
Single-component lipid nanoparticles for engineering SOCS1 gene-silenced dendritic cells to boost tumor immunotherapy†
Zexuan
Yu‡
a,
Mengtong
Wu‡
a,
Yingshuang
Huang
a,
Yishu
Wang
a,
Yijun
Chen
a,
Qiulin
Long
a,
Ziming
Lin
a,
Lingjing
Xue
a,
Caoyun
Ju
*a and
Can
Zhang
 *ab
*ab
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing 210009, P.R. China. E-mail: zhangcan@cpu.edu.cn; jucaoyun@cpu.edu.cn
bChongqing Innovation Institute of China Pharmaceutical University, Chongqing 401135, China
First published on 14th November 2022
Abstract
Dendritic cells (DCs) that can prime antitumor responses show great potential in tumor immunotherapy, whereas the unsatisfactory effect which can be ascribed in part to the high expression of inhibitory cytokines, such as the suppressor of cytokine signaling 1 (SOCS1), restricts their application. Thus, silencing these genes in DCs is essential for DC-based therapy. However, safe and effective delivery of siRNA to DCs still faces challenges. Herein, we designed single-component lipid nanoparticles comprising a solely cationic lipid (OA2) for introducing siRNA into mouse DCs in order to inhibit the immunosuppressive gene and boost the effector responses of DC-based therapy. Compared to other multi-component lipid nanoparticles, single-component lipid nanoparticles are theoretically easy-to-control and detective, which is beneficial for future translation. We showed that the application of OA2 lipid nanoparticles significantly downregulated the expression of SOCS1 in DCs over 50%, compared with the commercial lipofectine2000. Besides, the treatment of OA2 lipid nanoparticles had no influence on the antigen capture of DCs. Thus, we fabricated a SOCS1-downregulated DC vaccine pulsed with Ova antigen and demonstrated that the antigen presentation and pro-inflammatory factor secretion ability of DCs were improved due to the SOCS1 downregulation, leading to an ameliorated immunosuppressive tumor microenvironment and finally exhibiting potent tumor prevention and suppression in B16-Ova tumor-bearing mice. Single-component lipid nanoparticles, which provide an available vector platform for siRNA delivery to primary DCs, appear to be a potent tool to engineer DCs and in turn boost DC-based tumor immunotherapy.
1. Introduction
Dendritic cells (DCs) are professional antigen-presenting cells that can efficiently capture, process, and present antigens to induce antigen-specific lymphocyte responses, thus playing an important role in initiating and regulating tumor-specific immunity.1–3 Leveraging these naive characteristics, DCs have been fabricated as DC vaccines ex vivo by pulsing with tumor-specific antigens and stimulating to maturation for tumor immunotherapy in cancer patients.4–6 For example, the first FDA-approved therapeutic vaccine, Provenge (Sipuleucel-T), uses activated DCs and other blood mononuclear cells for the treatment of prostate cancer. Despite the superior safety of DC vaccines in patients, the unsatisfactory effect with a low clinical response rate has restricted their application.7 This can be ascribed mainly to the limited immunogenic function of DCs due to the intrinsic suppressive mechanisms during DC maturation,7 such as the expression of the suppressor of cytokine signaling 1 (SOCS1), a major negative-feedback factor of immune responses in immune cells.8–10 Therefore, downregulating the SOCS1 expression in DCs would be expected to provide an enhanced activation and maturation of DCs, thereafter resulting in boosted antigen-specific tumor immunity.11,12Small interfering RNA (siRNA) is a specific and powerful tool for the degradation of target mRNA and blocking its subsequent protein translation both in vitro and in vivo.13 To date, the FDA has approved five siRNA therapeutics in treating several genetic diseases, indicating its huge potential in gene therapy. Nevertheless, siRNA mediated gene silencing in DCs still faces challenges.14 The conventional methodology for gene silencing in DCs includes electroporation and viral transduction. Although both of them are valid, electroporation which requires special equipment usually causes high mortality rate of cells,15,16 while viral transduction possesses certain potential risks in inducing serious side effects.17 In contrast, non-viral vectors, such as lipid-based nanoparticles, attract more and more attention for delivering siRNA to DCs because of their safety and biocompatibility.18,19 The typical lipid-based nanoparticles as gene vectors usually comprise several functional lipids such as cationic lipids or ionizable lipids for binding siRNA and facilitating endo/lysosomal escape,20 neutral phospholipid and cholesterol for improving the stability or membrane fusion capacity,21,22 and PEGylated lipids for enhancing the in vivo stability.23,24 Despite the effectiveness of each component, the more the components, the greater the complexity in controlling the reproducibility and stability of lipid-based nanoparticles, which hinders their industrialization to some extent.25 That is, lipid-based nanoparticles with fewer components and comparable gene delivery ability might be more promising in future applications.
Herein, we developed single-component lipid nanoparticles (OA2 LPs) which are composed of a solely novel cationic lipid (OA2) via the thin-film extrusion method for delivering SOCS1-targeting siRNA (SOCS1 siRNA) to mouse bone marrow dendritic cells (BMDCs) (Fig. 1). We first evaluated the gene silencing effect mediated by OA2 LPs in DCs. Next, the influence of SOCS1-downregulation by OA2 LPs on the capture of the tumor antigen by DCs as well as the subsequent expression of SOCS1 in DCs after antigen capture were explored. Afterwards, SOCS1-downregulated DC vaccines were fabricated by pulsing with the model tumor antigen, ovalbumin (Ova), to gain Ova-specific immunity. We found that SOCS1-downregulated DC vaccines have an enhanced antigen presentation ability and pro-inflammatory factor secretion ability of DCs both in vivo and in vitro, resulting in potent antitumor immunity through modulating the tumor microenvironment including increased immune cell infiltration and enhanced activation of T cells, thereby leading to dramatic tumor prevention and suppression in melanoma B16-OVA tumor-bearing mice with prolonged survival. The study provides a safe, efficient and convenient vector platform for introducing siRNA into primary DCs, which can be further developed to boost the response rate of DC vaccines in the clinic.
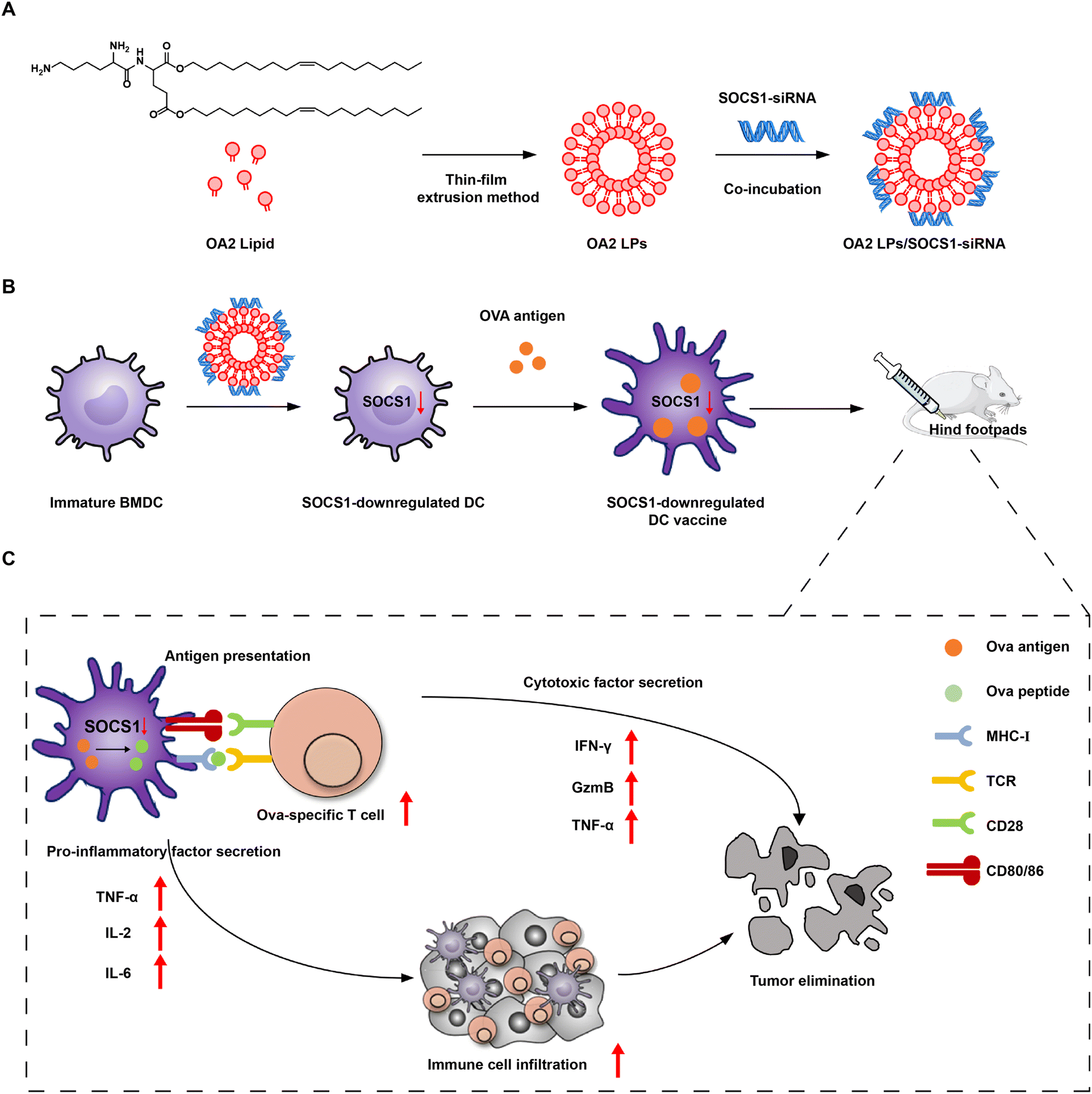 | ||
| Fig. 1 Schematic illustration of the preparation of SOCS1-downregulated DC vaccine via OA2 lipid nanoparticles (OA2 LPs) and the in vivo action in antitumor immunotherapy. (A) Preparation of OA2 LPs/SOCS1-siRNA. (B) Preparation of SOCS1-downregulated DC vaccine. (C) The in vivo antitumor immunotherapy of SOCS1-downregulated DC vaccine after injection into the hind footpads of melanoma B16-OVA tumor-bearing mice through enhancing pro-inflammatory factor secretion to modulate the immunosuppressive environment and boosting antigen presentation to improve the tumor killing effect of infiltrated antigen-specific T cells. | ||
2. Experimental
2.1. Materials
SOCS1-siRNA (sense strand: 5′-GCATCCGCGTGCACTTCCA-3′, antisense strand: 5′-TGGAAGTGCACGCGGATGC-3′), scrambled siRNA (NC-siRNA, sense strand: 5′-UUCUCCGAACGUGUCACGU-3′, antisense strand: 5′-AAGAGGCUUGCACAGUGCA-3′), FAM-labelled siRNA (Fam-siRNA), and Cy5-labelled siRNA (Cy5-siRNA) were purchased from RiboBio Co., Ltd (Guangzhou, China). Oleyl alcohol and L-glutamic acid were obtained from Aladdin Reagent Inc. (Shanghai, China), Boc–Lys(Boc)–OH was purchased from glbiochem Ltd (Shanghai, China). Other chemicals were acquired from Aladdin Reagent Inc. (Shanghai, China). All solvents were obtained from Shanghai Chemical Co. (Shanghai, China). Lipofectamine™ 2000 (Lipo2000, Invitrogen™) used as a positive control was purchased from Thermo Fisher Scientific Inc. (Massachusetts, USA) and used according to the manufacturer's instructions. Ovalbumin (Ova) was obtained from Sigma (MO, USA). Agarose, 6× nucleic acid loading buffer, LysoTracker Red, 5× protein sample loading buffer, phenylmethanesulfonyl fluoride (PMSF), and Hoechst 33342 were purchased from Sunshine Biotechnology Co., Ltd (Nanjing, China). Goldview was obtained from Sbsbio Inc. (Beijing, China). Antibodies used were acquired from BioLegend, Inc. (San Diego, USA). TRIzol® reagent (Invitrogen™) and RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific™) were purchased from Vazyme Biotech Co., Ltd (Nanjing, China). All primers were obtained from Sangon Biotech Co., Ltd (Shanghai, China). Granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from Peprotech (USA). The EasySep™ Mouse CD8+ T cell isolation kit, mouse IL-2, and CD3/CD28 beads were purchased from StemCell Technologies Inc. (USA). The LDH cytotoxicity assay kit was purchased from Beyotime Biotechnology (Shanghai, China).2.2. Mouse and cell lines
C57BL/6 mice (20–22 g, male) were purchased from Qinglongshan Animal Breeding Ground (Nanjing, China). OT-1 mice (18–20 g, male) were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). All animals were pathogen free and were allowed to access food and water freely. The Animal Care and Use Committee of the China Pharmaceutical University approved all of the animal experiments.The mouse melanoma cell line B16-Ova cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). B16-Ova cells were cultured in DMEM with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin–streptomycin. These cells were all cultured in an incubator (Thermo Scientific, USA) at 37 °C with a humidified atmosphere of 5% CO2.
2.3. Preparation and characterization of OA2 LPs/siRNA
The OA2 lipid was synthesized in three steps, and the detailed synthetic methods are shown in the ESI. The compound was confirmed via1H NMR, 13C NMR and high-resolution mass spectroscopy (HRMS). HRMS was recorded on a QSTAR XL Hybrid MS/MS mass spectrometer. 1H NMR and 13C NMR spectra were recorded on Bruker AV-300 or AV-500 instruments using CDCl3. Chemical shifts are presented as δ (ppm) units relative to the internal standard tetramethylsilane (TMS). Column chromatography separations were performed on silica gel (100–300 mesh).OA2 LPs were prepared via the thin-film extrusion method. In brief, OA2 lipid were dissolved in a mixture of CHCl3 and methanol (3/2, v/v), followed by evaporation to form a thin film. 5 mL of ultrapure water was then added to hydrate the film at 37 °C, followed by sequentially squeezing through 0.8 μm, 0.4 μm, and 0.2 μm membrane 11 times, respectively, to gain OA2 LPs. Afterwards, the OA2 LPs were incubated with siRNA for 30 min at different N/P ratios to form the OA2 LPs/siRNA complex. The N/P represents the molar ratio of nitrogen in OA2 LPs to phosphate in siRNA.
The morphology of OA2 LPs was observed using a high-resolution transmission electron microscope (HRTEM) with an operating voltage of 100.0 kV (JEM-2100F, Hitachi, Japan). The particle size and zeta potential of OA2 LPs and OA2 LPs/siRNA with the corresponding polydispersity index (PDI) were determined by a Zetasizer Nano ZS instrument (Malvern Instruments, U.K.). To investigate the stability of OA2 LPs/siRNA under transfection conditions, the particle sizes of OA2 LPs/siRNA in DC culture medium were measured at different time points for 24 h at 37 °C. Agarose gel electrophoresis assay was used for the evaluation of siRNA encapsulation by OA2 LPs with different N/P ratios. All samples containing 200 ng of siRNA were loaded onto 1% agarose gel for electrophoresis (100 V, 30 min). The gel was stained by Goldview and visualized on the gel imaging system (Tanon-3500, Shanghai, China).
2.4. Induction of BMDCs in vitro
BMDCs were induced from the bone marrow cells of male C57BL/6 mice using the modified Inaba method. In brief, the bone marrow cells (5 × 105 cells per mL) in the tibias and femurs were collected and cultured in a 12-well cell culture plate containing 2 mL of 1640 RPMI medium with 10% (v/v) FBS, 1% (v/v) penicillin–streptomycin, and 20 ng mL−1 GM-CSF. Then, the cells were cultured at 37 °C under a humidified atmosphere of 5% CO2 and the culture medium was changed on day 2, day 4 and day 5. On day 6, non-adherent and loosely adherent cells (BMDCs) were harvested.The cells were labelled with PE anti-mouse CD11c antibody and FITC anti-mouse CD86 antibody, and the expressions of CD11c and CD86 on BMDCs cells were determined using an Attune flow cytometer (Attune NxT, Thermo Fisher Scientific). The Inaba method allowed the harvesting of BMDCs with 91.3% purity and 33.3% maturity on day 6.
2.5. Evaluation of SOCS1 gene silencing effect in BMDCs
BMDCs were seeded into 12-well cell culture plates at a density of 2 × 106 cells per mL and cultured at 37 °C under a humidified atmosphere of 5% CO2. The cells were respectively incubated with Lipo2000/SOCS1-siRNA or OA2 LPs/SOCS1-siRNA at a dose of 2.5 μg of siRNA for 6 h. The cells were washed with PBS twice and re-suspended in fresh culture medium for another incubation of 48 h at 37 °C.For evaluation of SOCS1 mRNA levels in BMDCs, the total RNA was extracted by TRIzol® reagent according to the manufacturer's instructions. 2 μg of total RNA was transcribed into cDNA using RevertAid™ first strand cDNA synthesis kit. 0.2 μL of β-actin/SOCS1 forward primer, 0.2 μL of β-actin/SOCS1 reverse primer, 3 μL of DEPC water, 5 μL 2 × AceQ qPCR SYBR Green, and 1 μL cDNA were mixed together and subjected to quantitative RT-PCR assays (Applied Biosystems). The primers used in the qRT-PCR were 5′-Atggtagcacacaaccaggtggcagcc-3′ (SOCS1 forward), 5′-Tcatatctggaaggggaaggagctcaag-3′ (SOCS1 reverse), 5′-Ccaaccgcgagaagatga-3′ (β-actin forward), and 5′-Ccagaggcgtacagggatag-3′ (β-actin reverse), respectively.
For the analysis of SOCS1 protein levels, total protein was extracted from BMDCs. Protein samples with the same concentration were separated on 10% SDS-PAGE gel and then transferred onto PVDF membrane. After incubating with anti-SOCS1 or anti-β-actin monoclonal antibodies followed by goat anti-rabbit IgG H&L (ab150083, Abcam), the protein bands were visualized with the ECL system (Tanon 3500).
For the determination of cell apoptosis, the annexin V-FITC apoptosis detection kit (Vazyme) was used according to the manufacturer's instructions.
2.6. Cellular uptake and intracellular transport of OA2 LPs/siRNA
In the cellular uptake study, BMDCs were seeded into 12-well cell culture plates at a density of 2 × 106 cells per mL and cultured at 37 °C under a humidified atmosphere of 5% CO2. The cells were respectively incubated with Lipo2000/Fam-siRNA or OA2 LPs/Fam-siRNA at a dose of 2.5 μg of siRNA for 6 h. The cells were then collected for flow cytometry analysis and confocal laser scanning microscope (CLSM, Leica TCS SP5, Germany) observation.In the intracellular tracking study of OA2 LPs/siRNA, BMDCs were seeded into 12-well cell culture plates at a density of 2 × 106 cells per mL and cultured at 37 °C under a humidified atmosphere of 5% CO2. The cells were incubated with OA2 LPs/Fam-siRNA at a dose of 2.5 μg of siRNA for 15 min, 1 h, and 6 h, respectively, followed by observation by CLSM. The early endosomes, late endosomes, and lysosomes were obtained using the anti-EEA1 monoclonal antibody (ab09110, Abcam), anti-Rab7 monoclonal antibody (ab126712, Abcam), and LysoTracker Red separately. Fifteen minutes prior to observation, the nuclei were stained by Hoechst 33342.
2.7. Construction and characterization of SOCS1-downregulated DC vaccine
BMDCs were seeded into 12-well cell culture plates at a density of 2 × 106 cells per mL and cultured at 37 °C under a humidified atmosphere of 5% CO2. OA2 LPs/SOCS1-siRNA was added to the cells at a dose of 2.5 μg of siRNA and incubated for 6 h, followed by washing twice with PBS. 1 mL of fresh DC culture medium containing Ova (50 μg mL−1) was then added and incubated for another 6 h. After that, the cells were collected and washed twice with PBS to obtain the SOCS1-downregulated DC vaccine.Cy5-siRNA or Atto-488 Ova was used instead of SOCS1-siRNA or Ova antigen to determine the uptake of Ova or siRNA by BMDCs. The uptake of OVA or siRNA by BMDCs was determined by measuring the percentage of positive cells at 6 h via flow cytometry. The intracellular localization of siRNA and OVA antigen in BMDCs was observed by CLSM after incubation for 6 h.
2.8. Evaluation of pro-inflammatory factor secretion and antigen presentation of BMDCs in vitro
To study the secretion of the pro-inflammatory factor of BMDCs in vitro, LPS was used to stimulate the maturation of BMDCs after treatment with different BMDC formulations. The mRNA levels of TNF-α, IL-6, IL-2 were evaluated by q-PCR assays at 36 h after Ova uptake by BMDCs according to the standard protocol of q-PCR mentioned above.For the evaluation of the antigen presentation ability, the specific tetramer-positive T cells after treatment with different DC formulations was detected as follows. Different DC formulations were incubated with naive OT-1 T cells isolated from the spleens of OT-1 mice for 48 h at a ratio of OT-1 T cells to DCs of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then the cells were collected and stained by APC/Cyanine7 anti-mouse CD8a antibody (100714, biolegend) and PE anti-mouse H-2Kb bound to SIINFEKL antibody (141603, biolegend) for flow cytometry assay.
:1. Then the cells were collected and stained by APC/Cyanine7 anti-mouse CD8a antibody (100714, biolegend) and PE anti-mouse H-2Kb bound to SIINFEKL antibody (141603, biolegend) for flow cytometry assay.
The activation, proliferation, cytokine secretion, and cytotoxicity of OT-1 T cells after treatment with different DC formulations were measured to evaluate the stimulation of T cells by DCs. For the activation status of OT-1 T cells, the cells were stained by APC/Cyanine 7 anti-mouse CD8a (100714, Biolegend) and FITC anti-mouse CD69 (104505, Biolegend) for flow cytometry assay. For the proliferation assay, the cells were stained with CFSE at a final concentration of 2 μM at 37 °C for 15 min. Then, the cells were washed twice and resuspended in 1640 medium containing IL-2, followed by the addition of anti-CD3/CD28 beads at a bead-to-cell ratio of 1:1. After 48 h, the cells were collected and analysed by flow cytometry. For the cytokine secretion of OT-1 T cells, the cells were resuspended using 1640 complete medium containing 50 ng mL−1 PMA, 5 μg mL−1 BFA, and 1.5 μg mL−1 ionomycin. The cells were seeded into suspension 6-well cell culture plates at a density of 1 × 106 cells per mL, and cultured at 37 °C under a humidified atmosphere of 5% CO2 for 4 h. The cells were then stained with APC/Cyanine 7 anti-mouse CD8a followed by PE anti-mouse TNF-α, APC anti-mouse IFN-γ, and FITC anti-mouse GzmB for flow cytometry assay. The cytotoxic efficiency of OT-1 T cells was measured by quantifying the release of endogenous lactate dehydrogenase (LDH) from B16-Ova cells using the LDH cytotoxicity assay kit according to the manufacturer's instructions. The effective target cell (E/T) ratio was 10:1.
2.9. In vivo antigen presentation
To study the in vivo antigen presentation of SOCS1-downregulated DC vaccine, healthy male C57BL/6 mice were randomly divided into five groups (n = 4 per group) and subcutaneously injected with saline, DC (2 × 106), DC + Ova (2 × 106), DC-OA2/NC-siRNA + Ova (2 × 106), and DC-OA2/SOCS1-siRNA + Ova (2 × 106) per mice. The T cells of spleens were harvested using the EasySep™ Mouse CD8+ T cell isolation kit to analyse the proportion of antigen specific T cells (CD8+ Tetramer+) by flow cytometry. The cytotoxicity of isolated spleen T cells was assayed by the LDH cytotoxicity assay kit according to the manufacturer's instructions. The effective target cell (E/T) ratios were set as 25:1 and 50:1.
2.10. Remodeling the tumor microenvironment
Immune cell analysis in the tumor microenvironment was performed in B16-Ova tumor-bearing C57BL/6 mice. B16-Ova tumor cells (1 × 107 cells) were inoculated subcutaneously in the right flank of 6–8 week C57BL/6 mice to construct the tumor-bearing mouse model. When the average volume of the tumor reached approximately 50 mm3, the mice were randomly divided into five groups (n = 5 per group) and given four subcutaneous injections into the hind footpads on days 8, 12, 16, and 20 after tumor inoculation with the same formulations before mentioned. At day 24, the mice were sacrificed and single cell suspensions of tumors were collected from each group. The tumors were digested by collagenase IV for 45 min at 37 °C and treated with red blood cell lysis buffer to obtain single-cell suspension, which was then diluted to a concentration of 1 × 107 cells per mL in PBS.To study the infiltration and phenotype of CD11c+ DC cells in the tumor microenvironment, the cells were stained with separate anti-CD11c-PE, anti-CD45-Brilliant Violet 510TM, anti-CD86-FITC, anti-CD80-Alexa Fluor® 647, or anti-mouse MHC II-APC for 30 min, washed with PBS twice, and analysed by flow cytometry.
To study the infiltrated CD3+ CD8+ T cells in the tumor microenvironment, the cells were centrifugated in a gradient by 40 to 70% Percoll. Then, the cells were stained with separate anti-CD3-FITC, anti-CD8a-PE/Cyanine7, or anti-Ki67-PE for determination by flow cytometry. For cytokine secretion of tumor-infiltrating T cells, the cells were resuspended using 1640 complete medium containing 50 ng mL−1 PMA, 5 μg mL−1 BFA, and 1.5 μg mL−1 ionomycin. The cells were seeded into 6-well cell culture plates at a density of 1 × 106 cells per mL, and cultured at 37 °C under a humidified atmosphere of 5% CO2 for 4 h. The cells were then stained with anti-CD3-FITC and anti-CD8a APC/Cyanine7 followed by anti-TNF-α-PE, anti-GzmB-PE/Cyanine, and anti-IFN-γ-Brilliant Violet 510TM for flow cytometry assay. All flow cytometry data were analysed using the FlowJo software.
2.11. In vivo protective effect
To evaluate the protective effect of the SOCS1 gene silenced DC vaccine, healthy male C57BL/6 mice were randomly divided into five groups (n = 5 per group) and subcutaneously injected with saline, DC (2 × 106), DC + Ova (2 × 106), DC-OA2/NC-siRNA + Ova (2 × 106), and DC-OA2/SOCS1-siRNA + Ova (2 × 106) on days −14, −11, −8, and −4. B16-Ova cells (1 × 107) were then inoculated subcutaneously in the right flank of C57BL/6 mice after immunization on day 0. The changes in tumor size and body weight of the mice were monitored. Tumor size was calculated as (length × width2)/2.2.12. In vivo antitumor effect
The antitumor effect of the SOCS1 gene silenced DC vaccine was investigated in B16-Ova tumor bearing male C57BL/6 mice. When the average volume of the tumor reached approximately 50 mm3, the mice were randomly divided into five groups (n = 5 per group) and subcutaneously injected with saline, DC (2 × 106), DC + Ova (2 × 106), DC-OA2/NC-siRNA + Ova (2 × 106), and DC-OA2/SOCS1-siRNA + Ova (2 × 106) on days 8, 12, 16, and 20. The changes in tumor size and body weight of the mice were measured every two days. At day 24, mice were sacrificed and the tumor and normal tissues (heart, liver, spleen, lung, and kidney) were collected and weighed to calculate the tumor inhibition rate and the organ index. Additionally, the tumors and organs were dissected, embedded in paraffin, sectioned, and stained with haematoxylin and eosin (H&E) and/or TUNEL using routine methods. The sections were photographed using the microscope BX53 (Olympus). The serum samples were collected on day 24. Of which, IL-6, IL-10, and TNF-α concentrations were assayed using an ELISA kit (Elabscience), and the quantities of AKP, ALT, AST and BUN in the plasma were determined, respectively.The survival curve was recorded in another experiment every day over 50 days after tumor cell inoculation (n = 8 per group). Mice with tumors larger than 1000 mm3 at the longest axis were euthanized for ethical considerations.
2.13. Statistical analysis
The results are presented as means ± standard deviation (means ± SD) as indicated. The data were compared by the Student's t-test between two groups and ordinary one-way analysis of variance (ANOVA) for three or more groups. All statistical analyses were conducted using GraphPad Prism 8.0 (GraphPad Software Inc., CA, USA). The threshold of a statistically significant difference was defined as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns means no significance.3. Results and discussion
3.1. Preparation and characterization of OA2 LPs/SOCS1 siRNA
To endow the single-component lipid nanoparticles with similar functions of multi-component lipid nanoparticles, OA2 lipid was designed to contain a lysyl-head and two unsaturated oleyl tails connected by glutamic acid (Fig. 1A). The lysyl-head with two primary amines groups provides enough positive charge for binding siRNA and destroying endo/lysosomes in an acidic environment, while the unsaturated tails with lower phase inversion temperature than that of saturated alkyl chains further enhance the fusion ability with the endosomal membrane. Besides, the connected ester bond provided by glutamic acid can be degraded within cells by esterase, implying improved biocompatibility. Based on this, the OA2 lipid was successfully synthesized following the scheme in Fig. S1,† and characterized by 1H-nuclear magnetic resonance (1H-NMR, Fig. S2†), 13C-NMR (Fig. S3†), and high-resolution mass spectrometry (HR-MS) (Fig. S4†).After that, OA2 lipid nanoparticles (OA2 LPs) were prepared via the thin-film extrusion method, which showed an average particle size of 128.0 ± 0.1 nm and a zeta potential of 43.21 ± 1.24 mV (Fig. S5A†), with a uniformly spherical nanostructure as observed by transmission electron microscopy (TEM) (Fig. 2A). Next, OA2 LPs were co-incubated with SOCS1-siRNA to obtain the siRNA complex (OA2 LPs/SOCS1-siRNA). To ensure the steady loading of SOCS1-siRNA by OA2 LPs, the N/P ratio (referring to the number of nitrogen residues (N) in the OA2 lipid to phosphate (P) of siRNA) was optimized. With an increase of the N/P ratio, the particle size of OA2 LPs/SOCS1-siRNA plateaued at the N/P ratio over 3 (Fig. 2B), while the zeta potential gradually increased. Moreover, no siRNA band could be found when the N/P ratio was higher than 3 (Fig. 2C). Thus, the N/P ratio of 5 was chosen to fabricate the OA2 LPs/SOCS1-siRNA, which contained an average size of 129.1 nm and a zeta potential of +16.8 mV. And the TEM image indicated the larger particle size and rougher surface of OA2 LPs/SOCS1-siRNA than that of OA2 LPs, further suggesting the successful combination of siRNA by the OA2 LPs (Fig. S6†). In addition, the variation in the particle size of OA2 LPs/SOCS1-siRNA was measured after incubation under different conditions over time to evaluate its stability. It showed that OA2 LPs/SOCS1-siRNA remained stable in the presence of DC culture medium for at least 24 h (Fig. S5B†), which is beneficial for the uptake by DCs and subsequent gene silencing.
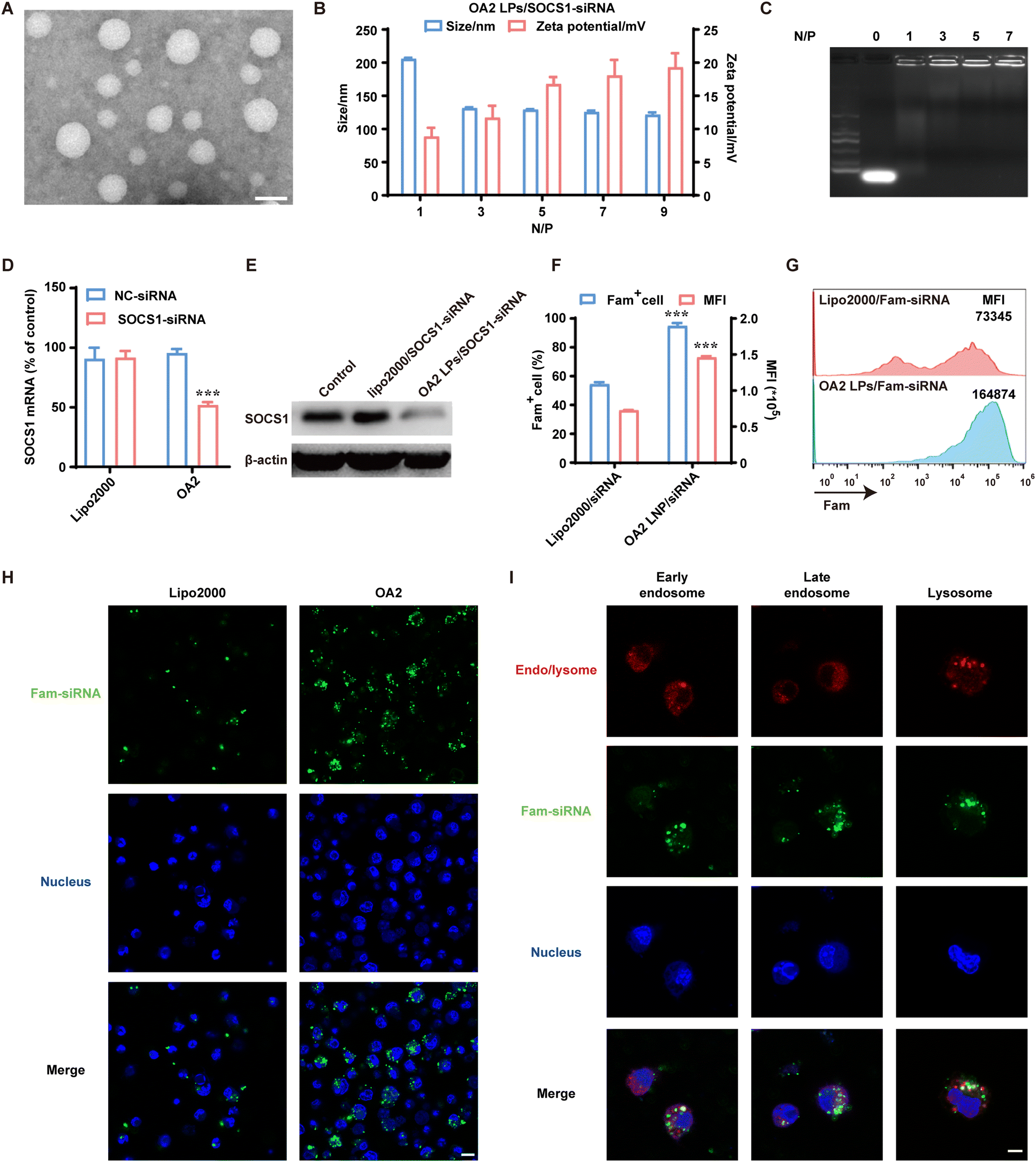 | ||
| Fig. 2 Preparation, characterization and gene silencing effect of OA2 LPs/SOCS1-siRNA. (A) TEM image of OA2 LPs. Scale bar: 50 nm. (B) Particle sizes and zeta potentials of OA2 LPs/SOCS1-siRNA at different N/P ratio (mean ± SD, n = 3). (C) Agarose gel electrophoresis analysis of OA2 LPs/SOCS1-siRNA at different N/P ratio (N/P = 0 stands for naked siRNA). (D and E) The expressions of SOCS1 mRNA (D) and protein (E) in BMDCs determined by q-PCR assay and western blot analysis, respectively. Mean ± SD, n = 3, ***P < 0.001, compared with Lipo2000/SOCS1-siRNA. (F and G) Cellular uptake of OA2 LPs/FAM-siRNA determined using the flow cytometry. Mean ± SD, n = 3, ***P < 0.001, compared with Lipo2000/FAM-siRNA. (H) The intracellular localization of FAM-siRNA (green) within BMDCs delivered by OA2 LPs or Lipo2000 using CLSM. The nuclei (blue) were stained with Hoechst 33342. Scale bar: 10 μm. (I) Subcellular distribution of OA2 LPs/FAM-siRNA (green) in BMDCs. The early endosome and late endosome were stained by EEA1 (red) and Rab7 (red), respectively. The lysosomes were stained by LysoTracker Red (red). The nuclei (blue) were stained with Hoechst 33342. Scale bar: 5 μm. | ||
In order to detect the gene silencing potential of OA2 LPs/SOCS1 siRNA in BMDCs, we first evaluated the cytotoxicity of OA2 LPs/SOCS1 siRNA on BMDCs with high purity (Fig. S7†). The results showed that the cell viability of BMDCs was over 90% after incubation with OA2 LPs/SOCS1-siRNA for 48 h, which was higher than that of BMDCs treated with the commercial lipofectin2000/siRNA complex (Lipo2000/SOCS1-siRNA) (Fig. S8†), indicating the possibility of OA2 LPs/SOCS1-siRNA as a siRNA delivery system to BMDCs due to its superior cell biocompatibility.
The gene silencing effect of OA2 LPs/SOCS1-siRNA in BMDCs was then evaluated. As verified by quantitative real-time polymerase chain reaction (q-PCR) assay, the level of SOCS1 mRNA in BMDCs transfected with OA2 LPs/SOCS1-siRNA was specifically decreased by approximately 50%, while scramble siRNA (NC-siRNA) delivered by OA2 LPs showed no gene silencing effect (Fig. 2D). Besides, limited inhibition on SOCS1 mRNA was found in the Lipo2000/SOCS1-siRNA group (Fig. 2D). Similar results in the expression of SOCS1 protein in BMDCs further indicated that the gene silencing effect induced by OA2 LPs (Fig. 2E).
For deeply understanding the improved gene silencing effect of OA2 LPs in PMDCs, we explored the cellular uptake and endosomal escape capabilities of OA2 LPs using fluorescent FAM-siRNA. After incubation with BMDCs for 6 h, OA2 LPs/FAM-siRNA resulted in a 95.19% positive rate of FAM+ BMDCs detected by flow cytometry, while that of Lipo2000/FAM-siRNA was only 54.79% (Fig. 2F). Moreover, higher mean fluorescence intensity (MFI) of BMDCs treated with OA2 LPs/FAM-siRNA indicated that more FAM-siRNAs were internalized by BMDCs, compared to that in the Lipo2000/FAM-siRNA group (Fig. 2F and G), which was further confirmed by the brighter green fluorescence of FAM-siRNA within BMDCs delivered by OA2 LPs than by Lipo2000 via confocal laser scanning microscopy (CLSM) images (Fig. 2H). These data suggested that OA2 LPs held the ability to enhance the uptake of siRNA by BMDCs. Upon internalization, OA2 LPs/siRNA must escape from the endo/lysosomes to gain access to the target mRNA. Thus, the subcellular distribution of OA2 LPs/siRNA in BMDCs was also monitored via CLSM (Fig. 2I). After incubation with OA2 LPs/FAM-siRNA for 15 min, the signal of FAM-siRNA (green) within BMDCs was mainly co-localized with the signal of EEA1 (red), an early endosome marker, indicating that OA2 LPs/FAM-siRNA entered early endosomes after endocytosis. When the incubation time was extended to 1 h, FAM-siRNA (green) partially co-localized with late endosomes indicated by Rab7 staining (red), a late endosome marker, indicating the increased escape of OA2 LPs from late endosomes. As time is prolonged, a great dissociation in the signals of FAM-siRNA (green) and LysoTracker red (red) occurred after 6 h of incubation, suggesting the efficient endosome escape of OA2 LPs/FAM-siRNA, which contributes to their resultant gene silencing effect.
Taken together, OA2 LPs can effectively deliver the loading siRNA into BMDCs and subsequent escape from the lysosomes, thus successfully downregulating the target genes. The inhibited negative feed-back SOCS1 genes in BMDCs endows BMDCs with the potential of improved activation and maturation.
3.2. Fabrication and characterization of SOCS1-downregulated DC vaccine
Having demonstrated the potent gene silencing effect induced by OA2 LPs in BMDCs, we further evaluated whether OA2 LPs could be applied to engineer BMDCs to obtain the SOCS1-downregulated DC vaccine. We first explored the influence of OA2 LPs uptake by BMDCs on the subsequent capture of the tumor antigen. After incubation with OA2 LPs/Cy5-siRNA for 6 h, fluorescent Atto-488 labelled Ova antigen as the model tumor antigen was incubated with BMDCs for another 6 h. Detected by flow cytometry, over 90% of BMDCs showed both Cy5 and Atto 488 fluorescence (Fig. 3A and B). Meanwhile, over 90% of Cy5+ BMDCs or Atto 488+ BMDCs was found in BMDCs treated with either OA2 LPs/Cy5-siRNA or Atto 488-OVA antigen alone (Fig. 3A and B). Similar results were observed from CLSM (Fig. 3C). These data suggested that the internalization of OA2 LPs/siRNA by BMDCs did not interfere in the subsequent uptake of the Ova antigen, implying that the SOCS1-downregulated DCs retained their antigen capture capability. | ||
| Fig. 3 Construction and characterization of SOCS1-downregulated DC vaccine. (A and B) Cellular uptake of OA2 LPs/Cy5-siRNA and Atto-488 OVA antigen in BMDCs by using flow cytometry. (C) Cellular uptake and intracellular localization of OA2 LPs/Cy5-siRNA (red) and Atto-488 Ova antigen (green) in BMDCs by using CLSM. The nuclei (blue) were stained with Hoechst 33342. Scale bar: 10 μm. (D) The expression of SOCS1 mRNA in BMDCs after the treatment with OA2 LPs/SOCS1-siRNA and Ova antigen determined by q-PCR assay (mean ± SD, n = 4). ns denotes no significant difference. (E) The expressions of CD80 and CD86 on BMDCs by using flow cytometry. | ||
In addition, whether the captured Ova antigen would affect the gene silencing effect of DCs remains unclear. Therefore, the level of SOCS1 mRNA was further verified by q-PCR assay after capturing the Ova antigen for 36 h by SOCS1-downregulated BMDCs. We found that there were no significant variations in the SOCS1 mRNA level before and after antigen capture (Fig. 3D), indicating that antigen stimulation would not reupregulate of SOCS1 due to the gene silencing effect mediated by OA2 LPs/SOCS1-siRNA.
Considering the non-interference of SOCS1-siRNA and Ova antigen in BMDCs, we thus fabricated the SOCS1-downregulated DC vaccine through sequential incubation with OA2 LPs/SOCS1-siRNA and Ova antigen for another 6 h. We showed that the expression of CD11c, a phenotypic marker of BMDCs, of SOCS1-downregulated DC vaccine (indicated as DC-OA2 LPs/SOCS1-siRNA + Ova in figures) was similar to that of untreated BMDCs (Fig. S9†). Furthermore, the expression of CD80 and CD86, the typical markers of DC maturation, of DCs or SOCS1-downregulated DCs pulsed with the Ova antigen was much higher than that of untreated DCs and SOCS1-downregulated DCs (Fig. 3E). These results suggested that SOCS1-downregulated BMDCs held the maturation ability of mouse DCs after antigen capture, indicating the potential in boosting antitumor immunity of the SOCS1-downregulated DC vaccine.
3.3. In vitro evaluation of SOCS1-downregulated DC vaccine for activating immunity
We next investigated the immune functions of the SOCS1-downregulated DC vaccine, including the production of pro-inflammatory factors and the antigen-presenting ability, which played crucial roles in boosted antitumor immunotherapy.26 The mRNA levels of pro-inflammatory factors including TNF-α, IL-6, and IL-2 in BMDCs stimulated by lipopolysaccharide (LPS) were detected by q-PCR assay (Fig. 4A). Due to the LPS stimulation, BMDCs indicated a significantly increased production of pro-inflammatory factors, compared with unstimulated BMDCs, confirming that mature BMDCs possess potent capability to secrete pro-inflammatory factors. Of note, the mRNA levels of TNF-α, IL-6, and IL-2 in the SOCS1-downregulated DC vaccine were the highest among all the groups, and nearly twice as high as that in the Lipo2000/SOCS1-siRNA + Ova treated BMDCs. These data suggested that SOCS1-downregulated DC vaccine achieved the enhanced production of pro-inflammatory factors due to the effective SOSC1 silencing mediated by OA2 LPs, implying the ability of reactivation of the tumor immunosuppressive microenvironment via secreting more pro-inflammatory factors to recruit more lymphocytes.27,28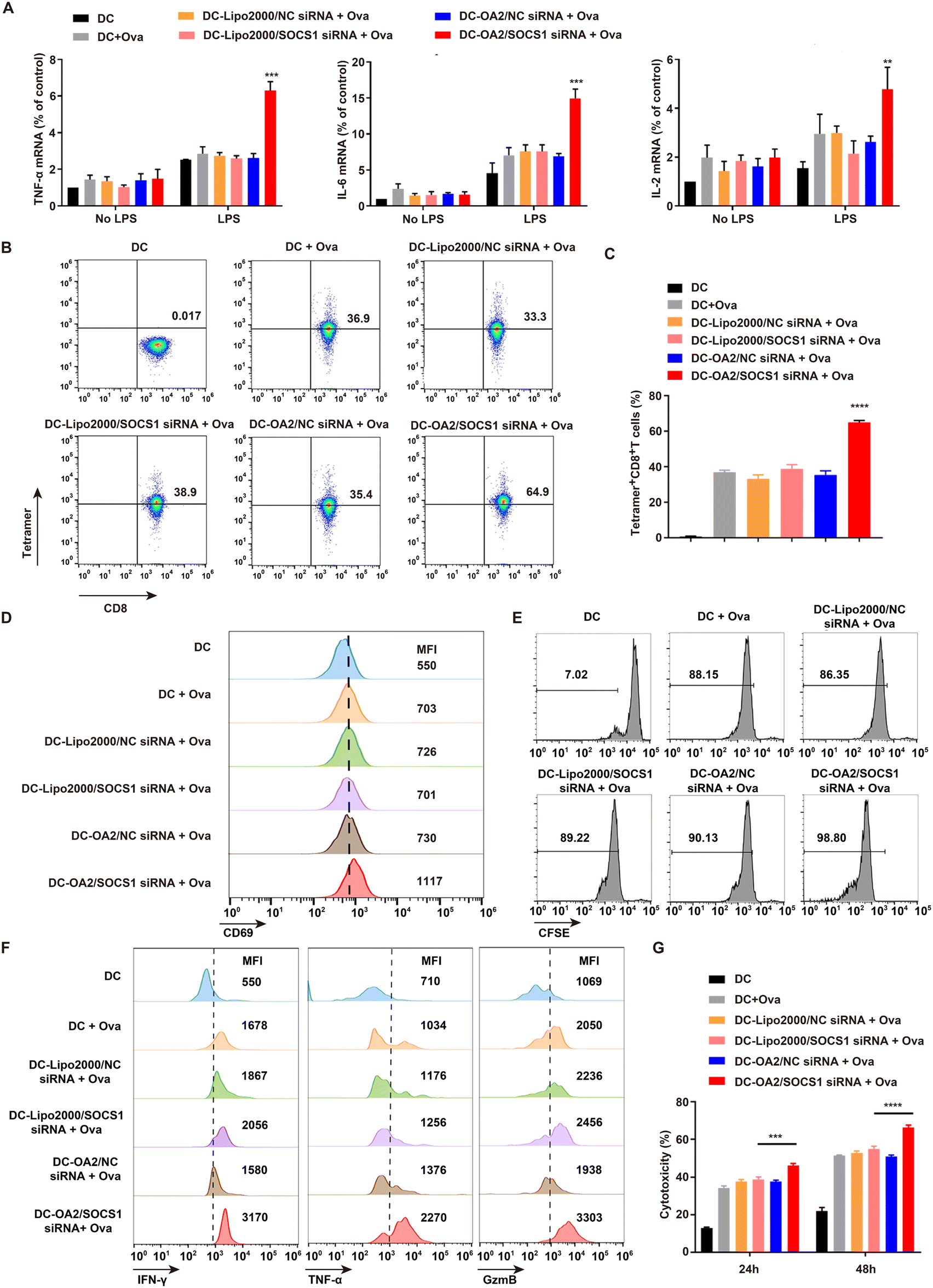 | ||
| Fig. 4 In vitro evaluation of functions of SOCS1-downregulated DC vaccine. (A) The mRNA levels of TNF-α, IL-6, and IL-2 in BMDCs after different treatment with or without LPS stimulation by q-PCR assay (mean ± SD, n = 4). (B and C) The percentage of Tetramer+ OT-1 T cells after 48 h-incubation with different DC formulations (mean ± SD, n = 3). (D) The expression of CD69 in OT-1 T cells after 48 h-incubation with different DC formulations (mean ± SD, n = 3). (E) The expansion of OT-1 T cells after 48 h-incubation with different DC formulations via CFSE assay. (F) Flow cytometry analysis of intracellular IFN-γ, TNF-α and GzmB in OT-1 T cells after 48 h-incubation with different DC formulations (mean ± SD, n = 3). (G) Cytotoxicity of OT-1 T cells after 48 h-incubation with different DC formulations against B16-Ova tumor cells as measured by release of lactate dehydrogenase (mean ± SD, n = 4). **P < 0.01, ***P < 0.001, ****P < 0.0001, compared with the DC-Lipo2000/SOCS1 siRNA + Ova group. | ||
Moreover, the antigen-presenting ability of SOCS1-downregulated DC vaccine, which determines the downstream T lymphocyte activation and antitumor immune responses,29 was explored through incubating the SOCS1-downregulated DC vaccine with OT-1 T cells for 48 h. We found that the percentage of Tetramer+ OT-1 T cells treated by the SOCS1-downregulated DC vaccine was 64.9%, significantly higher than in other groups (Fig. 4B and C). This indicated that SOCS1-downregulated DC vaccine successfully presented the Ova antigen to the OT-1 T cells as proved by the expression of MHC-I tetramer,30 which is probably ascribed to the effective inhibition of the immunosuppressive SOCS1 gene using OA2 LPs and thus interrupting intrinsic suppressive mechanisms of DCs.31
To further examine the activation effect of SOCS1-downregulated DC vaccine on OT-1 T cells, the activation, proliferation, inflammatory cytokine production, and cytotoxic activity of OT-1 T cells were analysed. We found the expression of CD69,32 the T cell activation marker, on OT-1 T cells co-cultured with SOCS1-downregulated DC vaccine was significantly upregulated by about 20% higher than other groups (Fig. 4D and Fig. S10†). Moreover, this DC vaccine markedly augmented the expansion of OT-1 T cells detected by the CFSE assay (Fig. 4E). Meanwhile, the production of cytotoxic factors including interferon (IFN-γ), tumor necrosis factor-α (TNF-α), and lytic enzyme granzyme B (GzmB) by OT-1 T cells was shown to be approximately 30% higher than in the other groups upon stimulation with the SOCS1-downregulated DC vaccine (Fig. 4F and Fig. S11†). In addition, after pre-treating with the SOCS1-downregulated DC vaccine, OT-1 T cells exhibited stronger cytotoxicity against B16-Ova cells of about 45% at 24 h and 64% at 48 h than in other groups at an effector cell/target cell (E/T) ratio of 10:1 (Fig. 4I). These results demonstrated that T cells could be effectively provoked by the SOCS1-downregulated DC vaccine according to their improved antigen-presenting ability.
Together, we have confirmed that SOCS1-downregulated DC vaccine is capable of producing pro-inflammatory factors and enhancing antigen-presenting ability, implying the capabilities of remodeling the tumor microenvironment and provoking tumor infiltrated T cells.
3.4. In vivo immune activation of SOCS1-downregulated DC vaccine
Since SOCS1-downregulated DC vaccine has shown an improved effect on cytokine release and T cell activation of BMDCs, we further detected whether it could take effect in vivo.Firstly, the in vivo antigen-presenting ability of SOCS1-downregulated DC vaccine was evaluated in normal mice (Fig. 5A), which were randomly divided into five groups and given four subcutaneous injections into the hind footpads on days 0, 4, 7, and 11 with one of the following formulations: (1) saline, (2) BMDCs (DC, 2 × 106 per mice), (3) untreated BMDCs pulsed with the Ova antigen (DC + Ova, 2 × 106 per mice), (4) OA2 LPs/NC siRNA-treated BMDCs pulsed with the Ova antigen (DC-OA2 LPs/NC-siRNA + Ova, 2 × 106 per mice), and (5) SOCS1-downregulated DC vaccine (DC-OA2 LPs/SOCS1-siRNA + Ova, 2 × 106 per mice). At day14, CD8+ T cells were isolated from the spleens of the mice after immunization for the investigation of Ova-specific CD8+ T cells. When the antigen is successfully presented to CD8+ T cells by DC vaccines, antigen-specific T cells would express a peptide-bound MHC molecular complex which can be detected by the MHC tetramer assay. As shown in Fig. 5B and C, there were 21.3% of tetramer-positive CD8+ T cells in mice immunized with the SOCS1-downregulated DC vaccine, while only 11.7% in mice immunized with the untreated DC pulsed with the Ova antigen, indicating the improved antigen presentation ability of DCs with SOCS1-downregulation. In contrast, DCs without Ova treatment exhibited a low ratio of tetramer-positive CD8+ T cells. Moreover, we found that the NC siRNA group showed similar antigen presentation ability with the DC + Ova group, suggesting that the carrier of OA2 LPs had no effect on DC function. We further determined the cytotoxicity of isolated CD8+ T cells from the spleen against the B16-Ova tumor cells at E/T ratios of 25:1 and 50:1 for 24 h (Fig. S12†). As E/T ratio increased, the cytotoxicity was gradually enhanced. Of these, the spleen CD8+ T cells of mice that received the SOCS1-downregulated DC vaccine exhibited the strongest cytotoxicity of about 45% at an E/T ratio of 50:1. These results indicate that the SOCS1-downregulated DC vaccine can effectively present tumor antigen to T cells and boost their antigen-specific cytotoxicity in vivo.
 | ||
| Fig. 5 In vivo immune activation of SOCS1-downregulated DC vaccine. (A) Schematic illustration of the experimental design of in vivo evaluation of antigen presentation. (B and C) Representative flow cytometric images (B) and quantitative analysis (C) of Tetramer+ CD8+ T cells in spleen (mean ± SD, n = 4). (D) Schematic illustration of the experimental design of in vivo evaluation of remodeling tumor microenvironment. (E and F) Representative flow cytometric images of tumor-infiltrated (E) CD45+ CD11c+ DCs and (F) CD80+ CD86+ DCs (mean ± SD, n = 5). (G) Representative flow cytometric images of tumor-infiltrated CD3+ CD8+ T cells (mean ± SD, n = 5). (H) Quantitative analysis of (E), (F) and (G). (I) Representative flow cytometric images of Ki67 expression of tumor-infiltrated CD3+ CD8+ T cells (mean ± SD, n = 5). (J) Flow cytometry analysis of intracellular GzmB, TNF-α and IFN-γ in tumor-infiltrated CD8+ T cells (mean ± SD, n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, compared with the DC + Ova group. | ||
Next, we investigated the ability of the SOCS1-downregulated DC vaccine to remodel the tumor immunosuppressive microenvironment in B16-Ova tumor bearing mice. These mice were randomly divided into five groups and given four subcutaneous injections into the hind footpads on days 8, 12, 16, and 20 after tumor inoculation with the same formulations before mentioned (Fig. 5D). We isolated and analysed the DC cells and CD8+ T cells from primary tumors at day 24 (Fig. 5E–J). Mice receiving SOCS1-downregulated DC vaccine showed the highest percentage of CD11c+ cells in CD45+ cells in tumors (Fig. 5E and H), as well as the expression of CD80, CD86, and MHC II (Fig. 5F and H, and Fig. S13†), the typical markers of mature DCs, suggesting the improved infiltration of DC cells in tumors and the promoted maturation induced by the SOCS1-downregulated DC vaccine. It can be ascribed to the enhanced production of pro-inflammatory factors by the SOCS1-downregulated DC vaccine to recruit more DCs into tumor sites and facilitate their maturation.31 Of these, mice treated with DC pulsed with Ova showed a similar ratio to mature DCs in the mice treated with NC group, both of which were lower than that receiving SOCSthe 1-downregulated DC vaccine, probably due to the expression of immune inhibitory factors that hampered the DC function in the tumor site. Besides, the mature DCs would initiate the activation of T cells. To verify this hypothesis, we further detected the percentage and functions of CD8+ T cells in tumors. The results showed that mice receiving the SOCS1-downregulated DC vaccine held a 2.4-fold higher CD8+ T cell percentage than that in the DCs pulsed with the Ova antigen group (Fig. 5G and H), implying that the downregulation of SOCS1 in DCs is critical for boosting the tumor-infiltration of CD8+ T cells. Similarly, the Ki67 expression of tumor-infiltrated CD8+ T cell in the group of the SOCS1-downregulated DC vaccine exhibited the highest ratio (Fig. 5I and Fig. S14†), indicating the promoted proliferation of tumor-infiltrated CD8+ T cell by the activation of the SOCS1-downregulated DC vaccine. Moreover, the capacity of tumor-infiltrating CD8+ T cells to secrete inflammatory cytokines, including IFN-γ, TNF-α, and GzmB was explored (Fig. 5J and Fig. S15†). We found that mice receiving theSOCS1-downregulated DC vaccine showed an improved percentage of IFN-γ+, TNF-α+, and GzmB+ tumor-infiltrating CD8+ T cells, presumably due to the effective activation of DC vaccine with SOCS1 downregulation. These results clearly illustrated the reinvigoration of the immunosuppressive tumor microenvironment by the SOCS1-downregulated DC vaccine by improving the immune cell infiltration and promoting their maturation and activation, which suggests a potential efficacy in tumor immunotherapy.
Overall, the engineered DC vaccines with SOCS1 downregulation exhibited improved capabilities of antigen presentation and T cell-activation in vivo, even in the immunosuppressive tumor microenvironment. That is, the effective inhibition of SOCS1 with negatively regulated function enhanced the immune functions of DCs, further verifying the successful delivery of SOCS1 siRNA to DCs via OA2 LPs.
3.5. In vivo immunization and safety evaluation of SOCS1-downregulated DC vaccine
In brief, if the vaccination of the SOCS1-downregulated DC vaccine can activate antigen-specific T cell response, the tumor infiltrated T cells can recognize the specific antigen-bearing tumor cells and thus kill them to prevent or treat the cancer. Thus, we first evaluated the prevention effect of the SOCS1-downregulated DC vaccine against melanoma. Before inoculation of B16-Ova tumor cells (1 × 107 per mice), normal mice were randomly divided into five groups and given four subcutaneous injections into the hind footpads on days −14, −11, −8, and −4 with one of the formulations mentioned above (Fig. S16A†). Then, the tumor volume was monitored every two days. As presented in Fig. S16B,† immunization with the SOCS1-downregulated DC vaccine significantly delayed the tumor growth as compared to other groups, and showed a tumor suppression rate of 74.41% at day 24, indicating that the SOCS1-downregulated DC vaccine has potential as a prophylactic vaccine. However mice receiving DCs and DC pulsed with Ova could only inhibit the tumor growth to some extent, indicating the necessity of inhibiting the expression of SOCS1 in DC-based therapy. Additionally, the body weights of mice receiving the SOCS1-downregulated DC vaccine showed no significant differences with that of other groups (Fig. S16C†).We next evaluated the antitumor efficacy of immunization of the SOCS1-downregulated DC vaccine in B16-Ova tumor-bearing mice. After inoculation of B16-Ova cells (1 × 107 per mice), the mice were randomly divided into five groups and given four intravenous injections into the hind footpads on days 8, 12, 16, 20 with one of the formulations mentioned above (Fig. 6A). Tumor growth and survival time were monitored after treatment. The SOCS1-downregulated DC vaccine-treated mice showed the slowest rate of tumor growth over 20 days, while other group-treated mice showed obvious tumor growth over time (Fig. 6B and C). Remarkably, mice treated with the SOCS1-downregulated DC vaccine exhibited a tumor suppression rate of 77.83%, and 60% of mice survived at day 40, while mice in other groups were sacrificed to tumor progression within 40 days (Fig. 6D). The histologic images of the tumor sections stained by TUNEL showed a massive cancer cell remission after treatment with the SOCS1-downregulated DC vaccine (Fig. 6E). These results confirmed the potent efficacy of the SOCS1-downregulated DC vaccine in tumor immunotherapy.
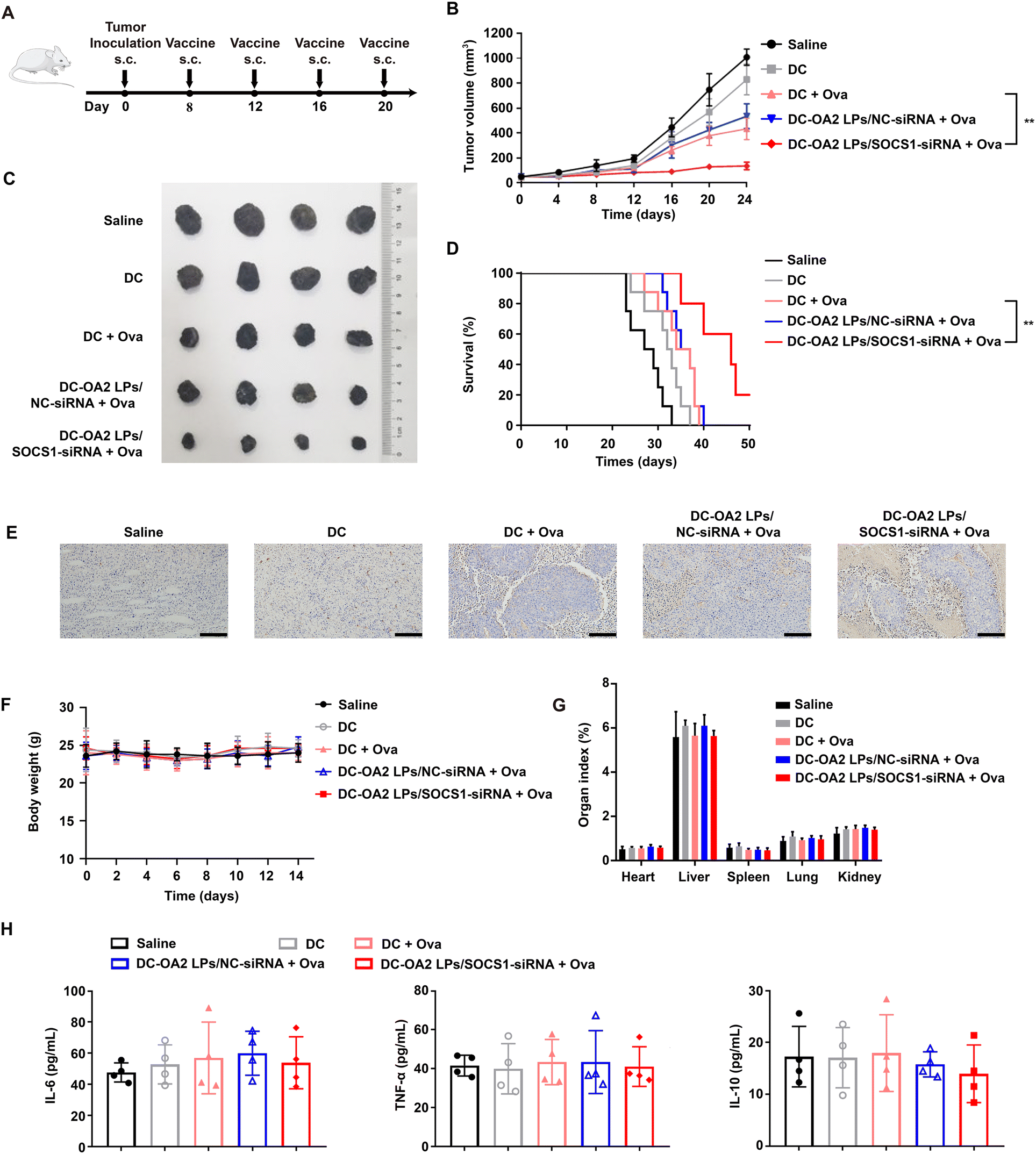 | ||
| Fig. 6 In vivo antitumor effect and safety evaluation of SOCS1-downregulated DC vaccine. (A) Schematic illustration of the experimental design of antitumor treatment. (B) The B16-Ova tumor growth curves after treatment with the different formulations (mean ± SD, n = 5). (C) Representative images of the isolated tumors of the mice after treatment at day 24. (D) Survival curves of mice receiving each treatment (n = 8). (E) Representative microscopic images of tumor sections stained by TUNEL. Scale bar: 100 μm. (F) The body weights of B16-Ova tumor-bearing mice after treatment over time (n = 6). (Mean ± SD, n = 5). (G) The main organ indexes of B16-Ova tumor-bearing mice after treatment at day 24 (mean ± SD, n = 5). (H) Quantification of IL-6, TNF-α, and IL-10 in the plasma of the mice after treatment with indicated formulations (mean ± SD, n = 4). **P < 0.01, ***P < 0.001, compared with the DC + Ova group. | ||
The body weights (Fig. 6F) and the main organ indexes (Fig. 6G) of mice receiving the SOCS1-downregulated DC vaccine remained stable, showing no significant differences with that of other groups. There was no substantial difference in the serum levels of IL-6, TNF-α and IL-10 between the SOCS1-downregulated DC vaccine and other groups, indicating that the SOCS1-downregulated DC vaccine did not cause a severe cytokine storm in mice (Fig. 6H). Meanwhile, no impairment of liver and kidney function was noted in mice treated with the SOCS1-downregulated DC vaccine (Fig. S17†). Also, no pathological changes were observed from the H&E staining images of the main organ sections (Fig. S18†).
Taken together, we have verified that the SOCS1-downregulated DC vaccine is safe and effective in treating B16-Ova tumors as both a prophylactic and a therapeutic vaccine.
4. Conclusions
In summary, we have successfully constructed a single-component lipid nanoparticle for effectively silencing the suppressive factor of SOCS1 in DCs. The SOCS1-downregulated DCs can be further pulsed with tumor antigens, which can serve as a tumor vaccine for remodeling the tumor environment and activating the antigen-specific T cell response, thus resulting in potent preventive and therapeutic effects in tumor immunotherapy, according with superior safety. This study provides a safe, effective, and convenient gene delivery platform for primary DCs and offers available engineered DCs which can be pulsed with specific tumor antigens for boosted tumor immunotherapy. Moreover, the single-component lipid nanoparticle with a simple composition takes advantage in further industrial transformation, which still needs further investigation.Author contributions
Z. X. Y.: Methodology, validation, and writing – original draft. M. T. W.: Methodology and investigation. Y. S. H.: Validation and writing – review & editing. Y. S. W.: Software and methodology. Y. J. C.: Software. Q. L. L.: validation. Z. M. L.: Methodology. L. J. X.: Writing – review and editing. C. Y. J.: Conceptualization, supervision, methodology, writing – review and editing, and funding acquisition. C. Z.: Supervision, funding acquisition, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81930099, 82130102, 92159304, 81773664, 82073785), the Natural Science Foundation of Jiangsu Province (BK20212011, China), Technology innovation project of Nucleic acid drug from National Center of Technology Innovation for Biopharmaceuticals (NCTIB2022HS01014), the “Double First-Class” University project (CPU2022QZ05, China), the 111 Project from the Ministry of Education of China and the State Administration of Foreign Expert Affairs of China (No. 111-2-07, B17047, China), and the Open Project of State Key Laboratory of Natural Medicines (No. SKLNMZZ202223, China). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University and approved by the Animal Ethics Committee of China Pharmaceutical University.References
- R. L. Sabado, S. Balan and N. Bhardwaj, Dendritic cell-based immunotherapy, Cell Res., 2017, 27, 74–95 CrossRef CAS PubMed
.
- K. Palucka and J. Banchereau, Cancer immunotherapy via dendritic cells, Nat. Rev. Cancer, 2012, 12, 265–277 CrossRef CAS
- O. P. Joffre, E. Segura, A. Savina and S. Amigorena, Cross-presentation by dendritic cells, Nat. Rev. Immunol., 2012, 12, 557–569 CrossRef CAS PubMed
- B. M. Carreno, V. Magrini, M. Becker-Hapak, S. Kaabinejadian, J. Hundal, A. A. Petti, A. Ly, W.-R. Lie, W. H. Hildebrand, E. R. Mardis and G. P. Linette, A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells, Science, 2015, 348, 803–808 CrossRef CAS
- W. Yang, G. Zhu, S. Wang, G. Yu, Z. Yang, L. Lin, Z. Zhou, Y. Liu, Y. Dai, F. Zhang, Z. Shen, Y. Liu, Z. He, J. Lau, G. Niu, D. O. Kiesewetter, S. Hu and X. Chen, In Situ Dendritic Cell Vaccine for Effective Cancer Immunotherapy, ACS Nano, 2019, 13, 3083–3094 CrossRef CAS PubMed
- S. Anguille, E. L. Smits, E. Lion, V. F. van Tendeloo and Z. N. Berneman, Clinical use of dendritic cells for cancer therapy, Lancet Oncol., 2014, 15, e257–e267 CrossRef CAS PubMed
- R. Yamanaka, J. Homma, N. Yajima, N. Tsuchiya, M. Sano, T. Kobayashi, S. Yoshida, T. Abe, M. Narita, M. Takahashi and R. Tanaka, Clinical Evaluation of Dendritic Cell Vaccination for Patients with Recurrent Glioma: Results of a Clinical Phase I/II Trial, Clin. Cancer Res., 2005, 11, 4160–4167 CrossRef CAS PubMed
- A. Yoshimura, T. Naka and M. Kubo, SOCS proteins, cytokine signalling and immune regulation, Nat. Rev. Immunol., 2007, 7, 454–465 CrossRef CAS PubMed
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Dendritic cells in cancer immunology and immunotherapy, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef CAS
- T. Hanada, H. Yoshida, S. Kato, K. Tanaka, K. Masutani, J. Tsukada, Y. Nomura, H. Mimata, M. Kubo and A. Yoshimura, Suppressor of Cytokine Signaling-1 Is Essential for Suppressing Dendritic Cell Activation and Systemic Autoimmunity, Immunity, 2003, 19, 437–450 CrossRef CAS PubMed
- E. Gilboa, Knocking the SOCS1 off dendritic cells, Nat. Biotechnol., 2004, 22, 1521–1522 CrossRef CAS
- L. Shen, K. Evel-Kabler, R. Strube and S.-Y. Chen, Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity, Nat. Biotechnol., 2004, 22, 1546–1553 CrossRef CAS PubMed
- J. Chen, A. Ellert-Miklaszewska, S. Garofalo, A. K. Dey, J. Tang, Y. Jiang, F. Clément, P. N. Marche, X. Liu, B. Kaminska, A. Santoni, C. Limatola, J. J. Rossi, J. Zhou and L. Peng, Synthesis and use of an amphiphilic dendrimer for siRNA delivery into primary immune cells, Nat. Protoc., 2021, 16, 327–351 CrossRef CAS PubMed
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Delivery materials for siRNA therapeutics, Nat. Mater., 2013, 12, 967–977 CrossRef CAS
- A. Michiels, S. Tuyaerts, A. Bonehill, J. Corthals, K. Breckpot, C. Heirman, S. Van Meirvenne, M. Dullaers, S. Allard, F. Brasseur, P. van der Bruggen and K. Thielemans, Electroporation of immature and mature dendritic cells: implications for dendritic cell-based vaccines, Gene Ther., 2005, 12, 772–782 CrossRef CAS PubMed
- E. L. J. M. Smits, S. Anguille, N. Cools, Z. N. Berneman and V. F. I. Van Tendeloo, Dendritic Cell-Based Cancer Gene Therapy, Hum. Gene Ther., 2009, 20, 1106–1118 CrossRef CAS PubMed
- J. E. Boudreau, B. W. Bridle, K. B. Stephenson, K. M. Jenkins, J. Brunellière, J. L. Bramson, B. D. Lichty and Y. Wan, Recombinant Vesicular Stomatitis Virus Transduction of Dendritic Cells Enhances Their Ability to Prime Innate and Adaptive Antitumor Immunity, Mol. Ther., 2009, 17, 1465–1472 CrossRef CAS
- D. J. A. Crommelin, P. van Hoogevest and G. Storm, The role of liposomes in clinical nanomedicine development. What now? Now what?, J. Controlled Release, 2020, 318, 256–263 CrossRef CAS PubMed
- F. Zahednezhad, M. Saadat, H. Valizadeh, P. Zakeri-Milani and B. Baradaran, Liposome and immune system interplay: Challenges and potentials, J. Controlled Release, 2019, 305, 194–209 CrossRef CAS PubMed
- F. Ding, H. Zhang, J. Cui, Q. Li and C. Yang, Boosting ionizable lipid nanoparticle-mediated in vivo mRNA delivery through optimization of lipid amine-head groups, Biomater. Sci., 2021, 9, 7534–7546 RSC
- E. Álvarez-Benedicto, L. Farbiak, M. M. Ramírez, X. Wang, L. T. Johnson, O. Mian, E. D. Guerrero and D. J. Siegwart, Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA), Biomater. Sci., 2022, 10, 549–559 RSC
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing, Nat. Mater., 2021, 20, 701–710 CrossRef CAS
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. Lin, S. Chen, J. K. Narayanannair and K. G. Rajeev, Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles, Mol. Ther.–Nucleic Acids, 2013, 2, e139–e146 CrossRef CAS PubMed
- P. R. Cullis and M. J. Hope, Lipid nanoparticle systems for enabling gene therapies, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Chemistry of lipid nanoparticles for RNA delivery, Acc. Chem. Res., 2021, 55, 2–12 CrossRef
- F. Veglia and D. I. Gabrilovich, Dendritic cells in cancer: the role revisited, Curr. Opin. Immunol., 2017, 45, 43–51 CrossRef CAS
- G. Dranoff, Cytokines in cancer pathogenesis and cancer therapy, Nat. Rev. Cancer, 2004, 4, 11–22 CrossRef CAS PubMed
- J. B. Haanen, Converting cold into hot tumors by combining immunotherapies, Cell, 2017, 170, 1055–1056 CrossRef CAS
- A. Gardner and B. Ruffell, Dendritic cells and cancer immunity, Trends Immunol., 2016, 37, 855–865 CrossRef CAS
- B. W. MacNabb, S. Tumuluru, X. Chen, J. Godfrey, D. N. Kasal, J. Yu, M. L. Jongsma, R. M. Spaapen, D. E. Kline and J. Kline, Dendritic cells can prime anti-tumor CD8+ T cell responses through major histocompatibility complex cross-dressing, Immunity, 2022, 55, 982–997 CrossRef CAS
- T. Kobayashi and A. Yoshimura, Keeping DCs awake by putting SOCS1 to sleep, Trends Immunol., 2005, 26, 177–179 CrossRef CAS PubMed
- D. Cibrián and F. Sánchez-Madrid, CD69: from activation marker to metabolic gatekeeper, Eur. J. Immunol., 2017, 47, 946–953 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01549h |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |