DOI:
10.1039/D2BM01673G
(Paper)
Biomater. Sci., 2023,
11, 916-930
Endogenous stimulus-responsive nitric oxide releasing bioactive liposome for a multilayered drug-eluting balloon
Received
13th October 2022
, Accepted 27th November 2022
First published on 8th December 2022
Abstract
Drug-eluting balloon (DEB) system has been widely utilized for percutaneous coronary intervention (PCI), treating atherosclerosis to overcome the limitations of cardiovascular stents. With the anti-proliferative drug, everolimus (EVL), nitric oxide (NO) plays a key bioregulator role to facilitate the angiogenesis of endothelial cells (ECs) and inhibit the cell proliferation of smooth muscle cells (SMCs) in the lesions of cardiovascular diseases. Due to the very short lifetime and limited exposure area of NO in the body, the continuous release and efficient delivery of NO must be carefully considered. In this respect, a liposome-containing disulfide bonding group was introduced as a delivery vehicle of EVL and NO with the continuous release of NO via successive reaction cycles with GSH and SNAP in the blood vessel without the need for exogenous stimulations. With a multilayer coating platform consisting of a polyvinylpyrrolidone (PVP)/EVL-laden liposome with NO (EVL-NO-Lipo)/PVP, we precluded the loss of the EVL-encapsulated liposome with NO release during the transition time and maximized the transfer rate from the surface of DEB to the tissues. The sustained release of NO was monitored using a nitric oxide analyzer (NOA), and the synergistic bioactivities of EVL and NO were proved in EC and SMC with angiogenesis and cell proliferation-related assays. From the results of hemocompatibility and ex vivo studies, the feasibility was provided for future in vivo applications of the multilayer-coated DEB system.
Introduction
Coronary artery disease (CAD) is the most common heart disease throughout the world and is characterized by the narrowing of blood vessels via cholesterol deposits or plaque buildup in the wall of the arteries that supply oxygen, blood, and nutrients.1 As a representative non-surgical treatment, percutaneous coronary intervention (PCI) is an angioplasty procedure that expands the narrowed lesion using a catheter with stents, balloons, and lasers.2,3 To date, various medical devices have been developed for PCI treatments, such as bare metal stents (BMSs), drug-eluting stents (DESs), balloons, bioresorbable vascular scaffolds (BVSs) and others.4,5 Among them, the method for the slow release of a proliferation inhibitor and immunosuppressant with stents was superior to bare metal stents in lowering the occurrence of major adverse cardiac problems. Despite the effective outcomes, DESs have critical problems, including potential risks from the fracture and deformation of the stents and late vascular reendothelialization, stent thrombosis, and restenosis from the degradation of permanent implanted foreign materials in the body.6–8 To overcome these limitations, drug-eluting balloons (DEBs) have been introduced and widely applied for PCI treatments. Different from DESs, DEBs were designed to avoid insertion of permanent foreign materials in the blood vessel. They enable the rapid release of a high concentration of the drug, which is sustained in the vessel wall for no longer than a week, thereby preventing the potential risks caused by DESs.9 The cytostatic drugs were coated onto the surface of the balloons, and the drugs were released to the lesion directly during balloon inflation for continuous therapeutic effects while preventing the proliferation of smooth muscle cells (SMCs) in the lesion, followed by removing the balloon catheter. In general, rapamycin and its analogues have been utilized widely as an antiproliferative and immunosuppressant drug in interventional treatments.10 As macrolide derivatives, two types of rapamycin, sirolimus (SRL) and everolimus (EVL), selectively inhibit the mammalian target of rapamycin (mTOR), regulating cell proliferation, autophagy and apoptosis by participating in multiple signaling pathways.11,12 In particular, EVL, a derivative of SRL, showed different pharmacokinetic characteristics from SRL, such as better absorption, greater bioavailability, faster steady-state blood concentration after administration, and faster elimination after withdrawal.13 To minimize the major drawbacks of DEBs, including drug loss during insertion to lesions and the low drug delivery rate to the tissue due to vigorous blood flow and short balloon inflation times (within 1 min), we previously suggested a multilayer-coated DEB system composed of a hydrophilic polymer, drug-encapsulated liposome, and hydrophilic polymer, serially.14 In brief, a biocompatible and biodegradable hydrophilic polymer, polyvinylpyrrolidone (PVP), was coated on the 1st and 3rd layers of the balloon catheter to facilitate drug release to the lesion in hydrophilic environments and inhibit drug loss during the transition time, respectively. Moreover, a `hydrophobic drug (EVL)-encapsulated biocompatible liposome was introduced to the 2nd layer of the balloon catheter to deliver the drug efficiently to the target region. Despite the strong therapeutic potential and stability of the multilayer-coated DEB system, there remain many disadvantages due to the use of a high drug dosage, such as toxicities to endothelial cells (ECs) and long coating time.
To reduce the drug amount while maintaining therapeutic effects in the multilayer-coated DEB system, nitric oxide (NO) is herein additionally considered owing to its synergistic effects with the drug.15 NO is a soluble gas that participates in many regulatory functions from the cardiovascular system to neuronal functions.16 In general, NO is synthesized in the vascular endothelium from L-arginine as a substrate in a catalyzed reaction with three types of nitric oxide synthase (NOS): neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS).17 In detail, NO is produced by the conversion of L-arginine into L-citrulline in the presence of molecular oxygen and various biological factors such as tetrahydrobiopterin (BH4), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD).18 These processes are catalyzed by various types of nitric oxide synthases (NOSs), especially eNOS in endothelial cells.19 In the biological system, NO has different roles depending on the concentration and environments.20 NO facilitates cell proliferation in endothelial cells, whereas it downregulates cell proliferation in smooth muscle cells.21,22 Although diverse functionalities of NO have been actively studied in vitro, it has limited therapeutic effects for systems in vivo due to their limitation of exposure area, and short half-lives derived from rapid thermal and photochemical degradation.23,24 To overcome these obstacles, various types of NO donors, including diazeniumdiolates, s-nitrosothiols, n-nitrosamines, and organic nitrates, have been reported over decades. However, there is still difficulty in controlling the long term release of NO in biological systems.25
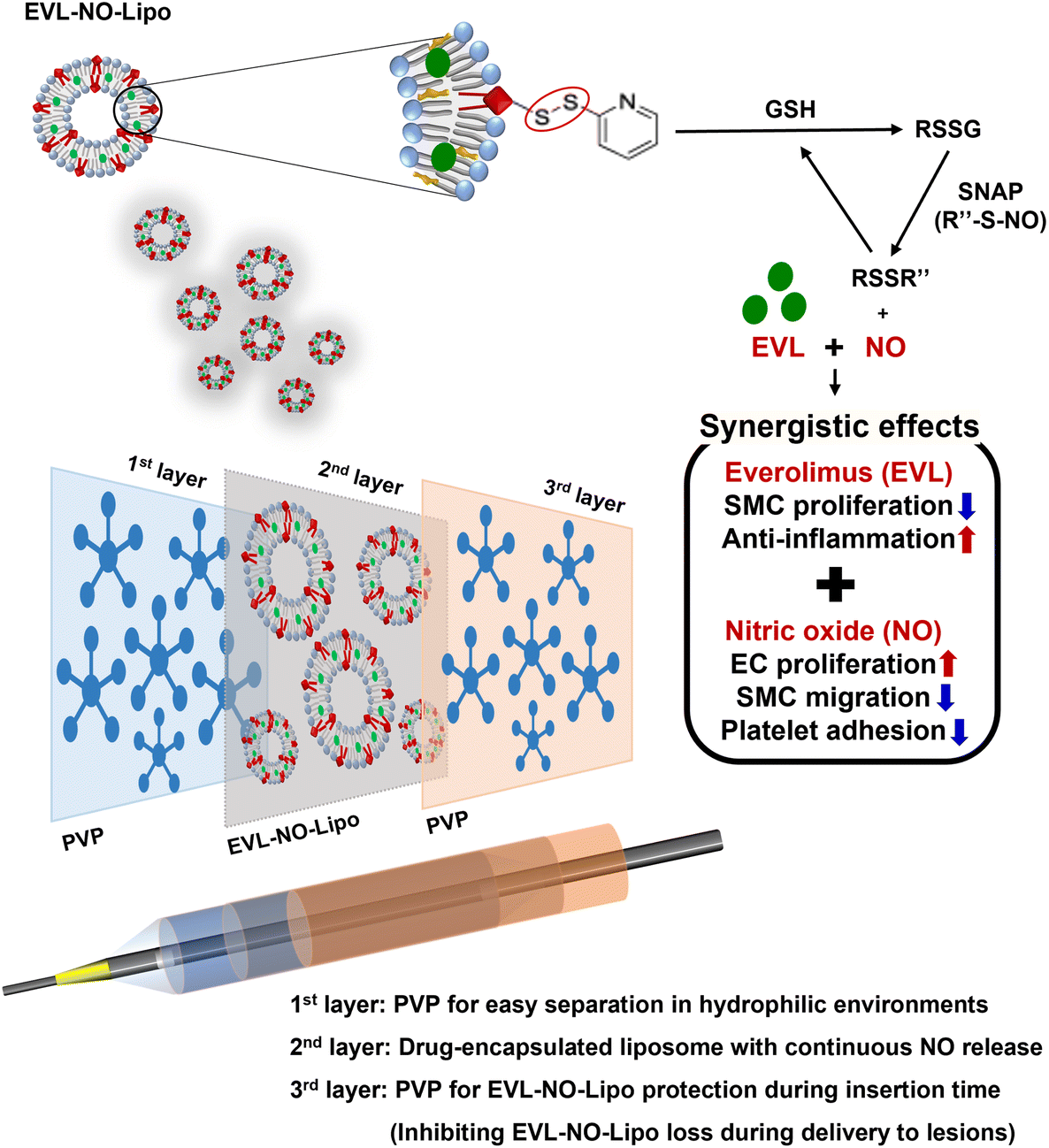
Inspired by the continuous flow of the glutathione (GSH) and NO donor, s-nitroso-N-acetylpenicillamine (SNAP), in the blood vessel, we introduced a system in this study in which NO can be consecutively released by reacting with GSH and SNAP.26 As shown in Fig. 1, a lipid containing the disulfide bonding group, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[PDP (polyethylene glycol)-2000] (DSPE-PEG (2000) PDP), was utilized as a material that enabled NO release laden with hydrophobic EVL. The disulfide bonding groups react with GSH to form RSSG, which generates NO by reacting with SNAP in the blood vessel.27 Continuously, the remaining RSSR compound can be transferred to RSSG by reacting with GSH. The repeated reaction cycle by GSH and SNAP results in the continuous release of NO in the presence of disulfide bonding groups (Fig. 1).
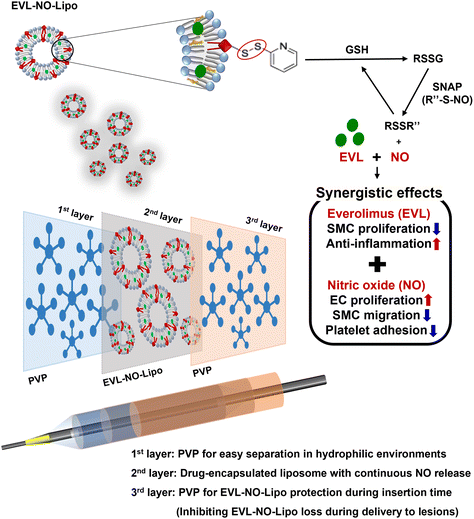 |
| | Fig. 1 Schematic illustration for multilayer coated drug-eluting balloon system with PVP/EVL encapsulated liposome with NO release (EVL-NO-Lipo)/PVP. | |
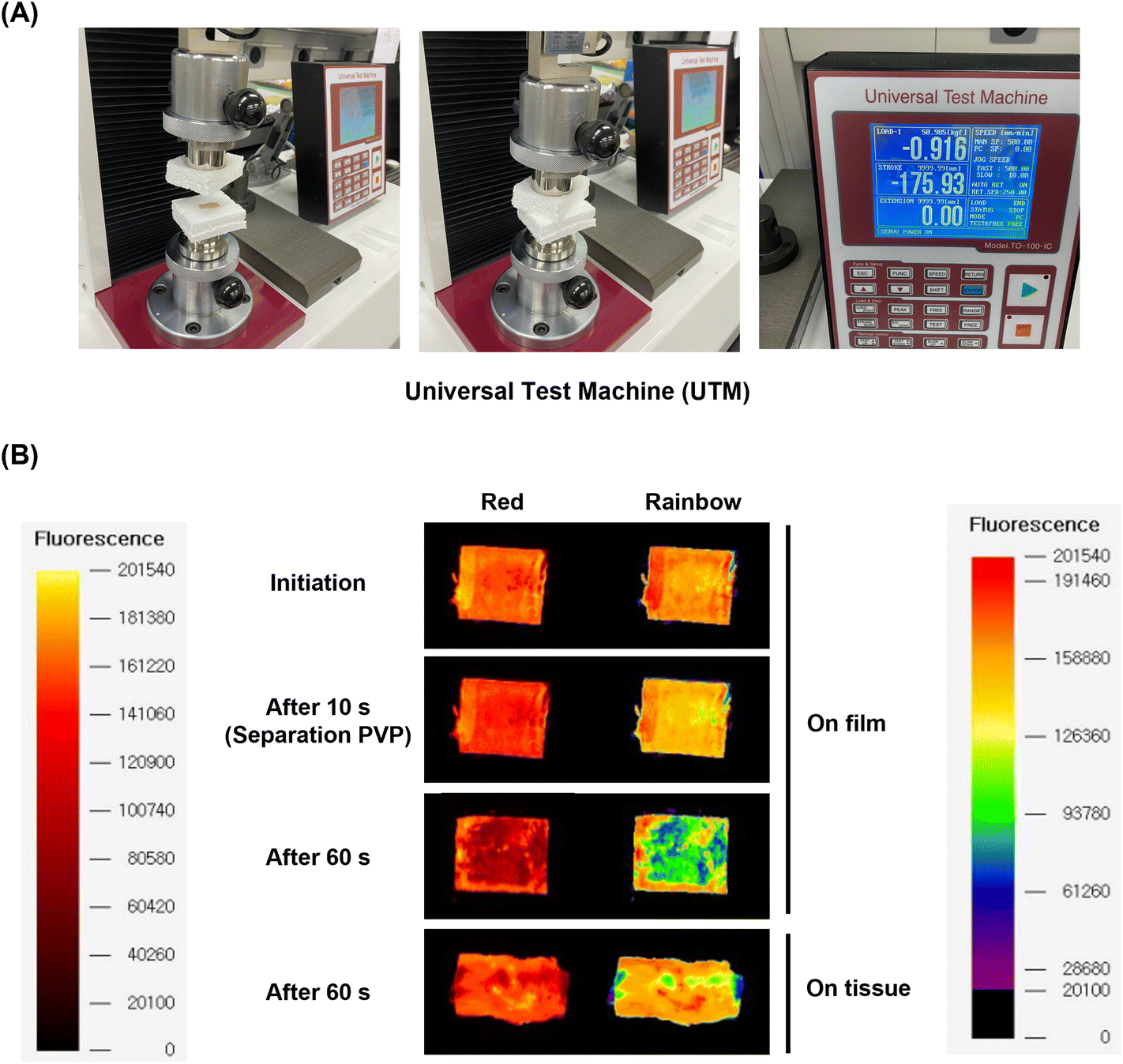
Herein, we propose a novel DEB system in a multilayer coating strategy that enables synergistic therapeutic effects with NO, while reducing the amount of cytostatic drug (EVL). The multilayer-coated DEB system, containing hydrophilic PVP, continuous NO releasing liposome with EVL, and hydrophilic PVP, was optimized. The synergistic properties of EVL and NO were investigated using various cell-based assays in human coronary artery endothelial cells (HCAECs) and human coronary artery smooth muscle cell (HCASMCs). The biocompatibility of the multilayer-coated DEB system was verified by hemocompatibility test, and the ex vivo study for the possibility of efficient drug transfer was simulated using a universal testing machine (UTM) in the condition for similar pressure of the vascular tissue for DEB treatments.
Results and discussion
Characterization of the EVL-laden liposome with nitric oxide (NO) release
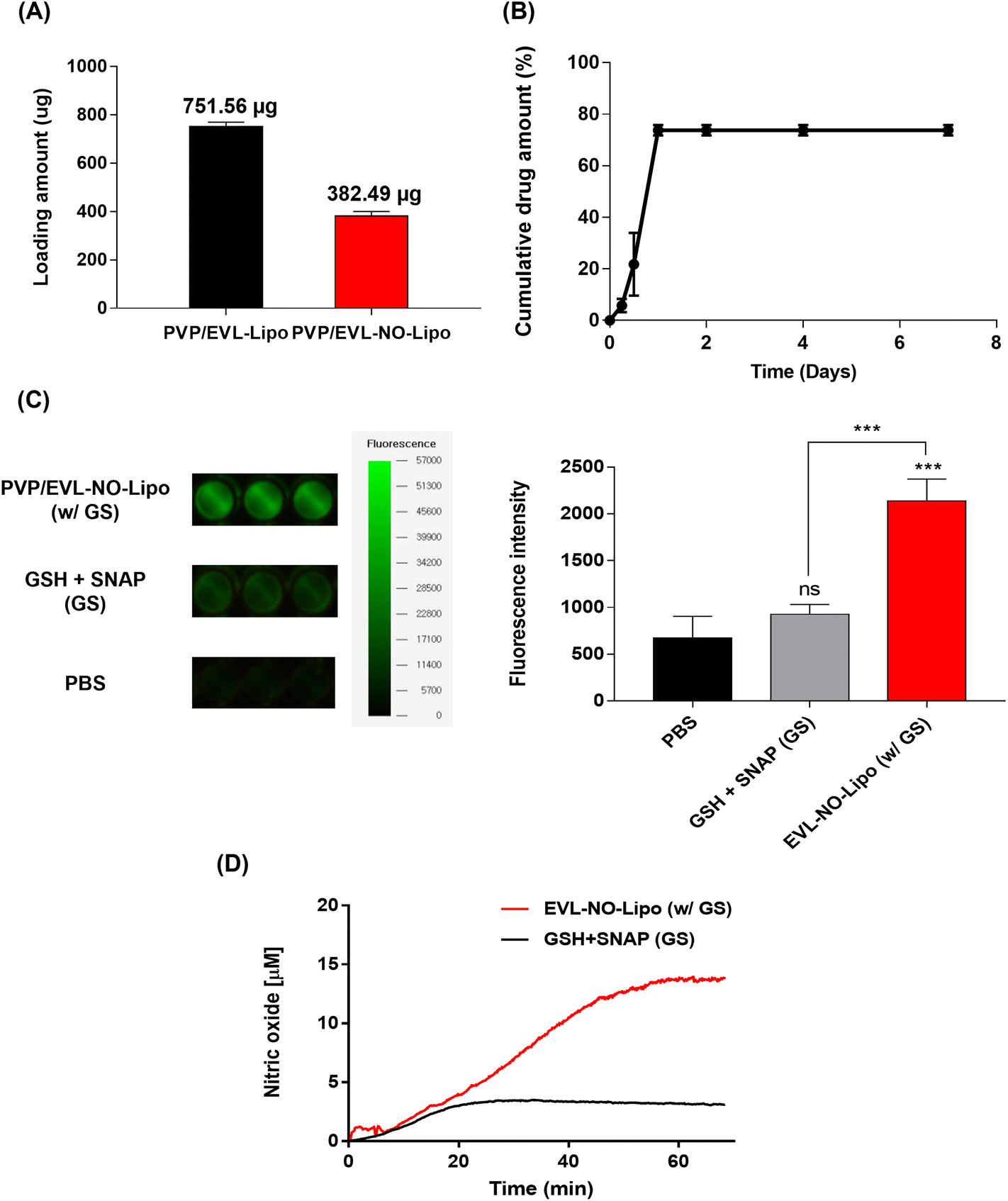
To comparatively analyze the synergistic properties of EVL and NO, two types of liposomes, EVL-encapsulated liposome (EVL-Lipo) and EVL-laden liposome with NO release (EVL-NO-Lipo), were synthesized using an optimum ratio of lipid and drug to maximize the encapsulation efficiency (Fig. 2A). The high drug encapsulation efficiencies of EVL-Lipo (71.7%) and EVL-NO-Lipo (76.0%) were achieved without surfactants via simple mixing during the thin-film hydration process for liposome formation (Fig. 2B). With the addition of the NO releasing lipid composition, the size and zeta potential are not significantly different from EVL-Lipo (Fig. 2C and D). In particular, a negative charge of around −30 mV indicated that the liposome maintained stability.28 Furthermore, TEM images showed a liposome structure with spherical types at a size range of around 100 nm (Fig. 2E). Next, the NO releasing property of EVL-NO-Lipo with the disulfide bonding group was monitored using a fluorescence-based DAF-FM assay. DAF-FM is a highly sensitive, photostable fluorescence probe to quantify NO release.29 With only GSH and SNAP, a small amount of fluorescence signal was shown due to the natural decomposition of the NO donor, SNAP. Conversely, GSH and SNAP with EVL-NO-Lipo displayed a significantly enhanced fluorescence signal (Fig. 2F), which demonstrated the release of NO from the disulfide bonding group of EVL-NO-Lipo, as described in Fig. 1. Moreover, the continuous releasing profile of NO by the disulfide bonding group with the addition of 10 μM GSH and SNAP was demonstrated using a nitric oxide analyzer (NOA) for 60 min under the condition that excludes the naturally decomposed SNAP-derived NO release with ethylenediaminetetraacetic acid (EDTA) as a stabilizer (Fig. 2G), as compared to only GSH and SNAP treatment as a control.30 Ten μM GSH and SNAP were utilized to simulate the blood vessel conditions.31,32 Different from other NO releasing platforms with exogenous stimulations, the sustained release of NO was achieved with successive cycles of NO release from the disulfide bonding group reacting with GSH and SNAP, which are naturally present in the blood vessel.33,34
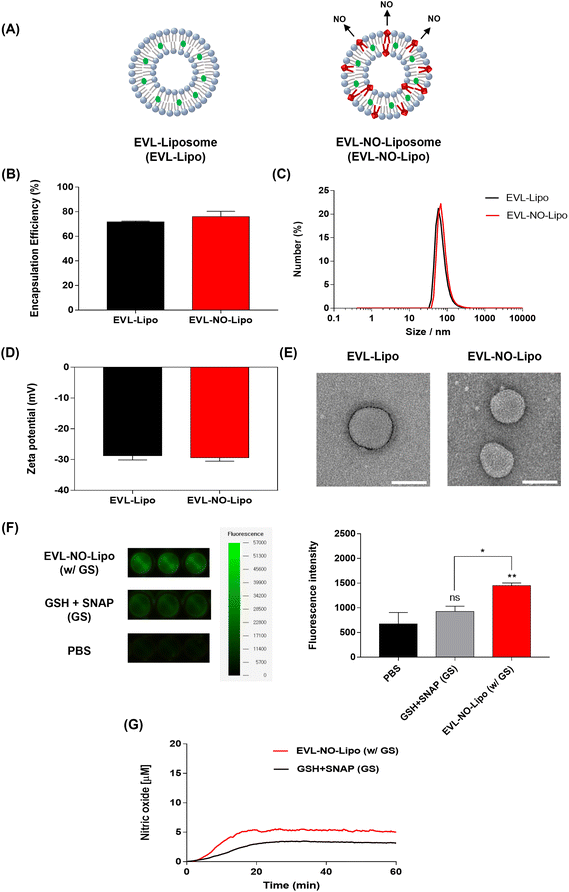 |
| | Fig. 2 Characterization of EVL-Lipo and EVL-NO-Lipo. (A) Two types of EVL-laden liposomes with and without the disulfide bonding group. (B) EVL encapsulation efficiency, (C) Size, and (D) Zeta potential for EVL-Lipo and EVL-NO-Lipo. (E) TEM images of EVL-Lipo and EVL-NO-Lipo (scale bars equal to 100 nm). (F) DAF-FM assays for NO releasing of EVL-NO-Lipo in the presence of GSH and SNAP, and quantitative analysis for fluorescence intensity from the DAF-FM assays. (G) NOA profile to prove continuous NO release (Values are presented as mean ± SD (n = 3)). | |
Surface analysis of the multilayer-coated balloon system
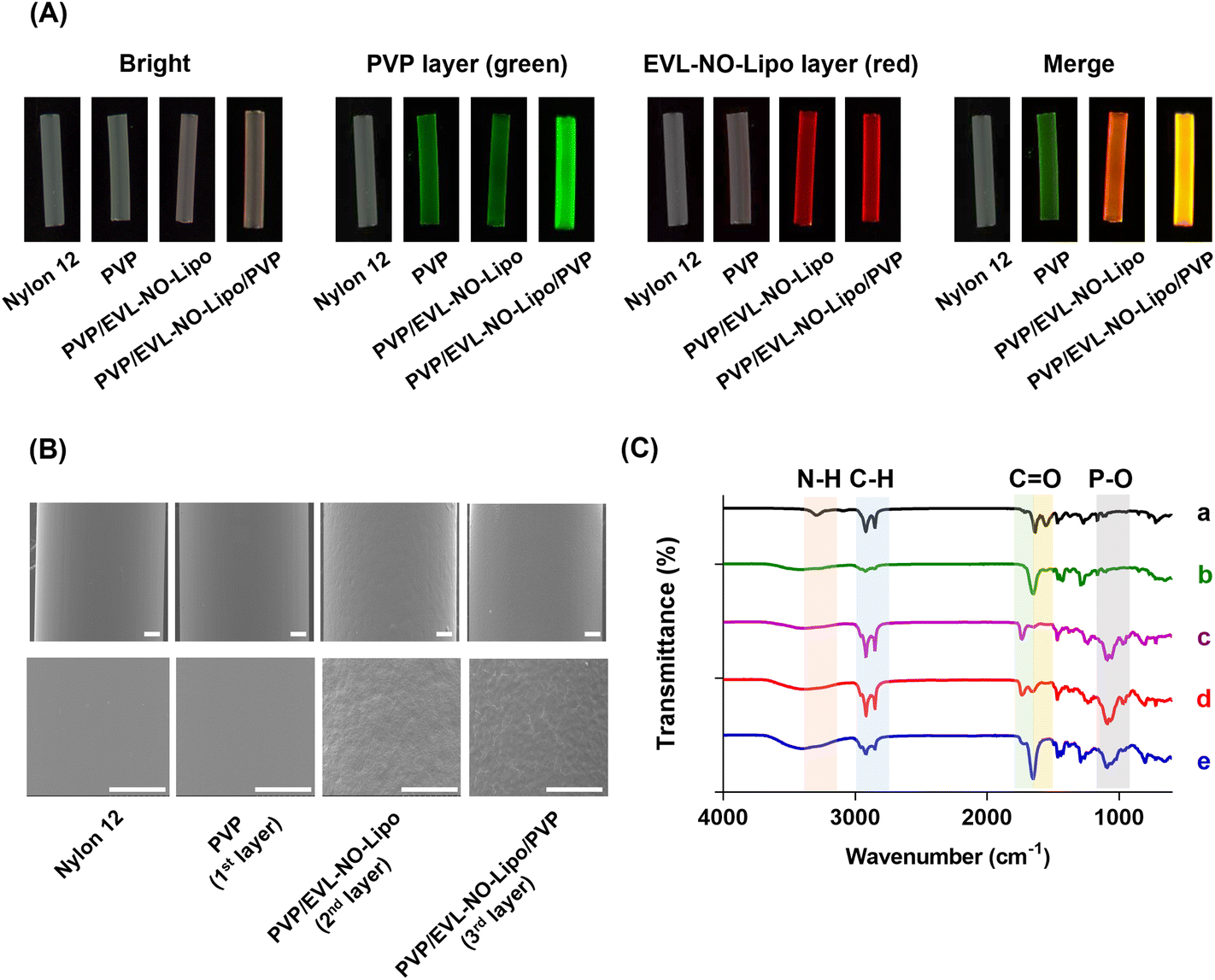
As an alternative for the balloon catheter, the Nylon 12 tube was used in this study to simulate the properties of the multilayer-coated balloon system. After multilayer coating of the Nylon 12 tube, the properties and morphology of the layers were analyzed with various methodologies, such as fluorescence microscopy, scanning electron microscopy (SEM), attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR), and X-ray photoelectron spectroscopy (XPS). 5(6)-Carboxyfluorescein-labeled PVP and Nile red-stained lipid were visualized with serial coating processes (Fig. 3A). With the accumulation of PVP by the additional coating processes, a significant green fluorescence signal was visualized after the coating of the 3rd layer compared to the coating of the 1st layer. Furthermore, a red fluorescence signal emerged after the coating of the 2nd layer with EVL-NO-Lipo. The differences in the surface roughness observed from SEM proved the multilayer coating of Nylon 12 tubes (Fig. 3B). With the coating of the 2nd layer with EVL-NO-Lipo, relatively rough surfaces appeared, which were covered by the coating of the 3rd layer with PVP. Additionally, changes in molecular interaction depending on the coating layers were analyzed using ATR-FTIR with the shift of peak broadness in the transmittance spectra (Fig. 3C). Upon coating the 1st PVP layers onto the tubes, the spectra of the HO bond (3352–3404 cm−1), C–H asymmetric stretching vibration (2955 and 1661 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide stretching vibration, and CH2 bending vibration (1424 and 1291 cm−1) were detected.35 After coating the 2nd layer with EVL-NO-Lipo, liposome-related peaks involving the CO ester bond (1736 cm−1) and the PO antisymmetric stretch (1232 cm−1) appeared. These liposome-related peaks decreased upon the coating of the 3rd layer with PVP with a clear appearance of the CO amide stretch (1657 cm−1).36 Additionally, the changes of the atomic elements and chemical compositions demonstrated the clear coating of each layer. As listed in Table 1, the differences in the atomic percentages of carbon (C), oxygen (O), nitrogen (N), and phosphorus (P) were evaluated depending on the serial coating with PVP, EVL-NO-Lipo, and PVP onto the Nylon 12 tubes. After the coating of the 2nd layer with EVL-NO-Lipo, the lipid-associated P2p value was newly detected with diminishing N1s value due to the complete covering of the 1st PVP layer. Moreover, the S2p value was evaluated due to the disulfide bonding group of DSPE-PEG (2000) PDP, which decreased with the overall coating with PVP in the 3rd layer. The relatively low intensity of S2p was derived from the low ratio of DSPE-PEG (2000) PDP in the liposome compositions (HSPC
O amide stretching vibration, and CH2 bending vibration (1424 and 1291 cm−1) were detected.35 After coating the 2nd layer with EVL-NO-Lipo, liposome-related peaks involving the CO ester bond (1736 cm−1) and the PO antisymmetric stretch (1232 cm−1) appeared. These liposome-related peaks decreased upon the coating of the 3rd layer with PVP with a clear appearance of the CO amide stretch (1657 cm−1).36 Additionally, the changes of the atomic elements and chemical compositions demonstrated the clear coating of each layer. As listed in Table 1, the differences in the atomic percentages of carbon (C), oxygen (O), nitrogen (N), and phosphorus (P) were evaluated depending on the serial coating with PVP, EVL-NO-Lipo, and PVP onto the Nylon 12 tubes. After the coating of the 2nd layer with EVL-NO-Lipo, the lipid-associated P2p value was newly detected with diminishing N1s value due to the complete covering of the 1st PVP layer. Moreover, the S2p value was evaluated due to the disulfide bonding group of DSPE-PEG (2000) PDP, which decreased with the overall coating with PVP in the 3rd layer. The relatively low intensity of S2p was derived from the low ratio of DSPE-PEG (2000) PDP in the liposome compositions (HSPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Chol:DSPE-PEG (2000) PDP = 5.5:4.0:0.5). The water contact angle (WCA) indirectly proves the changes of the coating layer through the differences in hydrophilicity according to the coating materials.37 PVP and liposome components decreased the contact angle, indicating their strong interaction with water molecules, which made the materials biocompatible by inhibiting the nonspecific interaction with proteins in the body.38 Specifically, the relatively higher hydrophilicity of the liposome made the contact angle smaller in the 2nd layer than in the 1st and 3rd layers with PVP (Table 1). As EVL-NO-Lipo was coated, the changes of WCA showed a similar tendency to that of coating EVL-Lipo.14
:Chol:DSPE-PEG (2000) PDP = 5.5:4.0:0.5). The water contact angle (WCA) indirectly proves the changes of the coating layer through the differences in hydrophilicity according to the coating materials.37 PVP and liposome components decreased the contact angle, indicating their strong interaction with water molecules, which made the materials biocompatible by inhibiting the nonspecific interaction with proteins in the body.38 Specifically, the relatively higher hydrophilicity of the liposome made the contact angle smaller in the 2nd layer than in the 1st and 3rd layers with PVP (Table 1). As EVL-NO-Lipo was coated, the changes of WCA showed a similar tendency to that of coating EVL-Lipo.14
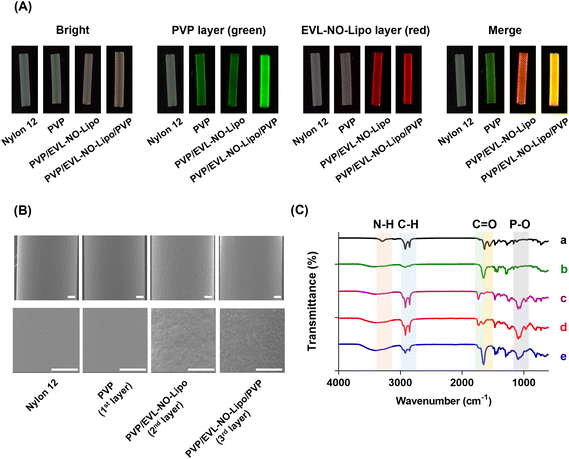 |
| | Fig. 3 Characterization of PVP, PVP/EVL-NO-Lipo, and PVP/EVL-NO-Lipo/PVP coated surfaces of Nylon 12 tubes. (A) Fluorescence images of the coated surfaces using FOBI. Green: 5(6)-Carboxyfluorescein-stained PVP, Red: Nile red-labeled liposome. (B) SEM images of the coated surfaces (Scale bars equal to 500 μm). (C) ATR-FTIR spectra for surfaces of Nylon 12 tube coated with PVP, PVP/EVL-No-Lipo and PVP/EVL-NO-Lipo/PVP. a. Bare Nylon 12 tube, b. PVP coated Nylon 12 tube, c. EVL-NO-Lipo only, d. PVP/EVL-NO-Lipo coated Nylon 12 tube, and e. PVP/EVL-NO-Lipo/PVP coated Nylon 12 tube. | |
Table 1 XPS atomic concentration and water contact angle (WCA) for the serial coating of Nylon 12 tube
| Sample |
XPS atomic concentration(%) |
Water contact angle (°) |
| C1s |
01s |
N1s |
P2p |
S2p |
| Nylon 12 |
74.31 |
15.5 |
10.19 |
— |
— |
83.47 ± 0.97 |
| PVP |
81.02 |
13.26 |
5.72 |
— |
— |
47.22 ± 0.04 |
| PVP/EVL-NO-Lipo |
81.12 |
15.37 |
1.93 |
1.27 |
0.32 |
23.02 ± 0.57 |
| PVP/EVL-NO-Lipo/PVP |
79.52 |
17.57 |
2.03 |
0.61 |
0.27 |
38.14 ± 0.51 |
EVL and NO release from the multilayer-coated system
To achieve the synergistic effects of EVL and NO while decreasing the drug amount by one-half, two types of multilayer-coated Nylon 12 tubes were manufactured with 760 μg EVL-encapsulated liposome and NO releasing liposome laden with 380 μg EVL, respectively. Both liposome formulations were lyophilized and rehydrated with 20% ethanol solutions to concentrate for efficient coating processes. Hydration using 20% ethanol does not affect the structure and stability of the liposome.39 The repeated coating with an ultrasonic spray coating instrument set the final concentrations of EVL at about 760 μg for EVL-Lipo and 380 μg for EVL-NO-Lipo, respectively (Fig. 4A). The drug releasing profile was monitored for 7 days to verify the role of multilayer coating on the DEB system. The large amounts of EVL were initially burst released due to efficient separation of the 1st PVP layer in the hydrophilic condition (Fig. 4B). Finally, about 80% of EVL was released from the coated surface. The NO release was also monitored using a fluorescence-based DAF-FM assay from the multilayer-coated Nylon 12 tubes (Fig. 4C). In the presence of only GSH and NO donor, a weak fluorescence signal was observed due to the natural decomposition of the NO donor, SNAP. With the coating of EVL-NO-Lipo, a relatively intense signal was generated because NO was released through the continuous reaction cycle by the disulfide bonding group in the liposome with GSH and SNAP. The continuous releasing profile of NO by the EVL-NO-Lipo coated Nylon 12 tube with the addition of 10 μM GSH and SNAP can be verified using a NOA over 60 min under the condition that inhibits the naturally originating SNAP-derived NO release using EDTA. After 40 min, the constant amounts of NO start to be released, because it needed time for EVL-NO-Lipo to be completely separated from the Nylon 12 tube (Fig. 4D). It revealed the sustained release of NO from the liposome containing the disulfide bonding group after coating onto Nylon 12 tube. The cumulative release amount of NO after 60 min was calculated through the integral value of the real time No release profile, and was estimated to be 222.6 μM.
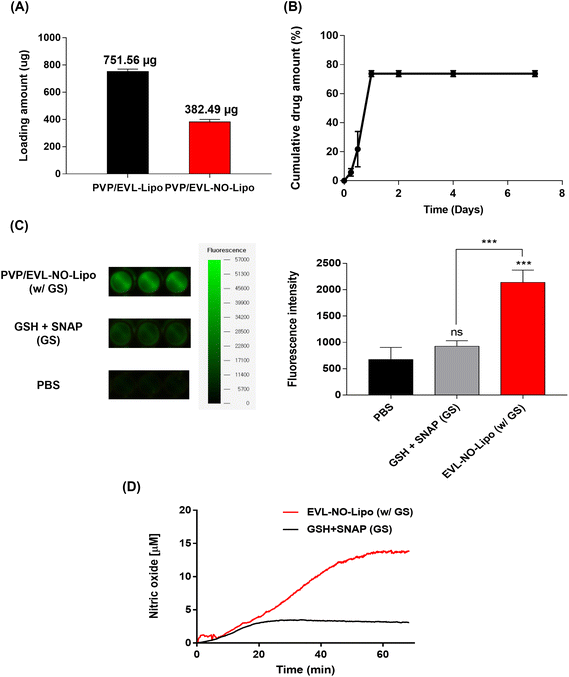 |
| | Fig. 4 Characterization for EVL and NO release on Nylon 12 tube. (A) Accumulated loading amount of EVL on PVP/EVL-Lipo and PVP/EVL-NO-Lipo. (B) EVL releasing profile of EVL-NO-Lipo coated Nylon 12 tube in the presence of PVP as the 1st layer. (C) DAF-FM assays for NO release of PVP/EVL-NO-Lipo coated Nylon 12 tube in the presence of GSH and SNAP, and quantitative analysis for fluorescence intensity from DAF-FM assays. (D) NOA profile to prove continuous NO release of the PVP/EVL-NO-Lipo coated Nylon 12 tube (Values are presented as mean ± SD (n = 3)). | |
In vitro biological activities of NO released from NO-Lipo
The angiogenesis-related tube formation assay was conducted using human coronary artery endothelial cells (HCAECs) to demonstrate the strong angiogenic property of NO released from NO-Lipo containing disulfide bonding group. NO is an endothelial survival factor, inhibiting apoptosis and accelerating proliferation of EC,40,41 with an increase in the expression of vascular endothelial growth factor (VEGF) and fibroblast growth factors (FGF) by upregulating the expression of nitric oxide synthase (NOS) in the body.41,42 Taken together, the exogenous disulfide bonding group is expected to facilitate the continuous NO release reaction with GSH and SNAP in the blood vessel. The results showed that the tube formation ability was significantly accelerated in the NO-Lipo in the presence of GSH (G) and SNAP (S) compared to the presence of only naturally decomposed NO donors (GS) (Fig. 5A). Moreover, various angiogenic-related parameters such as the total length, total branching length, number of nodes, and number of junctions, were quantified to verify the effects of NO derived from the disulfide bonding group of liposomes for tube formation at the indicated time in many ways (Fig. 5B). All parameters of tube formation exhibited the gradual increase with the introduction of NO-Lipo with GSH and SNAP. NO generally stabilizes hypoxia-inducible factor (HIF) 1α, which upregulates protein kinase G (PKG) signaling to stimulate secretion of VEGF and other angiogenesis-related growth factors.43,44 To prove this angiogenesis-related pathway by NO, the gene expression level of NO inducible angiogenic factors were comparatively analyzed depending on the use of disulfide bonding group with GSH and SNAP. As an exogenous NO donor, the disulfide bonding group-derived NO could activate HIF 1α, which resulted in the serial upregulation of PKG, VEGF, HGF, and CD31 (Fig. 5C). In general, both proliferation and migration of vascular smooth muscle cells (SMCs) contribute toward inducing lesion formation.45,46 Competitiveness between the migration of ECs and SMCs from the native vessel is also important for the successful treatment of vascular diseases.47 Different from the condition in EC, NO has been known to reversibly inhibit SMC migration independent of proliferation and cytotoxicity.48 After being cultured for 1 day, NO-Lipo with GSH and SNAP strongly inhibited the migration of HCASMC compared with the condition without GSH and SNAP (Fig. 5D).
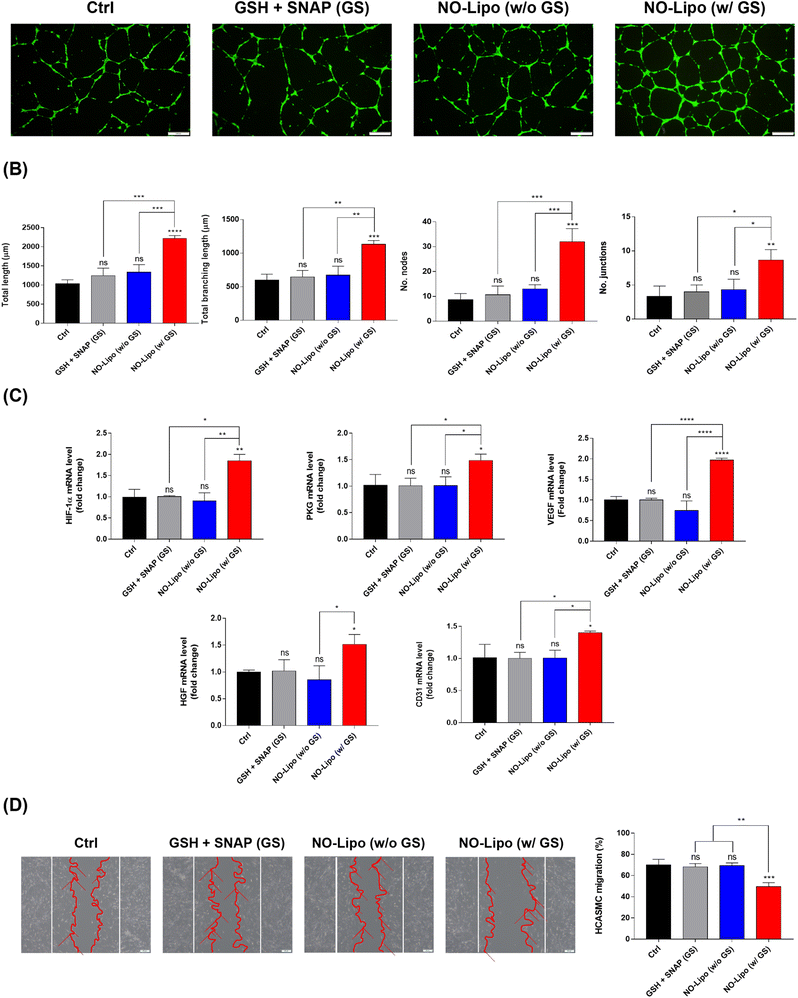 |
| | Fig. 5 Biological activities of NO release from No-Lipo. (A) Representative images of the angiogenesis effect by tube formation assay of NO-Lipo. (B) Analysis of the total length, total branching length, number of nodes, and number of junctions from the tube formation assay. (C) The gene expression levels of the representative angiogenic factors from the NO-Lipo treated human coronary artery endothelial cells (HCAECs). (D) Representative images of cell migration assays to evaluate the inhibitory effects of NO-Lipo for human coronary artery smooth muscle cells (HCASMCs) and calculated cell migration rates of various groups. (Values are presented as mean ± SD (n = 3) and statistical significance was obtained with one-way analysis of ANOVA with Tukey's multiple comparison post-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001)). | |
In vitro biological activities of EVL-Lipo and synergistic effects of EVL-NO-Lipo
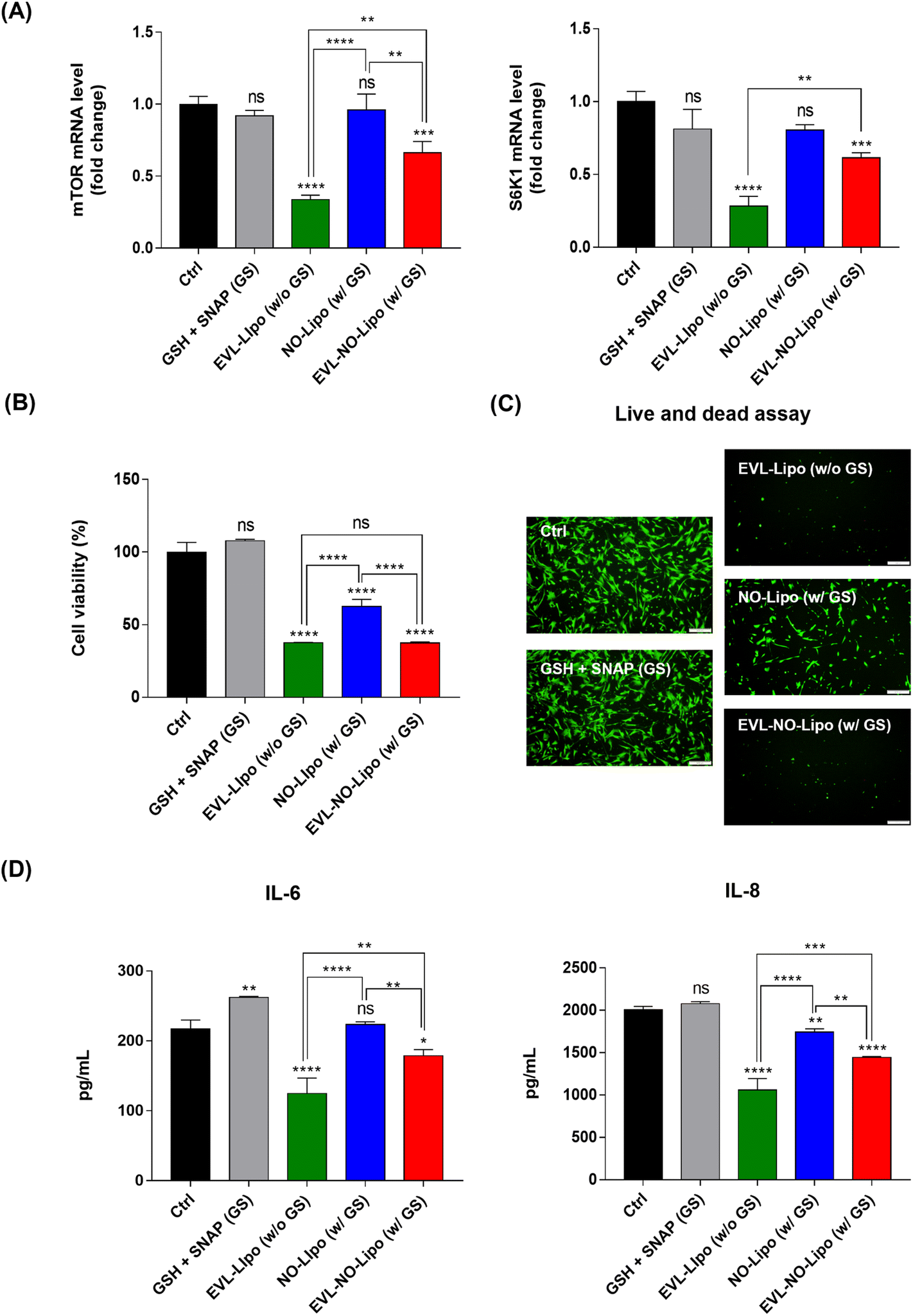
For many years, the FDA has approved the use of rapamycin and its derivates as immunosuppressants to inhibit organ transplant rejections. Especially, the anti-proliferative properties of EVL prompted the development of anti-cancer therapeutics.49 With similar bioactivities, the proliferation of SMC is reduced by inhibiting mTOR, which plays a critical role in regulating cell metabolism, proliferation, and survival in response to environmental stimulations using EVL.50 Serially, the inhibition of mTOR attenuates the phosphorylation of S6K1, resulting in significant downregulation of cell proliferation.51 To prove the occurrence of these processes, the gene expression levels of mTOR and S6K1 were compared in the SMC after incubation with EVL-Lipo and EVL-NO-Lipo. Attenuation of both factors was observed in the group with the EVL-containing group, whereas no suppression was shown by NO (Fig. 6A). These results demonstrated that the EVL-mediated anti-proliferation of HCASMC was induced mainly via inhibition of the mTOR and S6K1-related pathway. Furthermore, synergistic bioactivities of EVL and NO can be verified by showing the inhibitory effects of HCASMC proliferation.52 CCK-8 and live/dead assays were performed to investigate the proliferation inhibitory effects of EVL and NO to HCASMC after 24 h incubation. The group treated with only GSH and SNAP showed similar cell viability as the control group, which supported the weak and limited NO effects from decomposition of NO donors.53 However, the cell viability was reduced to about 60% by NO-Lipo (without EVL). Interestingly, only 30% HCASMC was viable after incubating with 380 μg EVL laden NO-Lipo (EVL-NO-Lipo), which indicated similar inhibitory effects as the group treated with 760 μg EVL encapsulated liposome (EVL-Lipo) (Fig. 6B). The similar trends are displayed in the fluorescence-based live/dead assay (Fig. 6C). These results were in good agreement with the excellent bioactivity in inhibiting HCASMC proliferation through continuous NO release, while reducing the amount of EVL that adversely affected endothelial cell activities using the liposome-containing disulfide bonding group. As shown in Fig. 6D, the production of pro-inflammatory cytokines, IL-6 and IL-8 was significantly reduced in the treatment with EVL and NO. A great anti-inflammatory effect was demonstrated via synergistic effects of EVL and NO due to the powerful anti-inflammatory and anti-proliferation effects of EVL and NO in SMC.54
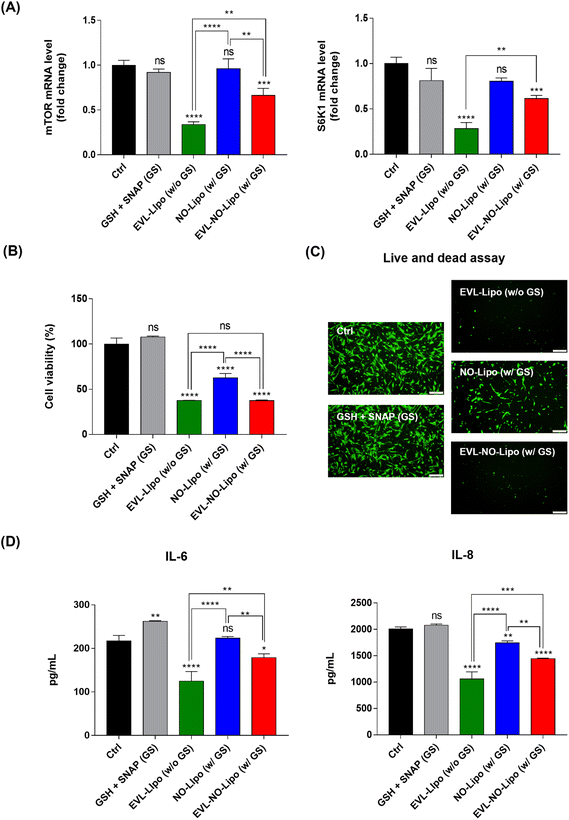 |
| | Fig. 6 Biological activities of EVL, and synergistic effects of EVL and NO from an EVL-NO-Lipo coated Nylon 12 tube. (A) The gene expression levels for the representative mTOR-related pathway of EVL. (B) Cell viability of HCASMC to prove the synergistic effects of EVL and NO. (C) Live/dead assay to visualize the proliferation inhibitory effects of EVL and NO. (D) ELISA to compare the expression of inflammation-related cytokine. (GSH + SNAP (GS), EVL-Lipo (760 μg EVL-encapsulated liposome), NO-Lipo (NO releasing liposome without EVL), EVL-NO-Lipo (380 μg EVL-laden liposome with NO release). Values are presented as mean ± SD (n = 3), and statistical significance was obtained with one-way analysis of ANOVA with Tukey's multiple comparison post-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001)). | |
Hemocompatibility study
In a PCI process to treat vascular diseases, protein adsorption has been known as the first obstacle to induce blood clots due to complex interactions between the blood and the surfaces of the catheter.55 In general, the coating surfaces with high hydrophobicity and roughness induce protein adsorption in the blood vessel. Along with albumin, the level of fibrinogen, as a major protein component of blood plasma, is an important factor to decide the compatibility for further applications of coating materials because fibrinogen accelerates platelet adhesion by transfer to fibrin with thrombin, which results in clot formation.56 Thus, the in vitro hemocompatibility of the coating materials was comparatively analyzed by evaluating the adsorption of albumin and fibrinogen onto multilayer-coated Nylon 12 tubes to prove the feasibility of a multilayer coating strategy for the DEB system. The BCA results showed the adsorption of both proteins did not increase with all coating materials (Fig. 7A). In addition, platelet adhesion was decelerated on the surface with PVP/EVL-NO-Lipo compared to only Nylon 12 tubes, which meant good hemocompatibilities of PVP and liposome as coating materials as indicated in SEM (Fig. 7B). The differences in platelet adhesion depending on the coating materials were additionally evaluated with lactate dehydrogenase (LDH) assay. Platelet adhesion onto the coated surfaces can be indirectly expected by the level of LDH because the LDH level increases with platelet activation.57,58 Compared to the bare Nylon 12 tube with hydrophobic surfaces, the level of platelet activation tended to gradually decrease on the surfaces coated with hydrophilic PVP and EVL-NO-Lipo (Fig. 7C) in agreement with the SEM images for platelet adhesion.
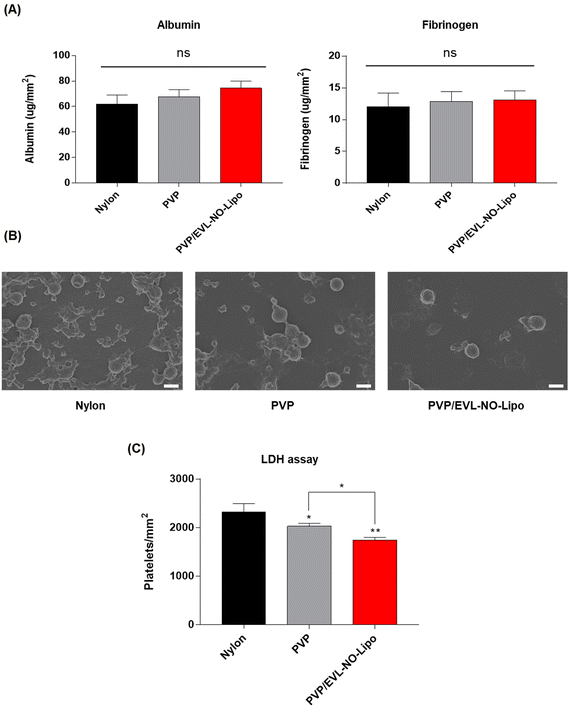 |
| | Fig. 7 Hemocompatibility test. (A) The level of protein adsorption of albumin and fibrinogen on various coating surfaces of Nylon 12 tube. (B) SEM images for platelet adhesion treated with various coating materials (scale bars equal to 4 μm). (C) LDH assay to measure platelet adhesion onto coated surfaces (Values are presented as mean ± SD (n = 3), and statistical significance was obtained with one-way analysis of ANOVA with Tukey's multiple comparison post-test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001)). | |
Ex vivo study for drug transfer on the tissue
To demonstrate the feasibility of future applications for the multilayer coating system, the drug transfer on the tissue from the multilayer-coated balloon system was simulated ex vivo using a UTM system customized from our lab.14 On the opposite site from the tissue of porcine aortic vessel, a multilayer-coated Nylon 12 film consisting of PVP, EVL-NO-Lipo, and PVP was attached. Both sites were compressed for 10 and 60 s with 0.9 bar, which is similar to the pressure on the vascular tissue during the ballooning treatment (Fig. 8A). During the initial 10 s, a red fluorescence signal from the Nile-red labeled liposome was maintained by the protection with the 3rd layer. After 60 s, the liposome layer was separated and transferred to the tissue (Fig. 8B). Because the catheter with DEBs was inserted into the lesion for 10 s and ballooning was performed for drug delivery during the additional 60 s in clinical trials, two time points were selected for monitoring the fluorescence changes.59 The Nile-red labeled liposome was transferred to the surface of the tissue, and the drug transfer rate was measured at 61.10%, which is expected to further increase in an environment where actual blood flow exists (Table 2).
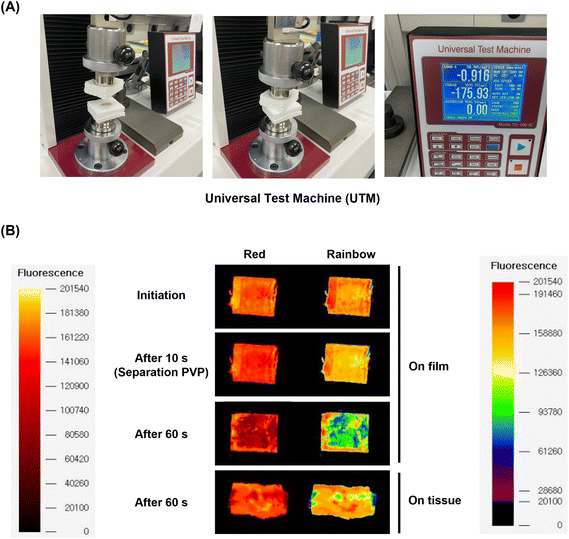 |
| | Fig. 8
Ex vivo study for EVL transfer onto tissue using UTM. (A) Customized UTM set-up to simulate drug transfer onto the tissue of porcine aortic arteries from a PVP/EVL-NO-Lipo/PVP-coated film. (B) Time-dependent fluorescence images of the film and tissue to confirm separation of the 3rd PVP layer and 2nd EVL-NO-Lipo layer (Red: Nile-red labeled liposome). | |
Table 2 Drug transfer rate from the PVP/EVL-NO-Lipo/PVP-coated film to the surface of the tissue with an ex vivo study
| Condition |
Drug transfer rate(%) |
Remained drug of film (%) |
Drug loss (%) |
| PVP/EVL-NO-Lipo/PVP |
61.10 |
20.60 |
18.31 |
Experimental
Materials
Nylon 12 tube and polyvinylpyrrolidone (PVP, average Mw: 3.5 kDa) were purchased from Misumi (Tokyo, Japan) and Acros Organics (Geel, Belgium), respectively. Hydro soy phosphatidylcholine (HSPC), cholesterol (Chol), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (mPEG2000-DSPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[PDP (polyethylene glycol)-2000] (DSPE-PEG (2000) PDP, referred as NO-Lipo) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 4-Amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). 5(6)-Carboxyfluorescein (5(6)-FAM), Nile red, glutathione (GSH), and S-nitroso-N-acetyl-DL-penicillamine (SNAP) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Everolimus (EVL) was provided by Osstem Cardiotech (Seoul, Korea). Phosphate-buffered saline (PBS) solution and fetal bovine serum (FBS) were purchased from Hyclone in GE Life Sciences (Marlborough, MA, USA). Trypsin EDTA, antibiotic-antimycotic, and Hoechst 33342 were obtained from Thermo Fisher Scientific (Waltham, MA, USA). A cell counting kit (CCK 8) was purchased from Dongin LS (Seoul, Korea). Human coronary artery endothelial cells (HCAEC), endothelial cell growth medium-2 (EGM-2), human coronary artery smooth muscle cells (HCASMC), and smooth muscle cell growth medium-2 (SMGM-2) were purchased from Lonza (Basel, Switzerland). Fibrinogen from human plasma and albumin from human serum were purchased from Sigma-Aldrich (St. Louis, MO, USA), and an enzyme linked immunosorbent assay (ELISA) kit for interleukin-6 (IL-6) and interleukin-8 (IL-8) was purchased from R&D Systems Inc. (Minneapolis, MN, USA). The porcine aorta vessel for ex vivo study was obtained from Daegu Gyeongbuk Medical Innovation Foundation (Daegu, Korea).
Preparation of EVL-NO-Lipo
Everolimus (EVL) was loaded in the nitric oxide (NO) releasing-liposome (NO-Lipo) by thin-film hydration method (Lipid:EVL = 20:1). Chloroform was used to dissolve the EVL and NO-Lipo components (HSPC:Chol:DSPE-PEG (2000) PDP = 5.5:4.0:0.5), which were subsequently dried for 15 min at 60 °C in a rotary evaporator with a constant vacuum condition. The thin film was resuspended in deionized water until the lipid membrane was completely hydrated. Then, tip sonication for 3 min and bath sonication for 15 min were serially used to create small unilamellar vesicles (SUVs). To isolate free drugs, liposome dispersion was centrifuged at 3000 rpm for 15 min. Due to the hydrophobicity of EVL, the free EVL aggregated and formed pellets. To concentrate the EVL-laden NO-Lipo (EVL-NO-Lipo) for efficient coating processes onto Nylon 12 tubes, the supernatant containing EVL-NO-Lipo was lyophilized (Laboratory floor model freeze-dryer, Ilshin FD8508, Korea) under vacuum for 24 h at 80 °C, and the EVL-NO-Lipo powder was rehydrated in 20% ethanol solution to adjust the final concentration to 2 mg mL−1.
Characterization of EVL-NO-Lipo
Utilizing dynamic light scattering (DLS, Zeta sizer, Malvern Panalytical, Malvern, UK), the size and surface charge of the EVL-NO-Lipo formulations were measured. The drug encapsulation efficiency (EE) is the ratio for the drug (EVL) encapsulated in EVL-NO-Lipo to the amount of drug used. To calculate the amount of EVL, 0.8 mL of methanol was mixed with 0.2 mL of EVL-NO-Lipo and sonicated with a tip sonicator for 7.5 min to completely break the liposome. The quantification of EVL was determined using high-performance liquid chromatography (HPLC, LC 1100, Agilent, Waldbronn, Germany) with an Agilent Eclipse column (5 micron, 4.6 × 150 mm C-18) at 50 °C. The mobile phase was composed of water, acetonitrile, and methanol (15:45:40), and flowed at a flow rate of 1.0 mL min−1. The profile was observed at a wavelength of 278 nm.
Ultrasonic spray coating
The coating materials were applied to the Nylon 12 tubes using an ultrasonic spray coating instrument (SoniCoater, Noanix Co., Cheongju, Korea). For efficient experiments, Nylon 12 tubes with the same composition as the balloon were utilized. The Nylon 12 tube was cleaned with ethanol and set on a mandrel prior to coating processes. For the 1st layer, PVP dissolved in tetrahydrofuran (THF, 1 mg mL−1) was sprayed in 100 cycles, and EVL-NO-Lipo dissolved in 20% ethanol (2 mg mL−1) were sequentially sprayed for the 2nd layer. To coat 380 μg of EVL for EVL-NO-Lipo, nine-repeated coating cycles were performed, whereas eighteen-repeated coating cycles were needed to coat 760 μg of EVL for EVL-Lipo. Finally, the 3rd layer with PVP solution in acetone (1 mg mL−1) was stacked on the EVL-NO-Lipo coated 2nd layer. All coating processes ran at a flow rate of 0.05 mL min−1 and voltage of 5 kV.
Drug and nitric oxide (NO) release
For mimicking the in vivo blood vessel environments, the sink condition solution, containing 0.05% Tween 20 in PBS solution, was set up at 37 °C shaking with 300 rpm to observe drug release from the multilayer-coated Nylon 12 tubes. The loading and total amounts of EVL were measured using HPLC. The standard curve was created in accordance with drug concentrations in the range of 0.5–1000 mg mL−1 to calculate the amounts of released EVL from the multilayer-coated Nylon 12 tubes. The cumulative amounts of drug were monitored for 7 days. The NO release property from EVL-NO-Lipo was observed with DAF-FM. The fluorescence intensity was measured using a fluorescence-labeled organism bioimaging instrument (FOBI, Neoscience, Seoul, Korea). Fifty μM DAF-FM solution was added to the EVL-NO-Lipo and EVL-NO-Lipo coated Nylon 12 tubes in 100 μL PBS solution containing 50 μM glutathione (GSH) and S-nitroso-N-acetylpenicillamine (SNAP) for 2 h at 37 °C. The time-dependent NO release behavior was detected using a nitric oxide analyzer (NOA; NOA 280i, Zysense, Frederick, Colorado, USA). The time-dependent releasing behaviors of NO from the EVL-NO-Lipo and EVL-NO-Lipo coated Nylon 12 tube were observed in PBS solution containing 10 μM GSH and SNAP during 60 min at 37 °C.
Analysis of surface properties
To visualize the multilayer-coated surface of the Nylon 12 tubes with PVP, EVL-NO-Lipo, and PVP, 5(6)-carboxyfluorescein (5(6)-FAM)-stained PVP (green) and Nile red-loaded NO-Lipo (red) were utilized. The fluorescence intensity was measured using FOBI (Neoscience, Seoul, Korea). The morphology of the cross-section of the coated films was observed by scanning electron microscopy (SEM, JEOL, Tokyo, Japan). Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR, Spectrum two, PerkinElmer, Waltham, MA, USA) was used to measure the IR transmittance of the coated surface at a range of 650 to 4000 cm−1. The ratio of the surface atomic concentration were evaluated with X-ray photoelectron spectroscopy (XPS, PHI5000 VersaProbe, Ulvac-PHI, Chigasaki, Japan). The hydrophilicity of the multilayer-coated surface was evaluated based on the water contact angle (WCA) of a water droplet with the films using optical bench-type contact angle goniometry (Crest Technology Co., Sterling, MA, USA). A drop of 10 μL of deionized water was put on the surface of the film coated with PVP, EVL-NO-Lipo, and PVP, and the WCA was confirmed within 10 s from the specimens.
Cell cultures
Human coronary artery endothelial cells (HCAECs) were cultured using endothelial cells growth medium-2 (EGM-2) containing 5% fetal bovine serum (FBS; GIBCO, NY, USA). Human coronary artery smooth muscle cells (HCASMCs) were cultured using smooth muscle cell growth medium (SmGM) with 5% FBS. Both cells were grown at 37 °C under humidified 5% CO2 atmosphere. The medium was changed every two or three days, and all assays were performed with HCAECs and HCASMCs at passage 7.
Tube formation assay
HCAECs (2.0 × 105 cells per well) were grown for 16 h in EGM-2 media containing 5% FBS on 24 well plates (Corning, NY, USA) coated with Matrigel. NO-Lipo was added to the cells with and without 10 μM GSH and SNAP. Calcein AM-stained living cells were visualized to monitor tube formation using fluorescence microscopy (CKX53, OLYMPUS, Japan). The ImageJ (Wayne Rasband, NIH, USA) plugin for the angiogenesis analyzer was employed to quantify the angiogenesis-related properties.
SMC migration
HCASMCs were cultured until confluent with a monolayer at the starting density of 3.0 × 105 cells per well in 6 well plates. A sterilized 1 ml pipette tip was used to scratch the cells on the center of the wells, followed by the treatment with NO-Lipo with and without GHS and SNAP after rinsing with PBS solutions. A microscope was used to visualize the migration of SMC after 24 h incubation. The ImageJ (Wayne Rasband, NIH, USA) plugin for the wound healing tool was utilized to assess the percentage of open area.
RT-qPCR for angiogenesis and mTOR-related factors
RNA was extracted from NO-Lipo treated HCAEC (3.0 × 105 cells per well) and EVL-Lipo treated HCASMC (1 × 105 cells per well) using the universal RNA extraction kit (Accuprep® Universal RNA Extraction Kit, Daejeon, K-3141, Bioneer) and TRIzol reagent (15596018, Thermo Fisher Scientific) following the manufacturer's instructions. A spectrophotometer (ND-1000; Thermo Fisher Scientific) was used to determine the RNA concentration. Using a PrimeScript RT Reagent Kit, RNA from each sample was reverse transcribed to complementary DNA. RT-qPCR was performed with a QuantStudio 3 real-time PCR (Applied Biosystems, Waltham, Massachusetts, United States) instrument and Power SYBR Green PCR Master Mix.
Enzyme-linked immunosorbent assay (ELISA)
The protein expression level of inflammation-related factors in HCASMC incubated with coating materials were analyzed using the Quantikine™ ELISA kit (R&D Systems, Minneapolis, MN, USA). The level of IL-6 and IL-8 was measured from cell conditioned media using ELISA after 2 h incubation of coating materials in the condition with and without GSH and SNAP to verify the synergistic effects of EVL and NO. ELISA was performed in accordance with the manufacturer's instructions.
Hemocompatibility test
The protein adsorption on the coated films was evaluated using standard curves of human serum albumin and human plasma fibrinogen. The coated Nylon 12 film was hydrated with PBS solution for 1 h at 37 °C, and then incubated with albumin solution (3.0 mg mL−1) and fibrinogen solution (0.2 mg mL−1) for 1 min. Following the manufacturer's instructions, the sample was washed with PBS solution and the amounts of protein were evaluated using a micro-BCA kit. For a platelet adhesion study, all coating materials were hydrated in 1 mL of PBS solution at 37 °C for 1 h, and then incubated with the concentrated platelet (5.0 × 104 platelets per μL, Nambu Blood Institute, Seoul, Korea). To immobilize the adhered platelet on specimens, 2.5% glutaraldehyde solutions were immersed and serial dehydration was performed with 50, 60, 70, 80, 90 and 100% ethanol. Afterward, platelet adhesion was visualized using field emission-scanning electron microscopy (FE-SEM; SIGMA, Carl Zeiss, Jena, Germany). In addition, the lactate dehydrogenase (LDH) assay was carried out to measure platelet adhesion to the coating materials (LDH cytotoxicity detection kit, TAKARA, Tokyo, Japan).
Ex vivo drug transfer study
Using a universal testing machine (UTM, TO-101, Test one, Busan, Korea), the ex vivo drug transfer rate was evaluated with porcine aortic arteries. In brief, a multilayer-coated film was attached to the surface of the test mold and the porcine aortic vessel was put on the opposite site. Then, two molds were compressed for 10 and 60 s with 0.9 bar pressure. After detachment from the mold, the tissue was immersed in chloroform and the multilayer-coated film was immersed in methanol to quantify the amount of EVL. The Nile red-labeled liposome was used to visualize the EVL-Lipo transfer onto tissues using FOBI.
Statistical analysis
Values are presented as mean ± SD (n = 3). GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) was performed all statistical analyses. One-way analysis of variance (ANOVA) and Tukey's multiple comparison post-test were used to compare group differences, and p values bellow 0.05 were regarded as statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Conclusions
In a previous study, we developed the multilayer-coated DEB system, containing PVP/EVL encapsulated liposome/PVP for efficient protection and transfer of drugs during DEB treatments. To overcome limitations derived from the high dosage of cytotoxic drugs, a strategy to reduce the drug amounts was suggested herein in a multilayer-coated DEB system. Continuous NO release, achieved by introducing liposome containing the disulfide bonding group in the presence of GSH and SNAP, was expected to show synergistic bioactivities with low amounts of drug. Moreover, the liposome was utilized to load hydrophobic EVL without crystallization in a hydrophilic condition. With bioactivities of NO, such as facilitating angiogenesis of EC and inhibiting migration of SMC in the lesions, biological effects on EC and SMC were demonstrated with half amounts of EVL. Moreover, changes of angiogenic related factors by NO and mTOR pathway associated factors by EVL were demonstrated at the gene level. With EVL-laden liposome releasing NO, multilayer-coated surfaces inhibited platelet adhesion due to the hydrophilic properties of the coating materials. Finally, the ex vivo study result with the UTM demonstrated the feasibilities of the PVP/EVL-NO-Lipo/PVP coated DEB system for future in vivo applications with drug protection and transfer abilities during a PCI treatment.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
This research was supported by the Basic Science Research Program (2020R1A2B5B03002344 and 2022R1I1A1A01068486) and Bio & Medical Technology Development Program (2018M3A9E2024579) through the National Research Foundation of Korea funded by the Ministry of Science and ICT (MSIT), and the Korea Medical Device Development Fund grant funded by the Korea government (the Ministry of Science and ICT, the Ministry of Trade, Industry and Energy, the Ministry of Health & Welfare, the Ministry of Food and Drug Safety) (202011A05-05), Republic of Korea.
References
- P. Libby and P. Theroux, Circulation, 2005, 111, 3481–3488 CrossRef PubMed.
- M. Alghrairi, N. Sulaiman and S. Mutashar, Sensors, 2020, 20, 1–23 CrossRef PubMed.
- S. Lee, B. W. Park, Y. J. Lee, K. Ban and H. J. Park, J. Tissue Eng., 2020, 11, 1–10 CAS.
- E. Lih, C. H. Kum, W. Park, S. Y. Chun, Y. Cho, Y. K. Joung, K.-S. Park, Y. J. Hong, D. J. Ahn, B.-S. Kim, T. G. Kwon, M. H. Jeong, J. A. Hubbell and D. K. Han, ACS Nano, 2018, 12, 6917–6925 CrossRef CAS PubMed.
- R. A. Byrne, G. W. Stone, J. Ormiston and A. Kastrati, Lancet, 2017, 390, 781–792 CrossRef PubMed.
- E. X. Han, J. Wang, M. Kural, B. Jiang, K. L. Leiby, N. Chowdhury, G. Tellides, R. G. Kibbey, J. H. Lawson and L. E. Niklason, J. Tissue Eng., 2021, 12, 1–8 CAS.
- D.-W. Jeong, W. Park, T. M. Bedair, E. Y. Kang, I. H. Kim, D. S. Park, D. S. Sim, Y. J. Hong, W.-G. Koh, M. H. Jeong and D. K. Han, Biomater. Sci., 2019, 7, 2499–2510 RSC.
- A. Hertault, F. Chai, M. Maton, J. Sobocinski, P. Woisel, B. Maurel, J. Lyskawa and N. Blanchemain, Biomater. Sci., 2021, 9, 212–220 RSC.
- W. Khan, S. Farah and A. J. Domb, J. Controlled Release, 2012, 161, 703–712 CrossRef CAS PubMed.
- T. Weichhart, M. Hengstschläger and M. Linke, Nat. Rev. Immunol., 2015, 15, 599–614 CrossRef CAS PubMed.
- U. Saran, M. Foti and J. F. Dufour, Clin. Sci., 2015, 129, 895–914 CrossRef CAS PubMed.
- W. Xu, M. Sasaki and T. Niidome, Pharmaceutics, 2022, 14, 492 CrossRef CAS PubMed.
- I. Bae, K. S. Lim, J. K. Park, J. H. Song, S. H. Oh, J. W. Kim, Z. Zhang, C. Park and J. T. Koh, Biomater. Res., 2021, 25, 1–8 CrossRef PubMed.
- H. I. Lee, W. K. Rhim, E. Y. Kang, B. Choi, J. H. Kim and D. K. Han, Pharmaceutics, 2021, 13, 614 CrossRef CAS PubMed.
- X. Tang, L. Chen, Z. Wu, Y. Li, J. Zeng, W. Jiang, W. Lv, M. Wan, C. Mao and M. Zhou, Small, 2022, 2203238, 1–13 Search PubMed.
- J. E. Barbato and E. Tzeng, J. Vasc. Surg., 2004, 40, 187–193 CrossRef PubMed.
- A. Cziráki, Z. Lenkey, E. Sulyok, I. Szokodi and A. Koller, Front. Pharmacol., 2020, 11, 1–20 CrossRef PubMed.
- C. Porrini, N. Ramarao and S. L. Tran, Biol. Chem., 2020, 401, 547–572 CrossRef CAS PubMed.
- A. Wijaya, Y. Wang, D. Tang, Y. Zhong, B. Liu, M. Yan, Q. Jiu, W. Wu and G. Wang, J. Mater. Chem. B, 2022, 10, 607–624 RSC.
- Y. Zhao, X. Ouyang, Y. Peng and S. Peng, Pharmaceutics, 2021, 13, 1917 CrossRef CAS PubMed.
- S. Guber, T. Ebrahimian, M. Heidari, N. Eliopoulos and S. Lehoux, Sci. Rep., 2018, 8, 1–11 CAS.
- S. J. Wimalawansa, Expert Opin. Pharmacother., 2008, 9, 1935–1954 CrossRef CAS PubMed.
- M. Kelm, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 273–289 CrossRef CAS PubMed.
- K. Gwon, W. Il Choi, S. Lee, J. S. Lee and J. H. Shin, Biomater. Sci., 2021, 9, 8160–8170 RSC.
- H. Feng, J. H. Kang, S. Qi, A. Kishimura, T. Mori and Y. Katayama, RSC Adv., 2021, 11, 34101–34106 RSC.
- Z. Luo, Y. Zhou, T. Yang, Y. Gao, P. Kumar and R. Chandrawati, Small, 2022, 18, 1–10 Search PubMed.
- A. Colletta, J. Wu, Y. Wo, M. Kappler, H. Chen, C. Xi and M. E. Meyerhoff, ACS Biomater. Sci. Eng., 2015, 1, 416–424 CrossRef CAS PubMed.
- D. Y. Wang, H. C. van der Mei, Y. Ren, H. J. Busscher and L. Shi, Front. Chem., 2020, 7, 1–15 RSC.
- G. Romano, M. Costantini, I. Buttino, A. Ianora and A. Palumbo, PLoS One, 2011, 6, e25980 CrossRef CAS PubMed.
- H. Qiu, P. Qi, J. Liu, Y. Yang, X. Tan, Y. Xiao, M. F. Maitz, N. Huang and Z. Yang, Biomaterials, 2019, 207, 10–22 CrossRef CAS PubMed.
- L. Hakuna, B. Doughan, J. O. Escobedo and R. M. Strongin, Analyst, 2015, 140, 3339–3342 RSC.
- H. Yu, S. Yu, H. Qiu, P. Gao, Y. Chen, X. Zhao, Q. Tu, M. Zhou, L. Cai, N. Huang, K. Xiong and Z. Yang, Bioact. Mater., 2021, 6, 1618–1627 CrossRef CAS PubMed.
- N. Hasan, J. Lee, H. J. Ahn, W. R. Hwang, M. A. Bahar, H. Habibie, M. N. Amir, S. Lallo, H. J. Son and J. W. Yoo, Pharmaceutics, 2022, 14, 22 CrossRef CAS PubMed.
- N. Sharma, A. K. Dhyani, S. Marepally and D. A. Jose, Nanoscale Adv., 2020, 2, 463–469 RSC.
- N. Ma, W. Liu, L. Ma, S. He, H. Liu, Z. Zhang, A. Sun, M. Huang and C. Zhu, e-Polym., 2020, 20, 346–352 CrossRef CAS.
- X. Zhao, C. Shi, X. Zhou, T. Lin, Y. Gong, M. Yin, L. Fan, W. Wang and J. Fang, Eur. J. Pharm. Sci., 2019, 138, 104994 CrossRef CAS PubMed.
- A. Martinez-Urrutia, P. Fernandez de Arroiabe, M. Ramirez, M. Martinez-Agirre and M. Mounir Bou-Ali, Int. J. Refrig., 2018, 95, 182–188 CrossRef CAS.
-
M. Bejaoui, H. Galai, F. Touati and S. Kouass, Dos. Forms, 2-21, IntechOpen, 99431.
- E. Pilch and W. Musiał, Int. J. Mol. Sci., 2018, 19, 3806 CrossRef PubMed.
- M. S. Joshi, P. J. Berger, D. M. Kaye, J. T. Pearson, J. A. Bauer and R. H. Ritchie, Clin. Exp. Pharmacol. Physiol., 2013, 40, 253–261 CrossRef CAS PubMed.
- S. Dimmeler and A. M. Zeiher, Cell Death Differ., 1999, 6, 964–968 CrossRef CAS PubMed.
- Y. N. Han, Y. J. Lee, K. J. Kim, S. J. Lee, J. Y. Choi, S. H. Moon and J. W. Rhie, Tissue Eng. Regener. Med., 2021, 18, 179–186 CrossRef CAS PubMed.
- W. Liu, Y. Yuan and D. Liu, Tissue Eng. Regener. Med., 2021, 18, 1035–1044 CrossRef CAS PubMed.
- M. Reinero, M. Beghetti, P. Tozzi, L. K. von Segesser, M. Samaja and G. Milano, J. Cardiovasc. Pharmacol. Ther., 2021, 26, 665–676 CrossRef CAS PubMed.
- X. Yang, Y. Yang, J. Guo, Y. Meng, M. Li, P. Yang, X. Liu, L. H. H. Aung, T. Yu and Y. Li, Mol. Ther. – Nucleic Acids, 2021, 23, 1136–1160 CrossRef CAS PubMed.
- Y. Zhuge, J. Zhang, F. Qian, Z. Wen, C. Niu, K. Xu, H. Ji, X. Rong, M. Chu and C. Jia, Int. J. Biol. Sci., 2020, 16, 2741–2751 CrossRef PubMed.
- R. K. Dubey, M. Rosselli, B. Imthurn, P. J. Keller and E. K. Jackson, Hum. Reprod. Update, 2000, 6, 351–363 CrossRef CAS PubMed.
- T. Matsumoto, M. Kojima, K. Takayanagi, K. Taguchi and T. Kobayashi, Am. J. Hypertens., 2020, 33, 793–803 CrossRef CAS PubMed.
- C. Vaury, P. Macaire, F. Goirand, B. Coudert, P. Guérard and A. Schmitt, J. Oncol. Pharm. Pract., 2021, 27, 235–237 CrossRef PubMed.
- S. K. Hwang and H. H. Kim, BMB Rep., 2011, 44, 506–511 CrossRef CAS PubMed.
- J. Ghosh and R. Kapur, Exp. Hematol., 2017, 50, 13–21 CrossRef CAS PubMed.
- R. Sarkar and R. C. Webb, J. Vasc. Res., 1998, 35, 135–142 CrossRef CAS PubMed.
- Z. A. Massy, D. Borderie, T. Nguyen-Khoa, T. B. Drüeke, O. G. Ekindjian and B. Lacour, Nephrol., Dial., Transplant., 2003, 18, 153–157 CrossRef CAS PubMed.
- D. Vitiello, P. E. Neagoe, M. G. Sirois and M. White, Cell. Mol. Immunol., 2015, 12, 40–52 CrossRef CAS PubMed.
- F. Batool, H. Özçelik, C. Stutz, P. Y. Gegout, N. Benkirane-Jessel, C. Petit and O. Huck, J. Tissue Eng., 2021, 12, 1–19 CAS.
- Y. K. Sung, D. R. Lee and D. J. Chung, Biomater. Res., 2021, 25, 1–10 CrossRef PubMed.
- B. Sivaraman and R. A. Latour, Biomaterials, 2010, 31, 832–839 CrossRef CAS PubMed.
- M. S. Williams, B. S. Coller, H. J. Väänänen, L. E. Scudder, S. K. Sharma and J. D. Marmur, Circulation, 1998, 98, 742–748 CrossRef CAS PubMed.
- M. Rockley, P. Jetty, G. Wells, K. Rockley and D. Fergusson, Syst. Rev., 2019, 8, 1–7 CrossRef PubMed.
Footnote |
| † These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |

 a,
Seung-Woon
Baek
abc,
Jun Yong
Kim
a,
Seung-Woon
Baek
abc,
Jun Yong
Kim