
Pharmaceutical liposomal delivery—specific considerations of innovation and challenges
Taoxing
Peng
ab,
Weihua
Xu
b,
Qianqian
Li
b,
Yang
Ding
*a and
Yongzhuo
Huang
 *bcd
*bcd
aState Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing, 210009, China. E-mail: dydszyzf@163.com
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 501 Haike Rd, Shanghai 201203, China. E-mail: yzhuang@simm.ac.cn; Fax: +86-21-2023-1981; Tel: +86-21-2023-1981
cNMPA Key Laboratory for Quality Research and Evaluation of Pharmaceutical Excipients, Shanghai 201203, China
dZhongshan Institute for Drug Discovery, Institutes of Drug Discovery and Development, Chinese Academy of Sciences, Zhongshan 528437, China
First published on 14th November 2022
Abstract
Liposomal technology has been widely used in the pharmaceutical field for the preparation of nano-sized drug delivery systems based on natural or synthetic lipids. Liposomes possess many attractive properties, such as easy processing, high biocompatibility, adaptable drug loading, and improved PK profiles. In recent decades, great efforts have been made in this field, and dozens of liposomal medicines have been marketed worldwide and many more are under preclinical or clinical investigations. Liposomes can enhance the aqueous dissolution and stability of the encapsulated drugs and modulate the in vivo fate of the drugs (e.g., prolonged half-life and increased drug accumulation in the pathological sites). Therefore, liposomal technology can improve the druggability of the candidates, enhance treatment efficacy and reduce side effects. This review discusses the prospects of liposomal delivery, including the specific considerations of innovation and challenges.
1. Introduction
Drug delivery systems (DDSs) refer to advanced dosage forms used in disease prevention and treatment.1 The design concept is to deliver the necessary amount of drugs to pathological sites at the appropriate time frame to achieve the maximum curative effect but with minimum side effects. In short, it can be called the “3R” delivery principle—right dose, right place, and right time.2 Drug delivery technology can improve the druggability of the candidates and make the drugs more potent and safer. As a case in point, liposomal drug delivery systems can be applied to various drugs to solve problems such as poor solubility and high side toxicity. By using liposomal technology, the side toxic effects of traditional chemo drugs can be significantly reduced, thereby improving the compliance of patients.3–5There is a massive market for DDSs, and liposomal drug research and development (R&D) is a heavily invested field for pharmaceutical enterprises. Apart from small molecule drugs, the application of liposomal technology has been expanded to biotechnological drugs. For example, nucleic acid drugs (e.g., small interfering RNA, siRNA,6 and messenger RNA, mRNA7) have been developing rapidly with continuous innovation, representing the third wave of modern drugs after small molecule drugs and antibodies.8 Due to the cell-impermeable nature of nucleic acid drugs, the treatment success largely depends on the vectors that can effectively transfer genetic material to the target cells.9,10 The liposome-based delivery is one of the most effective non-viral gene delivery methods.11,12
Liposomes provide a wide range of application prospects; some recent reviews have provided helpful information.12–14 There are still some unsolved challenges in this field and great efforts need to be made to meet the continuing demand in the R&D of new liposomal drugs. This article will focus on three aspects: the successful application, the confronting challenges, and the transition from concept to clinical application, to provide a summary of the technological advances and to discuss the challenges and what is the next with pharmaceutical liposomal delivery.
2. Overview of liposomal technology
Liposomes can be formed as closed vesicles with a bilayer structure when phospholipids or sphingolipids are dispersed in the aqueous phase, due to the self-assembly in which the hydrophobic tails of the lipids tend to gather, while the hydrophilic head is exposed to the aqueous phase.15 Liposomes can be used to deliver various kinds of drugs. The hydrophilic molecules can be encapsulated in the aqueous core, whereas hydrophobic drugs can be embedded in the bilayer.2.1 A timeline of liposomal formulations in the market
The development of liposomes dates to the 1960s.16 Alec Bangham, a British hematologist, first prepared liposomes, which, initially, were proposed as a biofilm model.17,18 Later, liposomes were explored as drug carriers. In 1988, the first pharmaceutical econazole liposomal product was born in Switzerland. In 1990, the first liposomal injection, amphotericin B liposome, was marketed.19 In 1995, the U.S. Food and Drug Administration (FDA) approved the first liposomal anticancer drug, doxorubicin liposome.3 To date, there are more than 20 liposomal medicines on the market, mainly anticancer drugs.20 A timeline of the major liposome products approved for marketing is shown in Fig. 1. To date, there have been dozens of marketed liposome-based therapeutics, together with an increasing number of many undergoing clinical trials in different therapeutic areas (Tables 1 and 2).12,21,22 The number of clinical trials in phase I, II and III was obtained by searching the clinicaltrial.gov database. Their applications involve various therapeutic areas, including cancer and anti-infection. In recent years, the official approvals of the liposomal combination CPX-351, inhaled amikacin liposomes, and mRNA cationic liposomes represent the breakthroughs in three directions: liposome-based precision combination therapy, an innovative combination of liposomal drugs and machinery, and liposome-based delivery of biomacromolecules.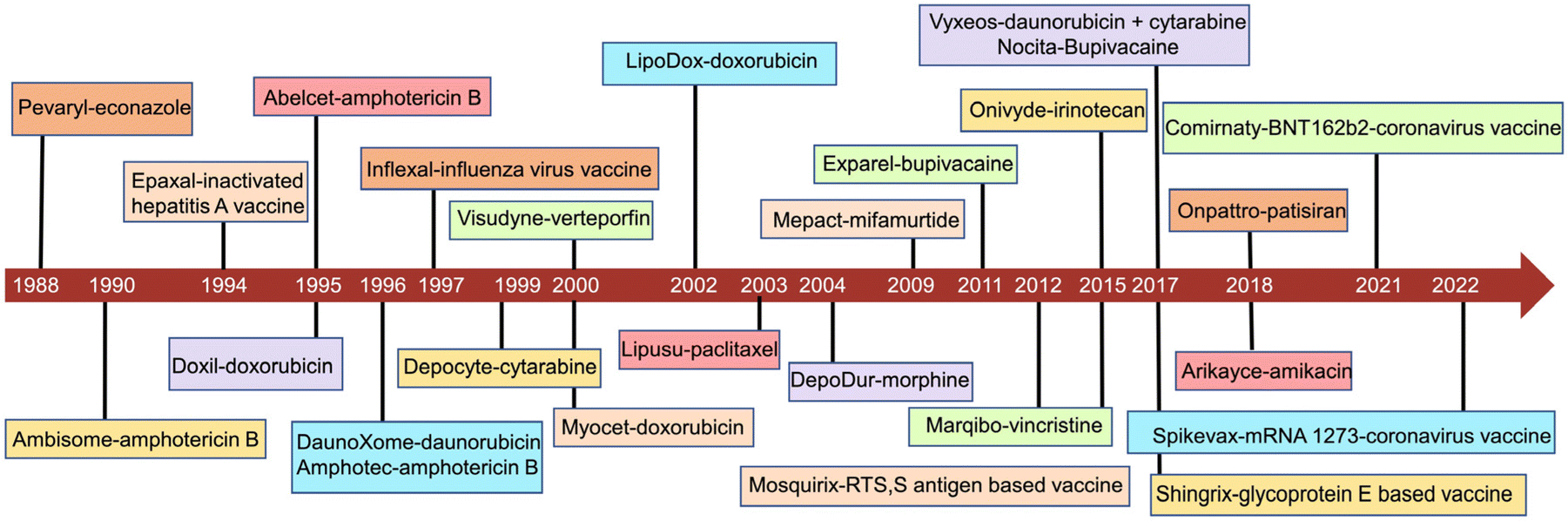 | ||
| Fig. 1 Timeline of the representative liposomal medicines. | ||
| Product name | Drug name | Therapeutic application | Approval |
|---|---|---|---|
| AmBisome | Amphotericin B | Anti-fungal agent | 1990 |
| Epaxal | Inactivated hepatitis A vaccine | Vaccine | 1994 |
| Doxil/Caelyx | Doxorubicin | Anti-cancer therapy | 1995 |
| Abelcet | Amphotericin B | Anti-fungal agent | 1995 |
| DaunoXome | Daunorubicin | Anti-cancer therapy | 1996 |
| Amphotec | Amphotericin B | Anti-fungal agent | 1996 |
| Inflexal V | Influenza virus vaccine | Vaccine | 1997 |
| Depocyte | Cytarabine | Anti-cancer therapy | 1999 |
| Visudyne | Verteporfin | Photodynamic therapy | 2000 |
| Myocet | Doxorubicin | Anti-cancer therapy | 2000 |
| LipoDox | Doxorubicin | Anti-cancer therapy | 2002 |
| Lipusu | Paclitaxel | Anti-cancer therapy | 2003 |
| DepoDur | Morphine | Analgesic | 2004 |
| Mepact | Mifamurtide | Anti-cancer therapy | 2009 |
| Exparel | Bupivacaine | Analgesic | 2011 |
| Marqibo | Vincristine | Anti-cancer therapy | 2012 |
| Onivyde | Irinotecan | Anti-cancer therapy | 2015 |
| Mosquirix | RTS,S antigen based vaccine | Vaccine | 2015 |
| Vyxeos | Daunorubicin/cytarabine | Anti-cancer therapy | 2017 |
| Nocita | Bupivacaine | Analgesic | 2017 |
| Shingrix | Glycoprotein E based vaccine | Vaccine | 2017 |
| Onpattro | Patisiran | RNAi | 2018 |
| Arikayce | Amikacin | Anti-fungal agent | 2018 |
| Comirnaty | Coronavirus vaccine | Vaccine | 2021 |
| Spikevax | Coronavirus vaccine | Vaccine | 2022 |
| Therapeutic areas | Product name | Drug name | Approval |
|---|---|---|---|
| Anti-cancer therapy | LEP-ETU | Paclitaxel | Phase II |
| Anti-cancer therapy | SPI-077 | Cisplatin | Phase II |
| Anti-cancer therapy | LE-SN38 | SN38 | Phase II |
| Anti-cancer therapy | CPX-1 | Irinotecan/floxuridine | Phase II |
| Anti-cancer therapy | S-ANNA | Anamycin | Phase III |
| Anti-cancer therapy | EndoTAG-1 | Paclitaxel | Phase III |
| Anti-cancer therapy | ThermoDox | Doxorubicin | Phase III |
| Anti-cancer therapy | Lipoplatin | Cisplatin | Phase III |
| Cardiovascular therapy | Liprostin | PGE-1 | Phase III |
| Vaccine | ARCoV | Coronavirus vaccine | Phase III |
| Vaccine | CVnCoV | Coronavirus vaccine | Phase III |
| Cancer vaccine | Stimuvax | Anti-MUC1 cancer vaccine | Phase III |
2.2 Liposome-based delivery technology
The delivery capacity of liposomes relies on their physicochemical properties, including phase transition temperature, membrane permeability and fluidity, particle size, and zeta potential.15 These properties are also important indicators for quality control in the R&D of liposome products. As shown in Fig. 2, liposomes can be divided into small unilamellar vesicular (SUV), large unilamellar vesicular (LUV), multilamellar vesicular (MLV), and multivesicular liposomes (MVL). Notably, surface modification is a common method for preparing various types of functionalized liposomes, including long-circulation liposomes, pH-sensitive liposomes, temperature-sensitive liposomes, and immunoliposomes.23–25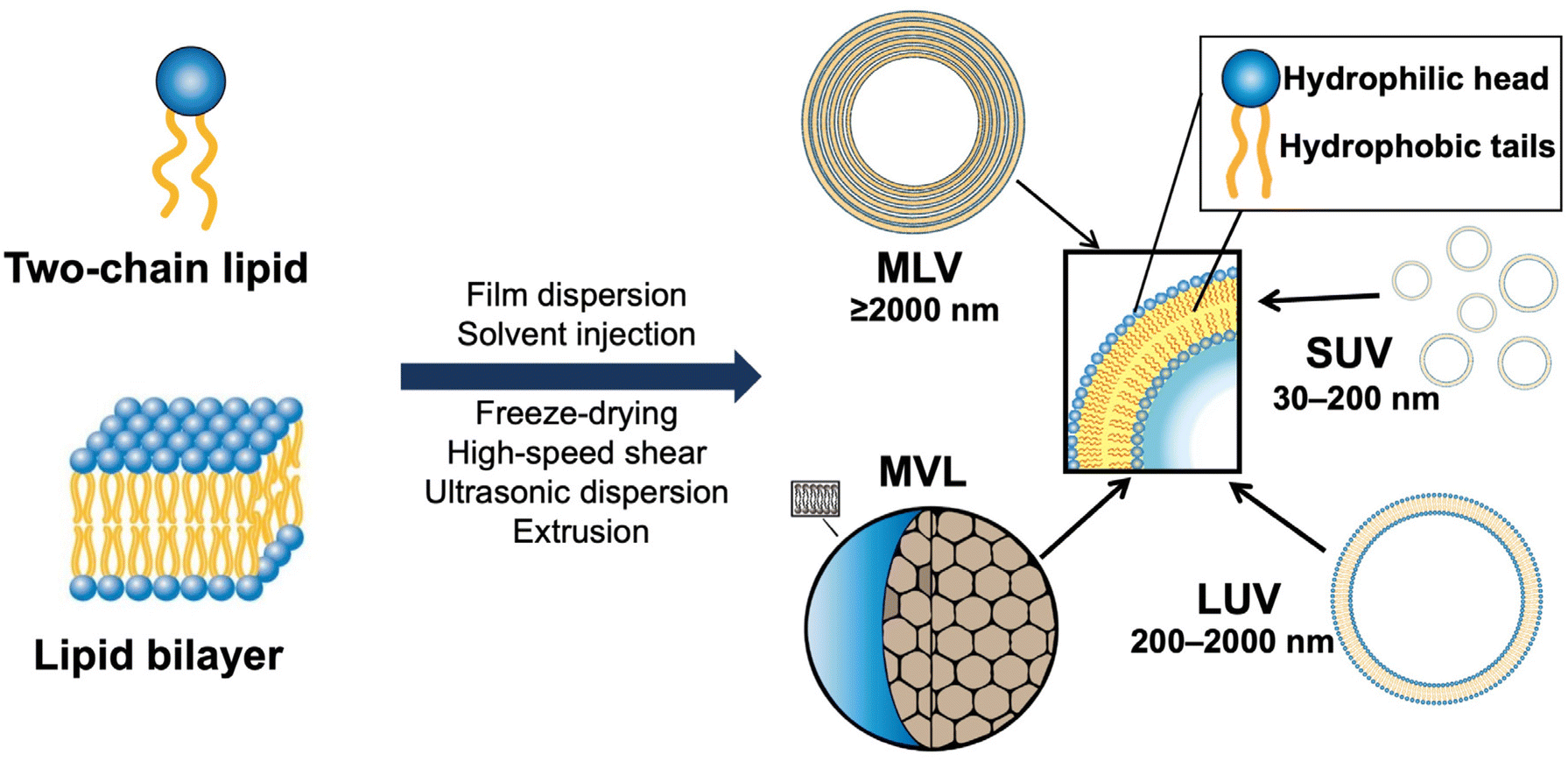 | ||
| Fig. 2 The structural classification of liposomes. | ||
Polyethylene glycol (PEG) has been widely used for surface modification in liposomes. Because of its enhanced surface hydrophilicity and steric hindrance of the PEG chains, PEG modification (aka PEGylation) can reduce the interaction between liposomes and serum proteins and increase the systemic circulation time of drugs. Above the phase transition temperature, the lipid layer would turn into a disordered state, thus leading to decreased colloidal stability and increased drug release. Dipalmitoylphosphatidylcholine (DPPC) and di-stearoyl phosphatidylcholine (DSPC) in a certain proportion are often used for preparing thermosensitive liposomes.25,26 Furthermore, physical methods (e.g., photoirradiation, alternating magnetic stimulation, and microwave irradiation) are usually applied for heating the target site where the thermosensitive liposomes accumulate.25
The interaction between liposomes and cells mainly includes adsorption, fusion, lipid exchange, leakage, endocytosis, and enzymatic digestion.27,28 For instance, liposomes may bind to the cell surface through electrostatic or ligand/receptor interaction, and the lipid components of liposomes and cell membranes can exchange with each other and fuse into the cells.12 Alternatively, the cells may ingest liposomes via phagocytosis, and the thus-formed phagosomes and lysosomes can fuse and develop into secondary lysosomes. As the liposomes disintegrate, the encapsulated drugs are released.
Because of their unique structural characteristics, liposomes can be used for the stable encapsulation of drugs with different physical and chemical properties, thereby improving drug stability and dissolution. Liposomes can also improve the pharmacokinetic profiles and biodistribution of the encapsulated drugs, and thus enhance the therapeutic index. It means that liposomes can deliver more drugs to the targeted tissues at the same dose compared to a conventional formulation, thereby reducing the drug accumulation in normal tissues, reducing adverse drug reactions, and improving the quality of life of patients.29
There is always a pursuit of higher treatment efficacy and safety in medical practice. As an advanced dosage form, liposomes have many incomparable advantages and provide huge opportunities in clinical translation.
3. Liposomal technology: innovation of clinical advantages
The clinical application of liposomes has a history of over 30 years. Due to the continuous demand for new medication in cancer therapy, liposomal formulations have become an R&D hotspot. The antitumor liposomal products mainly include anthracycline, camptothecin, and taxane derivatives, as summarized in Fig. 3. | ||
| Fig. 3 Schematic illustration of improved drug safety. | ||
3.1 Enhancing treatment efficacy
Amphotericin B is a commonly used drug for deep fungal infection by binding to ergocalciferol on the fungal cell membrane, which leads to the formation of pores, ion leakage, and ultimately fungal cell death. The conventional injection formulations generally include the solubilizing agent deoxycholic acid to increase its solubility, but often cause adverse reactions like fever, heart rhythm disorder, and acute erythrocyte hemolysis.30,31 After administration, amphotericin B is mainly excreted from the kidneys, with a narrow therapeutic index and severe nephrotoxicity.32The amphotericin B liposome (AmBisome) has a significantly higher therapeutic index and reduced nephrotoxicity,33 and can be used in patients with renal impairment or unacceptable toxicity precluding the use of amphotericin B. Cholesterol in AmBisome™ could effectively reduce the binding of amphotericin B to the membrane of normal cells.34 The negatively charged di-stearoyl phosphatidylglycerol (DSPG) could stably bind with the positively charged trehalose amine in amphotericin B. AmBisome can enhance therapeutic efficacy.19 Liposomal formulation alters the pharmacokinetic profile of amphotericin B, with a prolonged circulation time and high in vivo exposure, e.g., enhanced Cmax (22.9 μg mL−1) and AUC0–24 (171 μg h mL−1) after a 2 h infusion of liposomal amphotericin B (2 mg kg−1) than amphotericin B deoxycholate (Cmax: 1.4 μg mL−1, AUC0–24: 13.9 μg h mL−1).33 AmBisome is prone to accumulate in the reticuloendothelial organs such as the liver but has significantly reduced distribution in the kidneys (concentrationkidney: 0.87 μg g−1, 5 mg kg−1 d−1) compared to the conventional amphotericin B (concentrationkidney: 12.7 μg g−1, 1 mg kg−1 d−1), suggesting the reduced nephrotoxicity.35
Amphotericin B is a concentration-dependent antifungal drug, and a higher dose is beneficial for antifungal efficacy. AmBisome displayed prolonged post-antifungal effects for a duration of up to 12 hours.30 AmBisome showed MIC50 and MIC90 values of 1 and 2 μg mL−1, respectively. In contrast, the conventional amphotericin B showed MIC50 and MIC90 values of 4 μg ml−1 for both.36 The survival rate of AmBisome against multidrug-resistant Candida auris infection in mice was 4.5 times that of the conventional amphotericin B at 14 days post inoculation.36 Notably, inhaled administration of AmBisome has been under clinical investigation, as summarized in ref. 37, which suggests that prophylactic AmBiosome inhalation could benefit patients at high risk through reduced incidence of invasive pulmonary aspergillosis. AmBiosome has become a standard treatment in many infections, and given its higher dose tolerance, it is called as a replacement for the conventional amphotericin B for primary therapy for invasive fungal infections in clinical practice and research.38
3.2 Improving drug safety
Doxorubicin (DOX), an anthracycline, is potent in treating a variety of cancers. Yet, the main problem of doxorubicin in clinical application is the serious cardiac toxicity.3 Doxil is the first approved doxorubicin liposome. PEGylation can protect carriers from being recognized and phagocytized by the immune system, significantly reducing cardiotoxicity. Doxil is a PEGylated liposomal formulation and has a slower clearance from the circulation (0.04 l h−1 m−2) and a prolonged half-life (55 h) compared with conventional doxorubicin.39 Therefore, it brings forth a new problem: the increased probability of skin toxicity, exemplified by hand-foot syndrome.37 Different from Doxil, Myocet, a marketed non-PEGylated doxorubicin liposomal injection, shows the benefit of reducing skin and cardiac toxicities.40–42 In phase III clinical trials, both Myocet and Doxil exhibited similar responses (≈43%) and median survival rates (19 vs. 16 months; P = 0.79).43 Both Doxil and Myocet reduced the risk of cardiac toxicity,44 with the Cmax of doxorubicin in the heart decreased to 60% and 50% of doxorubicin solution, respectively.45 However, we cannot find a direct comparison study of cardiotoxicity between Doxil and Myocet. It is expected that they may have comparable cardiotoxicity due to their similar heart exposure. The clearance (5.1 ± 4.8 l h−1) of Myocet is much slower than that of conventional doxorubicin (46.7 ± 9.6 l h−1) but not as slow as that of PEGylated Doxil.46 As a result, the non-PEGylated doxorubicin had a lower rate of hand-foot syndrome than Doxil.44 Although the PEGylated doxorubicin liposomes exhibited higher plasma AUC than the non-PEGylated counterpart, paradoxically, the latter showed twice higher intratumor drug accumulation efficiency in a tumor-bearing murine model.47Irinotecan is a water-soluble camptothecin derivative and its metabolite SN38 is a DNA topoisomerase I inhibitor with potent anticancer activity. However, irinotecan has a suboptimal pharmacokinetic profile (e.g., rapid elimination) and acute toxicities (e.g., severe gastrointestinal reactions).48,49 The irinotecan PEGylated liposome (Onivyde) was approved by the FDA in 2015 for use in patients with metastatic pancreatic adenocarcinoma. In a phase III trial, the median overall survival in patients receiving Onivyde plus fluorouracil and folinic acid was 6.1 months, compared with 4.2 months in patients who received fluorouracil and folinic acid.50 Compared with free-form irinotecan, the dose-normalized PK of SN-38 (the active metabolite) after Onivyde treatment was characterized by lower Cmax (9.2 vs. 26.3 ng mL−1), prolonged terminal half-life (75.4 vs. 10.4 h), and higher AUC (710 vs. 229 ng h mL−1).51 The reduced peak concentration could alleviate the adverse reactions.
The traditional paclitaxel injection (Taxol) contains poor biocompatible solvents (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 blend of polyethoxylated castor oil and ethanol), which are highly irritating and may induce hypersensitivity. The incidence of paclitaxel hypersensitivity varies between 8 and 45% and premedication with glucocorticoids and H1 and H2 antagonists is needed for prevention.52 The first paclitaxel liposome product (Lipusu) has been marketed in China for the treatment of breast cancer, gastric cancer, and non-small-cell lung cancer, and has the benefit of relieving the adverse reactions.53 In the single-dose acute toxicity assays, the LD50 values of Lipusu (69.82 mg kg−1) were twice higher than that of Taxol (33.0 mg kg−1), and no hypersensitivity reactions were observed in the Lipusu group.54 Lipusu exhibited a superior safety profile over Taxol. In 2018, Lipusu accounted for about 63% of the paclitaxel market in China, with a sale of RMB 2.168 billion.55
:1 blend of polyethoxylated castor oil and ethanol), which are highly irritating and may induce hypersensitivity. The incidence of paclitaxel hypersensitivity varies between 8 and 45% and premedication with glucocorticoids and H1 and H2 antagonists is needed for prevention.52 The first paclitaxel liposome product (Lipusu) has been marketed in China for the treatment of breast cancer, gastric cancer, and non-small-cell lung cancer, and has the benefit of relieving the adverse reactions.53 In the single-dose acute toxicity assays, the LD50 values of Lipusu (69.82 mg kg−1) were twice higher than that of Taxol (33.0 mg kg−1), and no hypersensitivity reactions were observed in the Lipusu group.54 Lipusu exhibited a superior safety profile over Taxol. In 2018, Lipusu accounted for about 63% of the paclitaxel market in China, with a sale of RMB 2.168 billion.55
3.3 Precision combination therapy
Combination therapy is a common clinical practice in cancer management. The ideal combination can yield the maximum synergistic effect with the minimum doses (Fig. 4). The combination index (CI) method is used to identify an optimized ratio for the synergistic cytotoxicity evaluation (CI < 1, CI = 1, and CI > 1 respectively representing synergy, additivity, and antagonism).56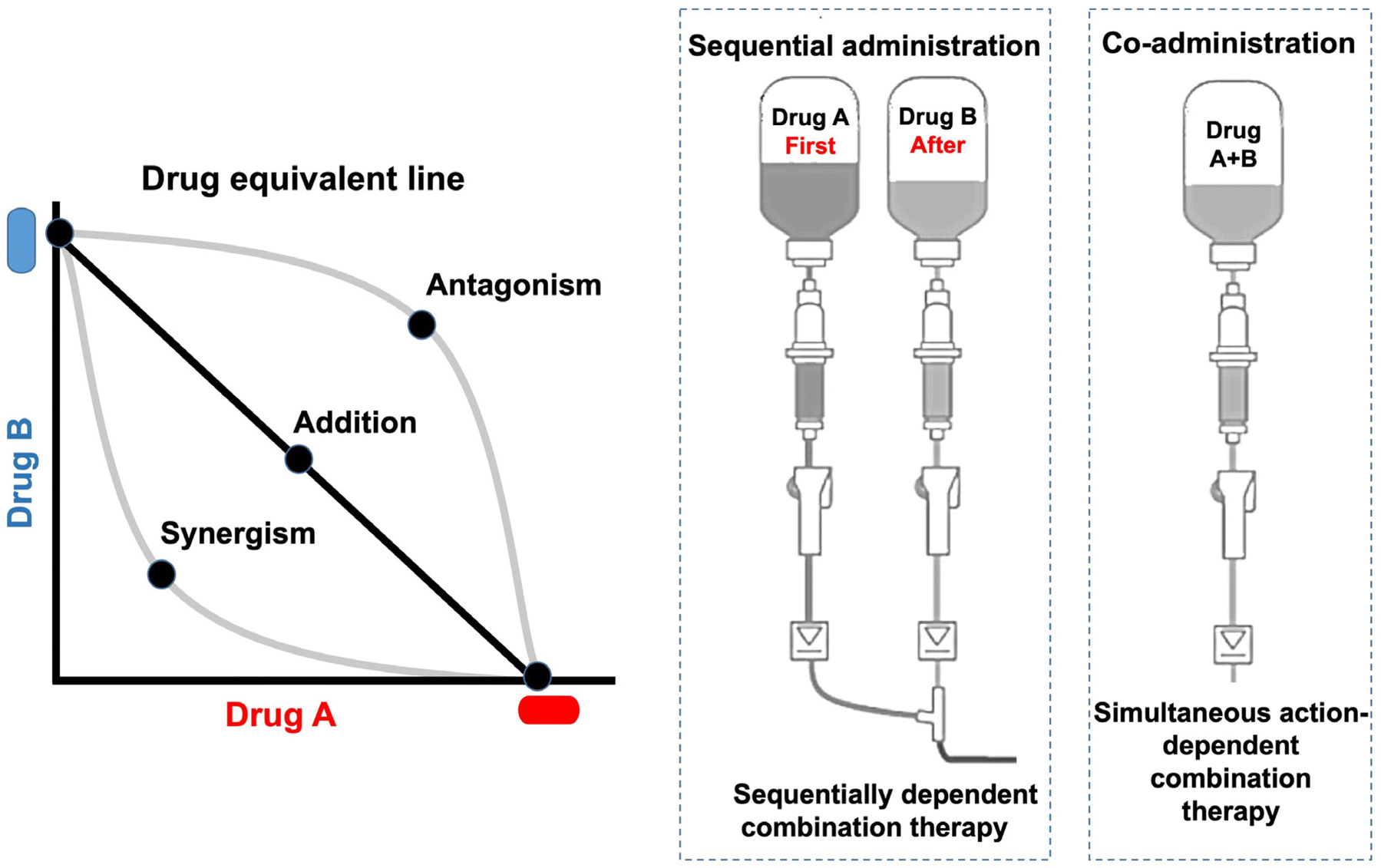 | ||
| Fig. 4 Schematic illustration of combination therapy. Combination therapy typically involves two patterns—sequential administration and co-administration. The latter one has drawn more and more attention for its simultaneous action on the targets. | ||
Currently, most combination regimens are developed on an empirical basis, and preclinical studies do not play a leading role; notably, they seldom yield synergistic effects but most are only additive.57 Although the optimized drug combination can be readily obtained by in vitro cell tests, clinical translation is difficult, which could partially account for the differences in the pharmacokinetics of individual drugs. The administration of combination drugs cannot be simultaneously delivered to tumor cells at a fixed-dose ratio via a regular formulation. The optimal efficacy relies on the synergistic dose ratio.
As a case in point, CPX-351 (Vyxeos) is a liposomal combination of daunorubicin and cytarabine at a 1:5 molar ratio, which is the first liposomal combination approved by the FDA to treat acute myeloid leukemia (AML). Compared with the co-administered free drugs, CPX-351 exhibited similar PK profiles of both drugs, and importantly, the molar ratio of the combination drugs in plasma can remain at a relatively stable level (Fig. 5A).58 The precise delivery of a fixed ratio is essential for achieving optimal treatment efficacy. The survival in the 1:5 ratio group was the highest among all the groups, in particular, yielding 3 times higher than in the 1:1 ratio group (Fig. 5B).59 A phase III study of CPX-351 (NCT01696084) showed that its median progression-free survival (PFS) was 9.5 months, compared to 5.9 months in the control group (cytarabine + daunorubicin therapy); furthermore, the complete remission rate of CPX-351 was 37%, while that of the control group was only 25%.60
 | ||
| Fig. 5 The liposomal combination of daunorubicin and cytarabine. (A) The plasma drug concentrations of CPX-351 in comparison with free drugs in mice. Inset: cytarabine: daunorubicin molar ratios in plasma after CPX-351 administration. (B) The survival rate of mice bearing leukemia tumor with multiple treatments with the liposomal combination containing various drug ratios. Reproduced with permission from ref. 58 and 59. Copyright © 2013 American Chemical Society; Copyright © 2009 Elsevier. | ||
The success of the above study inspired followers. For instance, CPX-1 liposome injection is a liposomal combination of irinotecan and floxuridine. A phase II study of the CPX-1 liposome injection in treating advanced colorectal carcinoma (NCT00361842) was completed in mid-2021. What is more, the clinical trial of liposomal injection of irinotecan and fluorouridine combination (LY01616) is underway for the treatment of advanced gastrointestinal cancers.61
3.4 Innovative administration route
Compared with single drugs, drug–device combination products have the advantages of enhancing drug efficacy and broadening the applicability. The successful cases include stents with drug coating, catheters with antibiotic coating, and wound patches containing antibacterial and anti-inflammatory drugs. The combination of advanced drug delivery systems and medical devices represents a new direction.Amikacin is an aminoglycoside antibiotic against a variety of nontuberculous mycosis (NTM). It needs to be administered intravenously and its use is limited due to the severe toxicity to hearing, balance, and renal function.62,63 Charge-neutral liposomes have been developed to deliver amikacin directly to the lungs. This amikacin liposome inhalation suspension (ALIS) is administered once a day using an eFlow atomization system (Fig. 6). ALIS could not only prolong the release and action of amikacin in the lungs but also reduce systemic exposure. In the lungs, the liposomes can facilitate the uptake by pulmonary macrophages infected with NTM.64,65 The FDA approved ALIS as an orphan drug and a qualified infectious disease product, rendering a 7-year market monopoly.
 | ||
| Fig. 6 Aerosol delivery of ALIS using the eFlow(R) technology. (A) Cryo-electron microscopy of ALIS liposomes. (B) Delivery of ALIS using PARI Lamira nebulizer. Liposome amikacin directly was delivered to the infection site in the lung, with 70% aerosol droplets in the respirable range (MMAD: 4.1–5.3 μm). Reproduced with permission from ref. 66. Copyright © 2021 Elsevier. | ||
By the application of medical devices, a new administration route for the drugs can be realized and thus change a therapeutic pattern. In addition, a self-administered device can improve the accessibility of the medication and the compliance of patients. The development of liposomes with an inhaled drug delivery mode assisted by medical devices is a promising method for lung diseases and could be promising for combating the Covid-19 pandemic.
4. Liposomal technology: the confronting challenges
There have already been dozens of marketed liposomal medications, but the R&D success in liposomal drugs is still a formidable challenge that involves the simultaneous optimization of several parameters to achieve a final liposomal formulation that is safe and effective.67 There are some unsolved fundamental issues in this field, which are discussed below.4.1 Clinical efficacy challenges of liposomal medicines
Liposomal technology is a landmark in the field of cancer nanomedicine. Taking a magic bullet-like action is the holy grail of cancer nanomedicine. In theory, nanomedicine can target the tumor cells while sparing the normal cells, thus enhancing therapeutic efficacy and reducing side toxicity. Liposomal doxorubicin is known to reduce cardiac toxicity because liposomes can diminish drug penetration through the continuous endothelial linings of blood vessels into the heart. However, the clinical observation has not demonstrated the expected benefits—the survival time of cancer patients receiving nanomedicines has not been significantly prolonged.3–5,14For example, PEGylated liposomal doxorubicin (PLD) did not exhibit enhanced efficacy compared with a traditional doxorubicin injection.39 This discouraging case is not alone in cancer nanomedicine, and a similar result was also observed in a study of comparative effectiveness of nanoparticle albumin-bound (nab)-paclitaxel vs. paclitaxel monotherapy, showing that the median overall survival was 11.2 months (nab-paclitaxel) and 10.8 months (paclitaxel).68
It remains unknown why the improved delivery does not lead to enhanced clinical efficacy. The improved delivery efficiency and therapy efficacy of PLD have been widely demonstrated in preclinical animal models.69 The selective delivery of PLD to metastatic breast carcinoma (bone metastases) was also found in the patients, showing a 10-fold higher intratumoral drug concentration compared with that in the adjacent normal tissue.70 But the cutting edge in delivery fails to translate into survival benefits.
What is more, cancer nanomedicines often cause a new problem—hand-foot syndrome, and its incidence increases significantly in the treatment with PLD. Clinical reports revealed that 40–50% of patients developed moderate to severe hand-foot syndrome.71,72 Considering the challenge in the clinical efficacy of liposomal drugs, we must rethink the current delivery strategy. A plausible reason could be the real bioavailability of the free drug in the target site. Doxorubicin must be sufficiently released for entering the nucleus where it takes action. Yet, little is known about this final step in the in vivo kinetics. The issue will be discussed in detail in section 4.4.
4.2 EPR challenges
The high permeability and retention effect (EPR) of solid tumors is associated with the rapid tumor growth that causes a defective and leaky tumor vascular endothelium and insufficient lymphatic drainage and blood flow; under this circumstance, the nanoparticles readily permeate and remain inside the tumor (Fig. 7).73,74 The nanoparticles can preferentially accumulate in the tumor via the EPR effect-based passive targeting delivery.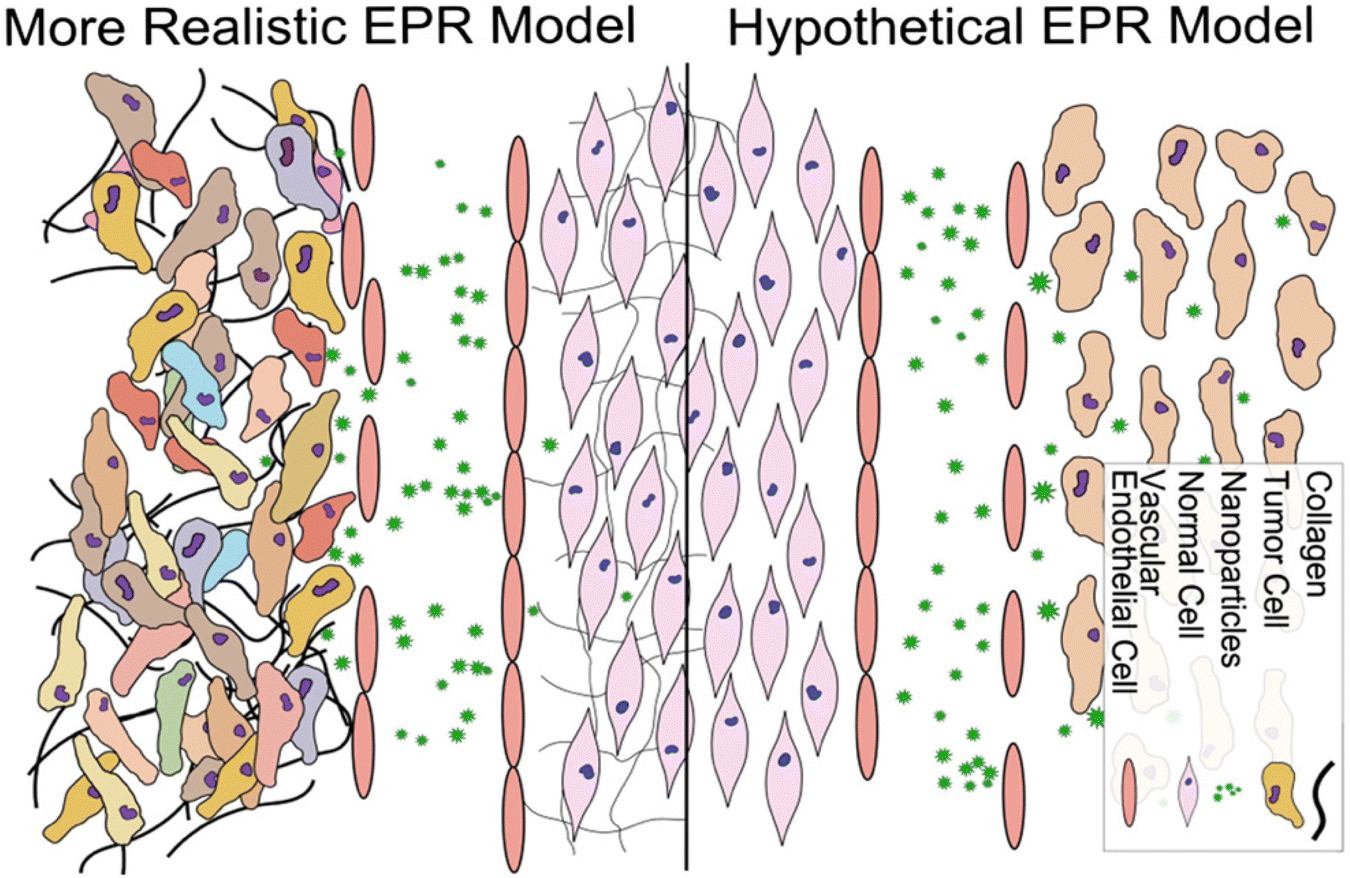 | ||
| Fig. 7 Conventional and more realistic depictions of EPR. Reproduced with permission from ref. 80. Copyright © 2014 Elsevier B.V. | ||
It has been well documented that long circulation is helpful when nanomedicines enhance tumor accumulation through the EPR effect compared with the free drug in the xenografted tumor models.75 Yet, due to the highly leaky vasculature in the subcutaneous flank tumor xenografts, such models are of limited value to evaluate the EPR effect. They probably provide an overstated impression on the EPR effect in nanoparticle delivery.76 But this is not the case in a spontaneous cancer model; a recent report showed neither long- nor short-circulating nanomedicines can enhance intratumor drug accumulation via the EPR effect in a transgenic spontaneous breast cancer model regardless of their size or composition.77 Understandably, spontaneous tumors develop slowly as compared to the xenografted ones that can develop within a few days; the rapidly growing tumors are characterized by an abnormal vascular structure and have more porous tumor blood vessels.78 Another study involving canine spontaneous tumor models revealed that the fast-growing carcinomas generally had higher liposome accumulation than the relatively slow-growing sarcoma, tracked by 64Cu-labeled liposomes.79
Therefore, it is a reasonable consideration that the EPR effect is not a general phenomenon in cancer patients, and instead, the EPR effect varies depending on the tumor type and status. Considering the heterogeneity of tumor histology, it is important to develop an effective method to screen out the patients who are sensitive to nanomedicine and can benefit from the EPR effect, e.g., by using non-invasive imaging of pre-dose imaging agent-labeled liposomes before treatment.80 For instance, radiolabeled liposomes can be used for cancer imaging as well as inflammatory lesion detection with good sensitivity and specificity, and are promising for in vivo diagnostics for screening those suitable for liposome-based therapy. It is time to call for precision nanomedicine: to give the right formulations to the right patients.
4.3 Targeting challenges of liposomal technology
To achieve the active targeting function, liposomes are typically modified with the functional molecules (e.g., sugars, peptides, and antibodies) to overcome the physiological delivery barriers (e.g., blood–brain barrier) via ligand/receptor interaction.81,82 Promising results have been commonly reported in preclinical studies, but there is a huge gap from preclinical to clinical application. Although the active targeting liposomal technology has been widely explored, it has not yet achieved successful clinical translation.83,84 An opinion is that the tumor cells are highly heterogenic and their surface characteristics are various even in the same tumor tissue, thus leading to insufficient active targeting efficiency. However, the active targeting delivery strategy in antibody–drug conjugates (ADC) has obtained breakthrough success; there have already been more than ten ADC products approved. In contrast, none of the antibody-modified liposomes (aka immunoliposomes) can survive through clinical trials. For example, anti-HER2 is a commonly used antibody component of the marketed ADCs (e.g., Trastuzumab deruxtecan); it demonstrates that the anti-HER2-mediated active targeting delivery strategy is workable. Anti-HER2 was also applied in an immunoliposome drug (MM-302) serving as a targeted delivery ligand, but the phase II clinical trial (HERMIONE) of MM-302 failed to meet the endpoint.85 Therefore, the heterogenic expression of the target receptors in the cancer cell surface may not be the major issue responsible for the failure of the immunoliposomes. A plausible account is that the cargo size matters. In ADC, typically the molecular weight ratio between the targeting ligand and the drug cargo is 40:1, while for immunoliposomes, the size of a liposomal sphere could be hundreds of times larger than the conjugated antibodies. In a figurative sense of the size difference, ADC is like a huge horse pulling a tiny cart, while immunoliposome is like a horse pulling a train. Therefore, there is a pressing need to elucidate the binding kinetics between immunoliposomes and cells via ligand/receptor interaction as well as the difference in the fluid dynamics of immunoliposomes in the human body and the experimental animals (typically, a mouse model). The knowledge will help understand the paradox.
Of course, many factors potentially affect the treatment outcomes in the patients. Further detailed investigations need to be carried out to illustrate the unknown issues.
4.4 In vivo fate challenges of liposomes
Compared with free drugs, liposomes sustain a highly complex fate. However, the biological fate of liposomes has not been fully understood. Currently, not much is known about the dynamic process of the “three elements” (i.e., drug, carrier, and material) and other auxiliary factors such as ligands, surfactants, and stabilizers.27 For example, the essential questions of when, where, and how drugs are released from liposomes in vivo remain still unanswered. A comprehensive interpretation of the biological fate will be of scientific significance for the design of liposomes and can accelerate the process of clinical transformation.86In an FDA guidance document (Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation), it is suggested that both free and total drugs should be described in a pharmacokinetic study. It means that the amounts of the free drug that is released from the liposome and the total liposome-encapsulated drug should be separated and determined.
Due to the limitation of the pharmaceutical analysis, it remains a formidable challenge for separating the released free drugs and the liposomal encapsulated drugs, as well as the subsequent quantitative determination. The traditional radiation and fluorescence labeling strategies cannot effectively distinguish the signals of released markers and carriers, resulting in great interference and uncertainty.87
The liposome-encapsulated drugs can be viewed as a specific form of “macro-prodrugs”, which remain inactive unless they are released and turn into a form of free molecules. Taking liposomal doxorubicin as an example, the free drug must enter the nucleus to intercalate within DNA base pairs for taking cytotoxic action (Fig. 8). In this sense, only the free intranuclear drug is bioavailable. But so far, the intratumoral, intracellular, and internuclear kinetics of liposomal drug release is little understood and remains in a “black box” status. The plasma concentration of liposomal drugs could hardly accurately reflect the actual concentration in the target (i.e., nucleus). A better understanding of the liposomal drug delivery process will be helpful for the rational design. However, it is even more difficult for separating the free drug and the encapsulated drug in a tissue or the target cells, and advanced bioanalytical methods are needed to be explored, too, to reach this goal.
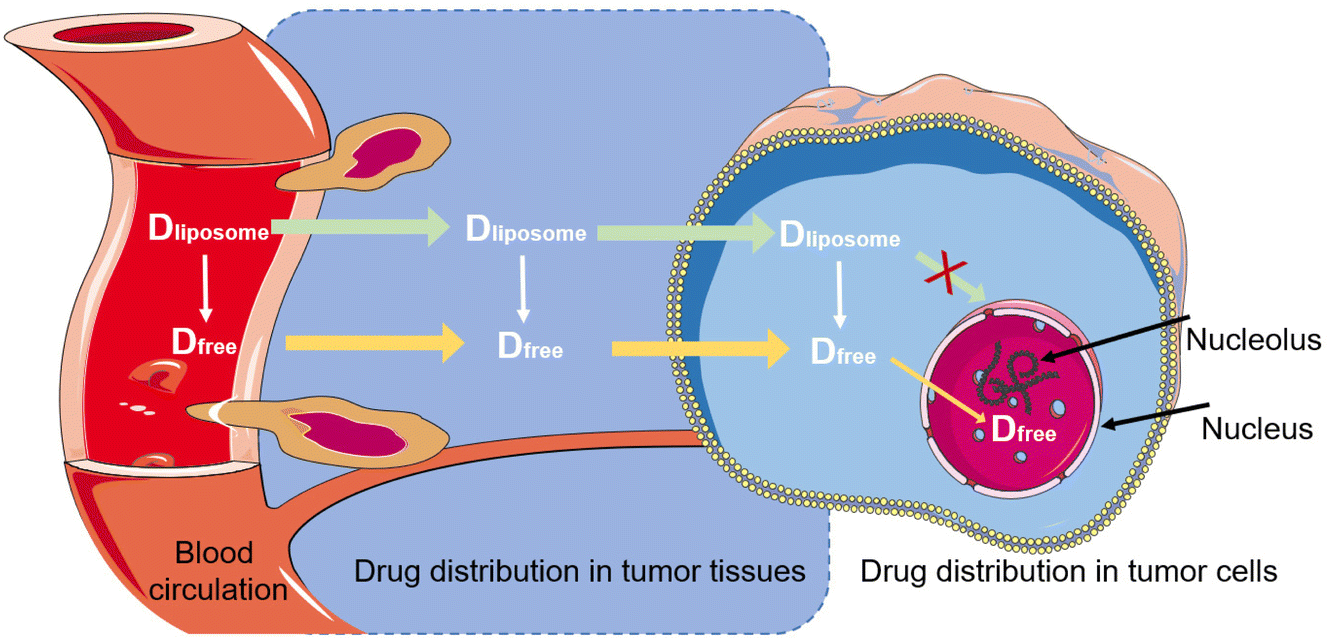 | ||
| Fig. 8 The illustration of the in vivo kinetics of liposomal drugs. | ||
4.5 Lipid excipient challenges
Pharmaceutical-grade phospholipids are the critical excipients of liposomes. The lipid materials significantly affect the physical and biopharmaceutical properties of liposomes. FDA industry guidance about liposomes has a strict requirement of lipid information provided for new drug applications. The high-quality requirement of lipid excipients leads to a high threshold for the R&D of liposomal drugs. The price of lipids is also costly. For example, for cation liposome-based mRNA delivery, the development of the industrialized production of lipids is the critical step in the R&D. The current patents related to a lipid-based delivery have created a high-tech barrier against the followers.4.6 Preparation technology challenges
A series of methods is available in liposome preparation, including injection method, film dispersion method, ultrasonic dispersion method, freeze-drying method, high-speed shear method, and extrusion method.15,88,89 Often, multiple methods are used in the process, requiring a multi-step operation. The complicated process renders difficulty in quality control between batches, such as particle size, size distribution, surface charge, and drug release behavior. In particular, the challenge of scaling up is a primary obstacle. The production equipment is usually customized by manufacturers, according to the needs of the specific liposome products and involves many mechanical engineering technologies.5. The transition from concept to clinical application
5.1 Microfluidics
Traditional liposomal preparation methods require multi-step complicated operations. Aggregation of the polydispersed particles occurs easily, resulting in batch-to-batch differences in the physical and chemical properties.To overcome the limitations of traditional methods, microfluidic technology in the field of continuous manufacturing has attracted more and more attention. As summarized in Fig. 9, microfluidic technology can control the fluid mixing and realize the controllable and repeatable preparation of liposomes.90,91 The prepared liposomes are uniform and the difference between batches is small.15,92 As a case in point (Fig. 10), the preparation of mRNA coronavirus vaccines relies on microfluidic technology.93
 | ||
| Fig. 9 Preparation technology of microfluidics and the advantages. | ||
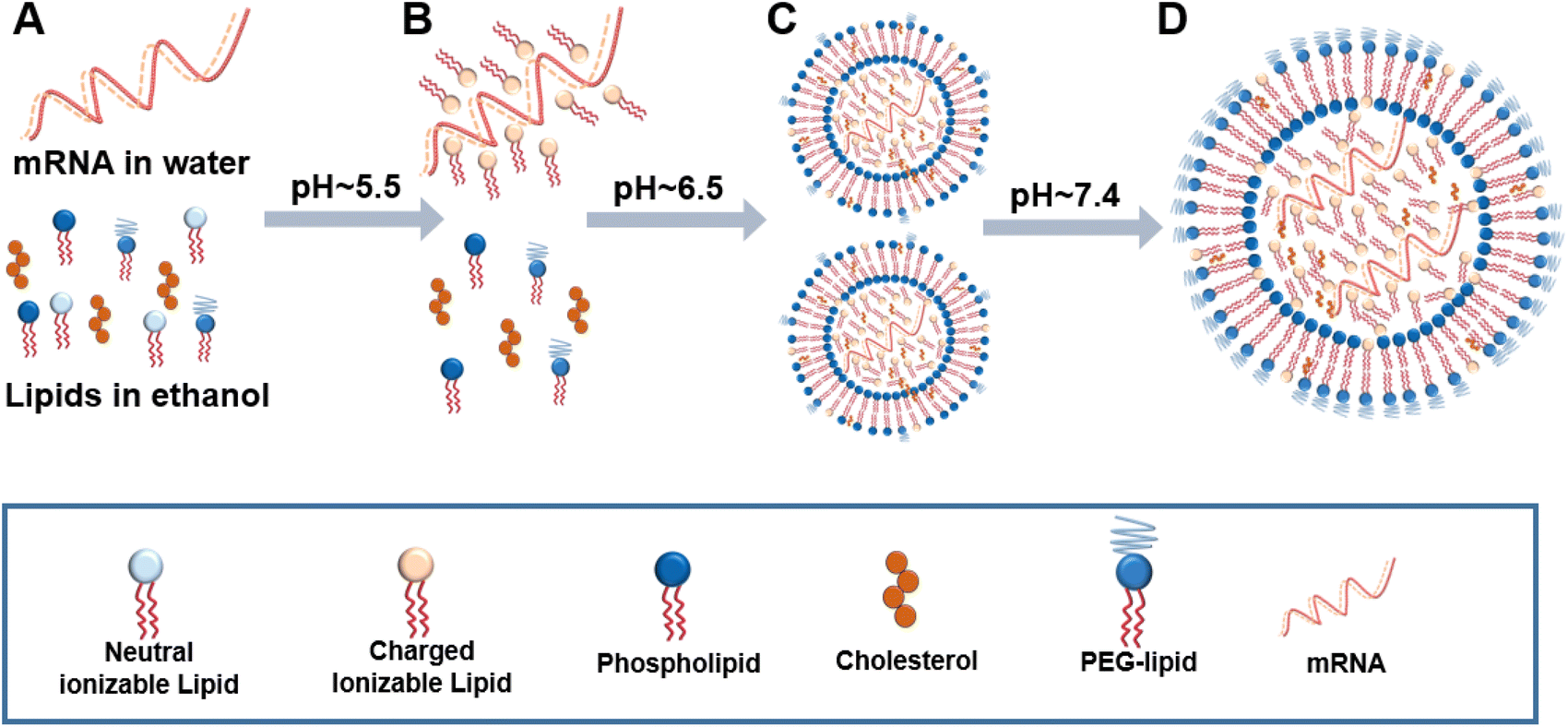 | ||
| Fig. 10 mRNA liposome preparation. (A) Rapid mixing of four lipids (ionizable lipid, DSPC, cholesterol, PEG-lipid) in ethanol and mRNA in an aqueous buffer (pH 4). (B) The ionizable lipid becomes protonated at pH 5.5, which is intermediate between the pKa of the buffer and that of the ionizable lipid. (C) The cationized lipid binds with mRNA, driving vesicle formation and mRNA encapsulation. (D) The elevating pH caused by dilution, dialysis, or filtration, results in the neutralization of the ionizable lipid, rendering it more hydrophobic and thereby driving vesicles to fuse and promoting further sequestration of the ionizable lipid with mRNA into the interior of the liposomes. | ||
However, the pharmaceutical application of microfluidic technology still mainly stays in laboratories.89 For the transition from the laboratory to clinical application, there are many problems to be solved, for example, how to accurately control the components, flow rate, and fluid dynamics.94,95 The output of the traditional microfluidic process is limited to milliliters per hour, unsuitable for mass production. A recent report revealed that a parallelized microfluidic device containing an array of 128 mixing channels can increase the production rate to 18.4 L h−1 by operating simultaneously.96 It is expected that a deeper understanding of the hydrodynamic theory of microfluidics and the liposome formation mechanism will promote its wide application. Microfluidic technology may provide a useful tool for building an automated continuous liposomal manufacturing platform and realizing the fast translation from academic research to industrial production.
5.2 Old drugs, new formulations
To cope with the patent challenge and market competition, new drug development via the 505(b)(2) application pathway is an important R&D strategy for pharmaceutical manufacturers. A 505(b)(2) application is usually used to get approvals for new delivery mechanisms, dosage forms, formulations, and new indications, which may benefit from faster and lower costs than the traditional process. Innovative liposomal formulations have been actively explored and there is a high market value for new liposomal products. For example, Doxil has achieved good market performance, reaching a sales peak of US $600 million in 2010.97 The development of liposome products by the 505(b)(2) pathway aims at overcoming the defects of the drugs, and improving the treatment efficacy or reducing side effects, thus offering practical benefits to patients. For instance, the paclitaxel cationic liposome EndoTAG-1 has been developed for neovascular targeting chemotherapy and under phase III clinical trial, with EMA and FDA orphan drug designations.98,99 EndoTAG-1 is characterized by a positive charge owing to the incorporation of cationic DOTAP in the lipid membrane. Tumor vessel endothelium and tumor cells possess highly negative surface charge. The cationic EndoTAG-1 thus can be preferentially adsorbed on the tumor neovascular endothelial cells, destroy tumor neovascularization, and effectively kill the cancer cells.100 In addition, EndoTAG-1 can also increase tumor microvessel leakage significantly by causing intratumoral vascular damage.101 The treatment also led to tumor-selective microthromboses, accompanied by an increase of platelet adherence and subsequent tumor microvessel occlusions.102 The neovascular targeting mechanisms by cationic liposomes are still largely unknown, though it is plausibly due to charge-dependent binding and uptake. The cationic lipid complexed camptothecin (EndoTAG-2) has also been in development.1035.3 Liposomal combination
In cancer therapy, drug combinations are generally used. The success of the CPX-351 liposome casts light on a new direction. Liposomes can encapsulate the combined drugs and deliver them to target tissues in a specific ratio to achieve the synergistic effect of combined chemotherapy.97,104 Combination therapy using liposomes with a fixed-dose ratio has attracted great attention in the pharmaceutical industry. The liposomal combination has many advantages such as enhanced efficacy and convenient use. In specific, combination therapy represents a useful method to overcome drug resistance, and liposomes can serve as an ideal carrier for codelivery of various kinds of combined drugs (e.g., cytotoxin/cytotoxin, cytotoxin/molecularly targeted drug, cytotoxin/sensitizing agent).105 For instance, a mannosylated liposome system was developed for codelivery of dihydroartemisinin and doxorubicin for treating the drug-resistant colon tumor, and this liposomal combination could enhance the intranuclear drug accumulation and cancer cell apoptosis, and promote autophagy.106 Liposomal technology provides a useful tool for synchronous drug delivery for achieving the synergistic effect in treatments.5.4 Liposomes for gene delivery
The success of gene therapy depends on the vectors that can effectively transfer genetic materials to the target cells and obtain sufficient gene expression in vivo with minimal toxicity. As the main components of liposomes, phospholipids determine the biocompatibility and efficacy of therapeutic liposomes. The quality of phospholipids affects the development of liposome products. The development of special functional phospholipids such as ionizable lipids has brought a breakthrough in the field. A successful case is a liposome-based system (ONPATTRO) developed by Alnylam to solve the problem of siRNA targeted delivery.107–109 ONPATTRO encapsulates the negatively charged siRNA drugs using ionizable cationic lipids DLin-MC3-DMA and PEG2000-C-DMG, and could significantly improve several clinical indicators of familial amyloid polyneuropathy.110,111 The successful application of cationic lipid carrier technology is of great significance in both technical innovation and clinical application, and opens a door for the development of siRNA drugs.Meanwhile, the development of nucleotide-based mRNA vaccines has been extensively promoted during the COVID-19 pandemic.10 Liposome-based carrier technology is the mainstream for mRNA delivery. A successful case is the lipid nanoparticle (LNP)-based mRNA anti-SARS-Cov-2 vaccines, in which an ionizable cationic lipid (apparent pKa < 6.5) is the essential component that can interact electrostatically and through hydrogen bonds with mRNA.112 The two major mRNA vaccine enterprises (Moderna and BioNTech) have both applied this delivery technology. Notably, there are many mRNA vaccines under clinical trials for various diseases including HIV (NCT05001373, NCT05217641), Zika virus (NCT04917861), seasonal influenza (NCT04956575), and ovarian cancer (NCT04163094). With the increasing value of nucleic acid drugs from early preclinical study to the clinic, the efficient, safe, and stable delivery of nucleic acid drugs by liposome and lipid carrier technology has been a hot spot.
6. Summary and perspective
Drug R&D has never been easy. It has been more than half a century since liposomes were discovered, and great advances have been observed in the medical application of liposomes for treating cancers and for anti-infection and analgesia. However, the clinical needs of liposome-based treatments are still unmet. Liposome technology is useful, and but may not fit for all drugs. Regarding what it can do and what it cannot do, researchers are still actively exploring its technical boundaries. So far, not much is known to predict the drug/lipid interaction for achieving rational design. The development of liposomal formulations still largely relies on trial and error.Although the resources and input have been mostly directed towards tumor drug delivery mediated by liposomal technology, it could hardly bring a revolutionary breakthrough in the short term. It is time to rethink what will be the next for pharmaceutical liposomal delivery. There are three essential issues that must be taken into account. First, the collaboration between pharmaceutical scientists and physicians is necessary to select a right drug for liposomal formulation development for a right disease application and to meet the clinical demand. Clinically oriented or clinically driven R&D should be emphasized. Second, it is important to explore the fundamental physical, chemical, and biological principles related to liposome preparation and therapy. A better understanding of the interaction between lipids and drugs, molecular dynamics in liposomal self-assembly, and the interplay among liposomes, body fluids, and cells will help develop efficient and safe liposome-based drug delivery systems. In addition, the illustration of the in vivo fate of liposomal drugs will provide helpful information for designing a treatment regimen with improved efficacy. Third, the application of special functional lipids can modulate the performance of liposome products. The development of novel lipids or lipid membrane additives could bring forth transformation to the field of liposome research. As a case in point, the LNP-based mRNA vaccines represent a breakthrough, in which ionizable phospholipids serve as the key element.
The application of advanced mechanical technology is useful to ease liposome preparation. Time and cost for the process can be reduced but with enhanced quality control. For example, microfluidic technology has been increasingly used in liposome manufacturing. Technological innovation will further promote the R&D of liposomal medicines.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are thankful to the National Key Research and Development Program of China (2021YFE0103100, 2021YFC2400600, China), NFSC (81925035) and Shanghai SciTech Innovation Initiative (19431903100, 18430740800) for the support. We also acknowledge the support by the projects of High-Level New R&D Institute (2019B090904008) and High-Level Innovative Research Institute (2021B0909050003) from the Department of Science and Technology of Guangdong Province.References
- S. S. Cai, T. Li, T. Akinade, Y. Zhu and K. W. Leong, Adv. Drug Delivery Rev., 2021, 176, 113884 CrossRef CAS PubMed.
- H. Wang and Y. Huang, Med. Drug Discovery, 2020, 6, 100024 CrossRef.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- Y. Barenholz, Curr. Opin. Colloid Interface Sci., 2001, 6, 66–77 CrossRef CAS.
- A. Alshehri, A. Grabowska and S. Stolnik, Sci. Rep., 2018, 8, 3748 CrossRef PubMed.
- H. Maroof, F. Islam, L. Dong, P. Ajjikuttira, V. Gopalan, N. A. J. McMillan and A. K. Lam, Cells, 2018, 7, 265 CrossRef CAS PubMed.
- Y. Weng, C. Li, T. Yang, B. Hu, M. Zhang, S. Guo, H. Xiao, X. J. Liang and Y. Huang, Biotechnol. Adv., 2020, 40, 107534 CrossRef CAS PubMed.
- J. Chen, Z. Guo, H. Tian and X. Chen, Mol. Ther.–Methods Clin. Dev., 2016, 3, 16023 CrossRef PubMed.
- D. Brain, A. Plant-Hately, B. Heaton, U. Arshad, C. David, C. Hedrich, A. Owen and N. J. Liptrott, Adv. Drug Delivery Rev., 2021, 178, 113848 CrossRef CAS PubMed.
- C. Zylberberg, K. Gaskill, S. Pasley and S. Matosevic, Gene Ther., 2017, 24, 441–452 CrossRef CAS PubMed.
- D. E. Large, R. G. Abdelmessih, E. A. Fink and D. T. Auguste, Adv. Drug Delivery Rev., 2021, 176, 113851 CrossRef CAS PubMed.
- Z. Gu, C. G. Da Silva, K. Van der Maaden, F. Ossendorp and L. J. Cruz, Pharmaceutics, 2020, 12, 1054 CrossRef CAS PubMed.
- N. Filipczak, J. Pan, S. S. K. Yalamarty and V. P. Torchilin, Adv. Drug Delivery Rev., 2020, 156, 4–22 CrossRef CAS.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef.
- A. D. Bangham, Nature, 1961, 192, 1197–1198 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660–668 CrossRef CAS PubMed.
- A. D. Bangham, M. W. Hill and N. G. A. Miller, in Methods in Membrane Biology, ed. E. D. Korn, Springer US, Boston, MA, 1974, ch. 1, pp. 1–68, DOI:10.1007/978-1-4615-7422-4_1.
- F. Meunier, H. G. Prentice and O. Ringden, J. Antimicrob. Chemother., 1991, 28(Suppl B), 83–91 CrossRef.
- G. Gregoriadis, Pharmaceutics, 2016, 8, 1–5 CrossRef.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef.
- G. M. Jensen and D. F. Hodgson, Adv. Drug Delivery Rev., 2020, 154–155, 2–12 CrossRef CAS.
- Y. Dou, K. Hynynen and C. Allen, J. Controlled Release, 2017, 249, 63–73 CrossRef CAS.
- Y. Fan and Q. Zhang, Asian J. Pharm. Sci., 2013, 8, 81–87 CrossRef CAS.
- L. Ho, M. Bokharaei and S. D. Li, J. Drug Targeting, 2018, 26, 407–419 CrossRef CAS.
- M. B. C. de Matos, N. Beztsinna, C. Heyder, M. Fens, E. Mastrobattista, R. M. Schiffelers, G. Leneweit and R. J. Kok, Eur. J. Pharm. Biopharm., 2018, 132, 211–221 CrossRef CAS.
- W. Wu and T. Li, Adv. Drug Delivery Rev., 2019, 143, 1–2 CrossRef CAS.
- H. Wang, M. Zheng, J. Gao, J. Wang, Q. Zhang, J. P. Fawcett, Y. He and J. Gu, Talanta, 2020, 208, 120358 CrossRef CAS PubMed.
- X. Meng, Z. Zhang, J. Tong, H. Sun, J. P. Fawcett and J. Gu, Acta Pharm. Sin. B, 2021, 11, 1003–1009 CrossRef CAS.
- N. R. Stone, T. Bicanic, R. Salim and W. Hope, Drugs, 2016, 76, 485–500 CrossRef CAS.
- J. D. Berman, G. Ksionski, W. L. Chapman, V. B. Waits and W. L. Hanson, Antimicrob. Agents Chemother., 1992, 36, 1978–1980 CrossRef CAS.
- F. Aversa, A. Busca, A. Candoni, S. Cesaro, C. Girmenia, M. Luppi, A. M. Nosari, L. Pagano, L. Romani, G. Rossi, A. Venditti and A. Novelli, J. Chemother., 2017, 29, 131–143 CrossRef CAS.
- I. Bekersky, R. M. Fielding, D. E. Dressler, J. W. Lee, D. N. Buell and T. J. Walsh, Antimicrob. Agents Chemother., 2002, 46, 828–833 CrossRef CAS PubMed.
- S. Jain, P. U. Valvi, N. K. Swarnakar and K. Thanki, Mol. Pharm., 2012, 9, 2542–2553 CrossRef CAS.
- K. Shimizu, M. Osada, K. Takemoto, Y. Yamamoto, T. Asai and N. Oku, J. Controlled Release, 2010, 141, 208–215 CrossRef CAS PubMed.
- J. Herrada, A. Gamal, L. Long, S. P. Sanchez, T. S. McCormick and M. A. Ghannoum, Antimicrob. Agents Chemother., 2021, 65, e00306-21 CrossRef.
- A. H. Groll, B. J. A. Rijnders, T. J. Walsh, J. Adler-Moore, R. E. Lewis and R. J. M. Bruggemann, Clin. Infect. Dis, 2019, 68, S260–S274 CrossRef CAS.
- L. Ostrosky-Zeichner, K. A. Marr, J. H. Rex and S. H. Cohen, Clin. Infect. Dis, 2003, 37, 415–425 CrossRef CAS.
- M. E. O’Brien, N. Wigler, M. Inbar, R. Rosso, E. Grischke, A. Santoro, R. Catane, D. G. Kieback, P. Tomczak, S. P. Ackland, F. Orlandi, L. Mellars, L. Alland, C. Tendler and CAELYX Breast Cancer Study Group, Ann. Oncol., 2004, 15, 440–449 CrossRef PubMed.
- S. Nagpal, S. Braner, H. Modh, A. X. X. Tan, M. P. Mast, K. Chichakly, V. Albrecht and M. G. Wacker, Eur. J. Pharm. Biopharm., 2020, 153, 257–272 CrossRef CAS.
- M. Marty, Breast, 2001, 10, 28–33 CrossRef.
- U. Kanwal, N. Irfan Bukhari, M. Ovais, N. Abass, K. Hussain and A. Raza, J. Drug Targeting, 2018, 26, 296–310 CrossRef CAS.
- G. Batist, G. Ramakrishnan, C. S. Rao, A. Chandrasekharan, J. Gutheil, T. Guthrie, P. Shah, A. Khojasteh, M. K. Nair, K. Hoelzer, K. Tkaczuk, Y. C. Park and L. W. Lee, J. Clin. Oncol., 2001, 19, 1444–1454 CrossRef CAS.
- A. Fukuda, K. Tahara, Y. Hane, T. Matsui, S. Sasaoka, H. Hatahira, Y. Motooka, S. Hasegawa, M. Naganuma, J. Abe, S. Nakao, H. Takeuchi and M. Nakamura, PLoS One, 2017, 12, e0185654 CrossRef.
- R. Luo, Y. Li, M. He, H. Zhang, H. Yuan, M. Johnson, M. Palmisano, S. Zhou and D. Sun, Int. J. Pharm., 2017, 519, 1–10 CrossRef CAS PubMed.
- C. E. Swenson, W. R. Perkins, P. Roberts and A. S. Janoff, Breast, 2001, 10, 1–7 CrossRef.
- R. L. Hong, C. J. Huang, Y. L. Tseng, V. F. Pang, S. T. Chen, J. J. Liu and F. H. Chang, Clin. Cancer Res., 1999, 5, 3645–3652 CAS.
- Y. N. Lamb and L. J. Scott, Drugs, 2017, 77, 785–792 CrossRef CAS PubMed.
- J. E. Frampton, Drugs, 2020, 80, 1007–1018 CrossRef CAS.
- S. S. Ur Rehman, K. Lim and A. Wang-Gillam, Expert Rev. Anticancer Ther., 2016, 16, 485–492 CrossRef PubMed.
- T. C. Chang, H. S. Shiah, C. H. Yang, K. H. Yeh, A. L. Cheng, B. N. Shen, Y. W. Wang, C. G. Yeh, N. J. Chiang, J. Y. Chang and L. T. Chen, Cancer Chemother. Pharmacol., 2015, 75, 579–586 CrossRef CAS PubMed.
- J. Boulanger, J. N. Boursiquot, G. Cournoyer, J. Lemieux, M. S. Masse, K. Almanric, M. P. Guay and Comité de l’évolution des pratiques en oncologie, Curr. Oncol., 2014, 21, e630–e641 CrossRef PubMed.
- Q. Chen, Q. Z. Zhang, J. Liu, L. Q. Li, W. H. Zhao, Y. J. Wang, Q. H. Zhou and L. Li, Zhonghua Zhongliu Zazhi, 2003, 25, 190–192 Search PubMed.
- H. Wang, G. Cheng, Y. Du, L. Ye, W. Chen, L. Zhang, T. Wang, J. Tian and F. Fu, Mol. Med. Rep., 2013, 7, 947–952 CrossRef CAS PubMed.
- A. Chonn and P. R. Cullis, Adv. Drug Delivery Rev., 1998, 30, 73–83 CrossRef CAS PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- D. A. Yardley, J. Controlled Release, 2013, 170, 365–372 CrossRef CAS PubMed.
- L. Ma, M. Kohli and A. Smith, ACS Nano, 2013, 7, 9518–9525 CrossRef CAS PubMed.
- P. Tardi, S. Johnstone, N. Harasym, S. Xie, T. Harasym, N. Zisman, P. Harvie, D. Bermudes and L. Mayer, Leuk. Res., 2009, 33, 129–139 CrossRef CAS.
- J. E. Lancet, G. L. Uy, J. E. Cortes, L. F. Newell, T. L. Lin, E. K. Ritchie, R. K. Stuart, S. A. Strickland, D. Hogge, S. R. Solomon, R. M. Stone, D. L. Bixby, J. E. Kolitz, G. J. Schiller, M. J. Wieduwilt, D. H. Ryan, A. Hoering, K. Banerjee, M. Chiarella, A. C. Louie and B. C. Medeiros, J. Clin. Oncol., 2018, 36, 2684–2692 CrossRef CAS PubMed.
- L. Pharma, First Patient Enrolled in Phase I Clinical Trial for the First Compound Liposome Formulation in China, Medication Designed to Improve Efficacy and Safety of Combined Chemotherapy, https://www.luye.cn/lvye_en/view.php?id=1934, (accessed July 2022).
- M. Bassetti, A. Vena, A. Russo and M. Peghin, Drugs, 2020, 80, 1309–1318 CrossRef CAS PubMed.
- R. Schiffelers, G. Storm and I. Bakker-Woudenberg, J. Antimicrob. Chemother., 2001, 48, 333–344 CrossRef CAS.
- M. Shirley, Drugs, 2019, 79, 555–562 CrossRef CAS PubMed.
- J. Zhang, F. Leifer, S. Rose, D. Y. Chun, J. Thaisz, T. Herr, M. Nashed, J. Joseph, W. R. Perkins and K. DiPetrillo, Front. Microbiol., 2018, 9, 915 CrossRef PubMed.
- Z. Li, W. Perkins and D. Cipolla, Eur. J. Pharm. Biopharm., 2021, 166, 10–18 CrossRef CAS.
- D. Guimaraes, A. Cavaco-Paulo and E. Nogueira, Int. J. Pharm., 2021, 601, 120571 CrossRef CAS.
- T. Luhn, S. Chui, A. Hsieh, J. Yi, A. Mecke, P. Bajaj, W. Hasnain, A. Falgas, T. G. N. Ton and A. Kurian, Ann. Oncol., 2018, 29, 1173–1185 CrossRef.
- V. Makwana, J. Karanjia, T. Haselhorst, S. Anoopkumar-Dukie and S. Rudrawar, Int. J. Pharm., 2021, 593, 120117 CrossRef CAS PubMed.
- Z. Symon, A. Peyser, D. Tzemach, O. Lyass, E. Sucher, E. Shezen and A. Gabizon, Cancer, 1999, 86, 72–78 CrossRef CAS.
- G. Liang, W. Ma, Y. Zhao, E. Liu, X. Shan, W. Ma, D. Tang, L. Li, X. Niu, W. Zhao and Q. Zhang, BMC Cancer, 2021, 21, 362 CrossRef CAS PubMed.
- D. Lorusso, A. Di Stefano, V. Carone, A. Fagotti, S. Pisconti and G. Scambia, Ann. Oncol., 2007, 18, 1159–1164 CrossRef CAS PubMed.
- H. Maeda, T. Sawa and T. Konno, J. Controlled Release, 2001, 74, 47–61 CrossRef CAS PubMed.
- T. Tanaka, S. Shiramoto, M. Miyashita, Y. Fujishima and Y. Kaneo, Int. J. Pharm., 2004, 277, 39–61 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- X. Luan, H. Yuan, Y. Song, H. Hu, B. Wen, M. He, H. Zhang, Y. Li, F. Li, P. Shu, J. P. Burnett, N. Truchan, M. Palmisano, M. P. Pai, S. Zhou, W. Gao and D. Sun, Biomaterials, 2021, 275, 120910 CrossRef CAS PubMed.
- A. Z. Wang, Sci. Transl. Med., 2015, 7, 294ed7 Search PubMed.
- A. E. Hansen, A. L. Petersen, J. R. Henriksen, B. Boerresen, P. Rasmussen, D. R. Elema, P. M. af Rosenschöld, A. T. Kristensen, A. Kjaer and T. L. Andresen, ACS Nano, 2015, 9, 6985–6995 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed.
- N. Dos Santos, C. Allen, A. M. Doppen, M. Anantha, K. A. Cox, R. C. Gallagher, G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, 2007, 1768, 1367–1377 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- T. Chen, B. He, J. Tao, Y. He, H. Deng, X. Wang and Y. Zheng, Adv. Drug Delivery Rev., 2019, 143, 177–205 CrossRef CAS.
- P. Shrimal, G. Jadeja and S. Patel, Chem. Eng. Res. Des., 2020, 153, 728–756 CrossRef CAS.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Adv. Drug Delivery Rev., 2018, 128, 54–83 CrossRef CAS.
- L. Capretto, D. Carugo, S. Mazzitelli, C. Nastruzzi and X. Zhang, Adv. Drug Delivery Rev., 2013, 65, 1496–1532 CrossRef CAS PubMed.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS.
- A. Forigua, R. L. Kirsch, S. M. Willerth and K. S. Elvira, J. Controlled Release, 2021, 333, 258–268 CrossRef CAS.
- M. D. Buschmann, M. J. Carrasco, S. Alishetty, M. Paige, M. G. Alameh and D. Weissman, Vaccines, 2021, 9, 65 CrossRef CAS.
- Y. Kim, B. Lee Chung, M. Ma, W. J. Mulder, Z. A. Fayad, O. C. Farokhzad and R. Langer, Nano Lett., 2012, 12, 3587–3591 CrossRef CAS PubMed.
- D. Liu, S. Cito, Y. Zhang, C. F. Wang, T. M. Sikanen and H. A. Santos, Adv. Mater., 2015, 27, 2298–2304 CrossRef CAS PubMed.
- S. J. Shepherd, C. C. Warzecha, S. Yadavali, R. El-Mayta, M. G. Alameh, L. Wang, D. Weissman, J. M. Wilson, D. Issadore and M. J. Mitchell, Nano Lett., 2021, 21, 5671–5680 CrossRef CAS PubMed.
- A. W. Tolcher and L. D. Mayer, Future Oncol., 2018, 14, 1317–1332 CrossRef CAS PubMed.
- A. Awada, I. N. Bondarenko, J. Bonneterre, E. Nowara, J. M. Ferrero, A. V. Bakshi, C. Wilke, M. Piccart and CT4002 study group, Ann. Oncol., 2014, 25, 824–831 CrossRef CAS PubMed.
- M. Su, L. Chen, E. Hitre, W. Lee, L. Bai, Z. Papai, S. Kang, M. Dvorkin, M. Lee, E. Ludovic, H. Choi, S. Oh, G. Bodoky, P. Artru, J. Hwang, I. Bazin, F. Bosc, J. Bachet, Z. Horváth, C. Chang and J. Lin, Ann. Oncol., 2019, 30(suppl. 4), iv23 CrossRef.
- S. Strieth, M. E. Eichhorn, B. Sauer, B. Schulze, M. Teifel, U. Michaelis and M. Dellian, Int. J. Cancer, 2004, 110, 117–124 CrossRef CAS PubMed.
- S. Strieth, M. E. Eichhorn, A. Werner, B. Sauer, M. Teifel, U. Michaelis, A. Berghaus and M. Dellian, Clin. Cancer Res., 2008, 14, 4603–4611 CrossRef CAS PubMed.
- S. Strieth, C. F. Nussbaum, M. E. Eichhorn, M. Fuhrmann, M. Teifel, U. Michaelis, A. Berghaus and M. Dellian, Int. J. Cancer, 2008, 122, 452–460 CrossRef CAS PubMed.
- M. E. Eichhorn, S. Luedemann, S. Strieth, A. Papyan, H. Ruhstorfer, H. Haas, U. Michaelis, B. Sauer, M. Teifel, G. Enders, G. Brix, K. W. Jauch, C. J. Bruns and M. Dellian, Cancer Biol. Ther., 2007, 6, 920–929 CrossRef CAS PubMed.
- L. D. Mayer, P. Tardi and A. C. Louie, Int. J. Nanomed., 2019, 14, 3819–3830 CrossRef CAS.
- M. Zhang, E. Liu, Y. Cui and Y. Huang, Cancer Biol. Med., 2017, 14, 212–227 CrossRef CAS.
- X. J. Kang, H. Y. Wang, H. G. Peng, B. F. Chen, W. Y. Zhang, A. H. Wu, Q. Xu and Y. Z. Huang, Acta Pharmacol. Sin., 2017, 38, 885–896 CrossRef CAS.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS.
- M. Yanez Arteta, T. Kjellman, S. Bartesaghi, S. Wallin, X. Wu, A. J. Kvist, A. Dabkowska, N. Szekely, A. Radulescu, J. Bergenholtz and L. Lindfors, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3351–E3360 CrossRef PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |