
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evolution of fluoride shuttle battery reactions and three-dimensional morphology changes of BiF3 microparticles in an ethylene carbonate-based liquid electrolyte†
Toshiro
Yamanaka
 *a,
Zempachi
Ogumi
a and
Takeshi
Abe
b
*a,
Zempachi
Ogumi
a and
Takeshi
Abe
b
aOffice of Society-Academia Collaboration for Innovation, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: yamanaka.toshiro.8n@kyoto-u.ac.jp
bGraduate School of Global Environmental Studies, Kyoto University, Katsura, Nishikyo, Kyoto 615-8510, Japan
First published on 12th August 2022
Abstract
Fluoride shuttle batteries (FSBs) use defluorination of metal fluorides and fluorination of metals, and they are considered as candidates of next-generation batteries with high energy densities. During FSB reactions of orthorhombic and cubic BiF3 (o-BiF3 and c-BiF3, respectively) in an ethylene carbonate-based liquid electrolyte, in situ Raman mapping and in situ laser scanning confocal microscopy (LSCM) for three-dimensional analysis were conducted almost simultaneously. As the potential of o-BiF3vs. Pb (EWE) was decreased to 0.4 V, desorption of F− started at the protrusions of o-BiF3 particles. After defluorination, EWE was increased to 0.6 V, and c-BiF3 appeared at protrusions of the surfaces of Bi. However, at the surfaces where o-BiF3 partially remained, o-BiF3 grew rather than c-BiF3. The apparent volumes of particles increased during defluorination and fluorination. The results are important for determining reaction mechanisms, and the results indicate the possibility of the use of ethylene carbonate-based liquid electrolytes.
Introduction
The development of next-generation batteries with high energy density is required for the establishment of a sustainable society with efficient use of energies and with low CO2 emission. Fluoride shuttle batteries (FSBs), in which fluoride or fluoride ions carry charges between an anode and a cathode, are considered as candidates for future batteries with high energy densities, and they have been extensively studied.1–24 Defluorination of metal fluorides and fluorination of the resultant metals described below are often used as battery reactions.| MFx + xe− → M + xF− (at the positive electrode, M being a metal) | (1) |
| M′ + yF− → M′Fy + ye− (at the negative electrode, M′ being a metal) | (2) |
It is important to study the evolution of FSB reactions and changes in the morphology of active materials during the reactions by using in situ methods in order to develop better electrolytes and electrodes. Raman spectroscopy has often been used for studies on battery reactions.28,29 We have conducted in situ Raman spectroscopy during battery reactions including FSB reactions.16,30–36 Recently, in situ Raman mapping was conducted almost simultaneously with in situ three-dimensional (3D) morphology analysis by laser scanning confocal microscopy (LSCM) and the method was found to be a powerful method for determining reaction mechanisms and rate-determining steps.36
In the present work, evolution of FSB reactions of orthorhombic and cubic BiF3 (o-BiF3 and c-BiF3), respectively (see Fig. 1(a) and (b)) microparticles partly embedded in a gold plating film (o-BiF3/gold and c-BiF3/gold, respectively, see Fig. 1(c)) in an ethylene carbonate-based electrolyte (tetramethylammonium fluoride (TMAF) dissolved in ethylene carbonate and diethyl carbonate (ECDEC)) was studied by in situ Raman mapping and in situ analysis of 3D morphology and colors by laser scanning confocal microscopy (LSCM). FSB reactions were found to proceed in the electrolyte, suggesting the possibility of future developments of ethylene carbonate-based electrolytes for FSBs with wide potential windows and with various breakthroughs. During fluorination after complete defluorination of o-BiF3, c-BiF3 appeared at protruded positions at the surfaces of pure Bi. On the other hand, o-BiF3 grew rather than c-BiF3 in the areas where Bi and partially remaining o-BiF3 coexisted, suggesting that o-BiF3 acted as nuclei for fluorination. The results also suggest that the rate-determining step of fluorination of pure Bi to form c-BiF3 is incorporation of F− from the electrolyte into Bi.
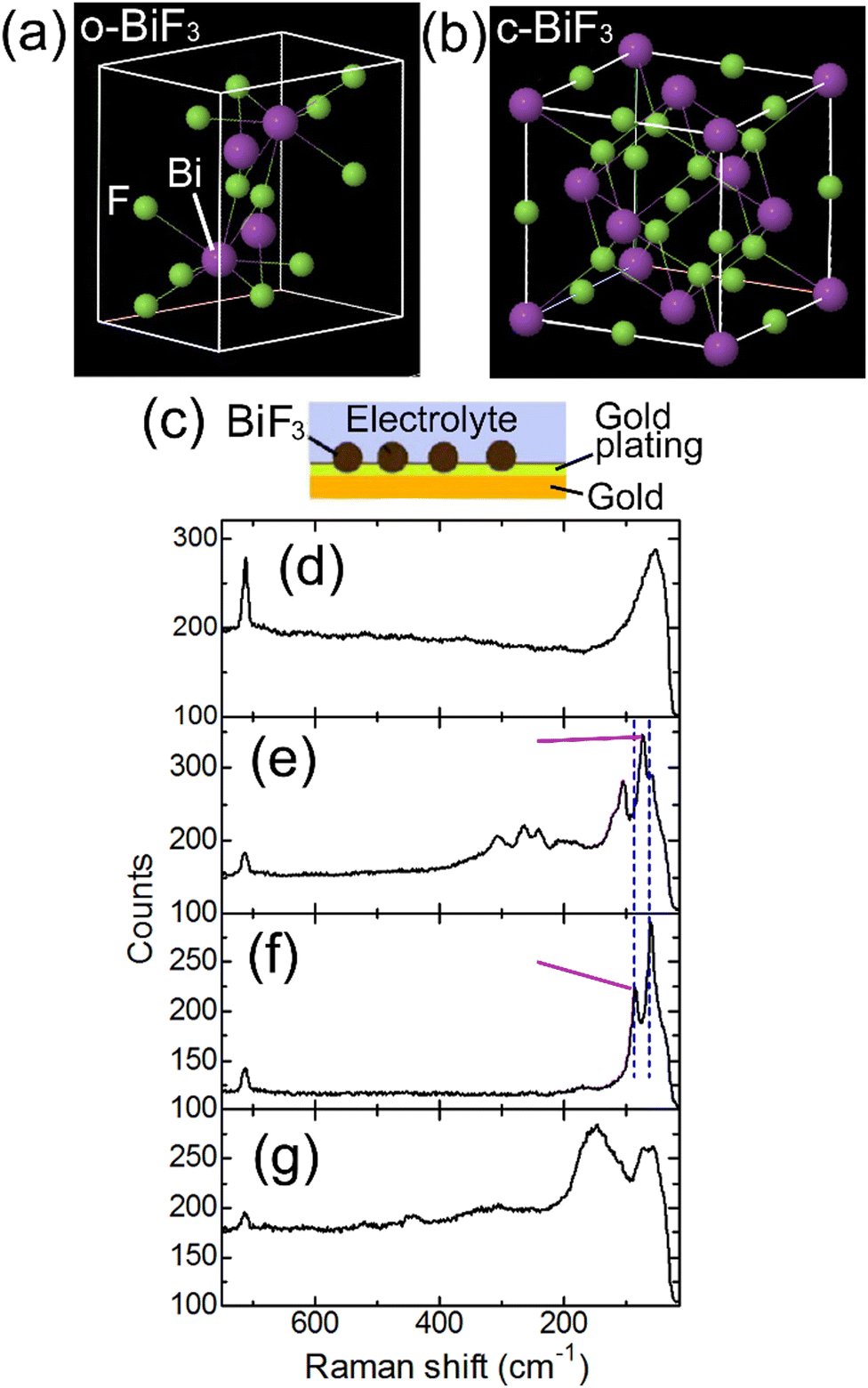 | ||
| Fig. 1 Structures of o-BiF3 and c-BiF3 and Raman spectra of o-BiF3/gold and c-BiF3/gold samples in electrochemical cells. (a) and (b) show structures of o-BiF3 and c-BiF3, respectively. (c) shows an illustration of an o-BiF3/gold or a c-BiF3/gold sample. (d) and (e) show Raman spectra obtained at the positions of the gold substrate and an o-BiF3 particle on an o-BiF3/gold sample in a cell before defluorination, respectively. (f) shows a Raman spectrum obtained at the position of a particle on the o-BiF3/gold sample in the cell after defluorination. (g) shows a Raman spectrum obtained at the position of a particle on a c-BiF3/gold sample in another cell before defluorination. | ||
Experimental
o-BiF3 microparticles were supplied by Fluorochem Ltd. c-BiF3 microparticles were prepared by annealing o-BiF3 microparticles at 150 °C in vacuum for 12 hours.34,35 Structures of o-BiF3 and c-BiF3 are shown in Fig. 1(a) and (b), respectively. The o-BiF3 and c-BiF3 particles were embedded in a gold plating film on a gold foil to make o-BiF3/gold and c-BiF3/gold, respectively (see Section S1 in ESI† and ESI of ref. 32 for details). Briefly, a gold foil (Nilaco Corporation, 99.95% pure, 0.2 mm in thickness) was put in a gold plating solution for deposition of 24 K gold (Marui Galvanizing Co. Ltd) in a glass tube. A small amount of o-BiF3 or c-BiF3 powder were then put in the solution. A platinum wire was inserted in the solution and a current was applied between the wire and the gold foil for deposition of gold. The amount of deposited gold corresponded to that of the gold plating film with a thickness of 2 μm without BiF3. The resultant o-BiF3/gold and c-BiF3/gold were washed with pure water and dried.An electrolyte was prepared in an Ar atmosphere in a glove box by dissolving about 0.08 M (saturation) of TMAF (Kumidas Co., Ltd) in ECDEC (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC = 1:1) (Kishida Chemical Co., Ltd). The ionic conductivity of the electrolyte was 4.2 × 10−3 S cm−1 at 25 °C (see Section S2 in ESI†).
:DEC = 1:1) (Kishida Chemical Co., Ltd). The ionic conductivity of the electrolyte was 4.2 × 10−3 S cm−1 at 25 °C (see Section S2 in ESI†).
Electrochemical cells were assembled in the glove box. The details of the cells are described in Section S1 in ESI† and ESI of ref. 36. The cell had a quartz window and a Pb wire as a counter electrode. The distance between the surface of the gold plating film and the quartz window was about 15 μm, and the gap between them was filled with the electrolyte. The cell was sealed with Kalrez and it was placed in an Ar flow during in situ Raman mapping and in situ LSCM measurements. The open circuit voltage (OCV) was typically 0.55 V. FSB reactions were induced by changing the potential of BiF3/gold vs. the Pb reference electrode (EWE).
In situ Raman mapping was conducted with an NRS-4500 Raman spectrometer (JASCO Corporation) and 532 nm laser light. It typically took 16 minutes for mapping of areas of 45 μm (150 points) × 50 μm (150 points).
In situ 3D analysis of morphologies of o-BiF3 and c-BiF3 microparticles at the surfaces of o-BiF3/gold and c-BiF3/gold samples was conducted by using LSCM (VK-X1000/1100, Keyence Corporation). Raman mapping and LSCM measurements were conducted by placing the cell on the sample stages of the Raman and LSCM apparatuses in turn without interrupting Ar flow and FSB reactions. Raman mapping was conducted for several areas (16 minutes for one area) and then the cell was transferred to the LSCM apparatus to analyze 3D morphologies for the areas. Accurate positioning after transfer of the cell between the two apparatuses took some time. Therefore, the timing of Raman mapping and that of LSCM measurements differed by 30 minutes to two hours.
Results and discussion
An electrochemical cell was constructed using an o-BiF3/gold sample. Fig. 1(d) and (e) show Raman spectra taken at the positions of the gold substrate and an o-BiF3 particle in the cell, respectively (see Fig. 1(c)). The peak at 715 cm−1 in Fig. 1(d) is assigned to the symmetric ring deformation mode of EC.37,38The peak at 52 cm−1 was not previously reported because this peak is out of the wavenumber ranges in previous papers, and it is thought to be Raman scattering from the electrolyte. The two clear peaks at 73 cm−1 and 105 cm−1 and small peaks from 150 cm−1 to 340 cm−1 in Fig. 1(e) are scattering from o-BiF3.39 After defluorination, Raman scattering of Bi appeared at 60 cm−1 and 85 cm−1 (Fig. 1(f)) as indicated by blue dashed lines.40–42 In the following, mappings of the areas of the peaks at 73 cm−1 (o-BiF3) and 85 cm−1 (Bi) indicated by red-violet lines are shown.
To induce defluorination of o-BiF3, EWE was decreased from OCV (0.55 V) to 0.2 V step by step as shown in Fig. 2(a), and the current shown in Fig. 2(b) appeared. Fig. 2(c) shows CCD (Charge-Coupled Device) images of o-BiF3/gold in the cell after keeping EWE at 0.4 V for 1 hour taken by a CCD camera of the Raman apparatus, Raman mapping of the areas of the peaks for o-BiF3 and Bi, and superposition of the CCD image and the mapping for Bi. The distribution of o-BiF3 coincides with that of dark areas in the CCD image, indicating that the dark areas are o-BiF3 particles. Bi had already appeared at some parts of the surfaces of o-BiF3 particles, indicating that defluorination had occurred at these parts, as shown by the mapping for Bi. After the o-BiF3/gold was kept at 0.4 V for 5 hours, defluorination further proceeded as shown in the mappings for Bi and o-BiF3 in Fig. 2(d), in which the areas of the peak for Bi increased at some positions and those for o-BiF3 at the corresponding positions decreased. In the CCD image in Fig. 2(d), contrasts of the particles became dark at the positions where defluorination proceeded, indicating that the dark contrasts correspond to Bi.
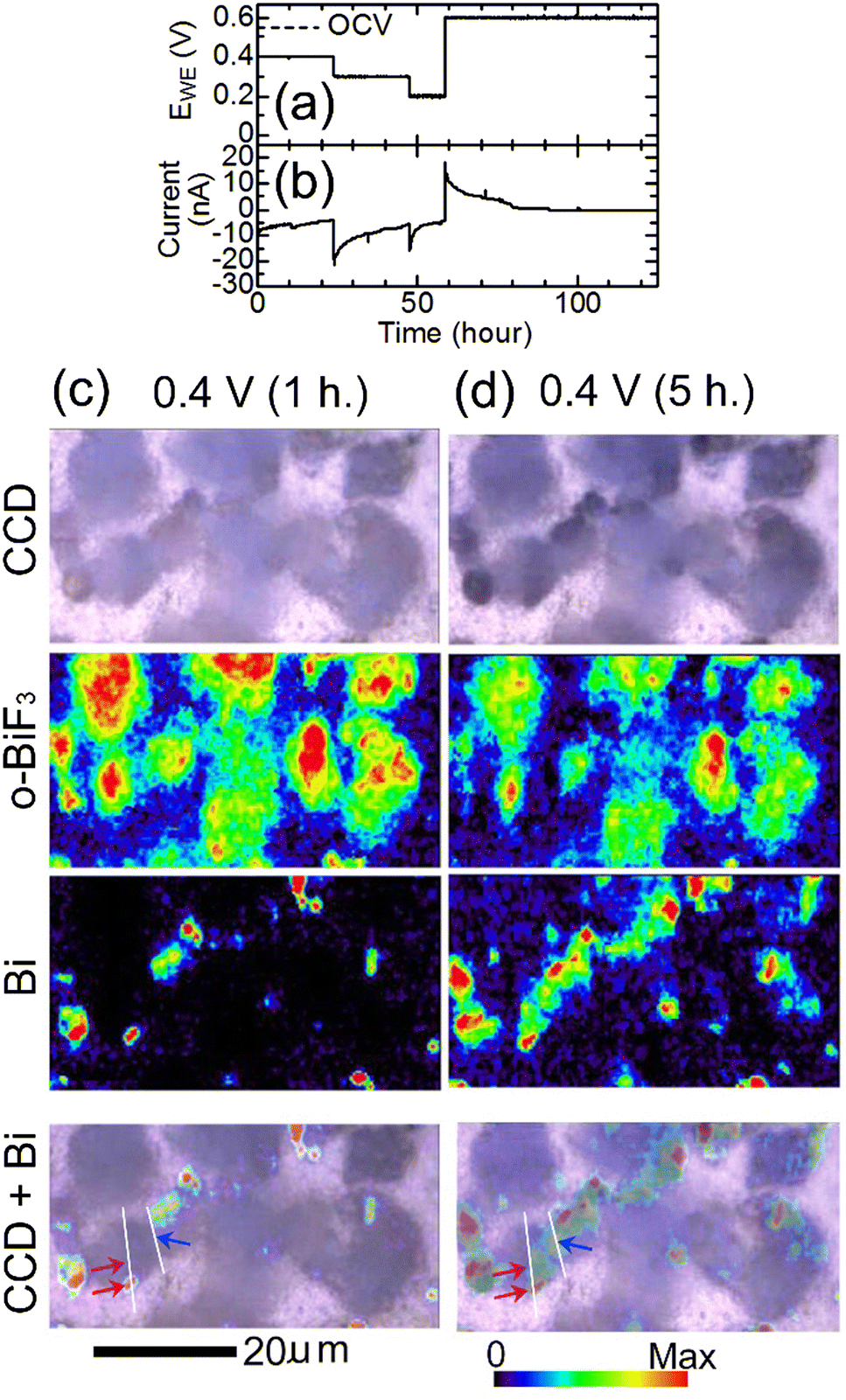 | ||
| Fig. 2 In situ Raman mapping of o-BiF3/gold during defluorination: (a) and (b) show the voltage applied to induce defluorination and the current that appeared, respectively. (c) and (d) show results after EWE was kept at 0.4 V for 1 hour and at 0.4 V for 5 hours, respectively. CCD images, mappings of areas of the peaks for o-BiF3 and Bi, and superposition of the CCD images and the mappings for Bi are shown. The red-violet and blue arrows in the superposition images indicate positions where defluorination started (nuclei of Bi appeared). Height profiles along white lines in the superposition images are shown in Fig. 3(c) and (d). | ||
Fig. 3 shows results of in situ LSCM measurements of the same area as that of Raman mapping shown in Fig. 2. Fig. 3(a) and (b) show results at OCV just before EWE was changed to 0.4 V and after EWE was kept at 0.4 V for 6 hours, respectively, the timings of which are close (within 1 hour difference) to those of Raman mapping shown in Fig. 2(c) and (d), respectively. The upper and lower panels of Fig. 3(a) and (b) show confocal CCD images and results of 3D analysis, respectively. In the confocal CCD images, all positions on the substrate surface and the protruding surfaces of particles in focus, resulting in clearer images than the CCD images taken by the Raman apparatus shown in Fig. 2(c) and (d) in which the focus was adjusted to the substrate surface and the protruding surfaces of the particles are therefore out of focus. In the confocal CCD image in Fig. 3(b), some parts of the particles, such as the positions indicated by blue and red-violet arrows, became darker than the corresponding positions in the image in Fig. 3(a), indicating that defluorination started at these positions. The 3D morphology in Fig. 3(b) did not change significantly compared with that in Fig. 3(a). Height profiles along lines A, B and C, D in Fig. 3(b), which include the positions indicated by the blue and red-violet arrows, respectively, are shown in Fig. 3(c) and (d), respectively. The positions indicated by the blue and red-violet arrows in Fig. 3(c) and (d) correspond to those in Fig. 3(b), respectively. It can be seen that there are protrusions at these positions, suggesting that defluorination by desorption of F− started at the positions with small radii of curvatures where electric fields are strong and that the rate-determining step of defluorination of large o-BiF3 particles by desorption of F− is extraction of F− from the surface of o-BiF3 into the electrolyte. Such phenomena are observed in defluorination of PbF236 and BiF335 in different electrolytes. Generally, an electric field at an interface between an electrolyte and the surface of an active material is strong when the radius of curvature of the surface is small, which promotes an electrochemical reaction, as is often observed by dendrite formation in electrodeposition of Li and Zn.43,44 Height profiles at additional different positions also suggest that defluorination started at protrusions (see Section S3 in ESI†).
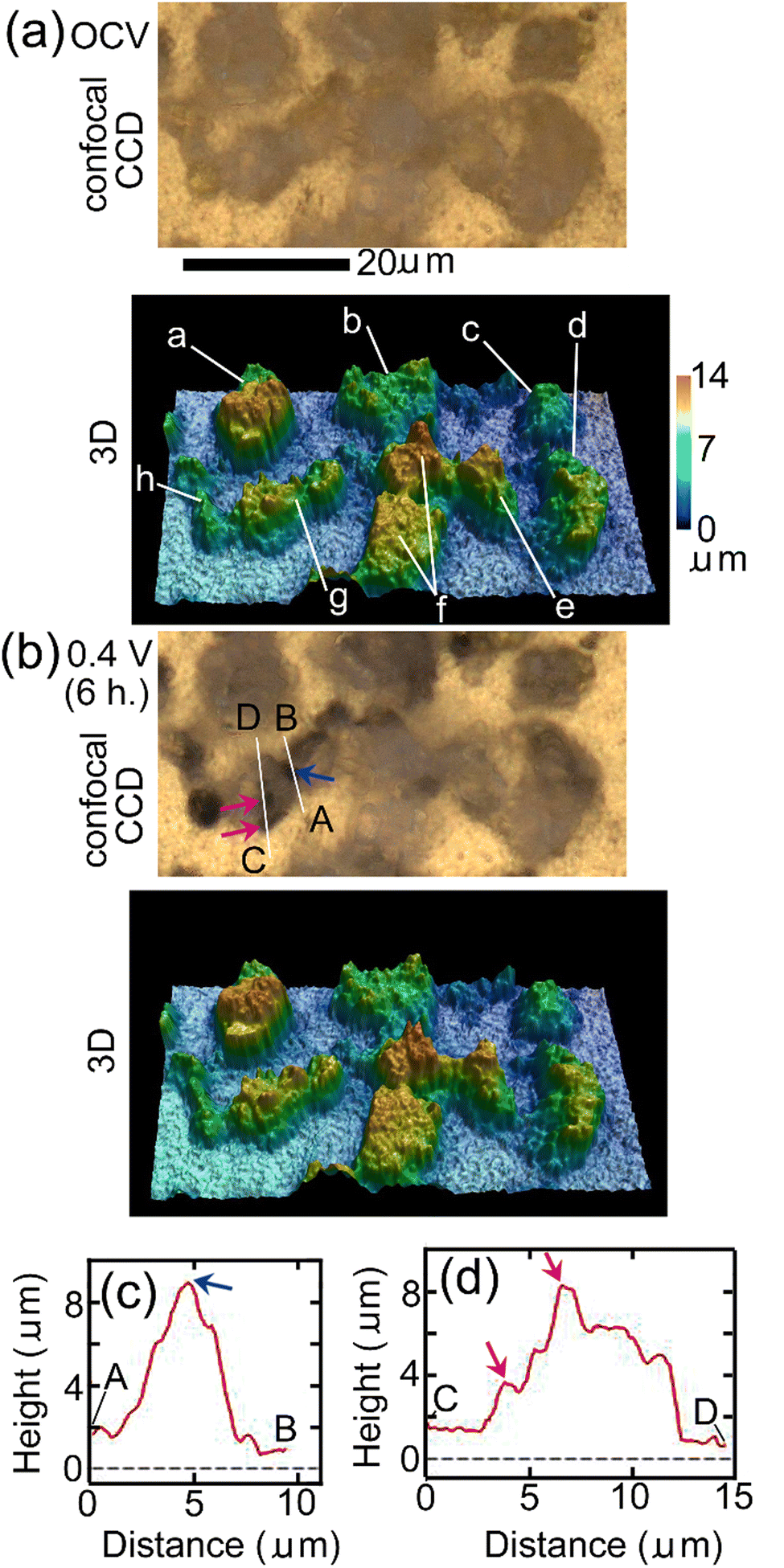 | ||
| Fig. 3 In situ 3D morphology analysis of o-BiF3/gold during defluorination: (a) and (b) show confocal CCD images and 3D morphology at OCV and after EWE was kept at 0.4 V for 6 hours, respectively. (a)–(h) Indicate particles of which apparent volumes above gold during defluorination and fluorination were analyzed as shown later. The contrasts of the positions indicated by blue and red-violet arrows in (b) became dark compared with those of corresponding positions in (a). (c) and (d) show height profiles along lines A, B and C, D in (b). Blue and red-violet arrows in (c) and (d) correspond to the positions indicated by blue and red-violet arrows in (b). | ||
E WE was decreased to 0.2 V to further promote defluorination. Fig. 4(a) shows CCD images, Raman mapping of the areas of the peaks for o-BiF3, c-BiF3 and Bi, and superposition of the CCD image and the mapping for c-BiF3 after o-BiF3/gold was kept at 0.2 V for 10 hours. The peak of c-BiF3 at 150 cm−1 was used for mapping for c-BiF3 (see the spectrum in Fig. 1(g) that was obtained at the position of a particle on a c-BiF3/gold sample in another electrochemical cell.) The contrasts of particles became dark and the areas of the peak for Bi increased, indicating that defluorination of the surfaces of the particles was almost completed. However, Bi and small amounts of remaining o-BiF3 co-existed on the particles indicated by c (only contours of the particle), d and e in the CCD image in Fig. 4(a), as shown by the mappings for o-BiF3 and Bi in Fig. 4(a). Then EWE was increased to 0.6 V to induce fluorination, and Raman and LSCM measurements were conducted during the fluorination processes. Fig. 4(b) and (c) show results after EWE was kept at 0.6 V for 18 hours and 50 hours, respectively. It can be seen that o-BiF3 grew at surfaces of particles c, d and e, where o-BiF3 and Bi co-existed before EWE was changed to 0.6 V. On the other hand, c-BiF3 grew at surfaces of particles at the center and the left side of which the surfaces were covered with only Bi before EWE was changed to 0.6 V (Fig. 4(a)). The results indicate that o-BiF3 acts as nuclei in fluorination of Bi to form o-BiF3.
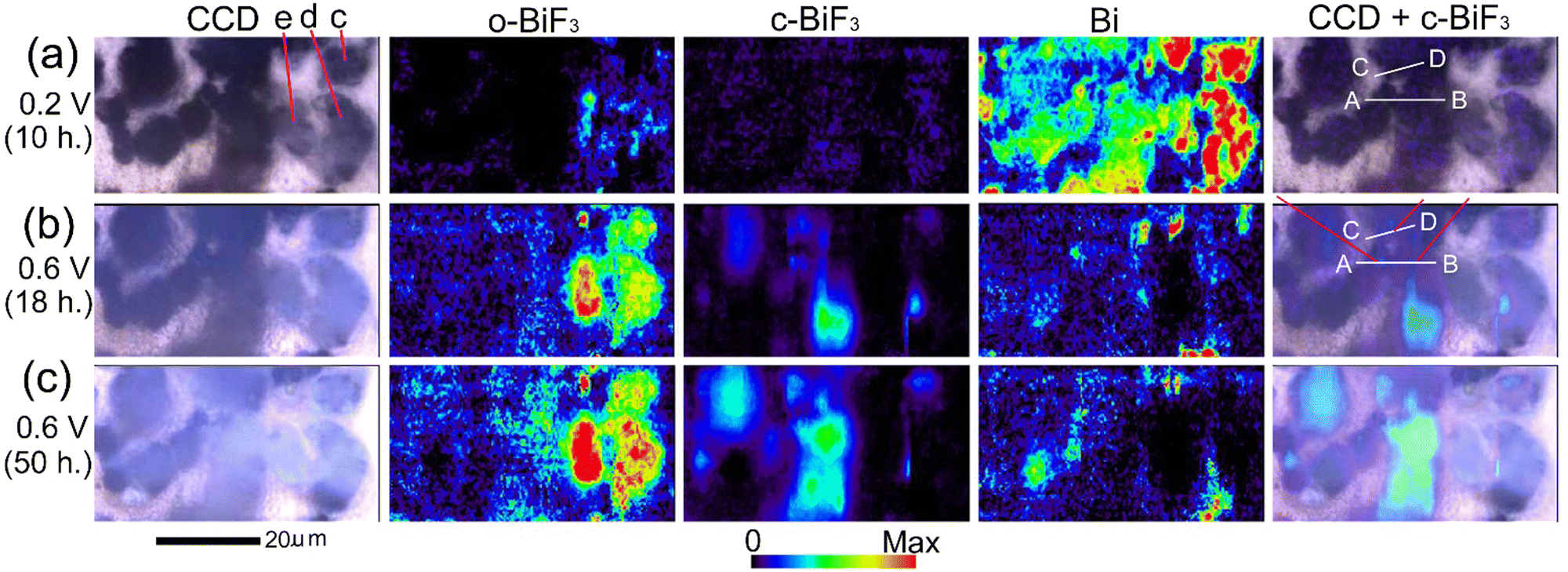 | ||
| Fig. 4 In situ Raman mapping of o-BiF3/gold during fluorination after defluorination: (a), (b) and (c) show results after EWE was kept at 0.2 V for 10 hours, at 0.6 V for 18 hours and at 0.6 V for 50 hours, respectively. CCD images, mappings of areas of the peaks for o-BiF3, c-BiF3 and Bi, and superposition of the CCD images and mappings for Bi are shown. The red-violet lines in the superposition image in (b) indicate positions where fluorination started (nuclei of c-BiF3 appeared). Height profiles along white lines AB and CD in the superposition images are shown in Fig. 5(d) and (e). | ||
Fig. 5(a)–(c) show results of in situ 3D analysis after EWE was kept at 0.2 V for 11 hours, at 0.6 V for 17 hours and at 0.6 V for 48 hours, respectively, the timings of which are close (within 2 hours difference) to those of in situ Raman mappings shown in Fig. 4(a)–(c), respectively. In the confocal CCD images in Fig. 5(b) and (c), contrasts of particles became brighter than those in Fig. 5(a), indicating fluorination. It can be seen that the apparent volumes of particles above gold increased during fluorination. (The apparent volume is the volume of a particle defined by the outermost surface of the particle that may have fine spaces (such as pores) inside. The real volume does not include the volumes of the fine spaces and cannot be calculated. Apparent volumes can be calculated by the software attached to the LSCM apparatus.) In the superposition of CCD images and mapping for c-BiF3 in Fig. 4, fluorination of the surface of particles at which only Bi existed is thought to have started at the positions indicated by red-violet lines. These positions correspond to the positions indicated by red-violet arrows in the confocal CCD image in Fig. 5(a). Height profiles along lines A, B and C, D that include the positions of the arrows in Fig. 5(a) are shown in Fig. 5(d) and (e), respectively. Red-violet arrows in Fig. 5(d) and (e) correspond to the positions indicated by the red-violet arrows in Fig. 5(a). It can be seen that there are protrusions at the positions, suggesting that fluorination starts at positions with small radii of curvatures where electric fields are strong. The rate-determining step of fluorination of Bi particles is thought to be incorporation of F− from the electrolyte to the surface of the particles.
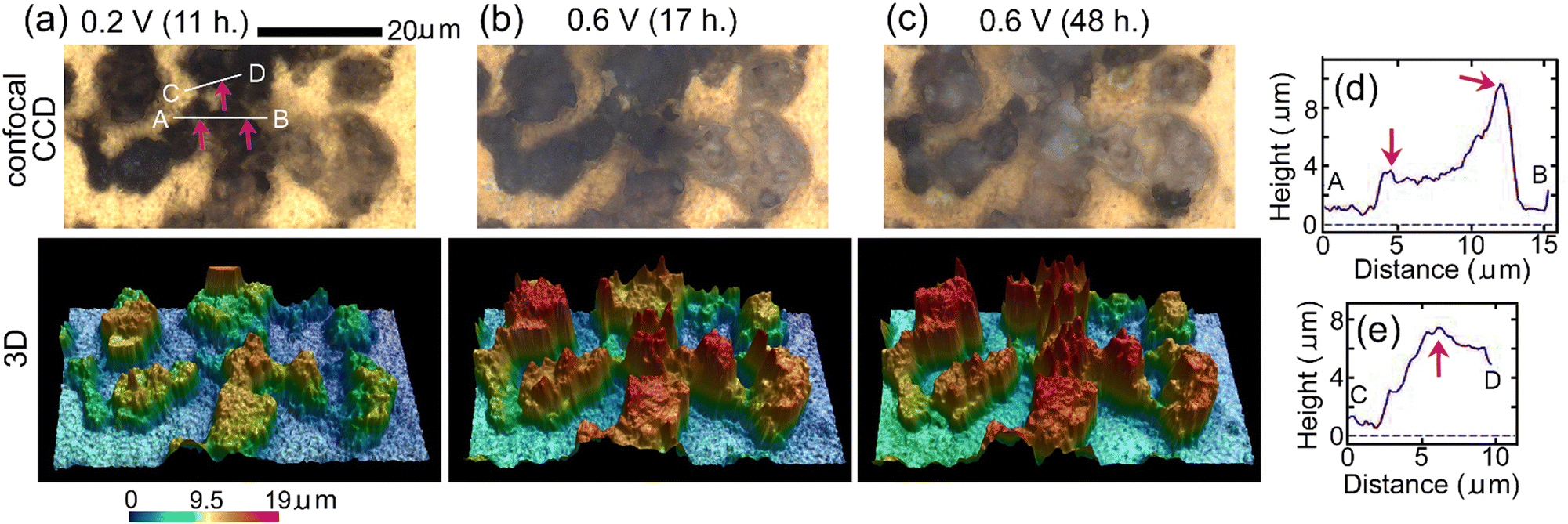 | ||
| Fig. 5 In situ 3D morphology analysis of o-BiF3/gold during fluorination after defluorination: (a), (b) and (c) show confocal CCD images and 3D morphology after EWE was kept at 0.2 V for 11 hours, at 0.6 V for 17 hours and at 0.6 V for 48 hours, respectively. The positions indicated by red-violet arrows in (a) correspond to the positions indicated by red-violet lines in Fig. 4(b) where nuclei of c-BiF3 appeared. (d) and (e) show height profiles along white lines A, B and C, D in (a), respectively. Red-violet arrows in (d) and (e) correspond to the positions indicated by red-violet arrows in (a). | ||
Fig. 6 shows changes in apparent volumes above gold of particles indicated by a–h in Fig. 3(a) during defluorination and fluorination processes shown in Fig. 2–5. It can be seen that apparent volumes of most of the particles increased slightly during defluorination, which is contrary to expectation. During subsequent fluorination, the apparent volumes above gold of the particles increased more clearly, being consistent with expectation. Thus, the apparent volumes of particles after a defluorination–fluorination cycle are larger than their initial values.
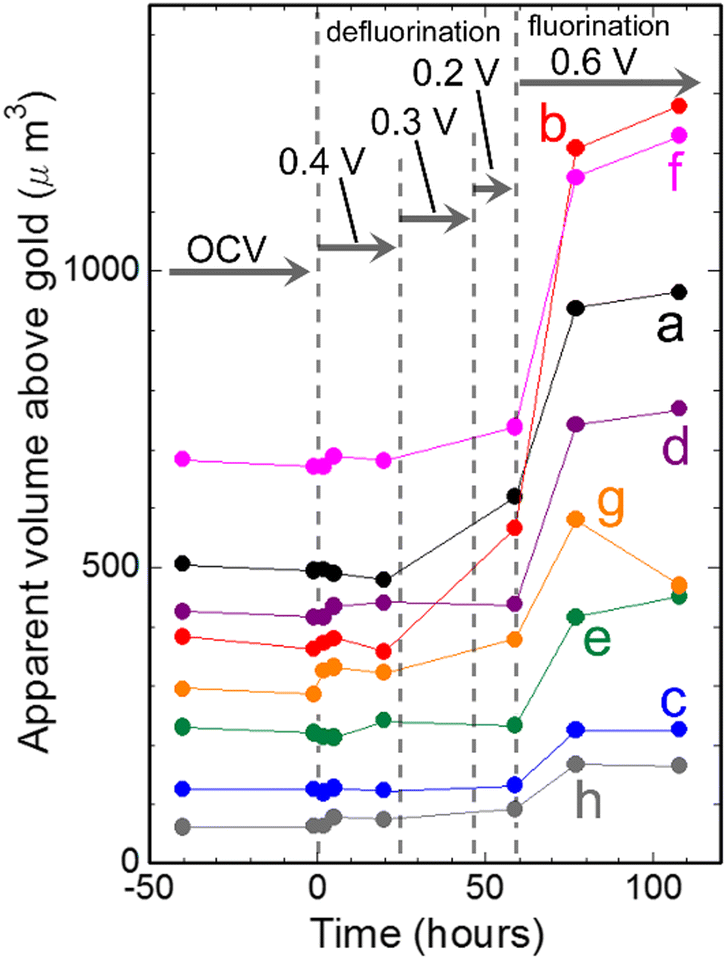 | ||
| Fig. 6 Changes in apparent volumes of particles indicated by a–h in Fig. 3(a) against time. | ||
Another cell was constructed using a c-BiF3/gold sample, and defluorination of c-BiF3 particles was studied. EWE was decreased from OCV (0.55 V) to 0.15 V step by step to induce defluorination as shown in Fig. 7(a), and the current shown in Fig. 7(b) appeared. The current at 0.4 V (−43 nA) is larger than that for o-BiF3 (−8.3 nA) (Fig. 2(b)). This may be due to a dark current of unknown side reactions because a higher rate of defluorination of c-BiF3 than that for o-BiF3 was not recognized on the basis of Raman measurements. The current after 2.5 hours in Fig. 7(b) is larger than the current from 22 to 60 hours in Fig. 2(b), which is partly due to the rapid decrease in EWE for c-BiF3 (As shown in Fig. 7(b), EWE decreased to 0.15 V within 5 hours.) compared with that for o-BiF3. (As shown in Fig. 2(b), EWE decreased to 0.2 V over a period of 47 hours.) Fig. 7(c) shows CCD images, Raman mapping of the areas of the peaks for c-BiF3 and Bi, and superposition of the CCD image and the mapping for Bi after c-BiF3/gold was kept at 0.4 V for 1 hour. The existence of Bi in the mapping for Bi indicates that the surfaces of particles had partially defluorinated. After keeping EWE at 0.15 V for 1 hour, the areas of the peaks for Bi and c-BiF3 increased and decreased, respectively, as shown by Fig. 7(d), indicating further defluorination.
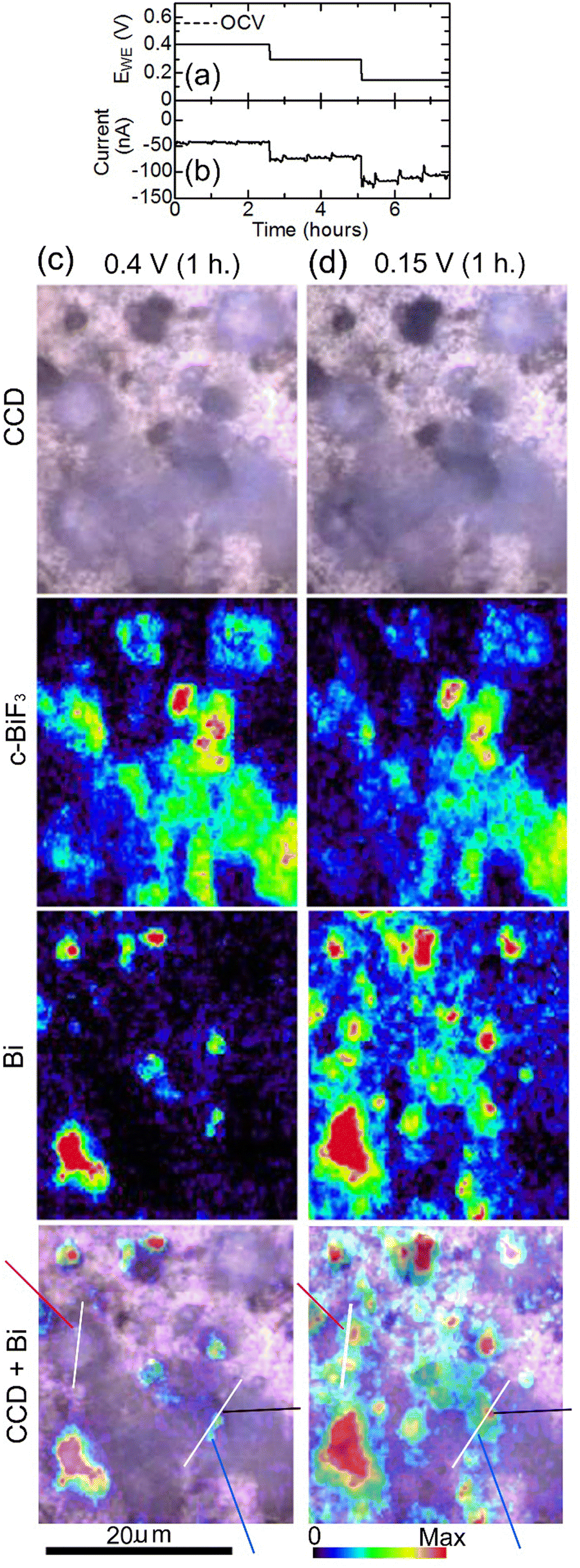 | ||
| Fig. 7 In situ Raman mapping of c-BiF3/gold during defluorination: (a) and (b) show the voltage applied to induce defluorination and the current that appeared, respectively. (c) and (d) show results after EWE was kept at 0.4 V for 1 hour and at 0.15 V for 1 hour, respectively. CCD images, mappings of areas of the peaks for c-BiF3 and Bi, and superposition of the CCD images and the mappings for Bi are shown. The blue, black and red-violet lines in the superposition images indicate positions where defluorination started (nuclei of Bi appeared). Height profiles along white lines in the superposition images are shown in Fig. 8(c) and (d). | ||
Fig. 8(a) and (b) show results of in situ 3D analysis conducted after EWE was kept at 0.4 V for 1.5 hours and at 0.15 V for 2.5 hours, respectively, of which timings are close (within 1.5 hours difference) to those of Raman mappings shown in Fig. 7(c) and (d), respectively. In Fig. 7(c), it can be seen that defluorination started at the positions indicated by blue, black and red-violet lines. Corresponding positions in the confocal CCD image in Fig. 8(a) are indicated by blue, black and red-violet arrows. Height profiles along lines A, B and C, D in Fig. 8(a) that include the positions indicated by the arrows are shown in Fig. 8(c) and (d), respectively. The positions indicated by blue, black and red-violet arrows in Fig. 8(c) and (d) correspond to those in Fig. 8(a). There are protrusions in these positions, suggesting that the rate-determining step of defluorination of c-BiF3 particles by desorption of F− is extraction of F− from the surface of c-BiF3 into the electrolyte. Height profiles at other positions were also obtained, and the results suggest that defluorination started at protrusions (see Section S4 in ESI†). Changes in apparent volumes above gold of particles indicated by a–g in Fig. 8(a) are shown in Fig. 8(e). Apparent volumes of particles surrounded by a white dashed curve in Fig. 8(a) were not analyzed because the morphologies of the particles changed too much during defluorination, and it was difficult to analyze the apparent volume of each of the particles. It can be seen that apparent volumes increased during defluorination.
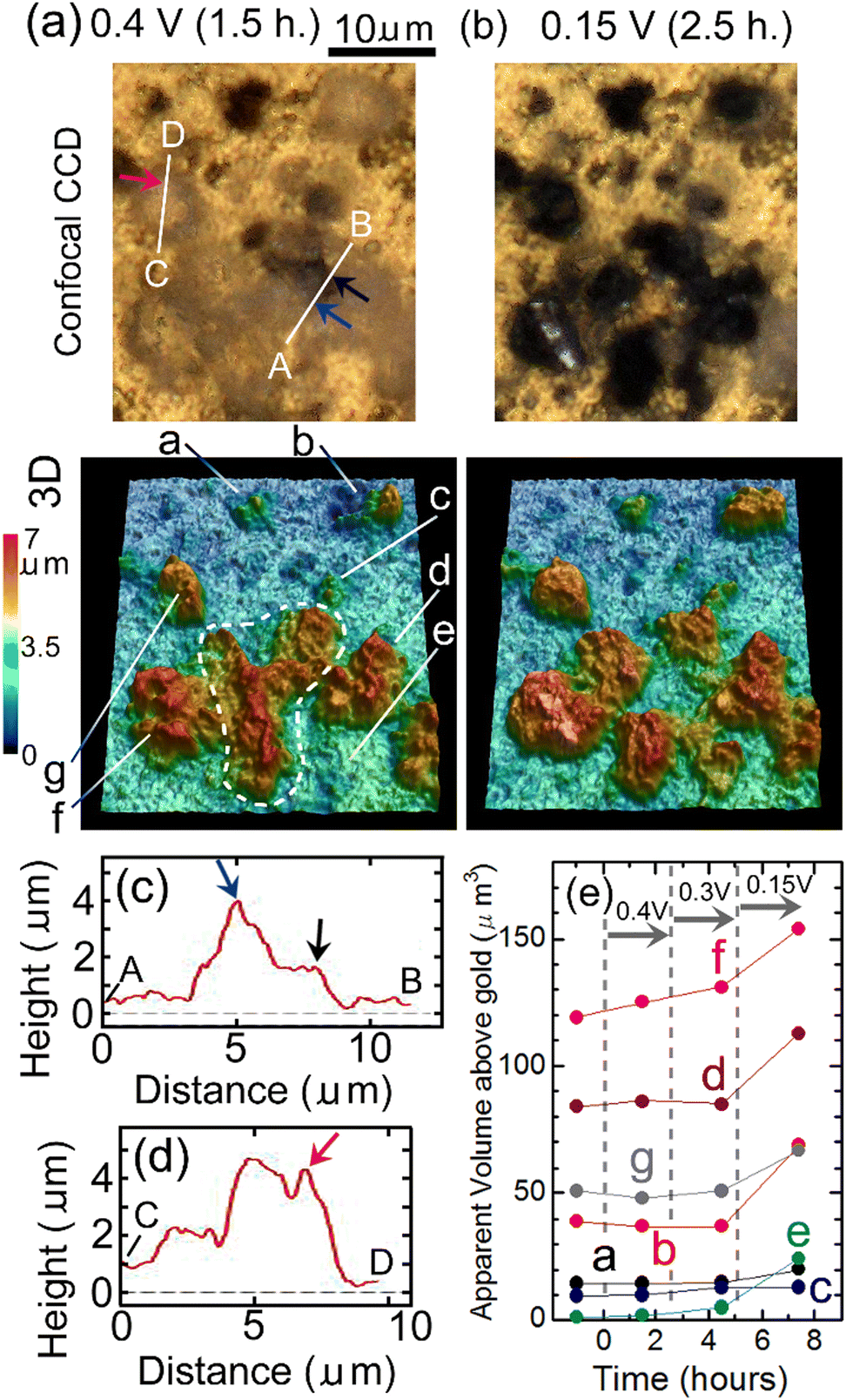 | ||
| Fig. 8 In situ 3D morphology analysis of c-BiF3/gold during defluorination: (a) and (b) show confocal CCD images and 3D morphology after EWE was kept at 0.4 V for 1.5 hours and at 0.15 V for 2.5 hours, respectively. The blue, black and red-violet arrows indicate positions where defluorination started (nuclei of Bi appeared). (c) and (d) show height profiles along lines A, B and C, D in (a), respectively. Blue, black and red-violet arrows in (c) and (d) correspond to the positions indicated by blue, black and red-violet arrows in (a). (e) shows changes in apparent volumes of particles indicated by a–g in (a) against time. Measurements of apparent volumes of particles surrounded by a white dashed curve were difficult due to large morphology changes. | ||
In Fig. 6 and 8(e), increases in apparent volumes can be seen during defluorination, which is contrary to expectation. However, clearer increases in apparent volumes were observed during defluorination of PbF2 in a CsF/LiBOB/tetraglyme electrolyte.36 Therefore, increases in apparent volumes of metal fluorides during defluorination in liquid electrolytes may not be rare phenomena. The density of the resultant Bi should be lower than the densities of initial o-BiF3 and c-BiF3, and structures of the resultant Bi may be dendritic. In the case of PbF2 in the Cs/LiBOB/tetraglyme electrolyte, fluorination was not clearly observed during oxidation because significant desorption of Pb (Pb → Pb2 + 2e−) occurred, resulting in decreases in apparent volumes of particles. On the other hand, in the present study, fluorination by direct reaction of F− and Bi was the main process during oxidation, and desorption of Bi during oxidation was less significant. Thus, increases in the apparent volumes during fluorination were clearly observed, as shown in Fig. 6. After a defluorination/fluorination cycle, the volumes increased compared with the initial values before the cycle. This leads to agglomeration of particles, which leads to loss of capacities of electrodes due to loss of surface areas of active materials when composite electrodes of BiF3 are operated in the present electrolyte. (Desorption of Pb during the fluorination process results in adsorption of Pb during the next defluorination process at, for example, the positions between particles, which also causes agglomeration.) The development of an appropriate fluoride–carbon complex may be needed to prevent agglomeration.
Ethylene carbonate-based electrolytes are used in lithium ion batteries, and wide potential windows of the electrolytes are attained by formation of SEI. It is thought that SEI should contain LiF to achieve good functions of SEI.27 LiF is produced from components of the electrolytes, e.g., Li+ and PF6−, during formation of SEI. The electrolyte used in the present study did not contain such effective components, and good SEI will not be obtained when the electrolyte is used for FSBs with negative electrodes (Al, Ce, etc.) unless some additives suitable for FSBs are developed. Results of cyclic voltammetry of the TMAF/ECDEC electrolyte (see Section S5 in ESI†) showed that the reduction current around −3 V at the 4th cycle was suppressed compared with that at the 1st cycle, but there was still significant reduction current at the 4th cycle, indicating that good SEI were not formed. Nevertheless, FSB reactions observed in the present study indicate the possibility of the use of ethylene carbonate-based electrolytes for FSBs to achieve wide potential windows in the future.
In lithium ion batteries, LiCoO2 (practical capacity: 160 mA h g−1) and graphite (372 mA h g−1) are used for positive and negative electrodes, respectively, and the voltage is about 3.7 V. If Cu (843 mA h g−1) positive and LaF3 (410 mA h g−1) negative electrodes are used with an ethylene carbonate-based electrolyte in an FSB, the cell voltage will be about 3 V, and the weight energy density of the FSB is expected to be about two-times higher than that of lithium ion batteries. However, in many reports of single electrode tests of FSB reactions with liquid electrolytes, the capacity and the cyclability are far from practical levels.9–11,15,17,18,21,22 In addition, there have been few reports of rechargeable FSB reactions in a liquid electrolyte using both positive and negative electrodes.6 Realization of FSBs with high energy densities and high cyclability comparable to those of lithium ion batteries requires many breakthroughs.
Conclusions
In situ Raman mapping and in situ LSCM were conducted almost simultaneously in the same areas of samples in turn during FSB reactions of o-BiF3 and c-BiF3 particles in an ethylene carbonate-based electrolyte in order to study changes in chemical distribution and 3D morphology of the particles. The rate-determining steps of defluorination of o-BiF3 and c-BiF3 particles are thought to be extraction of F− from BiF3 to the electrolyte. c-BiF3 formed in fluorination of Bi, and the rate-determining step in this process is thought to be incorporation of F− from the electrolyte to Bi. On the other hand, o-BiF3 grew rather than c-BiF3 during fluorination of surfaces where Bi and o-BiF3 coexisted, suggesting that o-BiF3 acted as nuclei for fluorination. The apparent volumes of particles increased during defluorination and subsequent fluorination. The increases in apparent volumes during defluorination of metal fluorides in liquid electrolytes are thought to be general phenomena. The results will lead to strategies to make better electrolytes and electrodes, and the results also indicate the possibility of using ethylene carbonate-based liquid electrolytes for FSBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is based on results obtained from a project, JPNP21006, commissioned by the New Energy and Industrial Technology Development Organization (NEDO).Notes and references
- F. Gschwind, G. Rodriguez-Garcia, D. J. S. Sandbeck, A. Gross, M. Weil, M. Fichtner and N. Hormann, J. Fluorine Chem., 2016, 182, 76–90 CrossRef CAS.
- M. A. Nowroozi, I. Mohammad, P. Molaiyan, K. Wissel, A. R. Munnangi and O. Clemens, J. Mater. Chem. A, 2021, 9, 5980–6012 RSC.
- M. A. Reddy and M. Fichtner, J. Mater. Chem., 2011, 21, 17059–17062 RSC.
- C. Rongeat, M. A. Reddy, T. Diemant, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2014, 2, 20861–20872 RSC.
- F. Gschwind, Z. Zao-Karger and M. Fichtner, J. Mater. Chem. A, 2014, 2, 1214–1218 RSC.
- K. Okazaki, Y. Uchimoto, T. Abe and Z. Ogumi, ACS Energy Lett., 2017, 2, 1460–1464 CrossRef CAS.
- M. A. Nowroozi, K. Wissel, J. Rohrer, A. R. Munnangi and O. Clemens, Chem. Mater., 2017, 29, 3441–3453 CrossRef CAS.
- H. Nakamoto, Z. Ogumi and T. Abe, Jp. Pat., JP6342837, 2018 Search PubMed.
- Z. Ogumi, T. Hirai, J. Yamaki, H. Horino, K. Okazaki and Y. Koyama, Jp. Pat., 2013-270415, 2013 Search PubMed.
- K. Konishi, K. Kono, T. Abe and Z. Ogumi, Jp. Pat., JP2017-10865, 2017 Search PubMed.
- H. Konishi, T. Minato, T. Abe and Z. Ogumi, J. Electroanal. Chem., 2018, 826, 60–64 CrossRef CAS.
- A. Grenier, A. G. Porras-Gutierrez, H. Groult, K. Beyer, O. J. Borkiewicz, K. W. Chapman and D. Dambournet, J. Mater. Chem. A, 2017, 5, 15700–15705 RSC.
- I. Mohammad, R. Witter, M. Fichtner and M. A. Reddy, ACS Appl. Energy Mater., 2018, 1, 4766–4775 CrossRef CAS.
- M. A. Nowroozi, S. Ivlev, J. Rohrer and O. Clemens, J. Mater. Chem. A, 2018, 6, 4658–4669 RSC.
- V. K. Davis, C. M. Bates, K. Omichi, B. M. Savoie, N. Momčilović, Q. Xu, W. J. Wolf, A. Michael, M. A. Webb, K. J. Billings, N. H. Chou, S. Alayoglu, R. K. McKenney, I. M. Darolles, N. G. Nair, A. Hightower, D. Rosenberg, M. Ahmed, C. J. Brooks, T. F. Miller III, R. H. Grubbs and S. C. Jones, Science, 2018, 362, 1144–1148 CrossRef CAS PubMed.
- T. Yamanaka, H. Nakamoto, T. Abe, K. Nishio and Z. Ogumi, ACS Appl. Energy Mater., 2019, 2, 3092–3097 CrossRef CAS.
- A. Celik-Kucuk, T. Minato, T. Yamanaka and T. Abe, J. Mater. Chem. A, 2019, 7, 8559–8567 RSC.
- A. Celik-Kucuk, T. Yamanaka, Y. Yokoyama and T. Abe, J. Electrochem. Soc., 2021, 168, 010501 CrossRef.
- S. Kobayashi, H. Nakamoto, D. Yokoe, A. Kuwabara, T. Abe and Y. Ikuhara, ACS Appl. Energy Mater., 2021, 4, 996–1003 CrossRef CAS.
- A. Celik Kucuk, T. Yamanaka and T. Abe, J. Mater. Chem. A, 2020, 8, 22134–22142 RSC.
- M. Kawasaki, K.-I. Morigaki, G. Kano, H. Nakamoto, R. Takekawa, J. Kawamura, T. Minato, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2021, 168, 010529 CrossRef CAS.
- T. Yamamoto, K. Matsumoto, R. Hagiwara and T. Nohira, J. Electrochem. Soc., 2021, 168, 040530 CrossRef CAS.
- K. Shimoda, T. Minato, H. Konishi, G. Kano, T. Nakatani, S. Fujinami, A. Celik-Kucuk, S. Kawaguchi, Z. Ogumi and T. Abe, J. Electroanal. Chem., 2021, 895, 115508 CrossRef CAS.
- X. Li, Y. Tang, J. Zhu, H. Lv, Y. Xu, W. Wang, C. Zhi and H. Li, Adv. Energy Mater., 2021, 11, 2003714 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- C. M. Julien and A. Mauger, AIMS Mater. Sci., 2018, 5, 650–698 CAS.
- L. Meyer, N. Saqib and J. Porter, J. Electrochem. Soc., 2021, 168, 090561 CrossRef.
- T. Yamanaka, H. Nakagawa, S. Tsubouchi, Y. Domi, T. Doi, T. Abe and Z. Ogumi, ChemSusChem, 2017, 10, 855–861 CrossRef CAS PubMed.
- T. Yamanaka, H. Nakagawa, S. Tsubouchi, Y. Domi, T. Doi, T. Abe and Z. Ogumi, Electrochim. Acta, 2017, 234, 93–98 CrossRef CAS.
- T. Yamanaka, T. Abe, K. Nishio and Z. Ogumi, J. Electrochem. Soc., 2019, 166, A635–A640 CrossRef CAS.
- T. Yamanaka, K. Okazaki, T. Abe, K. Nishio and Z. Ogumi, ChemSusChem, 2019, 12, 527–534 CrossRef CAS PubMed.
- T. Yamanaka, K. Okazaki, Z. Ogumi and T. Abe, ACS Appl. Energy Mater., 2019, 2, 8801–8808 CrossRef CAS.
- T. Yamanaka, A. Celik-Kucuk, Z. Ogumi and T. Abe, ACS Appl. Energy Mater., 2020, 3, 9390–9400 CrossRef CAS.
- T. Yamanaka, Z. Ogumi and T. Abe, J. Mater. Chem. A, 2021, 9, 22544–22554 RSC.
- Y. Yamada, F. Sagane, Y. Iriyama, T. Abe and Z. Ogumi, J. Phys. Chem. C, 2009, 113, 14528–14532 CrossRef CAS.
- M. Morita, Y. Asai, N. Yoshimoto and M. Ishikawa, J. Chem. Soc., Faraday Trans., 1998, 94, 3451–3456 RSC.
- V. Y. Kavun, E. I. Voit, R. M. Yaroshenko and V. K. Goncharuk, J. Non-Cryst. Solids, 2014, 401, 224–231 CrossRef CAS.
- L. Kumari, J. H. Lin and Y. R. Ma, J. Phys. D: Appl. Phys., 2008, 41, 025405 CrossRef.
- A. Reyes-Contreras, M. Camacho-Lopez, S. Camacho-Lopez, O. Olea-Mejia, A. Esparza-Garcia, J. G. Banuelos-Muneton and M. A. Camacho-Lopez, Opt. Mater. Express, 2017, 7, 1777–1786 CrossRef CAS.
- K. A. Trentelman, J. Raman Spectrosc., 2009, 40, 585–589 CrossRef CAS.
- Z. Hong and V. Viswanathan, ACS Energy Lett., 2018, 3, 1737–1743 CrossRef CAS.
- Z. Mu, Z. Guo and Y.-H. Lin, Energy Storage Mater., 2020, 30, 52–58 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ionic conductivity of the electrolyte, height profiles of particles, CV measurements of the electrolyte. See DOI: https://doi.org/10.1039/d2ya00184e |
| This journal is © The Royal Society of Chemistry 2022 |