
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High voltage and superior cyclability of indium hexacyanoferrate cathodes for aqueous Na-ion batteries enabled by superconcentrated NaClO4 electrolytes†
Xaver
Lamprecht‡
ab,
Philipp
Marzak‡
 ab,
Alexander
Wieczorek
a,
Nina
Thomsen
a,
Jongho
Kim
a,
Batyr
Garlyyev
a,
Yunchang
Liang
cd,
Aliaksandr S.
Bandarenka
*ab and
Jeongsik
Yun
*ab
ab,
Alexander
Wieczorek
a,
Nina
Thomsen
a,
Jongho
Kim
a,
Batyr
Garlyyev
a,
Yunchang
Liang
cd,
Aliaksandr S.
Bandarenka
*ab and
Jeongsik
Yun
*ab
aDepartment of Physics (ECS), Technical University of Munich, James-Franck-Straße 1, 85748 Garching, Germany. E-mail: bandarenka@ph.tum.de; jeongsik.yun@rist.re.kr; Tel: +49 (0) 89 289 12531
bCluster of Excellence E-Conversion, Schellingstraße 4, 80799 Munich, Germany
cMax Planck-EPFL Laboratory for Molecular Nanoscience and Technology, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
dInstitute of Physics (IPHYS), Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
First published on 5th August 2022
Abstract
Transition metal hexacyanometallates, so-called Prussian Blue Analogs (PBAs), have received massive attention as aqueous Na-ion battery electrode materials for large-scale energy storage systems not only due to their simple and low-cost synthesis procedures but also a superior cycling life. Still, aqueous Na-ion batteries based on PBA cathodes generally exhibit low energy density due to the relatively low redox potential and limitation of the electrochemical stability of aqueous electrolytes. It has been reported that the larger the empirical radii of the nitrogen-coordinated transition metal are, the higher the redox potential is obtained. In this context, sodium indium hexacyanoferrate appears to have a high potential to be a high-voltage cathode for aqueous Na-ion batteries. In this study, electrochemically deposited NaIn[Fe(CN)6] thin films demonstrated a promising performance as cathode materials for aqueous Na-ion batteries. Such electrodes showed a very high ‘mean half-charge potential’ of 1.57 V vs. RHE (at pH = 5.8, 1.0 V vs. SSC) and a specific capacity of 58 mA h g−1, making full use of the extended stability window of highly concentrated aqueous sodium perchlorate electrolyte (8 M). A strong dependence of the electrode stability on the employed electrolyte anion (ClO4− > NO3− > SO42−) and concentration was found, as well as on the anodic cut-off potential. These electrodes showed high stability with 75% capacity retention even after more than 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles for a reduced potential window. Previously found correlations for the formal electrode potentials of PBA-based cathode materials can be confirmed for NaIn[Fe(CN)6] as well: the quasi-reversible potential for intercalation/deintercalation of alkali metal cations appears to increase as the hydration energy of alkali metal cations becomes less negative. The nature of the nitrogen-coordinated transition metal present in various hexacyanoferrate-based PBAs appears to be involved in determining the electrode potential: the bigger empirical radii of these transition metals seem to correlate with the more positive electrode potentials.
000 cycles for a reduced potential window. Previously found correlations for the formal electrode potentials of PBA-based cathode materials can be confirmed for NaIn[Fe(CN)6] as well: the quasi-reversible potential for intercalation/deintercalation of alkali metal cations appears to increase as the hydration energy of alkali metal cations becomes less negative. The nature of the nitrogen-coordinated transition metal present in various hexacyanoferrate-based PBAs appears to be involved in determining the electrode potential: the bigger empirical radii of these transition metals seem to correlate with the more positive electrode potentials.
Introduction
Nowadays, many countries produce a significant share of their energy through renewable energy provision schemes.1,2 For reliable power supply with energy from renewable sources, effective energy storage systems (ESSs) are required to address the inflexible energy generation from intermittent resources, such as wind and solar energy.3,4 Among various ESS technologies, secondary batteries exhibit desirable properties for renewable energy applications.5 Compared to successful large-scale ESSs such as pumped hydroelectric or compressed air storage, rechargeable batteries have fewer geographic restrictions, less influence on the environment, better efficiency, and good scalability from micro- to grid-scale.6,7 In terms of energy density, Li-ion batteries are the primary ESS choice in electric vehicles and portable devices. However, despite their high energy density and well-developed manufacturing systems, Li-ion batteries would not be the best selection for grid-scale ESSs due to the high price and huge demand for Li. Furthermore, global power demand is estimated to increase to 30 TWavg by 2050.8 Consequently, the current energy provision infrastructure will not be sufficient to ensure worldwide energy supply. In order to scale up the deployment of renewable powers to the TW level, the affordability, abundance, and durability of materials for ESSs should be considered in the first place.9–11An alternative and promising candidate would be aqueous Na-ion batteries. Such batteries cannot compete with state-of-the-art Li-ion technology regarding energy density yet, but the abundance and low price of their prospective components make them attractive for large-scale applications.12 In addition, safety concerns such as the toxicity and flammability of organic solvents commonly used in Li-ion batteries will be fundamentally resolved by using water-based electrolytes.13
Transition metal hexacyanoferrates (TMHCFs), so-called Prussian Blue Analogs (PBAs), have aroused considerable attention as battery materials in the context of grid-scale applications due to their beneficial properties.14 Excellent cycle life, sufficiently large channels and interstitial sites in the crystal structure for Na+ transport and hosting, the capability of high rate charging/discharging, high round trip efficiency, and an inexpensive synthesis procedure using abundant elements make PBAs very attractive electrode materials.15–18 The general stoichiometry of this material class can be described by AMxTM[1][TM[2](CN)6], with AM representing intercalated alkali metal cations, whereas TM[1] and TM[2] are transition metals which are connected by cyanide ligands. TM[2], the C-coordinated transition metal, is iron in many cases, which is responsible for the faradaic response of PBA-based electrodes via its FeII/III redox activity, yielding specific capacities in the range of 50–80 mA h g−1.19–21
One important factor determining the energy density of a battery is the ‘mean half-charge potential’ (E1/2), which should be as positive as possible for a high-voltage cathode material while still allowing a reversible operation of the cell within the stability window of the aqueous electrolyte. Previous studies on nickel hexacyanoferrate (NiHCF),22 copper hexacyanoferrate (CuHCF),23 chromium(II) hexacyanoferrate (CrHCF),24 zinc hexacyanoferrate (ZnHCF)25 or vanadium hexacyanoferrate (VOxHCF)26 demonstrated a strong dependence of the respective FeII/III formal potential on the involved N-coordinated transition metal: E1/2 increases with larger ionic radii of TM[1].22,24,26 This makes indium hexacyanoferrate (InHCF)—a PBA derivative with an even greater ionic radius of TM[1] (indium) but yet a less studied compound—an interesting candidate for high-voltage aqueous battery cathodes.
To date, the intercalation chemistry of InHCF has been investigated in various aqueous electrolytes containing different cations, such as H+, Li+, Na+, K+, Mg2+, Ca2+, and Al3+.27–31 However, the influence of anionic components in the electrolytes on the electrochemical properties of InHCF cathodes has received less attention. It was shown in our previous works that the composition of electrolytes has a noticeable impact on the cycling stability and reversibility of various PBA electrodes.24,32 Furthermore, the concentration of electrolytes is another important aspect of designing high-performance PBA-based aqueous batteries.33 Extremely high salt concentrations in the solution expand the stability window of aqueous electrolytes.34–36 Using such highly concentrated electrolytes allows to operate cathode materials with the inherent redox activity close to or above the oxygen evolution reaction (OER, under standard conditions) at higher anodic cut-off potentials and thereby increases the specific capacity and energy density. To the best of our knowledge, the investigation of InHCF in highly concentrated aqueous Na-electrolytes is not reported yet.
In this work, we demonstrate a high-voltage PBA cathode, namely NaIn[Fe(CN)6] (InHCF), for aqueous Na-ion batteries and report our findings on the desired composition and concentration of aqueous electrolytes to maximize its electrochemical performance. As model systems, we utilized NaIn[Fe(CN)6] as thin film cathodes that were electrochemically deposited on gold substrates. The deposited InHCF electrodes exhibited considerably different cycling stability in 0.25 M NaClO4, 0.25 M NaNO3, and 0.25 M Na2SO4 aqueous electrolytes. Notably, an InHCF cathode demonstrated an extremely high cyclability (>75%) in 8 M NaClO4 electrolyte over 16000 cycles, whereas a sudden capacity drop was seen in 0.25 M NaClO4 already after the first 100 cycles. In addition, using 8 M NaClO4 electrolyte resulted in a significantly increased specific capacity by allowing a full de-sodiation of InHCF due to a more positive anodic cut-off potential compared to the more dilute electrolyte. However, extending the operational potential range is found to conflict with optimized cycling stability and Coulombic efficiency.
It should be noted that the InHCF thin films showed a significantly high ‘mean half-charge potential (E1/2)’ of 0.91 V vs. Ag/AgCl (SSC) (1.5 V vs. RHE, pH = 6.3) in 0.25 M NaClO4 and 1.0 V vs. SSC (1.57 V vs. RHE, pH = 5.8) in 8 M NaClO4 electrolytes, which is the most positive value reported so far to the best of our knowledge. This is in accordance with our previous studies and confirms the trend of the N-coordinated transition metal ionic radius on E1/2. It was furthermore found that the mean half-charge potential could be enlarged using alkali metal cations with more positive hydration energies except for K+.
Experimental
Electrochemical experiments were performed in a standard three-electrode configuration under an inert argon (Ar 5.0, Westfalen) atmosphere in an electrochemical glass cell. AT-cut Au quartz crystal wafers (Stanford Research Systems, USA, Ti adhesive layer, 5 MHz), and arrandee™ Au substrates (arrandee metal, Germany) served as the working electrodes. A platinum wire was used as a counter electrode, and an Ag/AgCl electrode (SSC, 3 M KCl, SI Analytics, “B 3420+”) was utilized as a reference electrode. A Bio-Logic VSP-300 potentiostat was employed for all experiments.The electrochemical deposition of NaIn[Fe(CN)6] was performed by means of cyclic voltammetry with a scan rate of 50 mV s−1. The aqueous deposition solution contained 2 mM K3[Fe(CN)6] (99%, Sigma-Aldrich), 2 mM In(III)Cl3 (≥99.9%, Carl Roth) and 0.25 M Na2SO4 (≥99%, Sigma-Aldrich). It has been advisable to use a two-step deposition procedure for NaIn[Fe(CN)6] thin films because possible anodic side reactions on the Au substrates apparently impaired the synthesis of the electrode material. First, the potential range was set to 0.4–1.05 V vs. SSC for 50 cycles. For the subsequent film growth, it was adjusted to 0.4–1.15 V vs. SSC for another 50 cycles. After the deposition, InHCF samples were dried in the electrochemical cell for 1 hour. The (de)intercalation of Na-ions from/into the NaIn[Fe(CN)6] thin films was investigated using cyclic voltammetry as well, with a lower potential limit of 0.4 V vs. SSC and an upper limit varied in the potential range of 1.15–1.55 V vs. SSC. The mass of electrode films electrodeposited onto Au quartz crystals was measured with a QCM200 electrochemical quartz crystal microbalance (EQCM, Stanford Research Systems, USA).
In order to investigate the influence of the electrolyte composition on the NaIn[Fe(CN)6] electrode performance, 0.25 M aqueous solutions of Na2SO4 (≥99%, Sigma-Aldrich), NaClO4 (>98%, Sigma-Aldrich) and NaNO3 (≥99%, Merck) were utilized as intercalation electrolytes. 8 M NaClO4 was used to improve the stability of the electrodes and to operate NaIn[Fe(CN)6] thin films with an extended potential window up to 1.55 V vs. SSC. The cation effect on InHCF electrodes was investigated by exchanging the intercalating species in 0.25 M AM2SO4 (AM = Li (99%, Alfa Aesar), Na (≥99%, Sigma-Aldrich), K (≥99%, Merck), Rb (99,8%, Alfa Aesar), Cs (≥99.99%, Carl Roth)) aqueous electrolyte.
Electrochemical impedance spectroscopy (EIS) was performed in the three-electrode configuration as described above. The amplitude of the probing signal was set to 10 mV and the scanned frequency range was between 2.5 kHz and 50 mHz with 10 points per decade in logarithmic spacing. The bias potential was varied in a range of 0.4–1.6 V vs. SSC, while only representative results obtained for 0.74 V, 1.02 V and 1.36 V vs. SSC are provided in this paper.
The composition of InHCF electrodes was investigated by X-ray photoelectron spectroscopy (XPS, SPECS, Germany). The X-ray source was an Al K-alpha anode (1486.61 eV, 12 kV, 200 W). Photoelectrons were detected with a PHOIBOS 150 2D CCD detector. XPS spectra were recorded for InHCF films in the states of charge corresponding to final potentials of 0.4 V, 1.2 V, and 1.6 V vs. SSC.
Scanning electron microscopy (SEM) measurements were performed with a NVision 40 (Zeiss, Germany) to investigate the surface morphology. For higher resolved imaging of the surface, atomic force microscopy (AFM) was conducted in tapping mode (scan rate 0.5 Hz) utilizing a multi-mode EC-STM/AFM device (Veeco Instruments, USA) equipped with RTESP-300 (Bruker, Germany) AFM tips. The Nanoscope IIID controller and the Nanoscope 5.31r1 software were utilized for the measurements, while the obtained data was analyzed with the WSxM software.37
Results and discussion
It is reported that PBAs can be synthesized either by the co-precipitation method or by electrochemical deposition.15–18,22,24,26,29 The electrodeposition of PBAs on a quartz crystal electrode facilitates good electrical contact between the deposited PBA films with the electrode and the growth of rigid films on the electrode.30,31 Furthermore, one can monitor the mass variations not only during the electrodeposition of PBA films but also during subsequent measurements, which enables us to calculate the mass of deposited films and mass changes during (de)intercalation with high precision in the order of nanograms.22 In our previous studies, electrochemically deposited PBA thin films displayed smooth and uniform surfaces and exhibited high specific capacities and rate capabilities.22,26,33 Therefore, InHCF electrodes were prepared by means of electrochemical deposition on Au quartz crystal substrates for this work as well.A tentative reaction formula for the electrochemical deposition of InHCF from a deposition solution containing Na+, In3+ and Fe(CN)63− precursors can be given as:38,39
| Na+ + In3+ + Fe(CN)63− + e− → NaIn[Fe(CN)6] | (1) |
Fig. 1(A) and (B) represent typical cyclic voltammograms (CVs) and the corresponding mass changes on the Au quartz crystal electrode for the InHCF deposition, respectively. Blue and black curves of the CVs and massograms in Fig. 1(A) and (B) result from the first and second deposition step, respectively. The anodic and cathodic peak currents gradually increase during potential cycling, which indicates the InHCF film formation. It was reported that the anodic and cathodic peaks originate from the reversible redox reaction of Fe-ions of InHCF films with simultaneous Na-intercalation and deintercalation.22,28,40 The deposited mass was calculated from the resonance frequency measured by EQCM using the Sauerbrey equation.41 The mass change by InHCF film deposition was monitored using an EQCM. As shown in Fig. 1(B), it was calculated that 90.8 μg cm−2 of the electrode material was deposited after the two-step deposition process. The mass loading of the active material equals to the total deposited mass of 90.8 μg cm−2 because of the absence of any other electrode components such as a binder and conductive additives. It should be noted that the provided CVs and EQCM curves represent the characteristic figures of InHCF deposition for all subsequent experiments, because the deposited mass of InHCF films is tunable with high precision, and the electrodeposition of InHCF films is highly reproducible. Next to the increasing mass loading, steady mass oscillations (see the inset figure of Fig. 1(B)) indicate Na-intercalation/deintercalation into/from the InHCF thin film. In other words, repeated cathodic and anodic potential sweeps cause not only InHCF thin film deposition, but also consecutive Na-intercalation and -deintercalation.
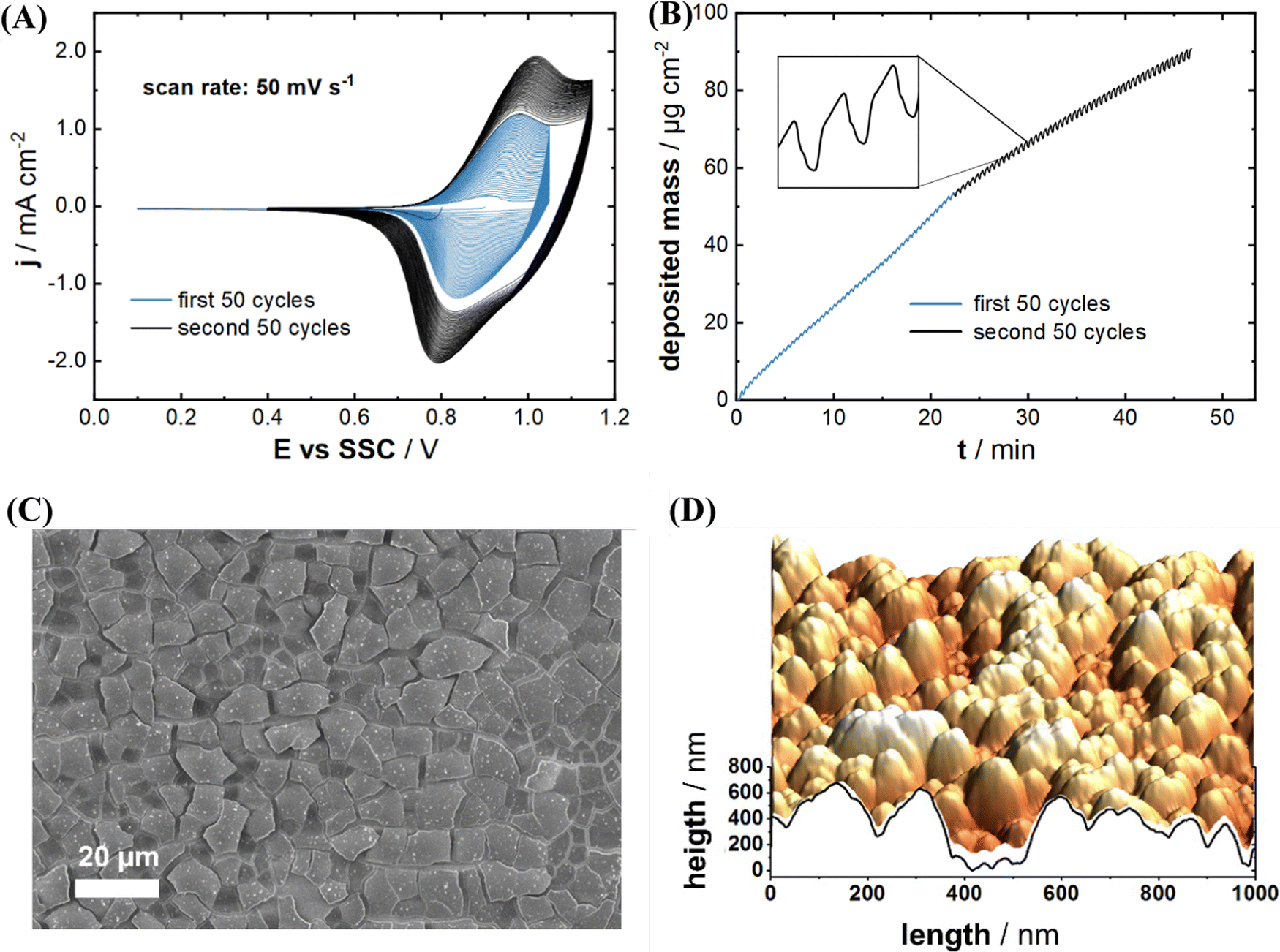 | ||
| Fig. 1 Electrodeposition and morphological characterization of NaIn[Fe(CN)6] (InHCF) thin films. (A) Representative CVs of the InHCF electrodeposition. (B) Real-time mass variation measurement of InHCF deposition. The blue and black lines in (A), (B) describe the current density and the deposited mass of InHCF films during the first and second deposition steps, respectively. (C) SEM image and (D) AFM image of electrodeposited InHCF thin films. | ||
The morphology of the electrochemically deposited InHCF thin films was investigated using SEM and AFM. Fig. 1(C) shows an exemplary SEM image of an electrodeposited InHCF thin film's surface. The surface shows squared features with a size in the order of 10 μm × 10 μm. The other surface visualization by AFM indicates that the surface has 300 nm of root mean square (rms) roughness (see Fig. 1(D)), which is rather rough compared to other electrodeposited PBA thin films with rms roughness of typically 10–20 nm for Na2Ni[Fe(CN)6], ∼100 nm for Na2Cu[Fe(CN)6] and ∼60 nm for Na2VOx[Fe(CN)6].22,26
It was reported that the voltammetric reversibility of Na+-(de)interaction, which is closely related to the energy efficiency of PBA electrodes, can be improved by carefully optimizing the electrolyte composition.22,24,33 We have shown that the Na+-(de)intercalation mechanism of some PBA materials, namely Na2Ni[Fe(CN)6], Na2VOx[Fe(CN)6], Na2Co[Fe(CN)6], and NaxMn[Mn(CN)6], involves quasi-reversible anion adsorption and desorption.22,26,33,42 The shape of CVs in Na-electrolytes usually does not change significantly if the anion is replaced. In the case of NaIn[Fe(CN)6], however, the shape of CV in 0.25 M NaClO4 electrolyte appears different from that in 0.25 M Na2SO4 and 0.25 M NaNO3 as shown in Fig. 2(A). Apparently, the nature of anions present in the electrolyte significantly affects the redox-behavior of the system. Characteristic redox peaks for (de)intercalation of Na-ions are shifted to more positive potentials in 0.25 M Na2SO4 and 0.25 M NaNO3 compared to those in 0.25 M NaClO4. The anodic currents at potentials more positive than 1.05 V vs. SSC and the presumably correlating cathodic currents are increased, too.
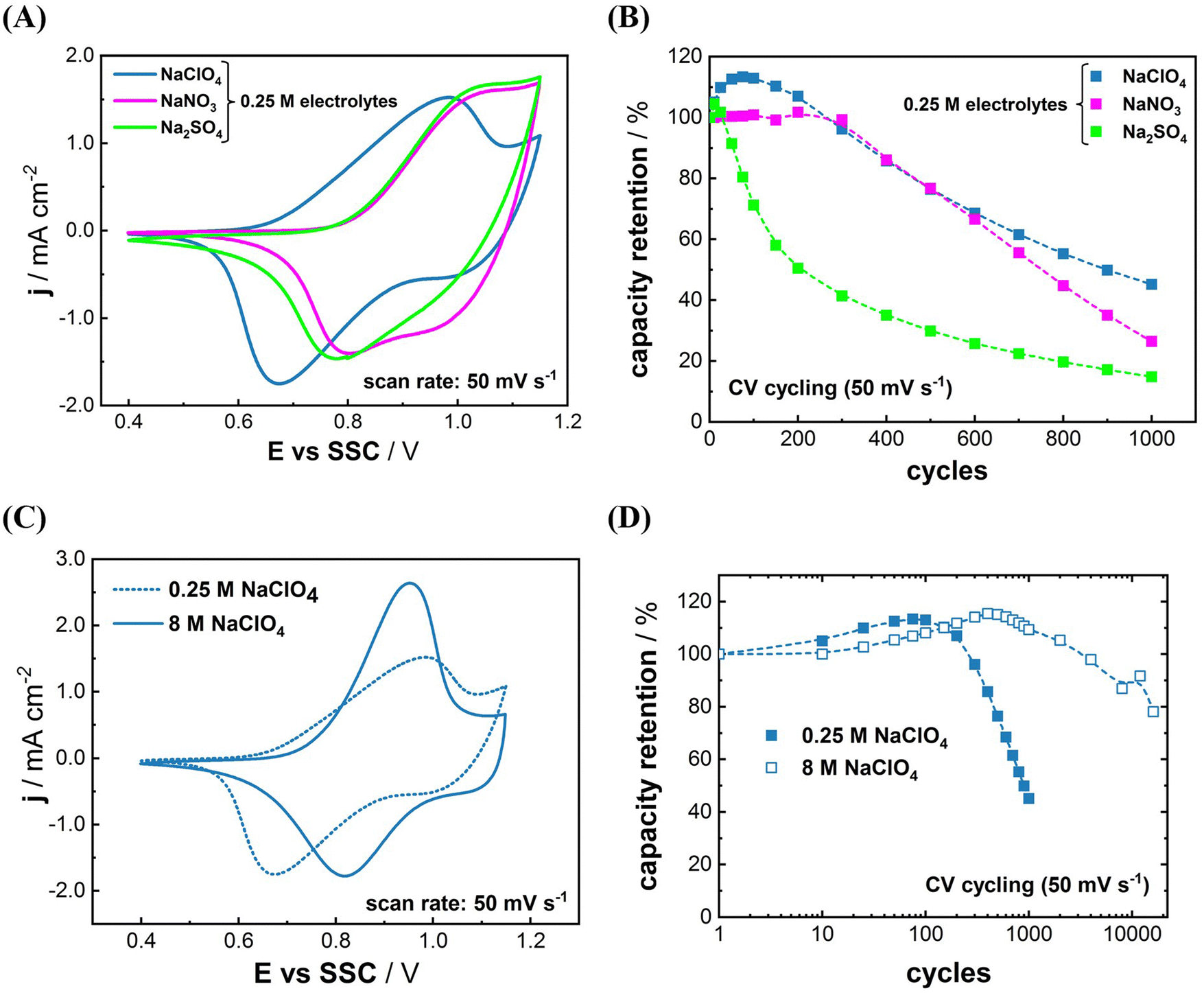 | ||
| Fig. 2 Influence of the electrolyte composition on cycling performance of InHCF thin films. (A) CVs of InHCF in 0.25 M Na+ electrolytes with varying anions (Na2SO4, NaNO3 and NaClO4), and (B) respective cycling stability in these solutions. (C) CVs and (D) long term cycling stability of InHCF in 0.25 M and 8 M NaClO4, respectively. The dashed lines in (B) and (D) serve as a guide to the eye for better representation of the measured data trends. | ||
Long-term cycling of NaIn[Fe(CN)6] electrodes further underlines the importance of the anion selection. In the case of NaClO4, an improved capacity retention was obtained as can be seen in Fig. 2(B). The anion-dependent stability trend of ClO4− > NO3− > SO42− is in accordance with previous findings for the PBA derivates, and gives further indication on an anion-chemisorption-induced degradation mechanism of such materials.32 Nevertheless, the cycling stability of NaIn[Fe(CN)6] in the 0.25 M Na+-electrolytes is yet poorer than for other PBA materials.15–18,24,33
Recently, the effect of highly concentrated electrolytes, namely water-in-salt electrolytes, has been vastly investigated.30,31,34–36 Extremely high concentrations can extend the electrochemically stable potential window of aqueous electrolytes even beyond 3 V. Tomiyasu et al. reported that the expansion of the stable potential window of aqueous electrolytes by strongly hydrating salts is ascribed to the weakened hydrogen bond network of water molecules, as well as the drastically lowered amount of free H2O.43 Consequently, the overpotential for OER is increased and the stable potential window of an aqueous electrolyte is extended. The potential range of Na+ deintercalation (charging) of the NaIn[Fe(CN)6] cathode overlaps with the onset of the oxygen evolution reaction. Thus, in order to fully exploit the capacity of the NaIn[Fe(CN)6] cathode, the use of water-in-salt electrolytes appears a promising strategy. Simultaneously, the solution's uptake capability for dissolved electrode constituents is strongly decreased due to an entire incorporation of H2O molecules in the salt ions’ solvation shells, which results in the stabilization of the active material.16,32
Many studies used expensive salts such as sodium triflate (NaOTF) and sodium (bis(trifluoromethane sulfonyl)-imide) (NaTFSI) to prepare superconcentrated electrolytes (>20 mol kg−1).44 In contrast, we utilized 8 M NaClO4 electrolyte for this work which is far cheaper and more abundant than 20 mol kg−1 of NaOTF and NaTFSI. Fig. 2(C) displays the difference between the electrochemical behavior of NaIn[Fe(CN)6] in 0.25 M and 8 M NaClO4 electrolytes. A more reversible CV was recorded in the 8 M NaClO4 electrolyte than that in the 0.25 M NaClO4 electrolyte, which is likely due to the increased ionic conductivity of the solution.45 Moreover, the long-term cyclability was greatly enhanced in the 8 M NaClO4 electrolyte (see Fig. 2(D)). Approximately 75% of the initial capacity of NaIn[Fe(CN)6] is retained in the 8 M NaClO4 after 16000 cycles, whereas only 45% of the initial capacity was retained after 1000 cycles in 0.25 M NaClO4. These results give further evidence, that highly concentrated electrolytes can effectively suppress the active material dissolution in aqueous systems.32
Fig. 3(A) compares CVs of NaIn[Fe(CN)6] cathodes in a 8 M NaClO4 aqueous electrolyte with different cut-off potentials. A quasi-reversible CV of the NaIn[Fe(CN)6] cathode could be obtained even with the cut-off potential as high as 1.55 V vs. SSC which is equal to 2.12 V vs. RHE (pH = 5.8). Here, violent water splitting reactions would usually take place in common aqueous electrolytes at standard conditions. Clearly, increasing the concentration of the electrolyte yields the desired effect. Considering the drastically improved stability of the cathodes (Fig. 2(D)), this effect likely goes hand in hand with the highly concentrated electrolytes suppressing the active material dissolution in aqueous systems.32 As apparent from Fig. 3(B), the larger operational potential range gives access to a much higher specific capacity of 58 mA h g−1 compared to 37 mA h g−1 for the lower anodic cut-off potential of 1.15 V vs. SSC (1.71 V vs. RHE). XPS analysis of the InHCF cathodes that were charged to three different cut-off potentials (0.4 V vs. SSC, green – 1.2 V vs. SSC, blue – 1.6 V vs. SSC, red) sheds light on the origin of the capacity increase. As shown in Fig. 3(C), this improvement results from a complete oxidation of all available FeII-sites upon the higher anodic cut-off potential, which is correlated with an entire de-sodiation of the NaIn[Fe(CN)6] cathode. The redox activity of FeII/III can be observed by a slightly increasing shoulder of the Fe 2p3/2 and 2p1/2 peak towards higher binding energies for higher potentials. Furthermore, the XPS peaks of Na-ions prove that the NaIn[Fe(CN)6] cathode could be fully de-sodiated if it is charged to 1.6 V vs. SSC. In contrast to the iron and sodium signals, the peak shape and position of the In 2p region remain virtually unchanged upon electrode oxidation and reduction, proving that indium is electrochemically inert in the investigated potential region and remains in oxidation state InIII.
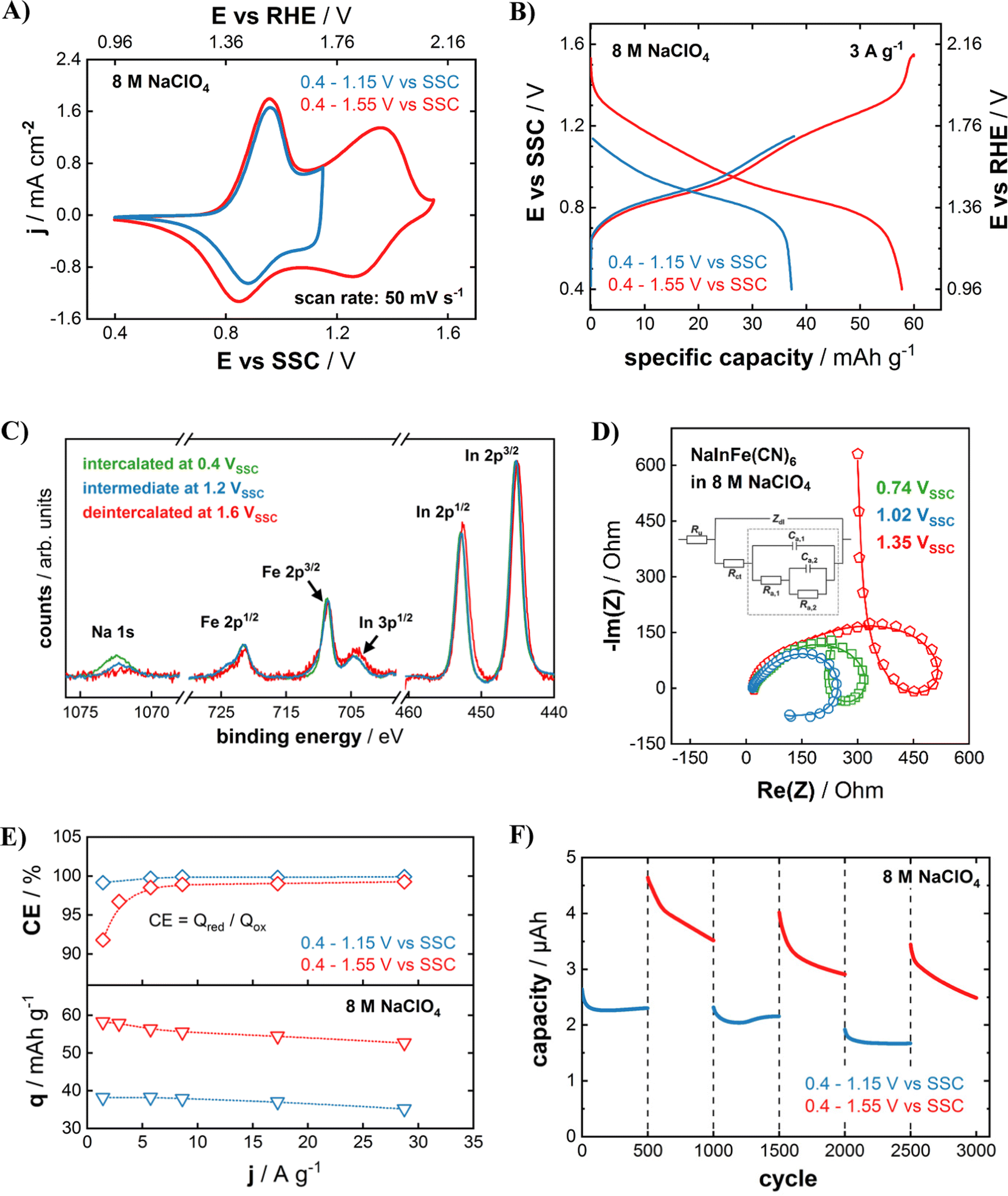 | ||
| Fig. 3 Impact of the anodic cut-off potential on electrochemical performance. (A) CVs and (B) galvanostatic charge–discharge curves for InHCF in 8 M NaClO4 for Ecut-off = 1.15 V (blue curves) and 1.55 V (red curves) vs. SSC. For a better correlation to the electrolyte stability window, the potentials are also given with respect to the RHE scale (pH = 5.8 for 8 M NaClO4). (C) XPS analysis of InHCF films in three different states of charge (cut-off potentials of 0.4 V, 1.2 V and 1.6 V vs. SSC). (D) EIS spectra of InHCF electrodes in 8 M NaClO4 at three different potentials (0.74 V, 1.02 V and 1.36 V vs. SSC). Open symbols are the obtained spectra and lines are the resulting fits. The inset figure represents the used equivalent circuit diagram for fitting the collected impedance spectra (see main text). (E) Obtained specific capacity q and Coulombic efficiency CE for different (dis)charging currents in galvanostatic cycling, as well as (F) cycling stability for the lower and higher Ecut-off. The dotted red and blue lines in (E) serve as a guide to the eye. | ||
Fig. 3(D) shows impedance spectra of InHCF electrodes measured at different potentials within the electrochemically active region. Loop-shaped spectra were obtained for all three investigated states of charge (SOC). The EIS spectra (open symbols) of InHCF electrodes were obtained at 0.74 V vs. SSC (squares), 1.02 V vs. SSC (circles), and 1.35 V vs. SSC (pentagons) and respective fits (solid lines) using an equivalent electric circuit (EEC) model. We have shown in several previous studies that loop-shaped impedance spectra indicate a complicated multistep reaction pathway in intercalation-type battery materials.22,26,42,46 In short, the quick FeII/III redox process results in the appearance of excess surface charges, which are compensated by intermediate anion ad/desorption from the electrolyte, until finally the slow Na+ transfer across the electrode–electrolyte-interface is concluded.22,42 The EEC model shown as an inset in Fig. 3(D) represents a physical model describing all involved physico-chemical processes. From the clear agreement of the fitted curves with the measured spectra, we conclude that the entire charge storage capability of InHCF in the investigated extended potential region is associated to Na+ de/intercalation reactions.
From the above considerations we propose the following tentative reaction equation for sodium deintercalation/intercalation in indium hexacyanoferrate electrodes:
| NaIn[FeII(CN)6] ↔ Na+ + e− + In[FeIII(CN)6] | (2) |
Yet, the obtained specific capacity of 58 mA h g−1 is lower than the theoretically expected value of ∼77 mA h g−1 (according to eqn (2)). However, the given stoichiometry in this study does not consider the certainly present crystal water, which would result in a lower theoretical value. Nevertheless, the experimentally obtained specific capacity is in reasonable accordance with other PBA cathode materials where only the FeII/III transition is electrochemically active.14 In both the reduced (Ecut-off = 1.15 V vs. SSC) and the extended potential range (Ecut-off = 1.55 V vs. SSC), NaIn[FeII(CN)6] cathodes exhibit a very promising rate capability easily sustaining up to almost 30 A g−1 with less than a by 10% diminished capacity (see Fig. 3(E)). However, the decreasing Coulombic efficiency from 99.2% for 30 A g−1 to 91.8% for 1.4 A g−1 for the extended operational window gives a strong indication of possible parasitic processes during the electrode oxidation. As the cut-off potential of 1.55 V vs. SSC (2.12 V vs. RHE, pH = 5.8) is much more positive than the theoretical onset of the oxygen evolution reaction (>0.9 V), we attribute the observed irreversibility to marginal water splitting reactions. Due to the strongly reduced OER reaction rate resulting from the highly concentrated NaClO4 electrolyte, its relative share is negligible for high dis/charging rates of the InHCF electrode, but gains significance once the absolute dis/charging current is reduced. As the OER is a surface-limited reaction, we expect a decreasing significance for higher mass loadings (i.e. film thicknesses).
Furthermore, we found that the extension of the operational potential range has detrimental effects on the cycling stability. As shown in Fig. 3(F), the electrode can be cycled in the reduced potential range without significant losses. However, once the cycling window is increased, the capacity quickly decreases over 500 cycles, while stabilizing again in the following 500 cycles in the reduced operational potential range. Concludingly, irreversible electrode degradation processes are triggered at potentials above 1.15 V vs. SSC. The investigation of exact mechanisms is beyond the scope of this study, however we could formulate a hypothesis that it is caused by either the adsorption of OH− or the emergence of H3O+ during the OER process, as both species have detrimental effects on the stability of PBA materials.32 Another possible degradation pathway could be the electrolysis of crystal coordination water within the InHCF lattice, which is indispensable for its structural integrity.14
Electrodeposited thin film PBA electrodes serve as an ideal model system to investigate the inherent electrochemical properties of this battery material class without non-active electrode components, such as conductive additives and the binder material, as well as effects stemming from the porous nature of the usually employed slurry-casted electrodes. Nevertheless, the mass loadings achieved by the thin film electrodeposition technique are usually too small for commercial viability. In order to scale up the mass loading of electrodeposited NaIn[Fe(CN)6] thin films to sub milligram level, we utilized conductive carbon cloth as a substrate for the electrodeposition. As shown in Fig. S1A (ESI†), the obtained CV scans during NaIn[Fe(CN)6] electrodeposition on the carbon substrate showed a continuous increase in cathodic and anodic peak currents similar to the deposition on Au quartz crystals. However, noticeably higher peak currents and more irreversible shape of CVs (larger peak separation) are observed compared to the thin film deposition on the planar Au-based substrate. Considering the good conductivity of the conductive carbon cloth, the higher peak separation is mainly ascribed to the altered mass and charge transport properties of the porous system. The resulting mass was approximately 483 μg cm−2, which exceeds the capability of Au quartz crystal to bear such weight.
When cycled in 8 M NaClO4 within the extended potential range (0.4–1.5 V vs. SSC) as shown in Fig. S1B (ESI†), the InHCF electrode with the high loading shows an excellent rate capability up to extremely high currents as the InHCF thin films electrode does. Over 50% of the initial capacity could still be obtained even at a high current of 26.4 A g−1, which corresponds to a rate of 455C. In general, a rate of 1C means that the electrode can be fully discharged in 1 hour. At the same time, the Coulombic efficiency is above 97% for the entire investigated current range from 0.26 A g−1 to 53 A g−1, yielding even better results than for the lower mass loading InHCF thin film on gold, as displayed in Fig. 3(E). Albeit an in-depth investigation of the electrochemical performance of the increased mass loading of InHCF on conductive carbon substrates is of interest, it is beyond the scope of this work. Nonetheless, the presented data clearly shows the relevance and promising nature of electrodeposited thin film PBA electrodes.
It has been frequently reported that PBA electrodes can accommodate various cations such as protons, alkali metal cations, alkali earth metal cations, and ammonium cation.19,20 Interesting phenomena of the intercalation chemistry of PBA electrodes are that their intercalation potentials can be tuned not only by modifying their structural composition but also according to the sort of intercalating species.22,24,26,29,33 For instance, nickel hexacyanoferrate and cobalt hexacyanoferrate exhibit drastic changes in the mean half-charge potential E1/2, depending on the involved alkali metal cations.22,47 In order to investigate the intercalation potential of different alkali metal cations, the InHCF electrodes were cycled in 0.25 M AM2SO4 electrolytes (AM = Li, Na, K, Rb and Cs). As expected, CVs of InHCF electrodes show different shapes, and the cathodic/anodic current peaks are in different potential ranges (Fig. 4(A)). The potentials of intercalation/deintercalation for alkali metal cations appear to shift to more positive values as the atomic number of the alkali metal cation increases in general. However, the E1/2 value for K+ (de)intercalation is smaller than that for Na+ (de)intercalation, which is in agreement with the literature.28 We have shown in previous works that different mean half-charge potentials according to the involved alkali metal cations can be correlated with their respective hydration energies.22Fig. 4(B) depicts the relation between hydration energies of alkali metal cations and electrode potentials of InHCF, NiHCF, and VOxHCF. The mean half-charge potentials of InHCF tend to slightly increase as the hydration energies of intercalating species become less negative except for potassium. The trend appears similar to the case of NiHCF, while the mean half-charge potentials of alkali metal cations for VOxHCF do not change much.
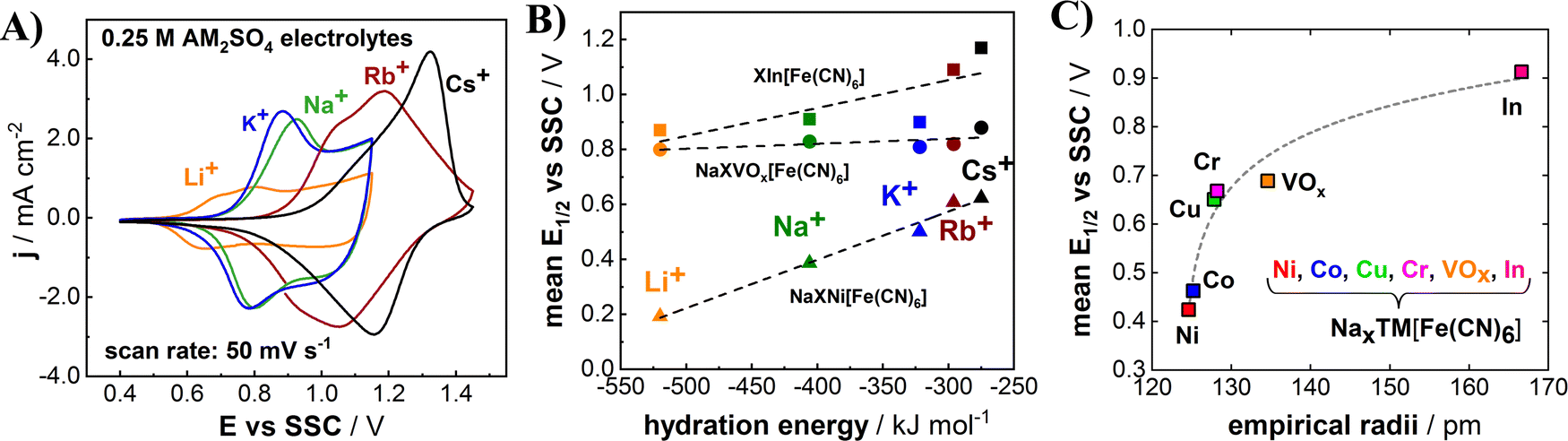 | ||
| Fig. 4 (A) CVs of InHCF and (B) its respective mean half-charge potentials (E1/2) compared to other PBA electrodes in aqueous 0.25 M AM2SO4 electrolytes (AM = Li, Na, K, Rb and Cs) in correlation to the respective AM hydration energies. (C) E1/2 of PBA-based electrodes for Na+ (de)intercalation as a function of empirical radii of involved nitrogen-coordinated transition metals. All values were obtained for 0.25 M Na+ electrolytes. The E1/2 for InHCF was determined for 0.25 M NaClO4 with a measured potential range of 0.4–1.15 V vs. SSC. The dashed lines in (B) and (C) are given as a guide for the eye. | ||
The mean half-charge potential of PBA-based electrodes can be also tuned by replacing one of the key elements in the structure—the N-coordinated transition metal TM[1]. It is reported that empirical radii of transition metals in PBA materials appear to have a certain correlation with the mean half-charge potential for Na+ intercalation as shown in Fig. 4(C).22,24,26 For the case of the herein investigated PBA based on the large indium cation at the N-coordinated crystal sites, the highest mean half-charge potential (0.91 V vs. SSC in 0.25 M NaClO4, potential range: 0.4–1.15 V vs. SSC) was obtained compared to other PBAs based on Ni, Co, Cu, Cr and VOx, which further supports the described empirical trend.
Conclusion
In this study we presented electrochemically deposited NaIn[Fe(CN)6] thin films as a promising cathode material for aqueous Na-ion batteries. The functionality of InHCF as an intercalation compound for aqueous Na-ion batteries was confirmed by means of cyclic voltammetry and galvanostatic cycling, as well as XPS and EIS. InHCF thin films yield a high and promising quasi-reversible potential (‘mean half-charge potential’) of up to 1.0 V vs. SSC (1.57 V vs. RHE, 8 M NaClO4, potential range between 0.4 V and 1.55 V vs. SSC) together with a specific capacity of 58 mA h g−1 at a charge-rate of 3 A g−1. We succeeded in operating the InHCF electrodes at potentials up to 1.55 V vs. SSC (2.12 V vs. RHE) even in an aqueous environment by using a highly concentrated electrolyte, namely 8 M NaClO4. Since the amount of free water in this electrolyte is significantly reduced, it also increases the stability of the InHCF electrodes by preventing active material dissolution. We obtained a specific capacity retention of 75% even after more than 16000 cycles of continuous (dis)charging, while operating the electrode potential up to 1.15 V vs. SSC. Nevertheless, irreversible parasitic processes during the charging process of the electrode likely occur for the higher anodic cut-off potential of 1.55 V vs. SSC (2.12 V vs. RHE) at low charging currents, which we ascribed to slow OER processes. These operational conditions also decreased the cycling stability of the electrode. Throughout our past research we investigated various members of the PBA material class and were able to identify a clear dependence of the respective electrode materials’ mean half-charge potentials (E1/2) on the empirical radii of the N-coordinated transition metals present in each compound: the higher the radius is, the more positive the mean half-charge potential will be. Confirming this empirical trend, the employment of the large indium cation in the PBA structure yielded a more positive quasi-reversible potential compared to other common PBA materials based on Ni, Co, Cu, Cr and VOx. Furthermore, similarly to other PBAs, InHCF showed versatile redox behavior and the capability of (de)intercalation when using different alkali metal cations as insertion species. The involved electrolyte anion was found to have a significant influence on the cycling stability, further adding evidence for an anion induced degradation mechanism for PBA materials.
Author contributions
Conceptualization: XL, PM, JY; formal analysis: XL, PM, AW, NT, YL, BG, JY; investigation: XL, PM, AM, NT, JK, BG, YL, JY; data curation & project administration: XL, PM, JY; resources, funding acquisition & supervision: ASB; writing, reviewing & editing: XL, PM, ASB, JY.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge the cluster of excellence “e-conversion” (German Research Foundation, DFG) for funding. J. Yun gratefully appreciates the financial support from Nagelschneider Stiftung.References
- D. Ginley, M. A. Green and R. Collins, MRS Bull., 2008, 33, 355–364 CrossRef CAS.
- J. P. Holdren, Science, 2007, 315, 737 CrossRef CAS PubMed.
- P. Denholm, E. Ela, B. Kirby and M. Milligan, The Role of Energy Storage with Renewable Electricity Generation, National Renewable Energy Laboratory, Golden, CO, 2010 Search PubMed.
- A. G. Ter-Gazarian, Energy Storage for Power Systems, Peter Peregrinus Ltd., London, UK, 1994 Search PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334(6058), 928–935 CrossRef CAS PubMed.
- G. L. Soloveichik, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 503–527 CrossRef CAS PubMed.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2(21), 7933–7947 RSC.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7(1), 19–129 CrossRef CAS PubMed.
- F. Beck and P. Rüetschi, Electrochim. Acta, 2000, 45(15–16), 2467–2482 CrossRef CAS.
- P. Ruetschi, J. Power Sources, 1993, 42(1–2), 1–7 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114(23), 11636–11682 CrossRef CAS PubMed.
- R. M. Wittman, M. L. Perry, T. N. Lambert, B. R. Chalamala and Y. Preger, J. Electrochem. Soc., 2020, 167(9), 090545 CrossRef CAS.
- B. Wang, Y. Han, X. Wang, N. Bahlawane, H. Pan, M. Yan and Y. Jiang, iScience, 2018, 3, 110–133 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2012, 159, A98–A103 CrossRef CAS.
- M. Pasta, C. D. Wessells, N. Liu, J. Nelson, M. T. McDowell, R. A. Huggins, M. F. Toney and Y. Cui, Nat. Commun., 2014, 5, 3007 CrossRef PubMed.
- H. W. Lee, R. Y. Wang, M. Pasta, S. W. Lee, N. Liu and Y. Cui, Nat. Commun., 2014, 5, 5280 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed.
- J. Qian, C. Wu, Y. Cao, Z. Ma, Y. Huang, X. Ai and H. Yang, Adv. Energy Mater., 2018, 8, 1702619 CrossRef.
- K. Hurlbutt, S. Wheeler, I. Capone and M. Pasta, Joule, 2018, 2(10), 950–960 CrossRef.
- Y. Xu, S. Zheng, H. Tang, X. Guo, H. Xue and H. Pang, Energy Storage Mater., 2017, 9, 11–30 CrossRef.
- J. Yun, J. Pfisterer and A. S. Bandarenka, Energy Environ. Sci., 2016, 9(3), 955–961 RSC.
- X. Y. Wu, M. Y. Sun, Y. F. Shen, J. F. Qian, Y. L. Cao, X. P. Ai and H. X. Yang, ChemSusChem, 2014, 7(2), 407–411 CrossRef CAS PubMed.
- R. Bors, J. Yun, P. Marzak, J. Fichtner, D. Scieszka and A. S. Bandarenka, ACS Omega, 2018, 3(5), 5111–5115 CrossRef CAS PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5(2), 1400930 CrossRef.
- B. Paulitsch, J. Yun and A. S. Bandarenka, ACS Appl. Mater. Interfaces, 2017, 9(9), 8107–8112 CrossRef CAS PubMed.
- J. Qiao, M. Qin, Y. Shen, J. Cao, Z. Chen and J. Xu, Chem. Commun., 2021, 57, 4307–4310 RSC.
- L. Chen, H. Z. Shao, X. F. Zhou, G. Q. Liu, J. Jiang and Z. P. Liu, Nat. Commun., 2016, 7, 11982 CrossRef PubMed.
- P. Marzak, M. Kosiahn, J. Yun and A. S. Bandarenka, Electrochim. Acta, 2019, 307, 157 CrossRef CAS.
- Z. Tong, R. Lian, R. Yang, T. Kang, J. Feng, D. Shen, Y. Wu, X. Cui, H. Wang, Y. Tang and C. S. Lee, Energy Storage Mater., 2022, 44, 497–507 CrossRef.
- Z. Tong, T. Kang, Y. Wan, R. Yang, Y. Wu, D. Shen, S. Liu, Y. Tang and C. S. Lee, Adv. Funct. Mater., 2021, 31, 210463 Search PubMed.
- X. Lamprecht, F. Speck, P. Marzak, S. Cherevko and A. S. Bandarenka, ACS Appl. Mater. Interfaces, 2022, 14(2), 3515–3525 CrossRef CAS PubMed.
- J. Yun, F. A. Schiegg, Y. Liang, D. Scieszka, B. Garlyyev, A. Kwiatkowski, T. Wagner and A. S. Bandarenka, ACS Appl. Energy Mater., 2018, 1, 123–128 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- I. Horcas, R. Fernández, J. M. Gomez-Rodriguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- V. V. Kurdakova, V. V. Kondrat’ev, O. V. Levin and V. V. Malev, Russ. J. Electrochem., 2002, 38, 1192–1199 CrossRef CAS.
- M. A. Malik, G. Horanyi, P. J. Kulesza, G. Inzelt, V. Kertesz, R. Schmidt and E. Czirok, J. Electroanal. Chem., 1998, 452, 57–62 CrossRef CAS.
- J. H. Sierra-Uribe, C. Islas-Vargas, A. Guevara-García, M. Oliver-Tolentino, G. Ramos-Sánchez, M. Galván and I. González, J. Electrochem. Soc., 2021, 168, 100511 CrossRef CAS.
- J. Kankare, Langmuir, 2002, 18, 7092–7094 CrossRef CAS.
- E. Ventosa, B. Paulitsch, P. Marzak, J. Yun, F. Schiegg, T. Quast and A. S. Bandarenka, Adv. Sci., 2016, 3, 1600211 CrossRef PubMed.
- H. Tomiyasu, H. Shikata, K. Takao, N. Asanuma, S. Taruta and Y. Y. Park, Sci. Rep., 2017, 7, 45048 CrossRef CAS PubMed.
- L. Jiang, L. Liu, J. Yue, Q. Zhang, A. Zhou, O. Borodin, L. Suo, H. Li, L. Chen and K. Xu, Adv. Mater., 2020, 32, 1904427 CrossRef CAS PubMed.
- W. Wu, S. Shabhag, J. Chang, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2015, 162(6), A803 CrossRef CAS.
- R. R. Gaddam, L. Katzenmeier, X. Lamprecht and A. S. Bandarenka, Phys. Chem. Chem. Phys., 2021, 23, 12926–12944 RSC.
- S.-M. Chen, J. Electroanal. Chem., 2002, 521, 29–52 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00130f |
| ‡ X. Lamprecht and P. Marzak contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |