
Unravelling the major factors in photo-oxidative stability of anthradithiophene derivatives†
Karl J.
Thorley
 *a,
Hoang
Le
b,
Yang
Song
b and
John E.
Anthony
ac
*a,
Hoang
Le
b,
Yang
Song
b and
John E.
Anthony
ac
aCenter for Applied Energy Research, University of Kentucky, Lexington, KY 40511, USA. E-mail: karl.thorley@uky.edu
bDepartment of Chemistry, Centre College, Danville, KY 40422, USA
cDepartment of Chemistry, University of Kentucky, Lexington, KY 40508, USA
First published on 30th September 2022
Abstract
Acenes are a set of polycyclic aromatic hydrocarbons commonly employed in opto-electronic devices, despite the fact that they can undergo chemical transformation under illumination which is detrimental to device performance. In this study, a series of anthradithiophenes bearing different substituents is investigated, considering the light absorption, concentration dependence, and reactivity with singlet oxygen to understand trends in photostability. Future molecular design can use these results to design more robust materials e.g. for solar conversion, or conversely more photoreactive materials for sensing applications.
Introduction
Acene derivatives are widely studied in the fields of organic electronics.1 Chemical modification can be used to affect molecular properties as well as bulk material properties through condensed phase intermolecular interactions.2 Inclusion of thiophene rings into the acene structure has been used as a staple structural modification, and anthradithiophene and its derivatives have been thoroughly explored as p-type semiconductor materials.3–9 Many applications of acenes involve absorption of light; there are a few examples of acene-containing materials acting as either donors10 or acceptors11 in bulk heterojunction solar cells, but perhaps a more realistic role in solar energy applications is their use as a photomultiplier layer for silicon solar cells due to their ability to undergo singlet fission.12 Elsewhere, solid state fluorescent acenes have been employed in waveguide materials.13 Unfortunately, light absorption often has the side effect of photodegradation of the acene material, which can be detrimental to device performance.14 While this has been well-documented for many acene materials, particularly the ubiquitous TIPS-pentacene, photostability of thiophene-containing acenes have only briefly been investigated, and knowledge of substituent effects is minimal. Photodimerisation15 is a potential degradation mechanism, first being observed in anthracene and tetracene, and substituents generally diminish the potential for photodimerisation through steric effects. Photo-oxidation to form endoperoxide species16 is a possible degradation route for functionalized acenes of all lengths. The driving force for both oxidation and dimerisation reactions is the formation of multiple Clar sextets, and the resulting stabilisation through increased aromaticity. Endoperoxide formation has been utilised in a positive manner in the areas of oxygen sensing materials,17 and since the reaction is often thermally reversible18 it can also be used as a delivery method for reactive oxygen species (ROS) in applications such as photodynamic therapy.19Photo-oxidation and endoperoxide formation can occur through two main mechanisms,20 demonstrated in Scheme 1. In both cases, the acene (or an additional photosensitiser) is excited by the absorption of incident light. The excited state can either transfer an electron to molecular oxygen to form ROS such as superoxide anion O2−, or can form singlet oxygen 1O2 by energy transfer through a triplet state. The reactive oxygen species then combine with a ground state acene molecule, which can either be a stepwise or concerted reaction pathway,21 which has been the subject of computational investigations.22,23 In some cases, the endoperoxide will be unstable, especially in acidic environments, and react irreversibly to give a quinone product.24 If the endoperoxide is stable, for example in the alkynylated acenes, the endoperoxide can thermally decompose to reform the original acene material by expelling oxygen.18 The singlet and triplet excited state molecules can also return to their ground states by emission or non-radiative decay rather than react with oxygen.
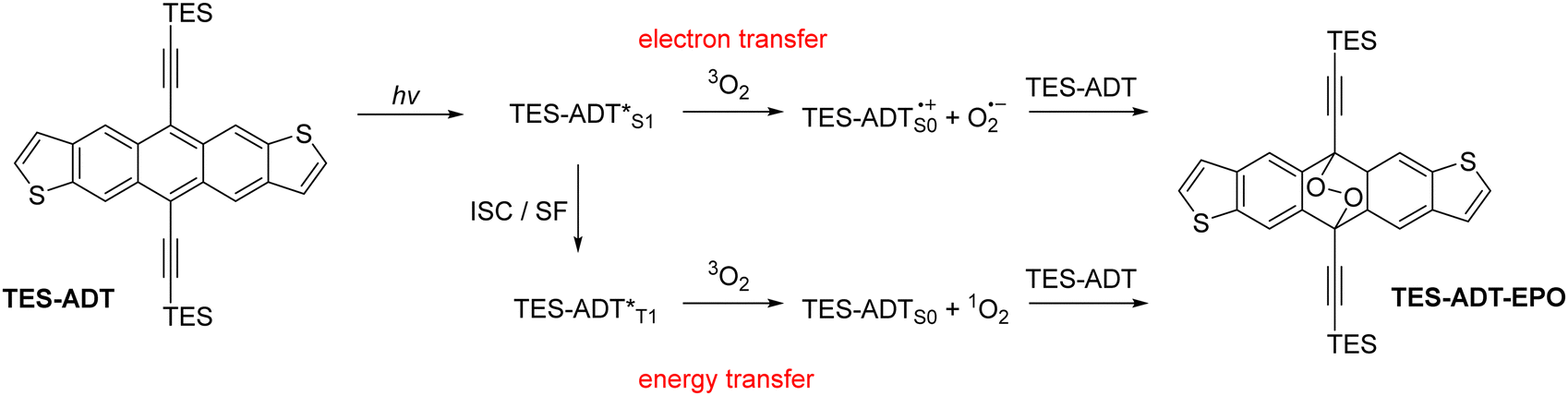 | ||
| Scheme 1 Photo-oxidation reactions of acene materials such as TES-ADT. ISC = intersystem crossing, SF = singlet fission. | ||
The photo-oxidation of acenes therefore has many contributing factors, and the present study aims to unravel which of these factors is the most influential. A more comprehensive understanding of the photoreactivity of acenes will enable the development of more stable materials for optical applications such as solar harvesting and photomultiplication, or conversely more photo-reactive materials for sensing or photo-medical applications.
Literature precedence and choice of chromophore
Photooxidation of anthracene and other acenes was discussed as long ago as the 1930s.25 More modern investigations of functionalized acene photostability have typically explored the extension of the polycyclic aromatic core, where increasing the number of fused rings generally results in less stable materials. Heterocyclic extensions (e.g. by thiophene rings) show a lesser impact on stability and increased conjugation than benzenoid extensions.26 Side chain substitutions, typically at the most reactive central acene rings, have been explored, in particular the comparison of ethynyl versus aryl groups; ethynylation extends conjugation whereas direct bonding between acene and aryl groups imparts some degree of steric stabilization.27 In arylethynyl acenes, electron donating and withdrawing effects on the aryl rings were shown to influence the reactivity of the acene with singlet oxygen.21 Other side chains that have been investigated include the alkyl ether side chains which greatly increase photoreactivity,24 making for good oxygen sensing materials. Polymers containing small acene monomers have been shown to be unstable towards photo-oxidation via triplet formation and singlet oxygen sensitization.28 Arrays containing multiple acenes can possess good photostability despite having poor stability towards singlet oxygen due to excited state deactivation.29For simple small molecule systems, the current understanding of acene stability towards photo-oxidation consists of two main conclusions; the low triplet energy of TIPS-pentacene enhances its photostability versus shorter acenes or pentacenes with other substituents,20 and that lowering the HOMO and/or LUMO energies can provide more photostability.21 A lower energy HOMO is thought to decrease the reactivity of the acene with reactive oxygen species, while a lower energy LUMO decreases the driving force for the generation of reactive oxygen species.
Surprisingly, substituent effects elsewhere on the acene core have not been thoroughly explored, despite the fact that they can be used to subtly adjust the molecular orbital energies and optical properties. Thus, we were interested to see how these substituents affected molecular photostability, and whether the HOMO and LUMO energies alone could be used to predict trends in photostability or whether other factors – such as the amount of light being absorbed or the reactivity with photosensitized singlet oxygen – would prove to be more influential.
In the present work, anthradithiophenes (ADT) were chosen to represent a family of acenes which generally do not photodimerise, and instead oxidise across only the central aromatic ring.26 The thiophene rings at the ends of the molecule are readily functionalized during the ADT synthesis, forming a range of related materials with varying orbital energies and absorption profiles. Alkynylation of the central ring of acenes has been a common approach to increasing photostability, as well as providing greater solubility in a range of solvents, and is therefore employed here.30 We recently showed that even the atomic substitution at the end of the ethynyl group affects this photostability,31 such that we include silyl and alkyl ethynyl substituents in our group of molecules. Fluorination has previously been shown to limit photoreaction,32 presumably through altering the molecular orbital energies by electron withdrawal. Therefore, we initially explored the photoreactivity of the anthradithiophene derivatives shown in Fig. 1, all of which were prepared as a mixture of syn and anti isomers, although this is not expected to influence the optical properties of the material.
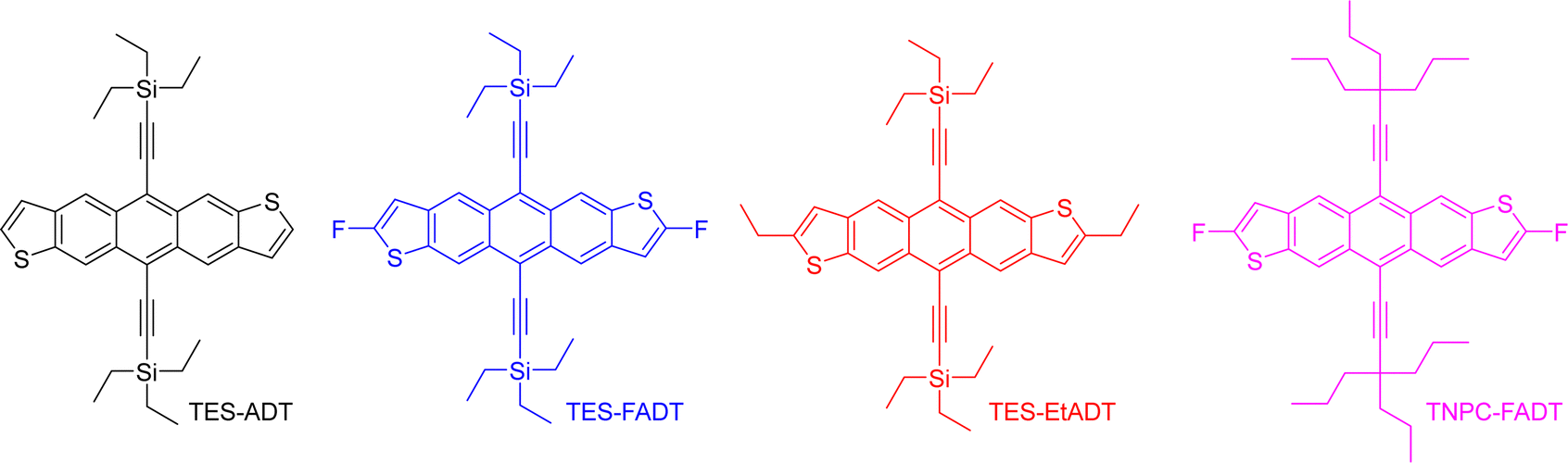 | ||
| Fig. 1 Chemical structures of anthradithiophene derivatives in our initial investigation. | ||
Results and discussion
General photostability: comparison of chromophores
Absorption spectra and extinction coefficients of the ADT derivatives were obtained in dilute solutions in chloroform. Hypsochromic shifts were present for all different substituents relative to the parent TES-ADT, where fluorination had the greatest effect. Time-dependent density functional theory (DFT) calculations revealed that the lowest excitations were purely HOMO−LUMO transitions for each derivative, and excitation wavelengths matched with experimental λmax within 5 nm. Slight changes in extinction coefficient were observed between derivatives, where fluorination increases the absorptivity at λmax the most. Computed oscillator strengths for the lowest energy excitation varied between 0.35 (TES-ADT) and 0.37 (TES-FADT) and integration of the experimental absorption bands resulted in transition dipole moments ranging from 2.0 (TNPC-FADT) to 2.3 (TES-FADT) Debye (Table S1, ESI†). All materials obeyed the Beer–Lambert law over the concentration ranges used in the present study (Fig. S12, ESI†).Further characterization was performed by cyclic voltammetry (Fig. S11, ESI†), where all ADTs exhibited fully reversible oxidation and reduction peaks. To aid the discussion, these potentials were used to estimate HOMO and LUMO levels (Table 1), based on a HOMO energy of −4.8 eV for a ferrocene standard. As might be expected, electron withdrawing fluorine substituents lower the HOMO energy, but the LUMO increases in energy resulting in the blue shifted absorption. This increase is surprising, and might be explained by the competing resonance electron donation and inductive electron withdrawal of fluorine substituents, which may be dependent on the substituent position. The electron donating ethyl groups in TES-EtADT increase both HOMO and LUMO energies, affecting the LUMO slightly more and resulting in only a 5 nm blue shift from TES-ADT. Electronic effects in TNPC-FADT result in a HOMO energy about the same as TES-ADT and a raised energy LUMO, as a result of electron donation from the alkyl ethynyl groups31 and electron withdrawal by fluorine. DFT calculated vertical ionisation potentials and electron affinities agreed well with these findings.
| E HOMO /eV | E LUMO /eV | λ max /nm | ε /M−1 cm−1 | S 0–T1c | |
|---|---|---|---|---|---|
| a Estimated from cyclic voltammetry potentials versus Fc/Fc+ (EHOMO = −4.8 eV). b Absorption maxima of UV-Vis spectra measured in CHCl3 at 298 K at a concentration of 10−5 M. c Calculated using optimally tuned ωB97XD/6-31G* in CHCl3 polarisable continuum. | |||||
| TES-ADT | −5.24 | −3.09 | 555 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
1.32 |
| TES-FADT | −5.35 | −3.07 | 525 | 31500 |
1.41 |
| TES-EtADT | −5.18 | −3.02 | 550 | 26300 |
1.36 |
| TNPC-FADT | −5.25 | −2.92 | 518 | 26500 |
1.44 |
Experimental triplet energies are not readily available for all materials, although TES-FADT and TES-ADT are reported to have an S0–T1 gap of around 1.1 eV by phosphorescence measurement.12 Vertical triplet energies calculated by TD-DFT using the Tamm–Dancoff approximation are slightly larger than this value, ranging from 1.32 eV (TES-ADT) to 1.44 eV (TNPC-FADT). Importantly all triplet energies are expected to be above 1 eV, meaning that the formation of singlet oxygen from ADT T1 states is energetically favourable, and both mechanistic routes (electron transfer or energy transfer) are possible.
Initial comparison of the photostability was probed using a white light source (LED desk lamp), mirroring a typical photostability experiment for new chromophores (Fig. 2). Each experiment was conducted with the same molecular concentration, rather than a normalized absorption. In each case, a decrease in absorption intensity was observed, with no new peaks arising in the visible region. Fluorination provided the most dramatic stabilisation of the ADT chromophore, as previously reported.32 Atomic substitution of the silicon atom with carbon decreased the photostability of the FADT chromophore, although TNPC-FADT was still more photostable than non-fluorinated derivatives. Alkylation also resulted in a more photostable material than TES-ADT despite its electron donating nature, hinting that photostability may not be related only to molecular orbital energies. All endoperoxides exhibit some degree of thermal reversibility,18 although this effect is much slower than the optical degradation of the ADT (approximately 10 times slower, Fig. S14 and S15, ESI†), and so has little effect on the observed photoreaction rates.
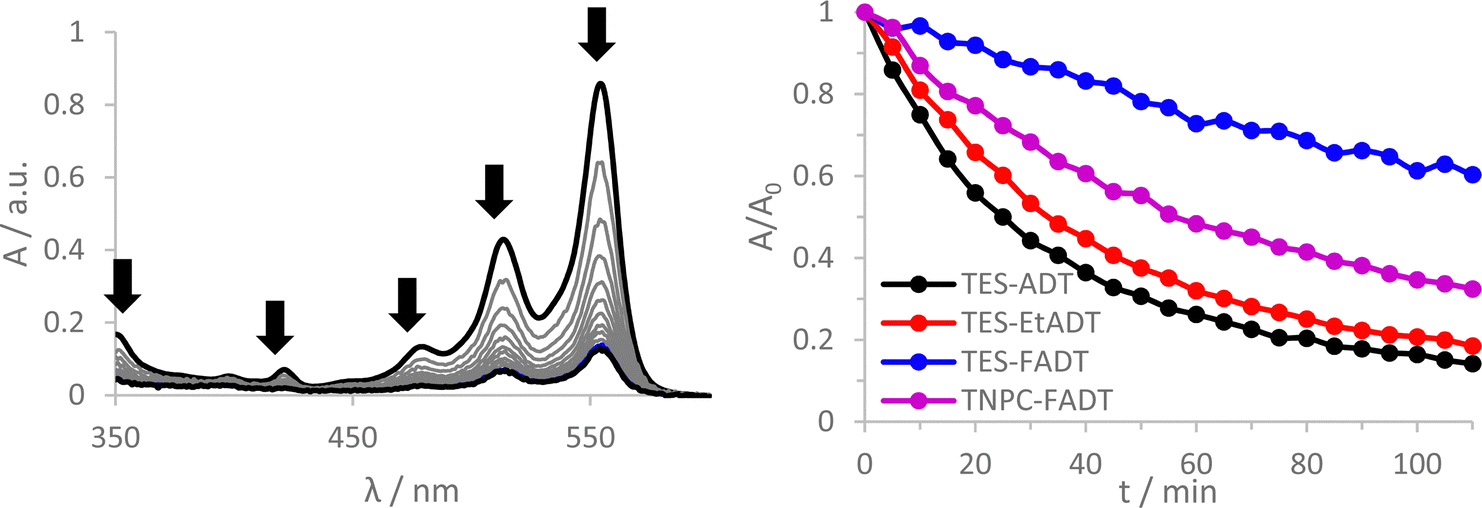 | ||
| Fig. 2 (left) Absorption spectra of TES-ADT in CHCl3 measured at timed intervals while under illumination from an LED desk lamp. (right) Normalised absorbance at λmax for ADTs over time, while under illumination from an LED desk lamp. | ||
One literature explanation of acene photostability is that it is the LUMO energy which drives the formation of reactive oxygen species.20 A higher LUMO energy should provide a greater driving force for electron transfer reactions of the excited state, but the unsubstituted TES-ADT actually has the lowest energy LUMO and the fastest rate of degradation. Likewise, the alternative 1O2 generation by the triplet state should be affected by the triplet energy level. From our computed values, TES-ADT also has the lowest energy triplet state and therefore smallest driving force for 1O2 sensitisation, yet is the least stable by experiment. A further molecular orbital energy argument is that a higher energy HOMO of the ground state ADT will result in faster reaction with the generated reactive oxygen.21 This also does not follow the observed reactivity, where TES-ADT would be expected to be less reactive than TES-EtADT and about the same as TNPC-FADT. Therefore many of the previous assumptions about orbital energies do not seem to clearly define the observed order of ADT photostability.
Spectral overlap
One potential explanation for the observed order of photostability is that the slight shifts in absorption between the different ADTs may affect how much light is absorbed by the chromophores, and therefore how much reactive oxygen can potentially be generated. The measured extinction coefficients (Table 1) vary slightly between derivatives, but when accounting for the width of the absorption bands, each ADT derivative generally absorbs a similar amount of light. Computed oscillator strengths, and measured transition dipole moments and half-width at half-maxima (Table S1, ESI†) are quite similar for each derivative.To investigate the absorbance from a specific light source, a range of LEDs with more focused spectral widths were used. The amount of light being absorbed by each material was estimated by integrating the product of the normalised light source intensity and chromophore extinction coefficient spectra. This accounts for the strength and width of the absorption bands, as well as overlap of the absorbance with the light source emission. These absorptivities were compared to the initial reaction rates, which were extracted from linear fits of absorbance over 10 min under illumination, where absorption was still above 90% of the initial value. Data is reported relative to TES-ADT with the equivalent light source.
With a blue LED, all derivatives show more light absorption than TES-ADT, particularly the fluorinated derivatives and their blue-shifted absorption. Despite this, the initial rates of all derivatives are comparable to or lower than TES-ADT. The rate of photodecomposition of TNPC-FADT is 1.5 times faster than TES-ADT, but the amount of light absorbed is 3 times higher. TES-FADT also absorbs around 3 times more light than TES-ADT, but its initial rate is 60% of TES-ADT. Similar trends are seen using a green LED, except that since the disparity in absorbance is now much less, TNPC-FADT shows a slower reaction rate: its absorbance is 1.7 times higher than TES-ADT while the relative initial rate is 85%.
Use of a white LED with its broader emission profile means that there is less variation in the amount of absorbed light versus TES-ADT. The three derivatives all react more slowly than TES-ADT, and the ordering of reactivity mirrors that seen with longer experiments with a different white light source in Fig. 2. In the case of TES-FADT, the integrated absorbance is almost exactly the same as TES-ADT. However, the initial rate of photo-oxidation is reduced drastically for TES-FADT, resulting in a chromophore that reacts at only 25% of the rate of TES-ADT.
Comparison of reaction rates of the same material with different light sources appears to show that when more light is absorbed, the initial rate is higher. However, the data in Fig. 3 are all relative to the absorbances and rates of TES-ADT. When comparing the different light sources, the absolute intensities of the LEDs are also important, which we were unable to establish reliably. The general trends do indicate that the light source is important, and that it might even change the relative ordering of the photostabilities of the molecular series. For example, if we had used only a white LED we would have concluded that TNPC-FADT is more stable than TES-EtADT. If we had used only a green LED, these two materials would have had very similar reaction rates, and if we had used only the blue LED the reactivities would have been reversed. With the consideration of light absorption, ambient light filters as used in photolithography facilities might aid safe handling of photoreactive acenes in laboratory settings.
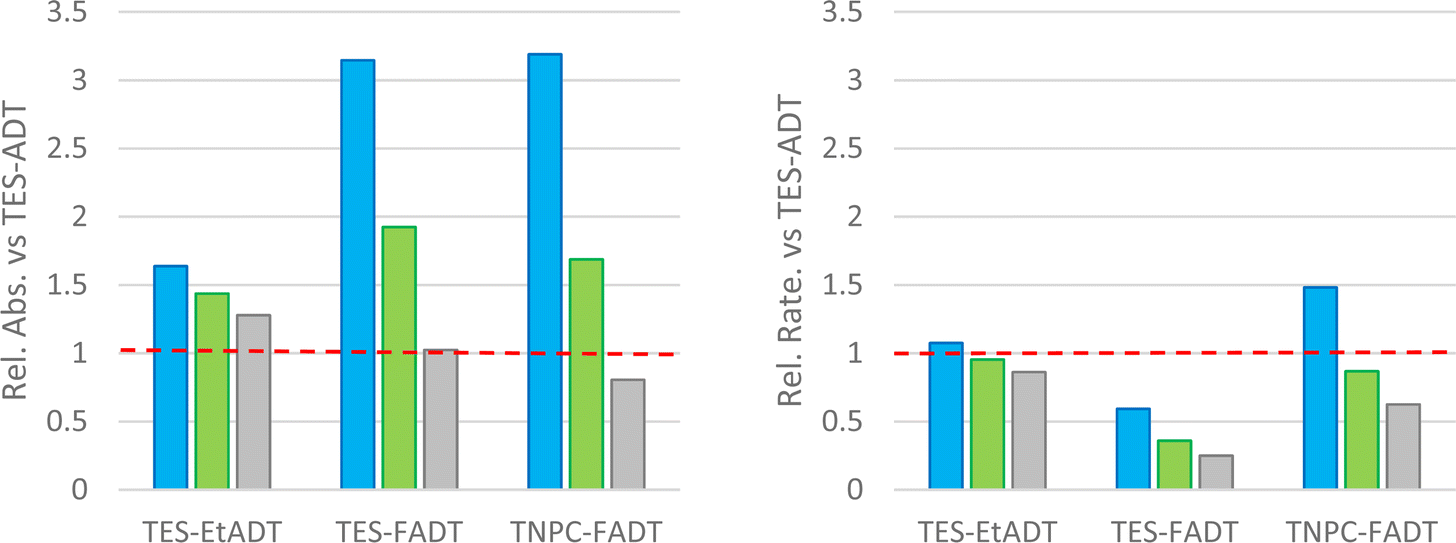 | ||
| Fig. 3 (left) Integrated absorbance of ADTs versus light source spectra relative to the parent TES-ADT. (right) Relative reaction rates for photo-oxidation of ADTs using different coloured light sources, estimated from linear fits of absorbance over 10 minutes where A/A0 > 0.9. All data relative to values determined for parent TES-ADT (depicted as the red line) with the same light source (blue, green, or white LED) and same concentration (3.3 μM in CHCl3). | ||
Concentration dependence
Yeates and co-workers have reported a concentration dependence of the photooxidation of silylethynyl-functionalized pentacene, where more concentrated solutions degrade more slowly.33 In the case of the ADTs investigated here, all show an opposite stability trend. More concentrated solutions become less stable, up to the limits that could be measured reliably by UV-Vis (Fig. 4 and Fig. S27–S30, ESI†). Measurement of initial reaction rates over this concentration range suggest a complex reaction order, where a plot of rate versus concentration fit best to a quadratic fit for TES-ADT (R2 = 0.992 compared to 0.977 for a linear fit) but a linear fit seems more adequate for the other derivatives over these short 10 minute experiments. The different behaviour of TES-ADT versus the other derivatives suggests that perhaps the terminally unsubstituted thiophene may influence the photostability in some way. We observed no evidence of any chemical reaction occurring at this position, through either NMR or absorption spectroscopy, or by mass spectrometry. Oligothiophenes with substituents adjacent to the sulfur atoms of the terminal rings have shown increased oxidative stability in transistor devices operated under air,34 whereas their unsubstituted counterpart's devices showed decrease in mobility of 70% after 100 days under similar conditions.35 These reported end group effects were not due to changes in molecular orbital energies or oxidation potential, which remained near constant between materials. Inclusion of substituents in the thiophene 2-positions seems an important design consideration for thieno-containing opto-electronic materials.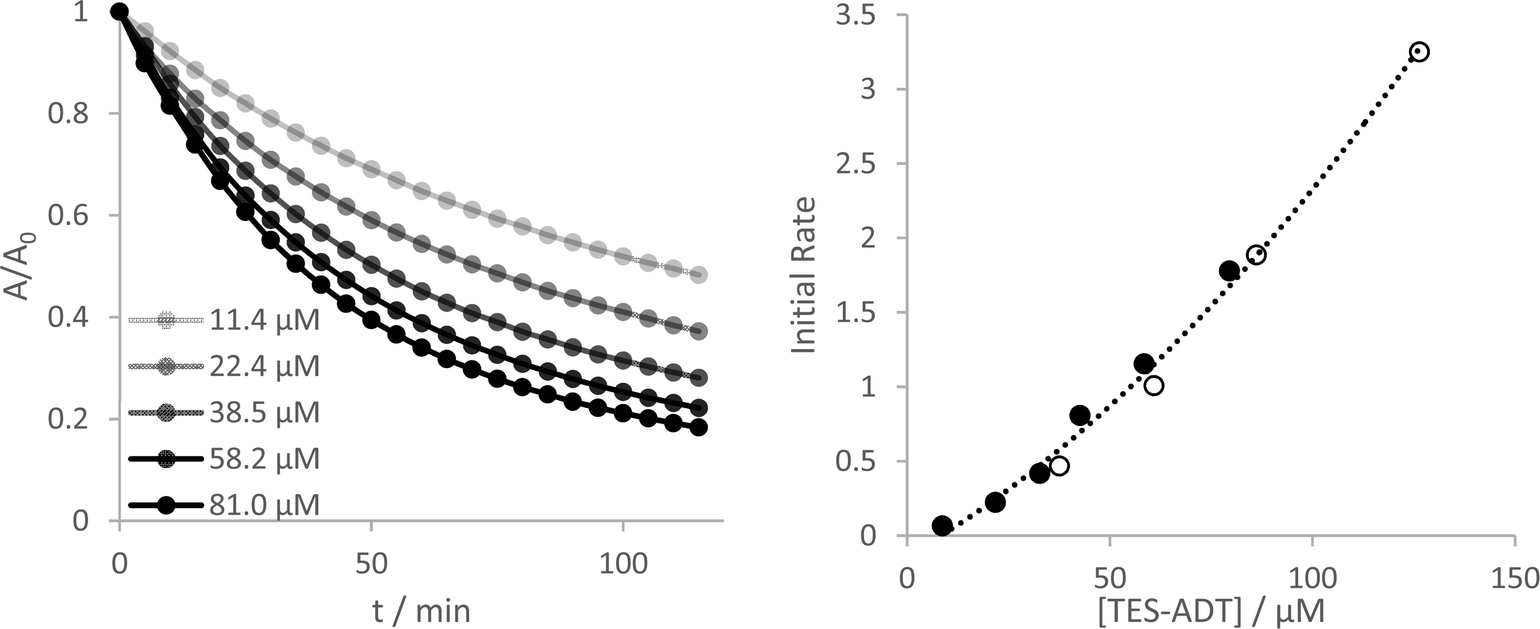 | ||
| Fig. 4 Normalised absorption at 555 nm over time of solutions of TES-ADT in CHCl3 at different concentrations (darker = more concentrated) under illumination by green LED. (right) Plot of initial rate versus initial concentration. Filled circles monitored the first vibronic peak (555 nm) while the open circles were from a repeat experiment using the second vibronic peak (515 nm) allowing higher concentrations without saturating the detector. | ||
Using the original two-hour experimental data, a plot of 1/A versus time provides the best linear fit of any of the rate laws data for all of the ADT derivatives (Fig. S18 and S19, ESI†), indicating a 2nd order reaction as the best approximation to this complex reaction mechanism. Consumption of ADT as the reaction proceeds directly influences the amount of reactive oxygen that can be produced which may not be a straightforward linear relationship.
The simplest explanation for the increasing reactivity with increasing concentration is the increased generation of reactive oxygen from the higher absorbance of the solution, as well as the increased likelihood of successful collisions between an ADT molecule and reactive oxygen before solvent quenching. A singlet fission process has been proposed to explain the opposite trend of decreasing reactivity with increasing concentration in TIPS-pentacene solutions.33 Singlet fission is a bichromophoric phenomenon whereby a singlet excited state on a single molecule is transformed into two triplet states on two molecules. This process has been observed in acenes including pentacene and ADT, made possible by the contingent factor that the triplet state is around half of the energy of the singlet state. Singlet fission has been studied in the solid state, and has also been observed in solution.36 As concentration increases, more molecules encounter one another, singlet fission becomes more likely to occur, and more triplet states can form. In the case of TIPS-pentacene, the triplet energy is around 0.8 eV37 and too low to sensitise 1O2 formation (∼1.0 eV). At the same time, singlet fission removes population of the S1 state and therefore hinders formation of superoxide by electron transfer. ADTs can undergo the same processes but have a higher triplet energy of around 1.3 eV, high enough to sensitise 1O2 formation. With increasing concentration, both TIPS-pentacene and our ADTs form an increased population of triplet states, except that in TIPS-pentacene these are inert and relax back to the ground state, while for ADTs increased amounts of 1O2 are formed resulting in increased EPO formation. The Ostroverkhova group have recently shown oxidation of functionalized tetracenes (whose triplet energies and overall photophysics are similar to TES-ADT) is more rapid from an entangled triplet–triplet state than from the S1 state,38 lending further evidence to the importance of the singlet fission process and photostability. A singlet fission process would lean the reaction mechanism towards singlet oxygen rather than other reactive oxygen species from electron transfer being responsible for the oxidation reaction. While singlet fission is not required to be able to explain the concentration dependence of ADT photostability, analogy with previous studies suggests it might contribute in some way.
The choice of solvent has been previously shown to affect the rate of photo-oxidation of TIPS pentacene, where the maximum oxygen concentration of the solvent was found to be the most important factor. We have also tested the solvent dependence of the photo-oxidation of the ADT derivatives in Fig. 1, where non-chlorinated or aromatic solvents greatly minimise the rate of reaction (Fig. S32, ESI†). Other chloroalkanes such as 1,2-dichloroethane exhibit similar rates of ADT oxidation as chloroform under otherwise identical conditions. These solvents are potentially unstable towards irradiation with homolytic bond fissure occurring, although this typically occurs with much higher energy photons than the LEDs used here produce. The photo-oxidation of anthracene was noted to be particularly fast in such solvents, whereas non-chlorinated solvents showed slow oxidation and competing dimerization.39 The reasoning given was due to increased triplet yield in chlorinated solvent, resulting in more efficient singlet oxygen production and therefore increased endoperoxide formation. We aim to conduct a more thorough investigation of these solvent effects with a range of materials in the near future. It is also worth noting that deuterated chloroform is by far the most common NMR solvent due to its low cost, simple solvent signal, and good solubility of a majority of organic molecules, but it may not be the best choice for characterizing particularly photosensitive materials, and in situ light experiments may be influenced by the choice of solvent. The choice of solvent does not appear to change the conclusions of this study; repeating the illumination experiments in toluene showed an overall slowing for all derivatives, but the order of reactivity remained unchanged (Fig. S31, ESI†).
NMR characterization
1H NMR spectroscopy provided vital information on the photodecomposition of the ADT derivatives. Despite the great increase in ADT concentration, the reactions proceeded surprisingly slowly, perhaps due to decreased light penetration into the sample. Nevertheless, using the light sources used for the UV-Vis experiments, the same product was produced regardless of irradiation wavelength. Literature precedence points towards endoperoxide formation rather than photodimerization, with new peaks appearing at 7.4, 7.5, 8.1 and 8.2 ppm (blue lines in Fig. 5) matching closely to other reported endoperoxides. ADT oxidation products were identified by inclusion of 0.1 equivalents of the well-known singlet oxygen sensitizer methylene blue40 and irradiation with red light. Methylene blue has been previously used in photosensitisation experiments to explore endoperoxide formation in acenes.26 The main absorption band of methylene blue is lower in energy than the ADT chromophores, meaning that methylene blue can be selectively excited by a red LED with the absence of any ADT absorption. Chemical shifts of peaks arising from in situ1O2 generation match those from direct irradiation of the ADT (Fig. 5b and c, blue lines), discounting the possibility of photodimerisation as a decomposition pathway. The other potential product is the ADT quinone caused by decomposition of the endoperoxide. Ethynyl substituents on the central ring tend to disfavour this decomposition route,21 which was ruled out by comparison of spectra of the photoreaction and the sparingly soluble ADT quinone used to synthesise our materials. 1H NMR also tells us that the endoperoxide is formed exclusively across the middle ring, given the product retains the symmetry of the parent ADT. The same photoproduct is also formed when TES-ADT solution in toluene was irradiated with white light (Fig. S43, ESI†). The NMR data for TES-ADT photoproducts are consistent with the other derivatives and with the expected endoperoxide product, discounting any chemical reaction at the unsubstituted 2-position of the thiophene ring. Upon leaving the NMR samples in the dark at room temperature, some of the endoperoxide reverted to the starting ethynyl ADT, showing that the reaction is (slowly) thermally reversible, as observed in UV-Vis experiments.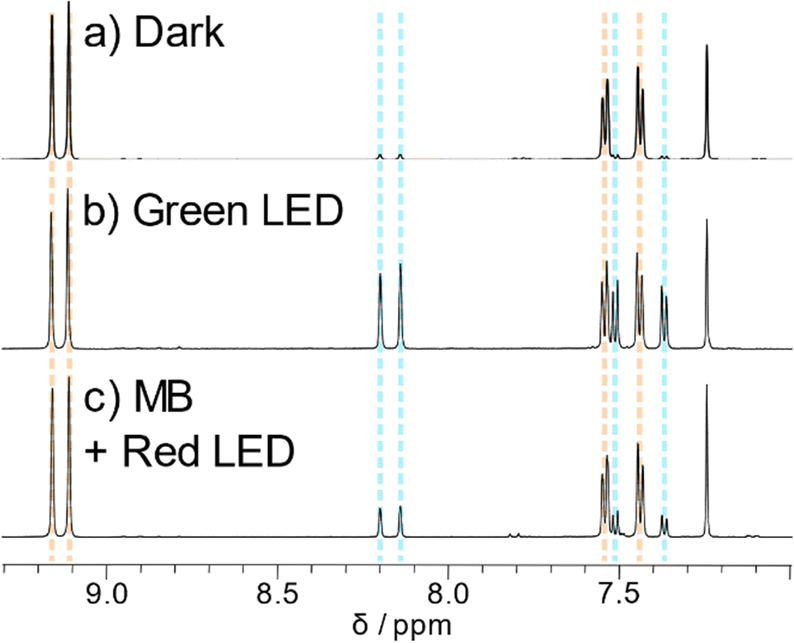 | ||
| Fig. 5 Aromatic region of 1H NMR spectra of TES-ADT (a) in the dark, (b) after white light irradiation and (c) with methylene blue and red light irradiation. All spectra measured in CDCl3 at 298 K. | ||
Reactivity of ADTs with 1O2: sensitisation by methylene blue
UV-Vis studies of the ADT/methylene blue mixture were used to discover trends in chemical reactivity between 1O2 and ground state ADT. Initial reaction rates from linear fits of absorbance are presented as the bar chart in Fig. 6. TES-FADT reacts the slowest with 1O2 while TES-ADT the fastest, mirroring their relative stabilities under white light illumination in Fig. 2. In comparison to the relatively good photostability of the TNPC-FADT under white light, its reaction in the presence of methylene blue is faster than expected, becoming less stable than TES-EtADT. In order to rationalize the observed reactivity of ADT derivatives with methylene blue sensitized singlet oxygen, we computed reaction paths using DFT (M06-2X/cc-pVDZ) by elongation of the carbon–oxygen bonds being formed at the central ring (Fig. S38–S40, ESI†). Reaction enthalpies (Fig. 6 dotted line, becoming less negative) and activation energies (Fig. 6 dashed line, becoming larger) both follow the same order: TES-ADT, TNPC-FADT, TES-EtADT, and TES-FADT. Thus, TES-ADT has the largest driving force for endoperoxide formation and also the lowest barrier for reaction to occur. The Bell–Evans–Polanyi principle can be applied to yield alternative activation energies (blue data points in Fig. 6) which are slightly larger than those from DFT reaction path calculations but show the same trends between derivatives. Comparing the computed thermodynamic data with the experimental relative reaction rates shows a good agreement, and the correlation with the computed activation energies suggests that there is little mechanistic difference between the differently substituted ADTs. This thermodynamic data provides a better guideline of reactivity than orbital energies alone for these systems.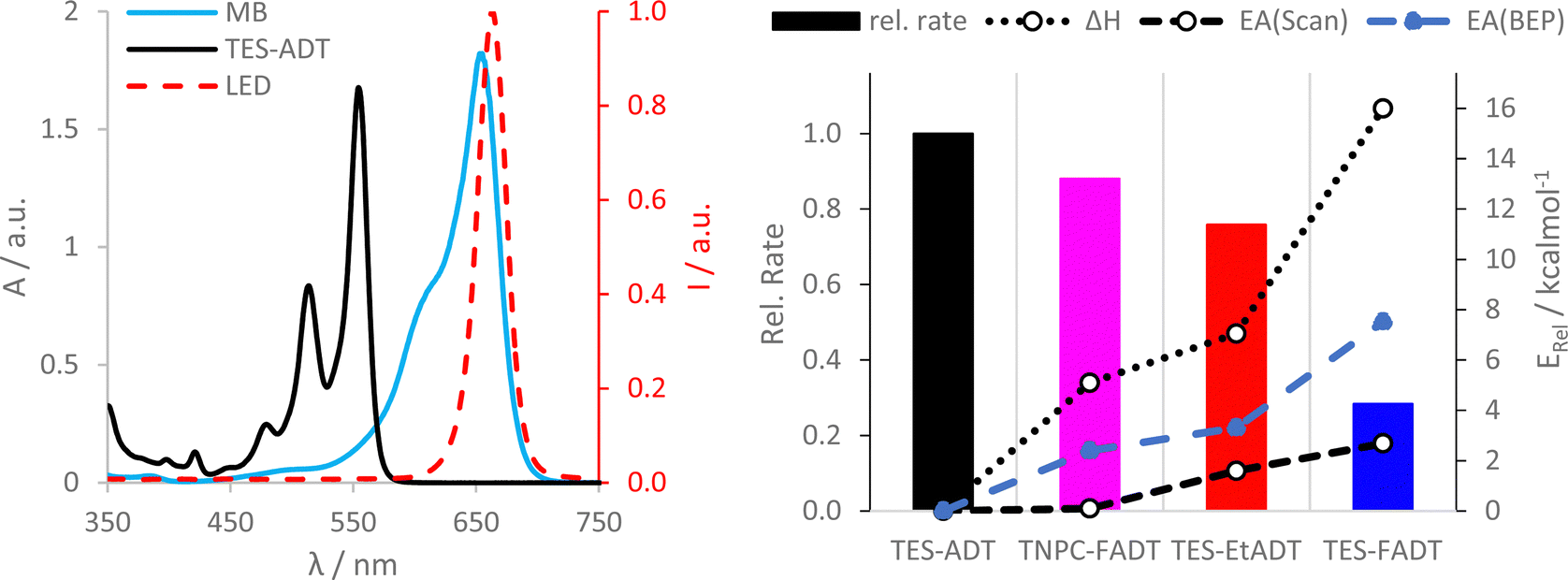 | ||
| Fig. 6 Photosensitised oxidation of ADT derivatives using methylene blue in CHCl3. (left) Absorption spectra of TES-ADT and methylene blue, and intensity spectrum of red LED. (bottom) Relative rate of endoperoxide formation of ADT derivatives (versus TES-ADT, left axis) and relative computed thermodynamic parameters (reaction enthalpy and activation energy versus TES-ADT, right axis). | ||
The reaction between TES-ADT and 1O2 was found to be concentration dependent (Fig. S41, ESI†). At constant TES-ADT concentration, increasing the amount of methylene blue (and therefore the amount of 1O2) results in a linear increase of initial reaction rate. For a constant methylene blue concentration, increasing the amount of TES-ADT also linearly increases reaction rate. This indicates that the (initial) rate is first order in ADT and first order in 1O2 with the dependence on ADT concentration being the stronger effect. In the case where 1O2 is being generated by the ADT itself, this might appear as an approximate 2nd order reaction. Fitting longer 2-hour experiments to a first order reaction (Fig. S36 and S37, ESI†) provides excellent fits for TES-FADT, TES-EtADT and TNPC-FADT, although TES-ADT deviates from linearity with a lower R2 of 0.97. This is in agreement with the observed differences in concentration dependence for TES-ADT versus the 2-functionalised derivatives (Fig. 4 and S27–S30, ESI†), and suggests any differences in concentration dependence come from the reaction of ROS with ground state TES-ADT rather than the generation of the ROS.
The relative rates of reaction of the different ADTs differ slightly from the direct excitation experiments, probably due to the differences in absorbed light and excited state dynamics, but this experiment suggests that perhaps the main factor in the photostability of acenes is the reactivity of the photogenerated reactive oxygen with ground state acene, a property that can be screened computationally.
Computational screening: what matters most?
From the previous discussion on the photo-oxidation mechanism, we have determined that a variety of factors are all important in determining photostability, namely the wavelength specific light absorption, concentration, and reaction enthalpies for endoperoxide formation. We were able to quickly screen numerous derivatives through DFT calculations to identify functionalized ADTs that could add to our knowledge of the photoreaction in question. For each material, we calculated the optoelectronic properties (ionization potential, electron affinity, and excitation to both singlet and triplet states, Fig. S52–S55, ESI†), and calculated the reaction enthalpy for endoperoxide formation (Fig. S56, ESI†).We selected two derivatives from this screening to use in photostability experiments. Cyano ADTs have previously been synthesized as acceptor materials in photovoltaics,41 while novel methoxy substituted ADTs were synthesised as outlined in the ESI.† TIPS-CNADT should have exceptionally low energy frontier molecular orbitals, and so the formation of reactive oxygen species should be minimised. However, the reaction of singlet oxygen with ground state TIPS-CNADT is predicted to be very favourable. In contrast, TES-OMeADT has high energy frontier orbitals and should therefore easily generate reactive oxygen, but the reaction enthalpy for endoperoxide formation is small in comparison to other ADT derivatives (despite a higher lying HOMO). Oxidation and reduction potentials measured by cyclic voltammetry (Fig. S11, ESI†) agreed with these computational estimates of molecular orbital energies.
From previous literature, we might expect that the cyano-ADT should be more stable due to its low orbital energies. Irradiation with white light (Fig. 7) shows the opposite; TIPS-CNADT is almost as reactive as TES-ADT, where the computed oxidation reaction enthalpies are similar to each other (−174 kJ mol−1 for TES-CNADT versus −171 kJ mol−1 for TES-ADT). On the other hand, the observed photostability of TES-OMeADT is more comparable to TES-FADT despite the high energy molecular orbitals. Instead, it is the low driving force for endoperoxide formation that determines the relatively good stability of the ether functionalized ADT. By integration of absorbance with respect to light source, TES-OMeADT does absorb less of the white light than TIPS-CNADT, but even at increased concentration TES-OMeADT remains vastly more stable. Comparison of the two derivatives by other means (e.g. sensitization or differing light source) is not feasible due to large differences in absorption profile. TIPS-CNADT (λmax = 583 nm) is red shifted from TES-ADT while TES-OMeADT is blue shifted (λmax = 535 nm). The results from this experiment were reproduced using toluene as the solvent (Fig. S34, ESI†). 1H NMR spectroscopy showed only one photoproduct in each case (Fig. S47 and S48, ESI†), strongly resembling the other endoperoxides. The surprising order of photostability indicates that the thermodynamics of endoperoxide formation are more important than individual orbital energies or absorptivities.
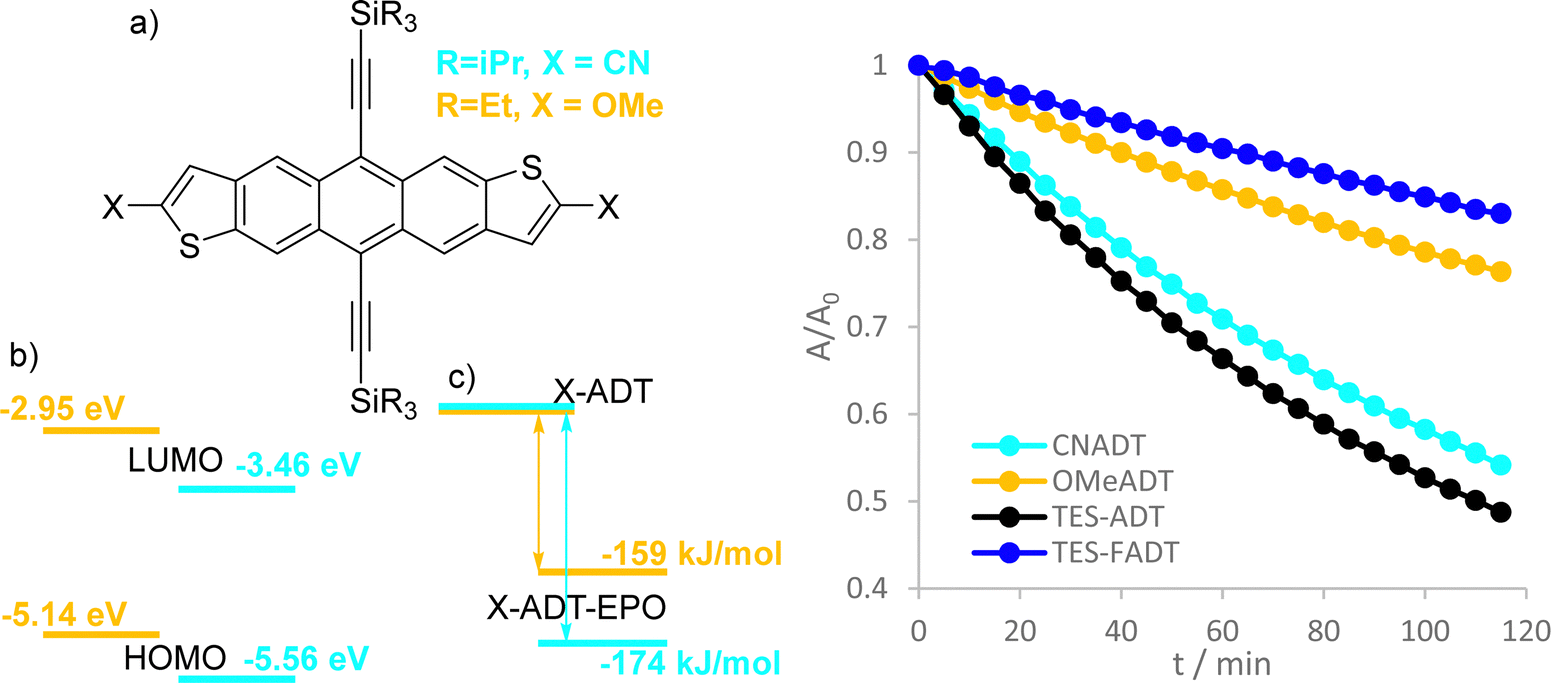 | ||
| Fig. 7 (a) Structure of TES-OMeADT (orange) and TIPS-CNADT (cyan), (b) molecular orbital energies estimated from cyclic voltammetry, (c) computed reaction enthalpies for endoperoxide formation, (d) normalised absorbance of TIPS-CNADT and TES-OMeADT in CHCl3 at 33 μM under irradiation by white LED. TES-ADT and TES-FADT (same conditions) are shown for comparison. | ||
We also computationally screened ADT cores with different substituents in various positions to determine any structure-property relationships for the endoperoxide formation reaction. The effects of the positions of electron donating and withdrawing substituents on frontier molecular orbital energies of acenes have previously been reported,42 whereas here we have concentrated on calculation of the reaction energetics. Removing the alkynyl groups made it plausible to run reaction path scans on these derivatives to estimate reaction enthalpies and activation energies by substituent identity and position (Fig. 8 and S57, ESI†).
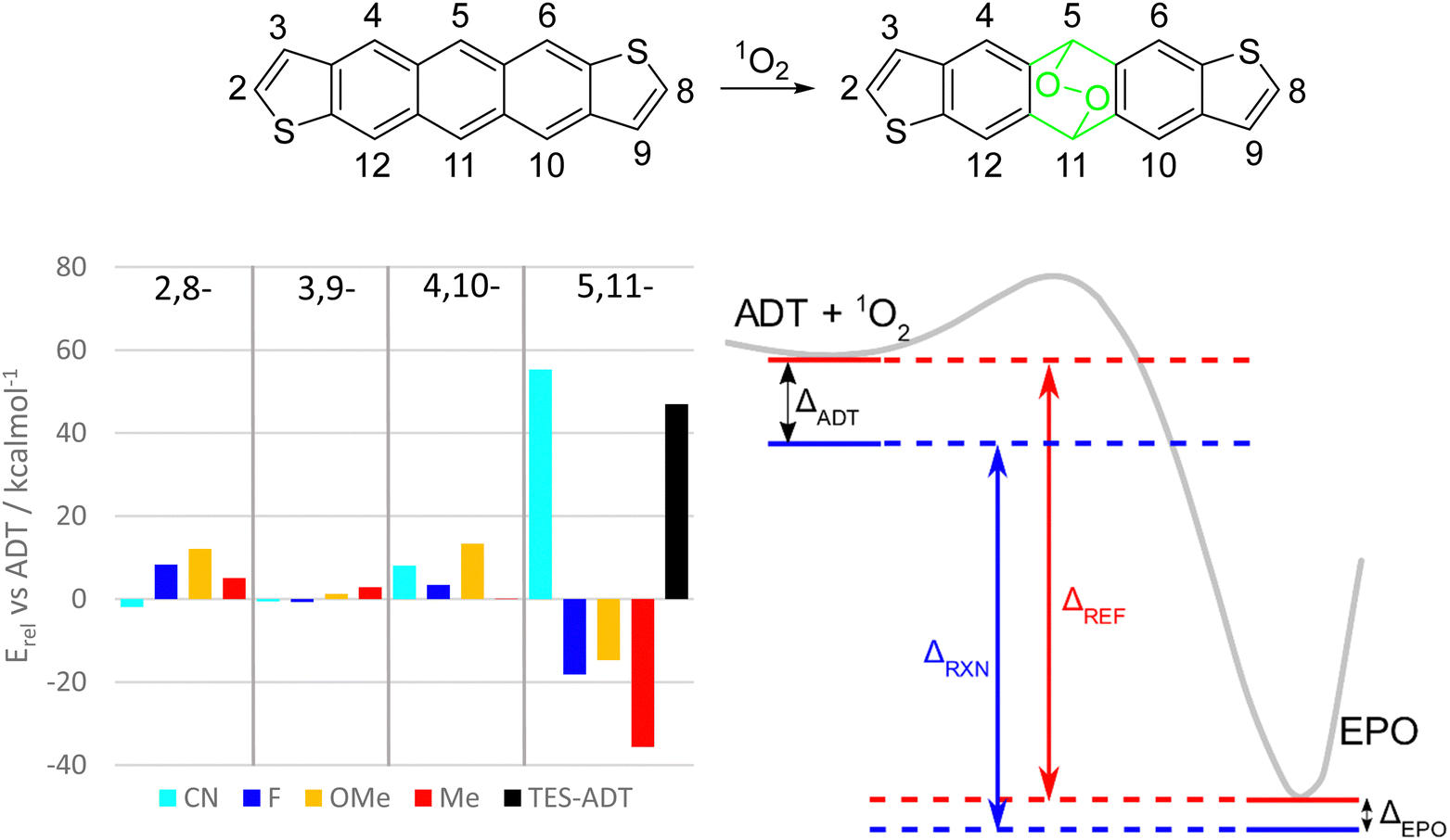 | ||
| Fig. 8 (left) Computed reaction enthalpies for endoperoxide formation from difunctionalised ADT derivatives with substituents in varying position around the acene periphery. All energies relative to unsubstituted ADT, where positive values indicate greater stability. Green atoms in the chemical structure denote those which are not part of the conjugated π-system. (right) Reaction path diagram indicating how relative stabilisation of ADT and EPO (black arrows) affects the overall reaction enthalpy (red and blue arrows). | ||
In general, any substituents at the 2,8-, 3,9-, or 4,10-positions tend to increase the activation barrier while decreasing the driving force for endoperoxide formation. Of these, the best position to functionalise to gain photostability depends on the substituent. For fluorine and alkoxy groups, the 2,8-positions on the thiophene rings should enhance the stability the most, whereas nitrile or alkyl groups would be more influential on the benzenoid ring at positions 4 and 10. The 3,9-position of the thiophene seems to have the smallest effect for all substituents. There do not seem to be any universal design strategies to altering the photostability of anthradithiophenes or other acenes. This may be in part due to role of the substituents in (de)stabilizing both the ADT and the endoperoxide simultaneously (Fig. 8, right). If a substituent is having a stabilizing effect on the ADT, it is likely that it also stabilizes the endoperoxide product, and thus the overall reaction enthalpy is dependent on whether these effects are stronger in the ADT or the endoperoxide. Using the terminology in Fig. 8, the reaction enthalpy of a given material ΔRXN can be expressed as the energy change of a reference reaction ΔREF (e.g. unfunctionalized ADT) minus the relative energies of the ADT and endoperoxides (ΔADT and ΔEPO). When the ADT is stabilized to a greater extent than the endoperoxide, as in the depiction in Fig. 8, the reaction enthalpy is decreased and the reaction less favourable.
Consideration of the change in reaction enthalpy as relative (de)stabilization of ADT and endoperoxide is quite analogous to the variation in HOMO, LUMO and optical gap observed in substituted acenes.42 In that case, the substituents either raised or lowered both the HOMO and LUMO together, and changes in optical gap originate from a greater effect on one orbital over the other. A simple comparison of the frontier orbital coefficients revealed which positions have greater influence over the HOMO or the LUMO. This analysis is not applicable to the present study since resonance and inductive effects influence the energies of all molecular orbitals within the molecule, as well as potentially altering the geometric structure of the acene core, and therefore the relative energies of the ADT and ADT-endoperoxide as a whole.
Substituents at the 5,11-positions have the most dramatic effects, since electronic (de)stabilization only affects the ADT starting material, while a loss of conjugation in the central ring largely removes these effects in the endoperoxide as shown by the green atoms in Fig. 8. For example, a substituent which stabilizes ADT by resonance effects at the central ring will lower the energy of the ADT side of the reaction path (ΔADT can be large), but the resulting endoperoxide will stay about the same (relative) energy as for the unfunctionalized ADT (ΔEPO is small). This leads to a smaller reaction enthalpy ΔRXN and less of a driving force for the reaction to proceed.
Inductive and resonance effects are both lost with this break in the π-system, but it appears the loss of extension of conjugation has the strongest effect, as seen with nitrile and alkynyl functional groups. Fluorine and alkoxy substituents at this position are expected to result in much less stable materials due to competing electron donation by resonance and electron withdrawal by induction. Nitrile groups in this position should greatly stabilize ADT towards photo-oxidation, even more so than alkynylation due to increased electron withdrawing ability. Tetracenes and pentacenes with similar cyano-substitution showed increased stability compared to the unfunctionalized parent acenes.43 Both nitrile and alkynyl groups extend the conjugated system of ADT by a few π bonds, which might be the key to their success in stabilizing against photo-oxidation. Comparison of total electronic energies of the different EPO isomers (see Table S2, ESI†) also suggest that only the nitrile groups serve as stabilizing groups.
A comparison of these findings with the substituent effects reported by Linker21 are interesting. In that study, the reaction rate was correlated with the HOMO energies of the arylethynyl derivatives with varying substituent. In general terms, an electron withdrawing group will lower the molecular orbital energies, while also providing an overall molecular stabilization effect. However, this will only be felt by the acene derivative, and not the endoperoxide due to the break in conjugation, resulting in a smaller reaction enthalpy. In this sense, reactivity trends for substituents in the 5,11 positions should also follow the molecular orbital trends (lower energy orbitals = more stable), but this may not be true in other positions where the endoperoxide can also be stabilized by the same substituents.
An apparent driving force for EPO formation is the formation of multiple Clar sextets. Despite this, there is no correlation between benzenoid aromaticity and reaction energy, as assessed by Nuclear Independent Chemical Shift (NICS) calculations (Fig. S58, ESI†). In the ADT materials, the middle ring that undergoes the cycloaddition is the most aromatic.44 The adjoining rings have NICSzz values around −10 ppm in ADT, while these rings appear to become less aromatic after endoperoxide formation. This is likely the consequence of global aromaticity in the acene versus local aromaticity in the endoperoxide. Comparison of the computed reaction enthalpies with the change in aromaticity for the ADT isomers (Fig. S58, ESI†) show no clear correlation. Therefore, while the Clar sextet may still be an explanation for the overall reaction driving force (i.e. ΔREF in Fig. 8), the substituent effects serve to adjust the relative energies of the ADT or EPO, and thus enhance or diminish the individual molecular reactivities.
Future design principles
From a range of experiments, we have investigated the factors that contribute to the photostability of anthradithiophene chromophores. These findings are likely to be applicable to other polycyclic aromatic hydrocarbons, particularly tetracenes which are photophysically quite similar, notwithstanding other contributing factors to photostability such as competing dimerisation reactions and the ability for singlet oxygen sensitisation according to triplet energies of the organic material. Previous studies suggested that molecular orbital energies are responsible for relative photostability trends, but when applied only to substituent effects, there is no clear correlation. Instead the reaction enthalpies for endoperoxide formation offer a better comparison to experimental photostability trends. In materials capable of singlet fission, the triplet energies may also play a larger role than the excited singlet state energies or LUMO level. Our experiments also show the roles of light absorption and concentration, while previous studies have shown solvent dependencies on photostability, so design of future photostability studies should take these factors into account to enable fair comparisons to be drawn. The importance of including any kind of substituent adjacent to sulfur in thiophene rings is also apparent, since TES-ADT was the least stable of the materials tested.In terms of molecular design, a first step in designing new chromophores can come from computational screening. Calculation of reaction enthalpies for the ground state reactions with singlet oxygen can indicate orders of reactivity which mirror the photostability quite well. This type of screening requires only a couple of geometry optimisations followed by comparison of total electronic energies. As well as the type of substituent, its position on the chromophore affects the reaction thermodynamics, which can be more easily obtained in silico than in the wet lab. Substituents on the positions of endoperoxide formation play the largest role in determining the reactivity towards reactive oxygen species (or in fact other dienophiles), while substituents at other positions may be used to augment these effects for more stable optoelectronic materials or more sensitive sensor materials. Through screening more and more materials, trends and explanations for the relative stabilities will become more apparent.
Conclusions
We have studied the photoreaction of various anthradithiophene derivatives resulting in the formation of endoperoxides. Excited state ADT reacts with molecular oxygen to form singlet oxygen or other reactive oxygen species, which then react with a ground state ADT molecule. Through sensitisation experiments and concentration dependence we found that this reaction should be first order with respect to ADT and first order with respect to singlet oxygen, which is itself related to the concentration of the absorbing material (ADT or an additional photosensitiser). Under different illumination conditions, the relative photostability trends between molecules could be altered, but the thermodynamics of the endoperoxide formation seemed to have the strongest influence. We have demonstrated that this can be easily predicted by computation, where a fine balancing act between substituent effects in the ADT versus the endoperoxide determines the relative photostability. This is especially important when substituents reside at the positions of endoperoxide formation, since conjugation effects are largely negated in the endoperoxide product, thus the substituent influences reaction enthalpies more than in other positions. Previous guidelines suggested low energy molecular orbitals minimised photoreaction, but our present study shows that these energies themselves are not that important, but rather the reaction thermodynamics play a greater role.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
JEA and KJT thank the National Science Foundation's DMREF program (DMR-1627428) for supporting the computational design of organic semiconductors.References
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- T. Salzillo, F. D’Amico, N. Montes, R. Pfattner and M. Mas-Torrent, CrystEngComm, 2021, 23, 1043–1051 RSC.
- M. Nakano, K. Niimi, E. Miyazaki, I. Osaka and K. Takimiya, J. Org. Chem., 2012, 77, 8099–8111 CrossRef CAS.
- D. Lehnherr, A. R. Waterloo, K. P. Goetz, M. M. Payne, F. Hampel, J. E. Anthony, O. D. Jurchescu and R. R. Tykwinski, Org. Lett., 2012, 14, 3660–3663 CrossRef CAS PubMed.
- B. Tylleman, C. M. L. Vande Velde, J.-Y. Balandier, S. Stas, S. Sergeyev and Y. H. Geerts, Org. Lett., 2011, 13, 5208–5211 CrossRef CAS PubMed.
- J.-Y. Balandier, F. Quist, S. Stas, B. Tylleman, C. Ragoen, A. Mayence, S. Bouzakraoui, J. Cornil and Y. H. Geerts, Org. Lett., 2011, 13, 548–551 CrossRef CAS PubMed.
- M. Mamada, H. Katagiri, M. Mizukami, K. Honda, T. Minamiki, R. Teraoka, T. Uemura and S. Tokito, ACS Appl. Mater. Interfaces, 2013, 5, 9670–9677 CrossRef CAS PubMed.
- O. D. Jurchescu, S. Subramanian, R. J. Kline, S. D. Hudson, J. E. Anthony, T. N. Jackson and D. J. Gundlach, Chem. Mater., 2008, 20, 6733–6737 CrossRef CAS.
- M. J. Sung, J. Hong, H. Cha, Y. Jiang, C. E. Park, J. R. Durrant, T. K. An, S. Kwon and Y. Kim, Chem. – Eur. J., 2019, 25, 12316–12324 CrossRef CAS.
- Y. Shu, Y. F. Lim, Z. Li, B. Purushothaman, R. Hallani, J. E. Kim, S. R. Parkin, G. G. Malliaras and J. E. Anthony, Chem. Sci., 2011, 2, 363–368 RSC.
- C. K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8(1), 1–12 Search PubMed.
- D. Tian and Y. Chen, Adv. Opt. Mater., 2021, 9, 2002264 CrossRef CAS.
- Y. S. Chung, N. Shin, J. Kang, Y. Jo, V. M. Prabhu, S. K. Satija, R. J. Kline, D. M. DeLongchamp, M. F. Toney, M. A. Loth, B. Purushothaman, J. E. Anthony and D. Y. Yoon, J. Am. Chem. Soc., 2011, 133, 412–415 CrossRef CAS.
- P. Coppo and S. G. Yeates, Adv. Mater., 2005, 17, 3001–3005 CrossRef CAS.
- W. Fudickar and T. Linker, J. Org. Chem., 2017, 82, 9258–9262 CrossRef CAS.
- V. Brega, Y. Yan and S. W. Thomas, Org. Biomol. Chem., 2020, 18, 9191–9209 RSC.
- W. Fudickar and T. Linker, Chem. – Eur. J., 2011, 17, 13661–13664 CrossRef CAS PubMed.
- J. Deckers, T. Cardeynaels, S. Doria, N. Tumanov, A. Lapini, A. Ethirajan, M. Ameloot, J. Wouters, M. di Donato, B. R. Champagne and W. Maes, J. Mater. Chem. C, 2022, 10, 9344–9355 RSC.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980–4986 CrossRef CAS.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed.
- S. H. Chien, M. F. Cheng, K. C. Lau and W. K. Li, J. Phys. Chem. A, 2005, 109, 7509–7518 CrossRef CAS.
- A. R. Reddy and M. Bendikov, Chem. Commun., 2006, 1179–1181 RSC.
- V. Brega, S. N. Kanari, C. T. Doherty, D. Che, S. A. Sharber and S. W. Thomas, Chem. – Eur. J., 2019, 25, 10400–10407 CrossRef CAS PubMed.
- A. Schönberg, Trans. Faraday Soc., 1936, 32, 514–521 RSC.
- J. Zhang, Z. C. Smith and S. W. Thomas, J. Org. Chem., 2014, 79, 10081–10093 CrossRef CAS PubMed.
- J. Zhang, S. Sarrafpour, T. E. Haas, P. Müller and S. W. Thomas, J. Mater. Chem., 2012, 22, 6182–6189 RSC.
- Y. W. Soon, H. Cho, J. Low, H. Bronstein, I. Mc Cullocha and J. R. Durrant, Chem. Commun., 2013, 49, 1291–1293 RSC.
- S. W. Thomas, Y. Yan, Z. A. Lamport and I. Kymissis, J. Org. Chem., 2020, 85, 12731–12739 CrossRef.
- M. M. Payne, S. R. Parkin, J. E. Anthony, C. C. Kuo and T. N. Jackson, J. Am. Chem. Soc., 2005, 127, 4986–4987 CrossRef CAS.
- K. J. Thorley, M. Benford, Y. Song, S. R. Parkin, C. Risko and J. E. Anthony, Mater. Adv., 2021, 2, 5415–5421 RSC.
- S. Subramanian, K. P. Sung, S. R. Parkin, V. Podzorov, T. N. Jackson and J. E. Anthony, J. Am. Chem. Soc., 2008, 130, 2706–2707 CrossRef CAS PubMed.
- L. Abu-Sen, J. J. Morrison, A. B. Horn and S. G. Yeates, Adv. Opt. Mater., 2014, 2, 636–640 CrossRef CAS.
- C. E. Mauldin, K. Puntambekar, A. R. Murphy, F. Liao, V. Subramanian, J. M. J. Fréchet, D. M. DeLongchamp, D. A. Fischer and M. F. Toney, Chem. Mater., 2009, 21, 1927–1938 CrossRef CAS.
- C. Videlot-Ackermann, J. Ackermann, H. Brisset, K. Kawamura, N. Yoshimoto, P. Raynal, A. el Kassmi and F. Fages, J. Am. Chem. Soc., 2005, 127, 16346–16347 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- R. Puro, J. D. B. Van Schenck, R. Center, E. K. Holland, J. E. Anthony and O. Ostroverkhova, J. Phys. Chem. C, 2021, 125, 27072–27083 CrossRef CAS.
- E. J. Bowen, Discuss. Faraday Soc., 1953, 14, 143–146 RSC.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS PubMed.
- M. J. Kendrick, A. Neunzert, M. M. Payne, B. Purushothaman, B. D. Rose, J. E. Anthony, M. M. Haley and O. Ostroverkhova, J. Phys. Chem. C, 2012, 116, 18108–18116 CrossRef CAS.
- B. M. Medina, J. E. Anthony and J. Gierschner, ChemPhysChem, 2008, 9, 1519–1523 CrossRef CAS PubMed.
- S. Katsuta, D. Miyagi, H. Yamada, T. Okujima, S. Mori, K. I. Nakayama and H. Uno, Org. Lett., 2011, 13, 1454–1457 CrossRef CAS PubMed.
- B. J. Lampkin, P. B. Karadakov and B. VanVeller, Angew. Chem., Int. Ed., 2020, 59, 19275–19281 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc02922g |
| This journal is © The Royal Society of Chemistry 2022 |