriA-TPA). In this case, additional CuII coordination, by one triazole unit, was observed in the solid state. Interestingly, this Cu-triazole bond could be replaced by coordination of an azide anion at the CuII(TPA) core, liberating the triazole unit (Fig. 12c). As mentioned earlier, TPA-based azidocopper(II) adducts are stable structural analogues of the biological end-on CuIIO2˙− intermediates. They are therefore convenient probes to investigate putative second coordination sphere stabilization via H-bonding. The electronic and vibrational properties of CuII(Hm-TriA-TPA)(N3) have thus been compared to that of the analogue CuII(Me3TPA)(N3−) adduct devoid of H-bonding units. Remarkably, UVvis and IR analyses reveal respectively a typical blue shift of the azido to CuII LMCT and an increase of the ν(N–N) stretching frequency, accounting for the stabilization of the azido adduct by the Ctriazole–H cavity.30,76–78
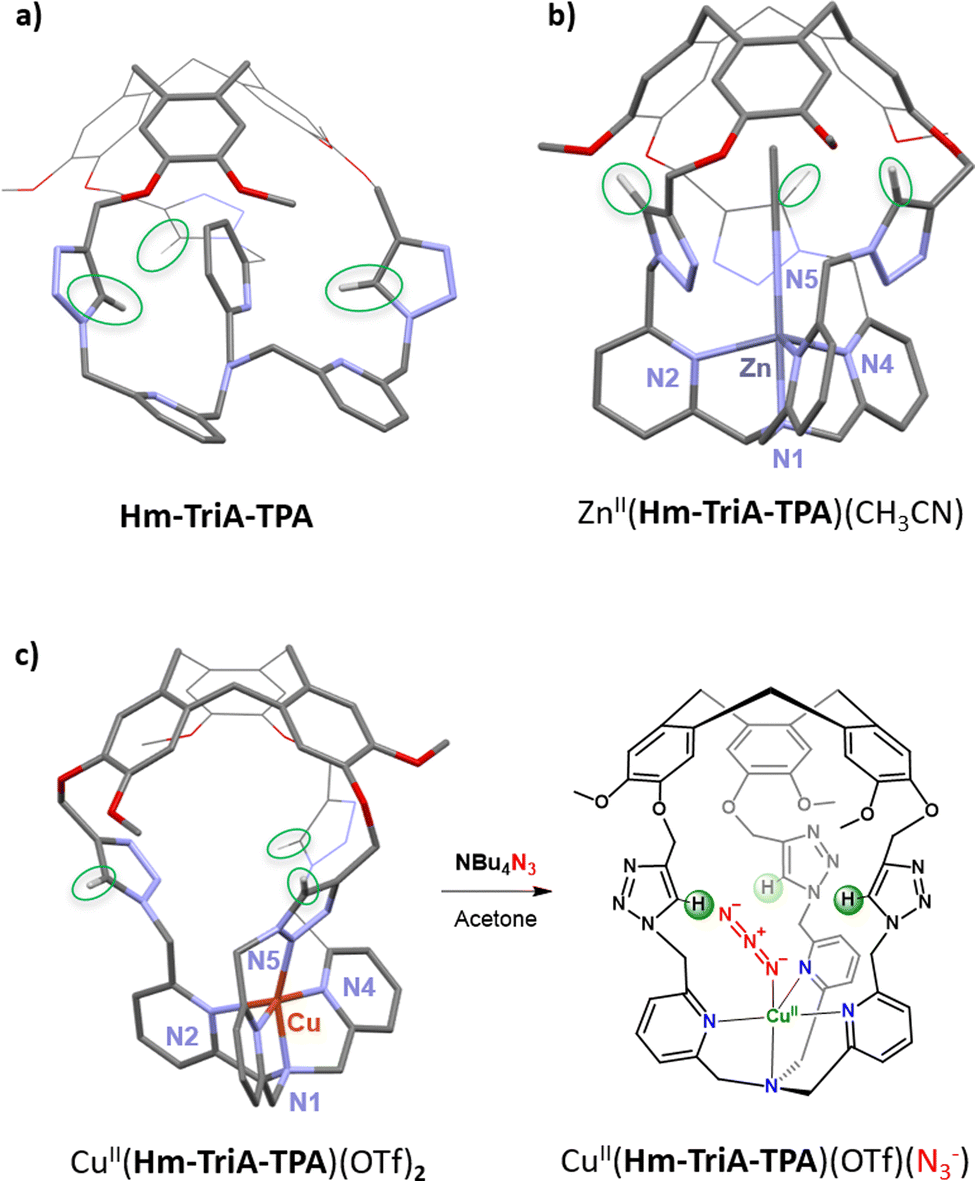 | ||
| Fig. 12 Views of the DFT structures of (a) the cage-ligand Hm-TriA-TPA and (b) its corresponding ZnII complex. (c) View of the XRD structure of CuII(Hm-TriA-TPA)(OTf)2 along with formation of the corresponding azido adduct upon addition of tetrabutylammonium azide. | ||
We were able to show that connecting both parts of a ditopic cage, via triazole linkers, can provide an easy route for confining bioinspired complexes in an H-bonding cavity. In addition to an endohedral functionalisation of the metal core, we confirmed that the Ctriazole–H bonds, found in the cavity, could engage H-bonding interactions at the second coordination sphere level. This approach therefore opens the way for further artificial catalysts displaying H-bonding cavities, allowing the stabilization of reactive metastable intermediates and/or substrate positioning.
Conclusions
With the development of both bioinspired catalysis and supramolecular chemistry, a series of well-defined synthetic cage structures have been designed to host enzyme-related artificial complexes. In this featured article, we have selected examples of caged complexes reflecting recent efforts in the field of bioinspired catalysis in a confined space. We have especially emphasized the confined catalysts based on the emerging hemicryptophane ligands developed in our group. All these systems have been designed to artificially reproduce some key features offered by the enzyme pockets, which have been clearly identified as follows.(1) Active site protection and reaction in complex media. The active metal core is usually deeply buried inside the enzyme architecture. This confers protection, in particular against intermolecular reactions between two complexes, resulting in the undesired formation of polynuclear species or degradation events. This insulation from the bulk solution also protects the metal core from its deactivation by external metal chelators found in the complex in vivo media at high concentrations (like biothiols). The ability of organic cage-complexes to avoid the formation of polynuclear O-bridged compounds has been exploited to reproduce (i) the reactivity and (ii) the 2-histidine-1-carboxilate facial triad of important O2 activating enzymes. Although the key role of the cage structure was clearly evidenced, O2 activation at caged bioinspired catalysts usually results in an intramolecular oxidation of the ligand. Developing new caged catalysts that are able to guide this reactivity toward the oxidation of external substrates with strong C–H bonds could therefore be considered as an important future direction. On the other side, hemicryptophane-based complexes have proven useful to obtain catalytically active catalysts operating in the presence of biological strong metal chelators. We therefore believe that this finding opens opportunities to develop artificial catalysts working in living cells for in vivo applications (bioconjugation reactions or drug development).
(2) Hydrophobic cavities for the improvement of the catalytic behavior and selectivity. Besides offering protection, the hydrophobic pocket of enzymes also allows for recognition events leading to highly efficient and selective transformations. Reactions performed by metalloenzymes indeed strongly benefit from the recognition of a specific substrate accompanied with a promoted product-release. Obtaining caged bioinspired catalysts that remain catalytically active is not an easy task. These systems should be robust enough to avoid cage disassembly and rigid enough to favor substrate recognition, but with a sufficient degree of flexibility allowing product release. Indeed, caged catalysts often suffer from inhibition by the product. As our knowledge on bioinspired hemicryptophane catalyst increases, we found that this cage structure offers a remarkable flexibility–rigidity balance. Bioinspired catalysts surmounted by the hemicryptophane cavity have been successfully applied in oxidation catalysis. The hydrophobic cavity was used to favor the accommodation of apolar substrates and promote the release of less polar products to avoid overoxidation side reactions. However, in the case of more recalcitrant substrates (like methane), further improvement of the catalytic efficiency, in terms of number of turnovers, is required to get closer to metalloprotein efficiency. In particular, our future efforts will be dedicated to combine highly stable capsules with more reactive bioinspired metal cores.
(3) Binding cavities. To precisely control the adjustment between the active site and its co-encapsulated substrate, the sole hydrophobic effect is not sufficient. For instance, catalysis at metalloprotein strongly benefits from their binding pockets that both interact with substrates and active sites. These non-covalent interactions guarantee substrate positioning and stabilization of highly reactive intermediates. Although a relatively large number of bioinspired complexes with discrete cavities have been reported over the past few decades, complexes equipped with a functionalized cavity remain rare. The field of confined bioinspired catalysis assisted by weak bonding cavities is in its infancy and remains mainly unexplored. Examples of H-bonding capsules able to stabilize structural (non-reactive) models of bio-relevant metal-based intermediates have been described.30,85 But switching to functional complexes displaying reactivity inside discrete H-bonding confined spaces is still particularly challenging. In this line, caged catalysts able to control substrate positioning, by mean of substrate-cavity weak bonding, appear as a major perspective to push the limits of current bioinspired confined catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from the French government under the France 2030 investment plan, as part of the Initiative d’Excellence d’Aix-Marseille Université – A*MIDEX (AMX-21-PEP-041). This work was supported by ANR-22-CE50-0009-01.References
- R. J. Martinie, J. Livada, W.-C. Chang, M. T. Green, C. Krebs, J. M. Bollinger and A. Silakov, J. Am. Chem. Soc., 2015, 137, 6912–6919 CrossRef CAS PubMed.
- R. Mehmood, V. Vennelakanti and H. J. Kulik, ACS Catal., 2021, 11, 12394–12408 CrossRef CAS.
- R. Breslow and L. E. Overman, J. Am. Chem. Soc., 1970, 92, 1075–1077 CrossRef CAS PubMed.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146–153 CrossRef CAS.
- T. Koike and E. Kimura, J. Am. Chem. Soc., 1991, 113, 8935–8941 CrossRef CAS.
- D. Vidal, G. Olivo and M. Costas, Chem. – Eur. J., 2018, 24, 5042–5054 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- J. Trouvé, P. Zardi, S. Al-Shehimy, T. Roisnel and R. Gramage- Doria, Angew. Chem., Int. Ed., 2021, 60, 18006–18013 CrossRef PubMed.
- C. Bravin, E. Badetti, G. Licini and C. Zonta, Coord. Chem. Rev., 2021, 427, 213558 CrossRef CAS.
- S. Scott, H. Zhao, A. Dey and T. B. Gunnoe, ACS Catal., 2020, 10, 14315–14317 CrossRef CAS.
- A. Robert and B. Meunier, ACS Nano, 2022, 16, 6956–6959 CrossRef CAS PubMed.
- J. Serrano-Plana, C. Rumo, J. G. Rebelein, R. L. Peterson, M. Barnet and T. R. Ward, J. Am. Chem. Soc., 2020, 142, 10617–10623 CrossRef CAS PubMed.
- K. Chen, M. Zangiabadi and Y. Zhao, Org. Lett., 2022, 24, 3426–3430 CrossRef CAS PubMed.
- D. Vidal, M. Costas and A. Lledo, ACS Catal., 2018, 8, 3667–3672 CrossRef CAS.
- I. Tabushi and Y. Kuroda, J. Am. Chem. Soc., 1984, 106, 4580–4584 CrossRef CAS.
- A. Parrot, S. Collin, G. Bruylants and O. Reinaud, Chem. Sci., 2018, 9, 5479–5487 RSC.
- S. C. Bete, C. Würtele and M. Otte, Chem. Commun., 2019, 55, 4427–4430 RSC.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- S. C. Bete and M. Otte, Angew. Chem., Int. Ed., 2021, 60, 18582–18586 CrossRef CAS PubMed.
- C. T. Mc Ternan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- B. Kauppi, K. Lee, E. Carredano, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Structure, 1998, 6, 571–586 CrossRef CAS PubMed.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- S. C. Bete, L. K. May, P. Woite, M. Roemelt and M. Otte, Angew. Chem., Int. Ed., 2022, 61, e202206120 CrossRef CAS PubMed.
- R. J. Jodts, M. O. Ross, C. W. Koo, P. E. Doan, A. C. Rosenzweig and B. M. Hoffman, J. Am. Chem. Soc., 2021, 143, 15358–15368 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- M. Morimoto, S. M. Bierschen, K. T. Xia, R. G. Bergman, K. N. Raymond and F. Dean Toste, Nat Catal., 2020, 3, 969–984 CrossRef CAS.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- C. Colomban, V. Martin-Diaconescu, T. Parella, S. Goeb, C. García- Simón, J. Lloret-Fillol, M. Costas and X. Ribas, Inorg. Chem., 2018, 57, 3529–3539 CrossRef CAS PubMed.
- T. Zhang, L. Le Corre, O. Reinaud and B. Colasson, Chem. – Eur. J., 2021, 27, 434–443 CrossRef CAS PubMed.
- Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed.
- S. Merget, L. Catti, G. M. Piccini and K. Tiefenbacher, J. Am. Chem. Soc., 2020, 142, 4400–4410 CrossRef CAS PubMed.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- K. D. Karlin, S. Kaderli and A. D. Zuberbühler, Acc. Chem. Res., 1997, 30, 139–147 CrossRef CAS.
- K. Komiyama, H. Furutachi, S. Nagatomo, A. Hashimoto, H. Hayashi, S. Fujinami, M. Suzuki and T. Kitagawa, Bull. Chem. Soc. Jpn., 2004, 77, 59–72 CrossRef CAS.
- E. W. Dahl, H. T. Dong and N. K. Szymczak, Chem. Commun., 2018, 54, 892–895 RSC.
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed.
- J. J. Liu, D. E. Diaz, D. A. Quist and K. D. Karlin, Isr. J. Chem., 2016, 56, 738–755 CrossRef CAS PubMed.
- S. Y. Quek, S. Debnath, S. Laxmi, M. van Gastel, T. Krämer and J. England, J. Am. Chem. Soc., 2021, 143, 19731–19747 CrossRef CAS PubMed.
- N. Le Poul, Y. Le Mest, I. Jabin and O. Reinaud, Acc. Chem. Res., 2015, 48, 2097–2106 CrossRef CAS PubMed.
- G. Izzet, J. Zeitouny, H. Akdas-Killig, Y. Frapart, S. Ménage, B. Douziech, I. Jabin, Y. Le Mest and O. Reinaud, J. Am. Chem. Soc., 2008, 130, 9514–9523 CrossRef CAS PubMed.
- G. Thiabaud, G. Guillemot, I. Schmitz-Afonso, B. Colasson and O. Reinaud, Angew. Chem., Int. Ed., 2009, 48, 7383–7386 CrossRef CAS PubMed.
- J. F. Torres, C. H. Oi, I. P. Moseley, N. El-Sakkout, B. J. Knight, J. Shearer, R. García-Serres, J. M. Zadrozny and L. J. Murray, Angew. Chem., Int. Ed., 2022, 61, e202202329 CrossRef CAS PubMed.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499–1503 CrossRef CAS PubMed.
- M. L. Singleton, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2010, 132, 8870–8871 CrossRef CAS PubMed.
- S. S. Nurttila, R. Zaffaroni, S. Mathew and J. N. H. Reek, Chem. Commun., 2019, 55, 3081–3084 RSC.
- R. Zaffaroni, N. Orth, I. Ivanovic-Burmazovic and J. N. H. Reek, Angew. Chem., Int. Ed., 2020, 59, 18485–18489 CrossRef CAS PubMed.
- D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS PubMed.
- J. Yang, B. Chatelet, V. Dufaud, D. Hérault, S. Michaud-Chevallier, V. Robert, J.-P. Dutasta and A. Martinez, Angew. Chem., Int. Ed., 2018, 57, 14212–14215 CrossRef CAS PubMed.
- B. Chatelet, L. Joucla, J.-P. Dutasta, A. Martinez, K. C. Szeto and V. Dufaud, J. Am. Chem. Soc., 2013, 135, 5348–5351 CrossRef CAS PubMed.
- D. Zhang, K. Jamieson, L. Guy, G. Gao, J.-P. Dutasta and A. Martinez, Chem. Sci., 2017, 8, 789–794 RSC.
- J. A. A. W. Elemans and R. J. M. Nolte, Chem. Commun., 2019, 55, 9590–9605 RSC.
- A. K. Surendran, G. L. Tripodi, E. Pluhařová, A. Y. Pereverzev, J. P. J. Bruekers, J. A. A. W. Elemans, E. Jan Meijer and J. Roithová, Nat Sci., 2022, e20220019 Search PubMed.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, G. Blondin, P. Dubourdeaux, M. Clémancey, J.-M. Latour, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- O. Perraud, A. B. Sorokin, J.-P. Dutasta and A. Martinez, Chem. Commun., 2013, 49, 1288–1290 RSC.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
- A. Santoro, J. S. Calvo, M. D. Peris-Díaz, A. Krężel, G. Meloni and P. Faller, Angew. Chem., Int. Ed., 2020, 59, 7830–7835 CrossRef CAS PubMed.
- E. Falcone, A. G. Ritacca, S. Hager, H. Schueffl, B. Vileno, Y. El Khoury, P. Hellwig, C. R. Kowol, P. Heffeter, E. Sicilia and P. Faller, J. Am. Chem. Soc., 2022, 144, 14758–14768 CrossRef CAS PubMed.
- P. Wu, ACS Chem. Biol., 2022, 17, 2959–2961 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- G. Qiu, P. Nava, A. Martinez and C. Colomban, Chem. Commun., 2021, 57, 2281–2284 RSC.
- T. G. Brevé, M. Filius, C. Araman, M. P. van der Helm, P. Hagedoorn, C. Joo, S. I. van Kasteren and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 9340–9344 CrossRef PubMed.
- Z. Zhu, H. Chen, S. Li, X. Yang, E. Bittner and C. Cai, Catal. Sci. Technol., 2017, 7, 2474–2485 RSC.
- T. V. Tran, G. Couture and L. H. Do, Dalton Trans., 2019, 48, 9751–9758 RSC.
- G. Qiu, P. Nava, C. Colomban and A. Martinez, Front. Chem., 2020, 8, 599893 CrossRef CAS PubMed.
- M. J. Hardie, Chem. Soc. Rev., 2010, 39, 516–527 RSC.
- J. Canceill, A. Collet, J. Gabard, G. Gottarelli and G. P. Spada, J. Am. Chem. Soc., 1985, 107, 1299–1308 CrossRef CAS.
- C. Colomban, B. Châtelet and A. Martinez, Synthesis, 2019, 2081–2099 CAS.
- D. Zhang, B. Bousquet, J.-C. Mulatier, D. Pitrat, M. Jean, N. Vanthuyne, L. Guy, J.-P. Dutasta and A. Martinez, J. Org. Chem., 2017, 82, 6082–6088 CrossRef CAS PubMed.
- G. Qiu, C. Colomban, N. Vanthuyne, M. Giorgi and A. Martinez, Chem. Commun., 2019, 55, 14158–14161 RSC.
- G. Qiu, D. E. Khatmi, A. Martinez and P. Nava, RSC Adv., 2021, 11, 13763–13768 RSC.
- G. Mukherjee, J. K. Satpathy, U. K. Bagha, M. Q. E. Mubarak, C. V. Sastri and S. P. de Visser, ACS Catal., 2021, 11, 9761–9797 CrossRef CAS.
- Z. Wojdyla and T. Borowski, Chem. – Eur. J., 2022, 28, e202104106 CAS.
- M. W. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- M. Bhadra, J. Yoon, C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2018, 140, 9042–9045 CrossRef CAS PubMed.
- D. E. Diaz, D. A. Quist, A. E. Herzog, A. W. Schaefer, I. Kipouros, M. Bhadra, E. I. Solomon and K. D. Karlin, Angew. Chem., Int. Ed., 2019, 58, 17572–17576 CrossRef CAS PubMed.
- M. A. Ehudin, A. W. Schaefer, S. M. Adam, D. A. Quist, D. E. Diaz, J. A. Tang, E. I. Solomon and K. D. Karlin, Chem. Sci., 2019, 10, 2893–2905 RSC.
- L. Vicens, G. Olivo and M. Costas, Angew. Chem., Int. Ed., 2022, 61, e2021149 CrossRef PubMed.
- G. Olivo, G. Capocasa, B. Ticconi, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2020, 59, 12703–12708 CrossRef CAS PubMed.
- G. Olivo, G. Farinelli, A. Barbieri, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2017, 56, 16347–16351 CrossRef CAS PubMed.
- G. De Leener, D. Over, C. Smet, D. Cornut, A. G. Porras-Gutierrez, I. López, B. Douziech, N. Le Poul, F. Topić, K. Rissanen, Y. Le Mest, I. Jabin and O. Reinaud, Inorg. Chem., 2017, 56, 10971–10983 CrossRef CAS PubMed.
- P. Aoun, N. Nyssen, S. Richard, F. Zhurkin, I. Jabin, B. Colasson and O. Reinaud, Chem. – Eur. J., 2022, e202202934 Search PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
- G. Qiu, D. Diao, L. Chaussy, S. Michaud-Chevallier, A. J. Simaan, P. Nava, A. Martinez and C. Colomban, Dalton Trans., 2022, 51, 10702–10706 RSC.
| This journal is © The Royal Society of Chemistry 2023 |