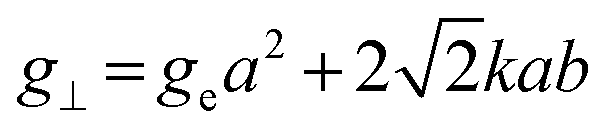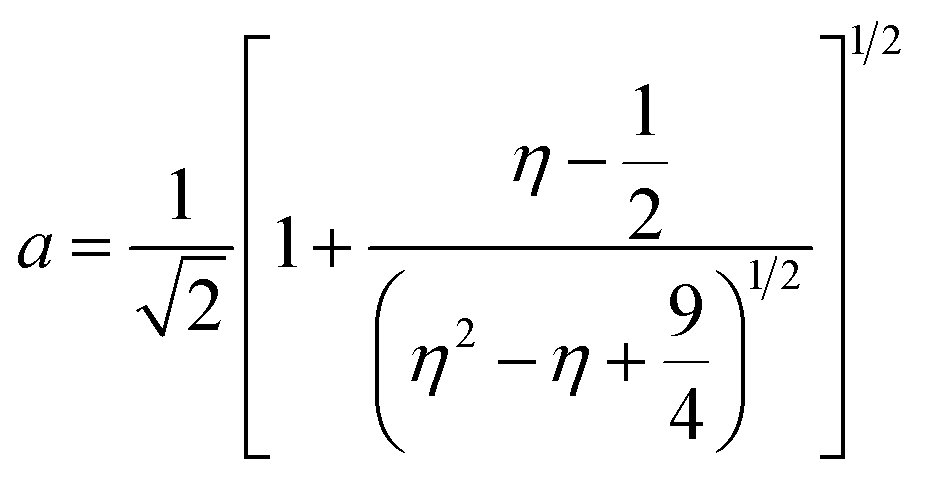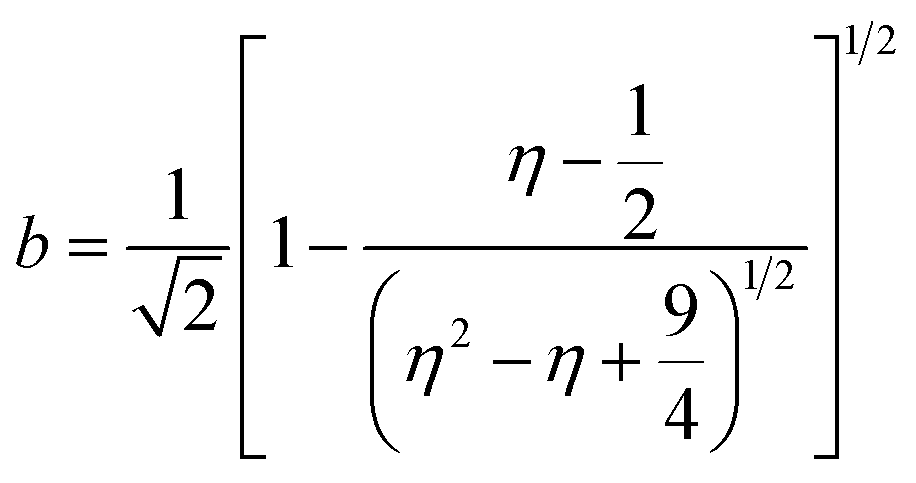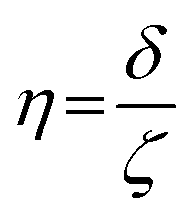DOI:
10.1039/D1TA09238C
(Paper)
J. Mater. Chem. A, 2022,
10, 5953-5961
Redispersion strategy for high-loading carbon-supported metal catalysts with controlled nuclearity†
Received
27th October 2021
, Accepted 21st November 2021
First published on 22nd November 2021
Abstract
Supported low-nuclearity metal catalysts integrating single atoms or small clusters have emerged as promising materials for diverse applications. While sophisticated synthetic methods provide a high level of nuclearity control in the subnanometre regime, these routes do not fulfil the requirements for translation into industrial practice of (i) effectiveness for high metal contents and (ii) facile scalability. Herein, we present a gas-phase redispersion strategy consisting of sequential C2H2 and HCl treatments to gradually disperse Ru, Rh and Ir nanoparticles supported on commercial activated carbon with metal content up to 10 wt% and initial average sizes of ≈ 1 nm into small clusters and eventually single atoms. Avoidance of nanoparticle surface overchlorination, which hinders C2H2 adsorption, is identified as key for the redispersion process, as demonstrated by the inefficacy of both C2H2–HCl cofeeding and inverse sequence (i.e., HCl first) treatments. Precise size control (±0.1 nm) is enabled by regulating the number of C2H2–HCl cycles. Detailed characterisation by X-ray absorption spectroscopy, electron paramagnetic resonance and time-resolved mass spectrometry reveals that the redispersion occurs via a layer-by-layer mechanism. Specifically, the migration of surface chlorinated metal species to the carbon support is induced by the C2H2 treatment, depleting accessible surface Cl atoms, while the subsequent HCl treatment rechlorinates the cluster surface. The strategy paves the way for the generation of high-density metal sites with tuneable nuclearity for tailored applications.
1. Introduction
Commercial heterogeneous catalysts frequently consist of supported highly dispersed metal species, targeting the maximal utilisation of the active component.1 Aiming at full participation of metal nuclei in the catalytic cycle, research efforts have been directed towards low-nuclearity species, ranging from supported clusters of at most a few tens of atoms to single-atom catalysts (SACs).2 Nanoscale engineering at the atomic level has demonstrated that the addition or removal of metal atoms to the active ensembles leads to distinct electronic properties and non-linear variation of reactivity patterns, often decisive to fulfil the catalytic cycle of the targeted application.2–5 However, the exploitation of the unique reactivity of low-nuclearity catalysts in industrial practice calls for the development of scalable synthetic methods with control over speciation and nuclearity in the subnanometre regime. To this end, a number of strategies has been developed for supported clusters with controlled nuclearity including selected-precursor deposition,6–8 support engineering,9 wet chemical reduction10 and dendrimer encapsulation.11 Nevertheless, these methods face several challenges to avoid formation of large clusters and nanoparticles (NPs), thermodynamically favoured over low-nuclearity species, at high metal loadings, which are necessary to achieve industrially-suitable density of active sites.2 Recently, remarkable progress has been achieved for the generation of high-loading single-atom catalysts (SACs). In particular, research efforts have been directed towards carbon supports, owing to their well-known ability to stabilise metal sites and tune their coordination environment.12 Pioneering studies have reported advanced synthetic methods to stabilise isolated atoms with high metal content either (i) during the synthesis of the heteroatom-doped carbon supports via pyrolysis of organic precursors (10–16 wt%)13 or functionalised graphene quantum dots (40 wt%),14 or (ii) by wet chemistry methods exploiting existing hosts (18 wt%).15 These strategies rely on generating suitable anchoring sites via the introduction of heteroatoms, defects and/or vacancies, but their applicability has not been demonstrated on practical supports,16 which would be highly desirable to expand their versatility.
Redispersion methods of metal NPs on practical supports constitute a compelling alternative, simultaneously offering a potential route to precisely tune metal species nuclearity in the subnanometre regime until full atomic dispersion is achieved.17 Several studies have demonstrated the efficacy of halogen-mediated strategies, namely chlorination, oxychlorination and halohydrocarbon-mediated treatments, generating MOxCly or MCly species that redisperse via interaction with the support.18 State-of-the-art studies have reported encouraging results on the efficacy of these strategies on metal loadings up to 5 wt% by synergistically coupling halohydrocarbons with compounds presenting lone pair electrons (e.g., CO, NO or O2), promoting the redispersion of the metal clusters by weakening the surface atom bonds to the cluster bulk via electron transfer.19–21 These findings hold promise for exciting prospects, calling for the exploration of other classes of promoters for halogen-mediated strategies, among which mildly-reducing compounds emerge as potentially effective candidates to cleave bonds in metal clusters.22 To this end, a promising pair may be identified in HCl and C2H2, on the basis of literature-reported Ru NPs supported on N-doped carbon that were atomically redispersed under C2H2 hydrochlorination conditions.23 Nevertheless, in that case competitive particle agglomeration was observed, resulting in lack of control over the redispersion process. To date, this remains a critical challenge that, if resolved, could pave the way towards a scalable strategy for the generation of high loading metal catalysts on commercial supports with tuneable nuclearity.
Herein, we present a scalable redispersion strategy, consisting of sequential C2H2 and HCl treatments, to synthesise Ru, Ir and Rh catalysts with metal loading up to 10 wt% on N-doped and commercial activated carbons with controlled metal nuclearity in the subnanometre regime down to atomic dispersion. Fine control over the metal species nuclearity is enabled by regulation of the number of C2H2–HCl cycles. The inefficacy of both the C2H2–HCl cofeeding and inverse sequence (i.e., HCl first) approaches demonstrates the beneficial impact of avoiding NP surface overchlorination, which prevents the adsorption of C2H2. By combination of microscopic, spectroscopic and spectrometric analyses, the redispersion mechanism is revealed to be a layer-by-layer process in which C2H2 prompts the migration of chlorinated metal species from the nanoparticle surface onto local defects in the carbon support, while HCl chlorinates the newly exposed metal atoms. This work presents a potentially industrially-amenable route to generate carbon-supported metal species with tuneable nuclearity for tailored catalytic applications.
2. Experimental
2.1 Metal NP synthesis
Commercial activated carbon (AC, Norit ROX 0.8) was ground and sieved (particle size 0.4–0.6 mm). The N-doped carbon support (NC) was prepared in a two-step synthesis, consisting of an oxidative polymerisation of aniline and a subsequent carbonisation step.24 Aniline (50 mmol, Acros, 99.5%) was dissolved in deionised water (40 cm3, pH 0.4; adjusted by hydrochloric acid, 1.25 M, Sigma Aldrich, >37%) at room temperature, cooled to 277 K, and subsequently added to a precooled solution (277 K) of ammonium persulfate (50 mmol, Acros, 98%) in deionised water (20 cm3). After vigorous manual stirring (5 min), the mixture was kept at room temperature for 24 h to complete the polymerisation process. The formed polyaniline was thoroughly washed with deionised water for neutralisation, dried in vacuum (393 K, heating rate 5 K min−1, hold time 12 h), and afterwards treated at 1073 K (heating rate 5 K min−1, hold time 1 h, static N2). The obtained N-doped carbon, having a N content of 9.7 wt% and a total surface area of SBET = 517 m2 g−1, was ground and sieved (particle size 0.4–0.6 mm). All metal-based catalysts were prepared via an incipient wetness impregnation method with a nominal metal loading of 10 wt%. The desired metal precursor, RuCl3·xH2O (ABCR, 99.9%, 40.0 wt% Ru), IrCl3·xH2O (ABCR, 99.9%, 65 wt% Ir) or RhCl3·xH2O (Acros Organics, 99.9%, 38 wt% Rh), was dissolved in water (1.5 cm3 g−1) and the obtained solution was added dropwise to the selected carbon carrier (AC or NC). Subsequently, the samples were dried (473 K, heating rate 5 K min−1, hold time 12 h, static air) to yield the nanoparticle catalysts, denoted as 10M/AC or 10M/NC. The prereducing treatment was carried out at 573 K (heating rate 10 K min−1, hold time 3 h), flowing pure H2 (PanGas, purity 5.0) at a total volumetric flow of FT = 40 cm3 STP min−1.
2.2 Redispersion treatment
The redispersion treatment was conducted at atmospheric pressure in a continuous-flow fixed-bed micro-reactor (Scheme S1†). The gases C2H2 (PanGas, purity 2.6), HCl (Air Liquide, purity 2.8, anhydrous), Ar (PanGas, purity 5.0, internal standard) and He (PanGas, purity 5.0, carrier gas), were fed using digital mass-flow controllers (Bronkhorst) to the mixing unit, equipped with a pressure indicator. A quartz micro-reactor (10 mm inner diameter) was loaded with the catalyst (Wcat = 0.1 g, particle size 0.4–0.6 mm) and placed in a homemade electrical oven. A K-type thermocouple fixed in a coaxial quartz thermowell with the tip positioned in the centre of the catalyst bed was used to control the temperature during the redispersion treatment. Prior to treatment, the catalyst was heated in a He flow to the desired temperature (T = 473 K) and allowed to stabilise for at least 30 min before the gas mixture was fed (total volumetric flow, FT = 15 cm3 STP min−1). The sequence for each cycle consisted of (i) exposure to C2H2 atmosphere for 1 h (40 vol% C2H2, 44 vol% He and 16 vol% Ar), (ii) He flushing for 5 min (84 vol% He and 16 vol% Ar), (iii) exposure to HCl atmosphere for 1 h (44 vol% HCl, 40 vol% He and 16 vol% Ar) and (iv) He flushing for 5 min (84 vol% He and 16 vol% Ar). The treated samples are denoted as 10M/C X where M: metal, C: “AC” or “NC”, X: # of C2H2–HCl treatment cycles and y: “a” if the cycle terminated after the C2H2 treatment. Time-resolved mass spectroscopy (MS) analysis of the reactor outlet stream was performed with a Pfeiffer Vacuum Thermo-Star GDS 320 T1 MS to monitor the relative concentration of produced vinyl chloride (C2H3Cl, VCM) with m/z 62 and m/z 64. In line with the natural isotopic abundance of 35Cl and 37Cl, m/z 62 and m/z 64 were detected in a characteristic 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
2.3 Characterisation
Inductively coupled plasma optical emission spectrometry (ICP-OES) was conducted using a Horiba Ultra 2 instrument equipped with photomultiplier tube detection. The solids were dissolved in a 3:1-mixture of HNO3 and H2O2 under sonication until the absence of visible solids. Powder X-ray diffraction (XRD) was measured using a PANalytical X'Pert PRO-MPD diffractometer with Cu-Kα radiation (λ = 1.54060 Å). The data was recorded in the 10–70° 2θ range with an angular step size of 0.017° and a counting time of 0.26 s per step. Scanning transmission electron microscopy (STEM) with a high-angle annular dark-field (HAADF) detector was performed on an aberration-corrected HD2700CS (Hitachi) microscope operated at 200 kV. High-resolution transmission electron microscopy (HRTEM) was performed on a double-corrected microscope JEM-ARM300F (GrandARM, JEOL), which was operated at an acceleration potential of Uacc = 300 kV (electron gun: cold-field emitter; ΔE ≈ 0.35 eV). Static volumetric chemisorption of acetylene was performed in a Micromeritics 3Flex Chemi instrument at 303 K. Energy-dispersive X-ray spectroscopy was conducted in a Talos F200X instrument with an FEI SuperX detector at 200 kV. Image frame times ranged from 20 to 40 μs (512 × 512 px). Samples were prepared by dipping the copper grid supporting a holey carbon foil in a suspension of the solid in ethanol and drying in air. The Ir 4f and Rh 3d X-ray photoelectron spectroscopy (XPS) spectra of the Ir and Rh samples were acquired on a Physical Electronics Quantera SXM instrument using monochromatic Al-Kα radiation, generated from an electron beam operated at 15 kV, and equipped with a hemispherical capacitor electron-energy analyser. The samples were analysed at an electron take-off angle of 45° and a constant analyser pass energy of 55 eV. Owing the Ru 3d core levels superimposing the C 1s core level, the Ru speciation was analysed based on Ru 3p XPS spectra.25 The Ru 3p XPS spectra of the Ru samples were acquired on a Physical Electronics Quantum 2000 instrument, where the samples were analysed at an electron take-off angle of 45° and a constant analyser pass energy of 46.95 eV. Both spectrometers were calibrated for the Au 4f7/2 signal at 84.0 ± 0.1 eV. The Ru 3p and Rh 3d XPS spectra were fitted by mixed Gaussian–Lorentzian and the Ir 4f XPS spectra were fitted by Functional Lorentzian component profiles after Shirley background subtraction. The selected peak positions are based on literature data and fixed with an error of ±0.2 eV. The detailed fitting parameters are given in the ESI.† The elemental concentrations were quantified based on the measured photoelectron peak areas (C 1s, N 1s, O 1s, Cl 2p, Ru 3p, Ir 4f and Rh 3d), using the respective photoionisation cross sections as relative sensitivity factors.26 X-ray absorption fine structure (XAFS) measurements were carried out at the SuperXAS beamline of the Swiss Light Source.27 The incident photon beam was provided by a 2.9 T superbend magnet and subsequently collimated using a Pt-coating (for the Ru K edge). The rejection of higher harmonics and focusing were achieved with a Rh-coating (for Ir L3 edge) and a Pt-coating (for Ru K and Rh K edges), respectively. The beamline was calibrated using the respective metal foils. The sample area illuminated by the X-ray beam was 0.5 mm × 0.2 mm. All spectra were recorded in transmission mode at room temperature. The extended X-ray absorption fine structure (EXAFS) spectra were acquired with a 1 Hz frequency (0.5 s per spectrum) and then averaged over 5 min. The resulting raw data were processed using the ProQEXAFS software,28 and the EXAFS spectra were analysed using the Demeter software.29 Hydrogen temperature-programmed reduction analyses (H2-TPR) were performed in a Micromeritics Autochem II 2920 analyser equipped with a thermal conductivity detector (TCD). Peaks in the TCD signal are assigned based on reference values in the literature.30,31 Continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy experiments were performed at 10 K on a Bruker Elexsys E500 spectrometer equipped with an Oxford helium (ESR900) cryostat operating at X-band frequencies (≈9.4 GHz) using a ER4122SHQE Bruker EPR Resonator. All CW EPR spectra were acquired (spectrometer settings: microwave frequency = 9.4 GHz, center field = 330 mT, sweep width = 200 mT, modulation frequency = 100 kHz, microwave power = 207 μW, power attenuation = 30 dB, conversion time = 327.68 ms, time constant = 81.92 ms), while all measured g-factors were offset-corrected against a known standard (i.e., free radical 1,1-diphenyl-2-picrylhydrazyl). The EPR spectra were simulated on Matlab using the Easyspin software.32 The positions of spectral features in EPR spectra are defined by the principal values of the g-tensor: a single g-factor for the isotropic case, gxx, gyy and gzz for the orthorhombic case, gII and g⊥ for the axial case. Specifically, for a low spin d5 ion in a tetragonally distorted site (like signal Ru–Cl), it is possible to quantitatively relate the principal values of the g-factor with crystal field parameters through the following equations:33| | | gII = ge(a2 − b2) − kb2 | (1) |
| | 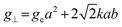 | (2) |
| | 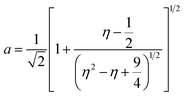 | (3) |
| | 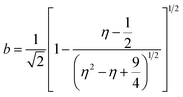 | (4) |
| | 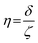 | (5) |
where ge: the free electron g-factor (ge = 2.0023), k: the orbital reduction factor, δ: is the splitting of the t2g(0) and t2g(±1) energy levels induced by the tetragonal crystal field and ζ: the one-electron spin–orbit coupling constant, which can be assumed independent on structural changes in the RuIII environment. η is therefore directly proportional to the crystal field splitting. The increase of η with the number of C2H2–HCl treatment cycles agrees with our interpretation as discussed in the results and discussion section (vide infra, Section 3.2.1). The best fit was obtained for k = 1.
3. Results and discussion
3.1 Controlled redispersion of carbon-supported Ru NPs
3.1.1 Development of the redispersion strategy.
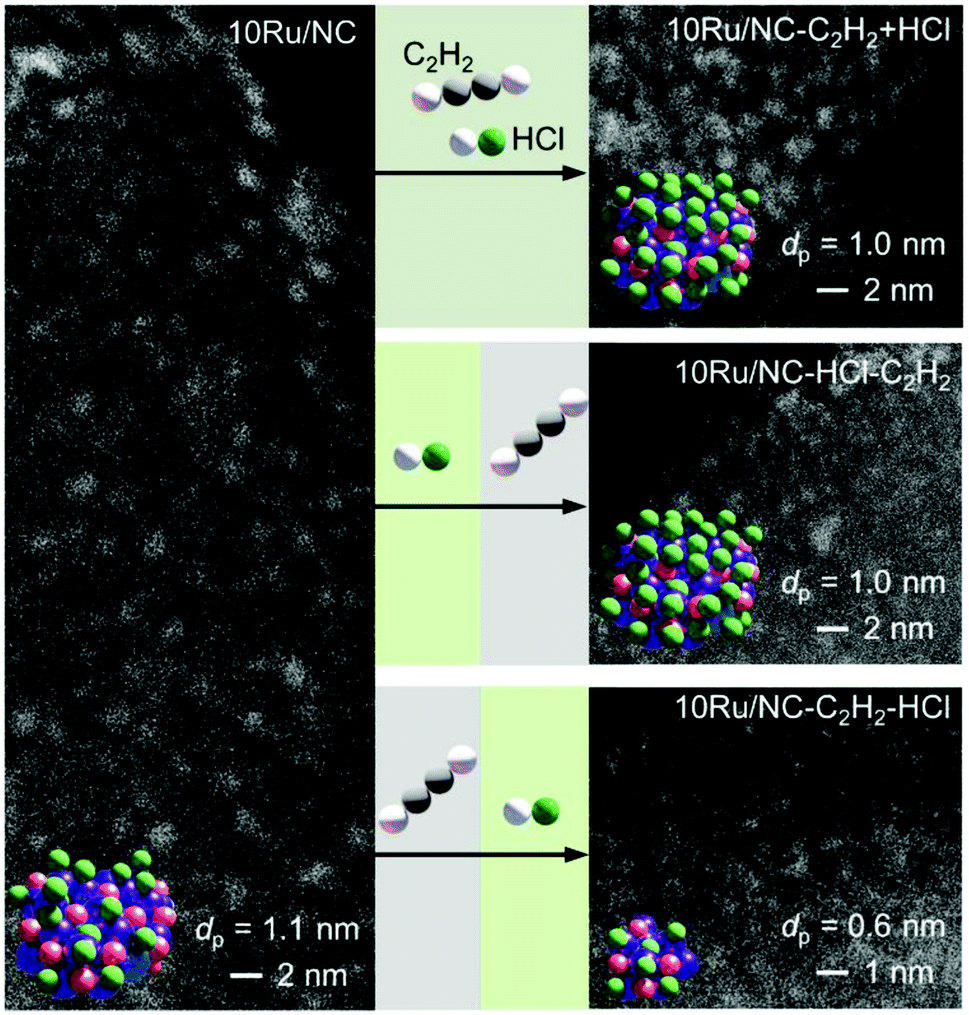
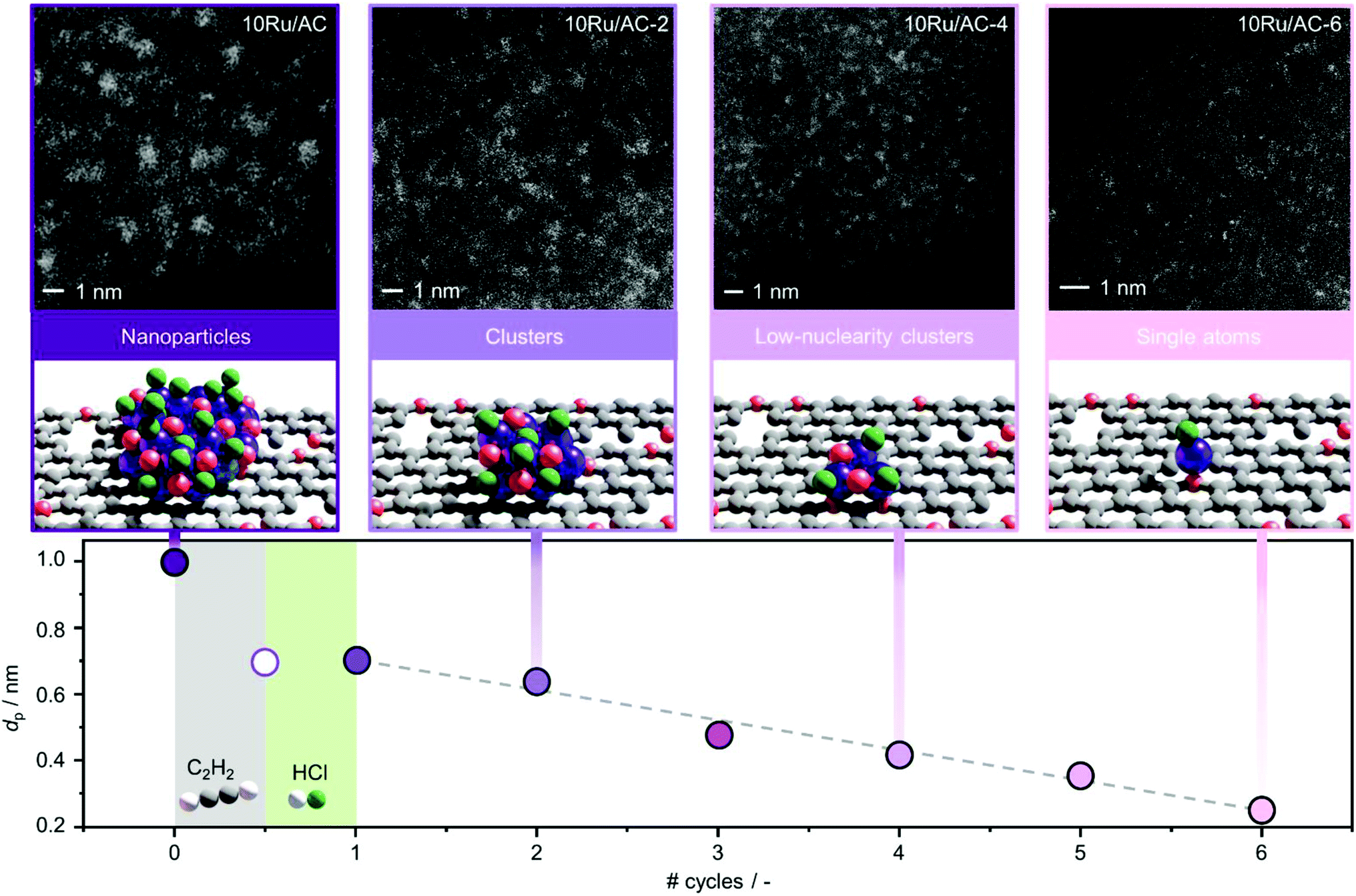
Carbon-supported Ru NPs were first synthesised by simple incipient wetness impregnation of AC and NC with aqueous solutions of RuCl3·xH2O (nominal metal loading of 10 wt% confirmed by ICP-OES, Table S1†) and thermally activated at 473 K (see experimental section, sample code: 10Ru/AC and 10Ru/NC, respectively). In an effort to overcome the current limitations in the effectiveness of halogen-mediated redispersion methods, a potential solution may consist in promoting the effect of the selected halogen compound with a mildly-reducing agent.18 To this end, C2H2 and HCl were identified as a promising pair, prompted by the previously-reported redispersion of NC-supported Ru NPs into single atoms induced by simultaneous exposure to both gases (cofeeding) that is, yet, ineffective for Ru NPs supported on commercial AC due to the weaker anchoring properties of O-functionalities compared to N-functionalities.34 However, owing to lack of control over the redispersion driving force, the cofeeding approach could not lead to atomic dispersion of NC-supported Ru NPs upon increasing the metal content (from 5 wt% to 10 wt%, Fig. 1 and S1†), even with prolonged treatment durations (from 12 to 50 h, Fig. S1†). To overcome this limitation, a new approach was explored aiming to (i) decouple, (ii) control and (iii) maximise the effect of each gas in the process. Therefore, the Ru NPs were sequentially exposed to HCl and C2H2 atmospheres and vice versa. The treatment conditions were selected to achieve equivalent exposure to each molecule as experienced when C2H2 and HCl were cofed. While treatment with HCl first did not affect the size of the Ru NPs, exposure to C2H2 first resulted in partial redispersion. This observation reveals the central role of avoiding competitive adsorption of the two gases, which presumably leads to surface overchlorination preventing adsorption of C2H2,35 in gaining control over the process. Furthermore, the partial redispersion observed in the latter case hints at a surface layer removal of Ru species, suggesting that multiple C2H2–HCl treatment cycles may be required to fully disperse NPs into isolated atoms. On the basis of these findings, a new strategy was developed, consisting of cyclic exposure of Ru NPs to alternating (1 h) C2H2 and HCl atmospheres. Strikingly, not only does this strategy achieve atomic dispersion on both NC and commercial AC, as visualised in HAADF-STEM analysis (Fig. 2, S1 and S2†), but it also enables gradual and controlled NP redispersion, as reflected in the evaluation of the average cluster size at different stages of the treatment (Fig. 2, sample code: 10M/C-X where M: metal, C: “AC” or “NC”, X: # of C2H2–HCl treatment cycles and y: “a” if the cycle terminated after the C2H2 treatment). In stark contrast to the previously-reported methods, which could modulate the reduction in the NP size in the nanometre regime,36 the presented strategy enables finely-resolved control over the redispersion process to the subnanometre regime, tuning the metal nuclearity by regulating the number of C2H2–HCl treatment cycles.
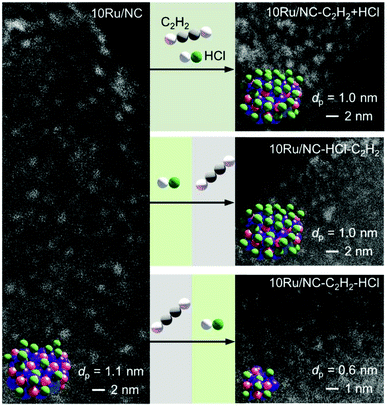 |
| | Fig. 1 HAADF-STEM images of 10Ru/NC, in fresh and treated forms, indicating the average metal particle size (dp). The graphical representation of the different samples (colour code: purple = Ru, red = O and green = Cl) show the effect of different redispersion treatments: C2H2–HCl cofeeding for 12 h (10Ru/NC-12h, ineffective in the redispersion of the Ru NPs) and sequential HCl and C2H2 atmosphere for 6 h each (10Ru/NC-HCl-C2H2) likely resulting in overchlorinated Ru NPs as well as sequential C2H2 and HCl atmospheres for 6 h each (10Ru/NC-C2H2–HCl) leading to partial redispersion. Treatment conditions: Wcat = 0.1 g, FT = 15 cm3 STP min−1, T = 473 K, cofeeding: 40 vol% C2H2, 44 vol% HCl, 16 vol% Ar, C2H2 exposure: 40 vol% C2H2, 44 vol% He, 16 vol% Ar and HCl exposure: 44 vol% HCl, 40 vol% He, 16 vol% Ar. | |
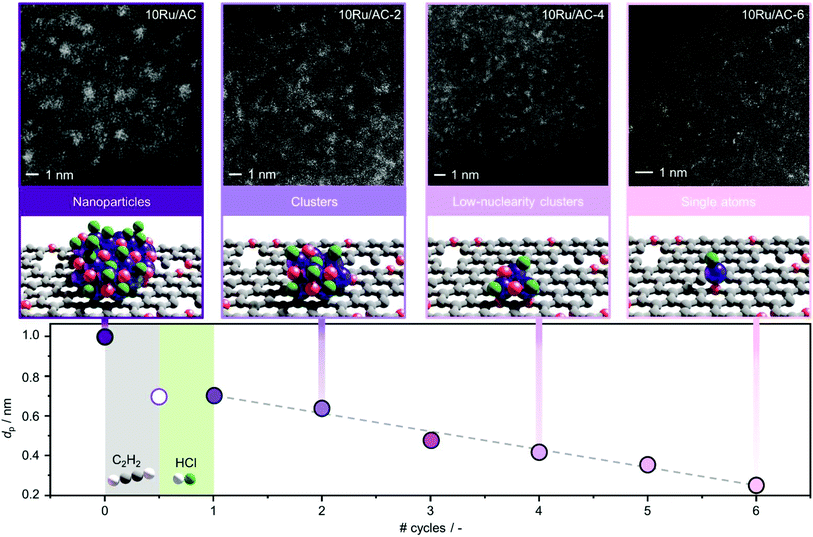 |
| | Fig. 2 Average metal particle size (dp) of 10Ru/AC, in fresh and treated forms, as a function of the number of C2H2–HCl treatment cycles and HAADF-STEM images of selected samples, visualising the control over nuclearity enabled by the redispersion treatment. One cycle consists of exposure to C2H2 atmosphere followed by exposure to HCl atmosphere, for 1 h each. Treatment conditions: Wcat = 0.1 g, FT = 15 cm3 STP min−1, T = 473 K, C2H2 treatment: 40 vol% C2H2, 44 vol% He, 16 vol%, Ar, HCl treatment: 44 vol% HCl, 40 vol% He, 16 vol% Ar. | |
3.1.2 Role of Cl surface atoms of fresh nanoparticles in the redispersion process.
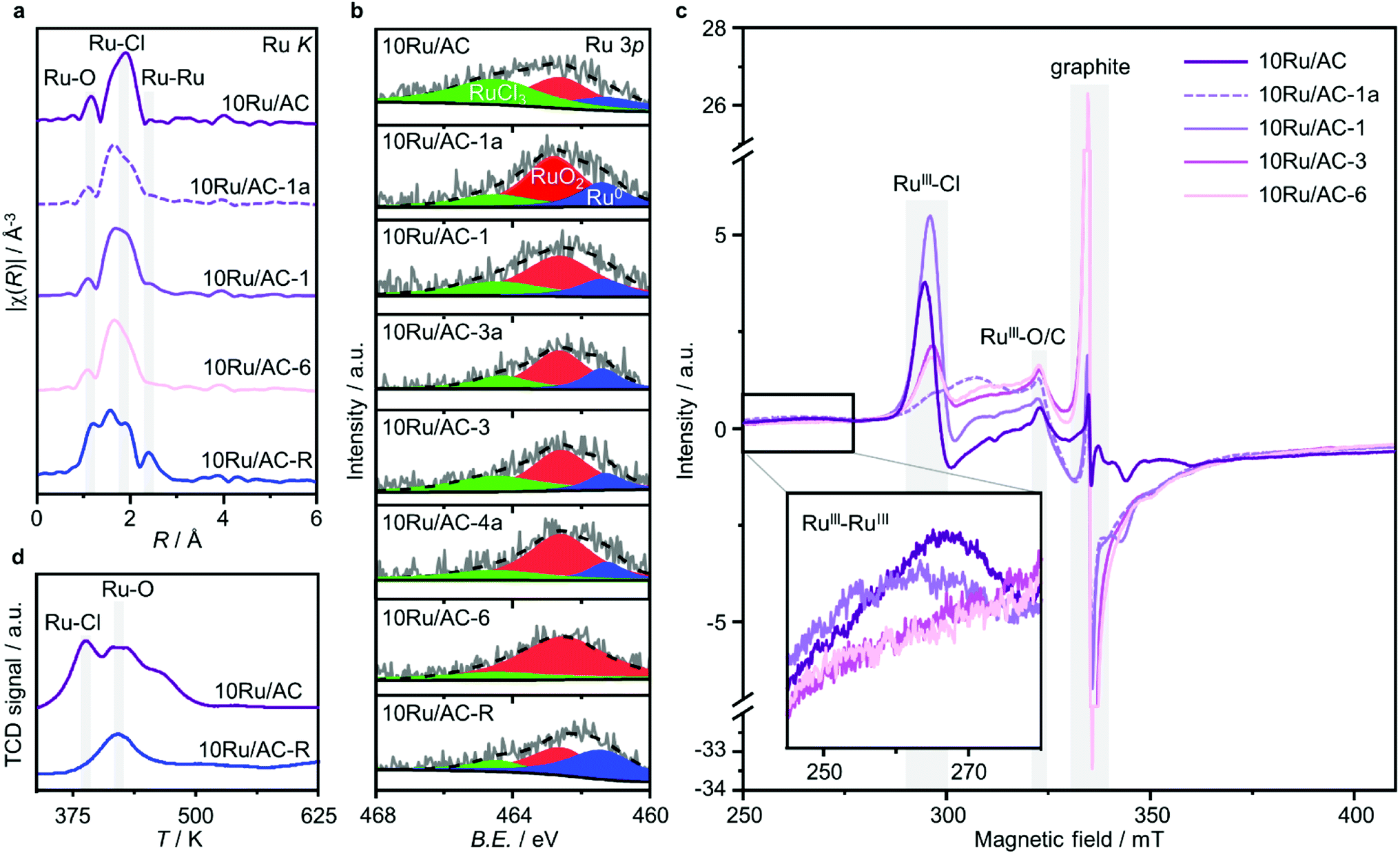
Owing to the small size of Ru clusters, below the resolution of XRD (≤1 nm, Fig. S3†), their structural characterisation mainly relies on EXAFS (Fig. 3a and S4, Tables S2 and S3†). Interestingly, no pronounced metallic contributions were observed, suggesting a metal oxychloride architecture, RuOxCly, where Ru atoms are bridged by O atoms. To explore the role of Cl ligands in the redispersion process, the Ru NPs were prereduced under a H2 atmosphere, 10Ru/AC-R, keeping the temperature lower than 673 K to remove surface Cl atoms while preventing RuOx reduction to metallic Ru.34 Accordingly, the chlorination degree was markedly reduced (Ru–Cl CN = 2.0 and CN = 0.9 in 10Ru/AC and 10Ru/AC-R, respectively) and the sample acquired an enhanced metallic character CN = 0.2 and CN = 1.2 in 10Ru/AC and 10Ru/AC-R, respectively). Owing to the limitation of EXAFS in distinguishing between Ru–O and Ru–C bonds, more accurate information on the impact of the prereducing treatment on RuOx species was acquired by H2-TPR analysis of 10Ru/AC and 10Ru/AC-R, showing that RuOx species were substantially preserved (Fig. 3d). Notably, the removal of surface Cl atoms inhibited the NP redispersion (Fig. S5†), indicating their pivotal role in the process by forming mobile RuOxCly species.18,23
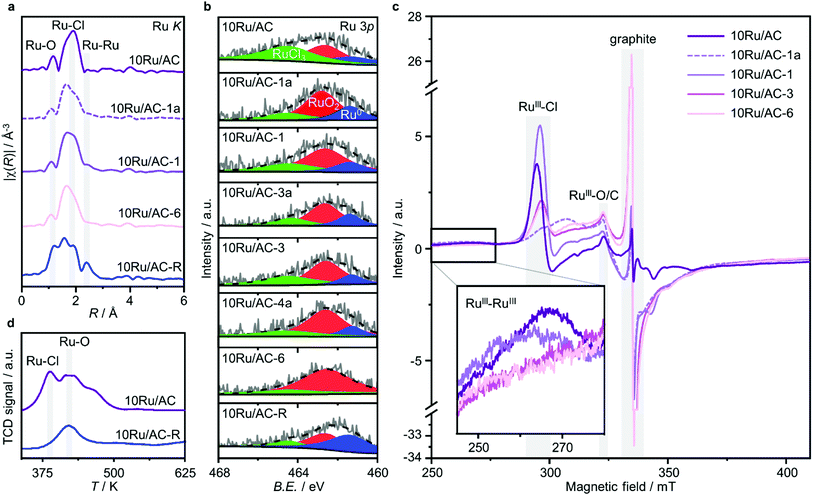 |
| | Fig. 3 Characterisation of 10Ru/AC, in fresh and treated forms, resolving the roles of C2H2 and HCl in the redispersion process. (a) Ru K edge spectra showing progressively stronger Ru–O/C and weaker Ru–Cl interactions, (b) Ru 3p XPS spectra evidencing increasingly prominent oxidised character and (c) EPR spectra exhibiting (i) three different contributions to the RuIII signal, showing increasing anisotropy and weaker RuIII–RuIII interactions over the course of the treatment, as well as (ii) a graphite signal evidencing a progressively increase in local defect on graphitic carbon phases in the support. (d) H2-TPR profiles of 10Ru/AC and 10Ru/AC-R, showing significant loss in the Ru–Cl peak upon reduction, in line with the metallic Ru character detected in both the Ru K edge and Ru 3p XPS spectra of the reduced sample. | |
3.1.3 Evolution of the metal nuclearity.
Insights on the generated cluster speciation were gained by means of XPS and XAS analyses, conducted on 10Ru/AC samples after consecutive C2H2–HCl treatment cycles. In line with the XANES results (Fig. S6†), the Ru 3p XPS spectra progressively assume a more oxidised character (Fig. 3b, Table S4†). Owing to the oxychloride architecture of the Ru clusters, where Ru–Ru interactions are weak, information on their nuclearity from EXAFS relies on other coordination shells. Remarkably, two distinct Ru–O/C fingerprints are identified upon fitting the EXAFS spectra of 10Ru/AC in both fresh and treated forms (Fig. S7, Table S2†): (i) short-bonding Ru–O/C contributions at R = 1.83 Å ascribable to RuOxCly species in the clusters,37 and (ii) long-bonding Ru–O/C contributions at R = 2.2 Å likely reflecting metal-support interactions, possibly of carbide nature.38 The evolution of the metal nuclearity over the course of the treatment thus manifests in progressively decreasing short-bonding Ru–O/C interactions (CN = 0.9 and CN = 0.5 in 10Ru/AC and 10Ru/AC-6, respectively), contrasted by more prominent long-bonding Ru–O/C interactions (CN = 2.4 and CN = 4.7 in 10Ru/AC and 10Ru/AC-6, respectively). Additionally, in agreement with XPS analysis and the weaker adsorption of HCl on Ru single atoms compared to NPs,35 the redispersed Ru species present a lesser chlorinated character (Ru–Cl CN = 2.0 and CN = 1.5 in 10Ru/AC and 10Ru/AC-6, respectively).
3.2 Redispersion mechanism
3.2.1 Contributions of C2H2 and HCl to the redispersion process.
To probe the role of each gas in the redispersion process, a comparative analysis was conducted between prolonged exposure (12 h) of 10Ru/AC solely to C2H2 and HCl. Remarkably, HAADF-STEM analysis revealed that only C2H2 promotes the process, leading to partial redispersion (≈0.4 nm), while exposure to HCl leaves the NP size unaltered (Fig. S5†). However, it should be noted that atomic dispersion could not be achieved with sole C2H2, likely limited from depletion of the originally present surface Cl atoms. Consistently, analysis of the Ru K edge EXAFS spectra show stronger long-bonding Ru–O/C interactions in 10Ru/AC-1a compared to 10Ru/AC (CN = 4.1 and CN = 2.4, respectively), complemented by weaker short-bonding ones (CN = 0.5 and CN = 0.9, respectively) and a simultaneous loss in Ru–Cl coordination (CN = 1.4 and CN = 2.0, respectively), which is partially recovered upon exposure to HCl (CN = 1.9 in 10Ru/AC-1) while both the long-bonding and the short-bonding Ru–O/C interactions remain substantially unaffected (CN = 4.1 and CN = 0.5 in 10Ru/AC-1a and CN = 4.2 and CN = 0.6 in 10Ru/AC-1, respectively). Accordingly, comparison of the Ru 3p XPS spectra of 10Ru/AC-1 and 10Ru/AC-1a, as well as of 10Ru/AC-3 and 10Ru/AC-4a (i.e., at later stages of the redispersion treatment), indicates formation of new chlorinated Ru species on the surface of the metal cluster occurring via oxidative Cl-addition under HCl atmosphere. Conversely, comparison of the Ru 3p XPS spectra of 10Ru/AC-1a and 10Ru/AC, as well as of 10Ru/AC-4a and 10Ru/AC-3, shows a mildly reducing effect of C2H2, mainly resulting in the removal of the surface Cl atoms in the metal cluster.
These results were further corroborated by EPR analysis, simultaneously providing additional insights on the progressively stronger interaction of the Ru species with the support. Owing to the non-Kramers nature of Ru0 and RuIV species, EPR only detects RuIII species, presenting a low-spin d5 configuration that gives rise to one unpaired electron (s = 1/2). These can be fitted by a weighted sum of four contributions (Fig. 3c and S8, Table S5†): (i) a broad anisotropic signal with axial g-factor ascribed to RuIII mainly coordinated by Cl (weak-field coordinating atoms) in an tetragonally distorted geometry (g⊥ = 2.265 and gII = 1.95, RuIII–Cl signal), (ii) another anisotropic signal with rhombic g-factor attributed to RuIII mainly coordinated by O and C (strong-field coordinating atoms) (gxx = 2.08, gyy = 1.99, gzz = 1.865, RuIII–O/C signal), (iii) a very broad signal, assigned to RuIII ions interacting in relatively large clusters (g ≈ 2.15, RuIII–RuIII signal), as well as (iv) an additional sharp signal with isotropic g-factor, typical of delocalised electrons in graphitic carbon structures (g = 2.0048, graphitic carbon signal).39,40 In agreement with EXAFS and XPS analyses, the intensity of the RuIII–Cl signal in 10Ru/AC dramatically decreases upon exposure to C2H2, 10Ru/AC-1a, regaining prominence upon exposure to HCl, 10Ru/AC-1. Compared to 10Ru/AC, the RuIII–Cl signal in 10Ru/AC-1 is stronger, evidencing chlorination of an increased number of metal sites resulting from the higher dispersion degree and lower anisotropy, reflected by a shift towards higher magnetic field and indicative of a lower chlorination degree in the redispersed metal sites. After the first cycle, the cluster anisotropy gradually decreases over consecutive treatments, resulting in a stronger crystal field induced by higher coordination with the O or C functionalities in the support (i.e., increasing η, see experimental section, Fig. S9a†). The signal intensity progressively reduces in favour of increasing intensity of the RuIII–O/C signal (Fig. S9b†). In line with this, RuIII–RuIII interactions are no longer detected in the spectra of either 10Ru/AC-3 or 10RuAC-6. Intriguingly, the stronger interaction of the Ru species with the support also reflects in the more prominent graphitic carbon signal, ascribed to local defects in graphitic carbon structures that originate from exposure to C2H2 and presumably serve as anchoring sites for the mobile RuOxCly species (Fig. 3d and S9c†). Owing to the known ability of Ru to form graphitic carbon layers,23 HRTEM analysis was conducted to verify the accessibility of the metal sites, but no evidence of encapsulation of Ru clusters is visible (Fig. S10†), which is consistent with the similar surface metal content in 10Ru/AC and 10Ru/AC-6 detected in XPS (Table S6†). Notably, the EPR spectra of 10Ru/AC-R, prereduced under a H2 atmosphere, and 10Ru/AC-R-6, where the redispersion is inhibited, exhibit similar graphitic carbon signals (Fig. S8†), further corroborating the important role of the local defects in the process.
3.2.2 C2H2 adsorption as descriptor for the redispersion process.
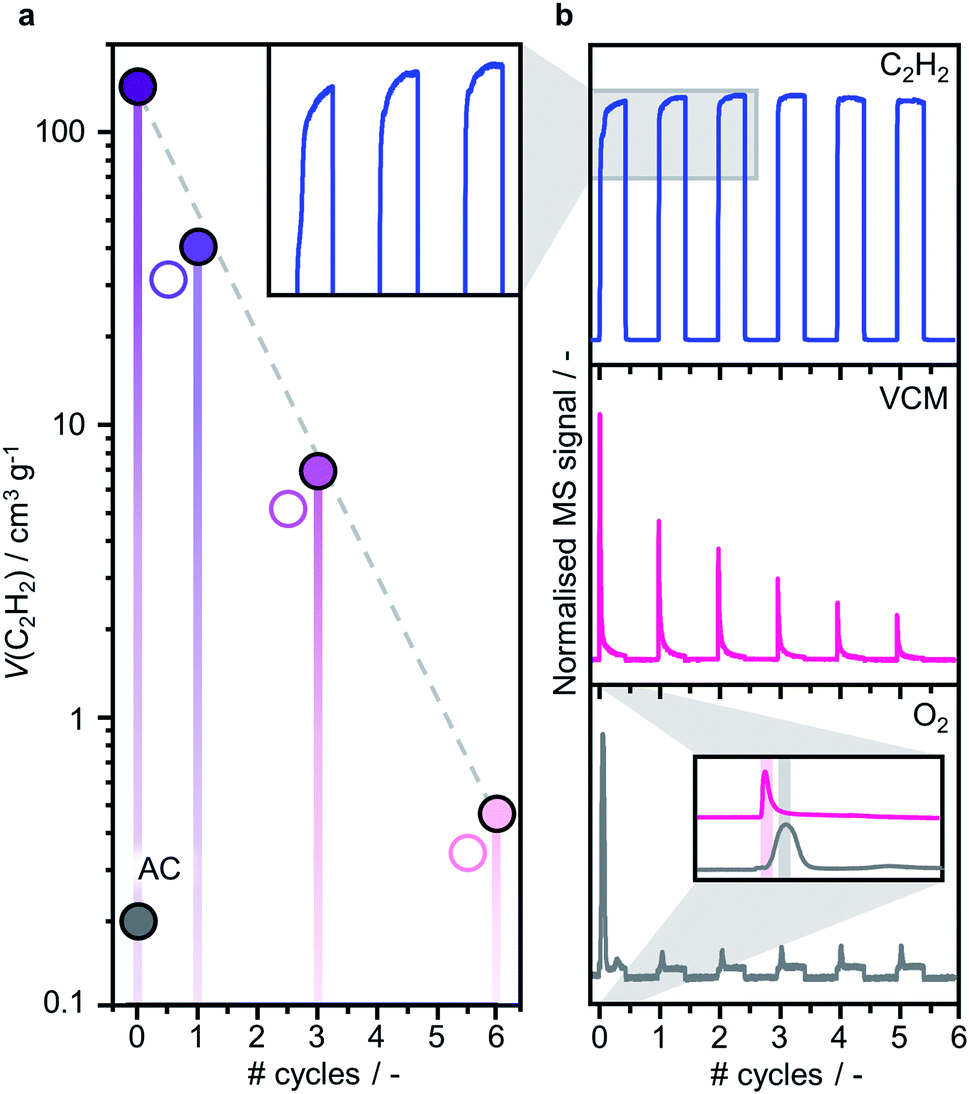
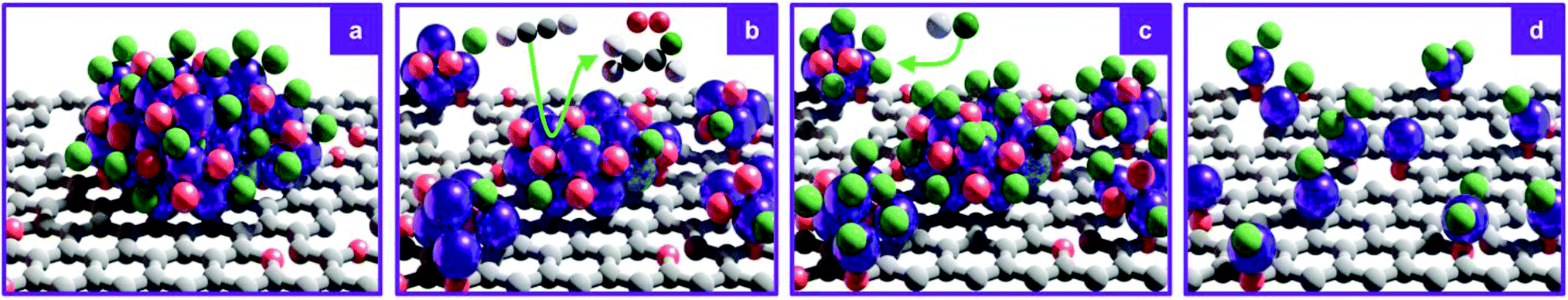
Consistent with the reported superior catalytic activity of RuOxCly NPs compared to Ru single atoms in the C2H2 hydrochlorination reaction for VCM production,23 the C2H2 adsorption capacity, identified as a key descriptor for the performance,41 decreases with increasing Ru dispersion (also reflected in decreasing VCM evolution during the treatment, vide infra, Fig. 4). Accordingly, the C2H2 adsorption capacity only marginally increases upon exposure to HCl, which leaves the cluster size unaltered, presumably due to the desorption of some residual acetylene molecules. To explore the dynamics of the redispersion process, the reactor outlet stream was analysed by time-resolved MS. During the C2H2 treatments, the C2H2 signal rapidly rises and gradually stabilises, indicating initial adsorption and subsequent conversion into VCM. Intriguingly, the sharp maximum in the VCM signal is followed by a delayed, broader O2 signal whose maximum occurs after VCM is almost fully depleted. Following the O2 evolution, no more VCM is detected, in line with the steady-state C2H2 signal, clearly indicating complete exhaustion of the accessible Cl atoms initially present on the cluster surface, which will require exposure to HCl for rechlorination. These findings enable delineation of a layer-by-layer redispersion mechanism, consisting of the following steps: (i) the C2H2-induced removal of surface Cl atoms via VCM evolution, (ii) the generation of undercoordinated RuOxCly species migrating from the original Ru cluster onto defects in the carbon support, possibly promoted by the exothermicity of the C2H2 hydrochlorination reaction, (iii) the coupling of undercoordinated surface O atoms presumably originating from RuOxCly species detachment (leading to the formation and evolution of O2) and (iv) the rechlorination of cluster surface upon exposure to HCl (Fig. 5). The high aptitude of RuOx species to be chlorinated agrees with previous density functional theory simulations on carbon-supported RuOx NPs.35 Furthermore, adsorption energies of C2H2, −189 kJ mol−1, and HCl, −255 kJ mol−1, computed by density functional theory,35 explain the importance of avoiding competitive adsorption in the process. Specifically, the dominant adsorption properties of HCl over C2H2 suggest that in the presence of both gases the evolution of VCM would be followed by immediate rechlorination, inhibiting the redispersion process by hindering the adsorption of C2H2 and thus the formation of the mobile RuOxCly species.
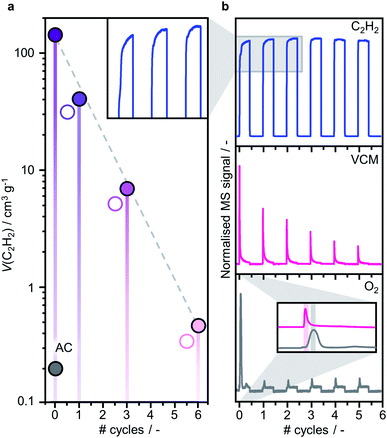 |
| | Fig. 4 (a) C2H2 adsorption capacity determined by chemisorption analysis of 10Ru/AC, fresh and after consecutive C2H2–HCl treatment cycles, and (b) time-resolved MS product analysis, showing the evolution of peaks associated to C2H2, VCM and O2 during the C2H2–HCl treatment cycles. The decreasing trend in the C2H2 adsorption capacity of the samples agrees with the C2H2 signal in the product analysis, exhibiting progressively shorter time required for stabilisation (as shown in the inset). | |
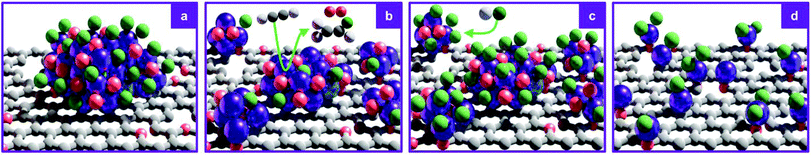 |
| | Fig. 5 Schematic representation of the C2H2-induced redispersion mechanism of (a) RuOxCly NPs supported on carbon. (b) C2H2 reduces the NP surface, evolving VCM, depleting accessible surface Cl atoms, and O2 while promoting Ru redispersion into smaller clusters or atoms. (c) Exposure to HCl leads to oxidative Cl addition, rechlorinating the cluster surface. (d) The alternating treatment is effective until full atomic dispersion is achieved. Colour code: purple = Ru, red = O, green = Cl, grey = C and white = H. | |
3.3 Extrapolation to other metals
Encouraged by the controllability and scalability, we explored the effectiveness of the redispersion strategy for supported Ir and Rh catalysts. Remarkably, not only was atomic dispersion achieved in both cases at 10 wt% loading, but the same fine control over nuclearity was enabled by regulation of the number of C2H2–HCl treatment cycles (Fig. 6, S11 and S12†). As for the Ru NP catalysts, no prominent metallic character was observed for either Ir or Rh NPs in either XPS or XAS (Fig. S13 and S14, Tables S3, S7 and S8†), likely due to the high affinity of these metals for Cl (M–Cl CN = 5.1 and CN = 4.4 in 10Ir/AC and 10Rh/AC, respectively). However, in contrast to 10Ru/AC, no short-bonding M–O/C contributions could be identified, suggesting a metal chloride architecture and further corroborating the key role of mobile metal chloride species in the redispersion process. In line with the XPS analysis, showing dechlorination and a more oxidic character (Fig. S13, Tables S7 and S8†), the metal-support interaction with the support becomes more prominent in the atomically redispersed species (Ir–O/C CN = 0.7 and CN = 1.6 in 10Ir/AC and 10Ir/AC-6, respectively, and Rh–O/C CN = 2.5 and CN = 4.0 in 10Rh/AC and 10Rh/AC-6, respectively), while the M–Cl interactions decrease (Ir–Cl CN = 3.6 in 10Ir/AC-6 and Rh–Cl CN = 1.6 in 10Rh/AC-6). These results hold promise for the general applicability of the redispersion strategy to a broad range of metals.
 |
| | Fig. 6 Successful extrapolation of the redispersion treatment to supported Ir and Rh catalysts. HAADF-STEM images of 10Ir/AC and 10Rh/AC, in fresh and treated forms, together with average particle size (dp). Atomic dispersion is visualised in 10Ir/AC-6 and 10Rh/AC-6. | |
4. Conclusions and outlook
In summary, our study demonstrated a redispersion strategy, comprising alternating C2H2 and HCl treatments, to synthesise high-loading (10 wt%) Ru, Ir and Rh catalysts on carbon supports with controlled metal nuclearity in the subnanometre regime. Precise size control (±0.1 nm) is enabled by regulating the number of C2H2–HCl cycles. The avoidance of NP surface overchlorination, which hinders C2H2 adsorption, is identified as key for the remarkable efficacy of the C2H2–HCl alternating strategy, as demonstrated by inhibition of the redispersion process resulting from both the C2H2–HCl cofeeding and inverse sequence (i.e., HCl first) approaches. Detailed characterisation, including XAS, EPR and time-resolved MS analyses revealed that the metal NP redispersion follows a layer-by-layer process in which C2H2 promotes the migration of surface MOxCly/MCly species to the carbon support while HCl regenerates them via chlorination of the newly-formed surface MOx/M species.
For future endeavours, the C2H2–HCl alternating redispersion approach offers a compelling synthetic route to finely tune metal nuclearity at high loadings and on practical supports, one of the most challenging aspects in nanoscale engineering of catalytic materials. Such control will be central to (i) derive structure–performance relationships at high density of active sites, where metal site proximity may give rise to enhanced catalytic performance,42 and (ii) implement the acquired knowledge in the design of supported metal catalysts with defined nuclearity for large-scale, targeted applications. To this end, the modulation of the coordination environment of the redispersed metal species constitutes an additional synthetic parameter that will require dedicated studies for optimal catalytic behaviour. Finally, the scale-up amenability of the presented redispersion protocol holds promise for high throughout synthesis, to ultimately produce commercial supported low-nuclearity metal catalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation. The authors thank the Scientific Center for Optical and Electron Microscopy at the ETH Zürich, ScopeM and the SuperXAS beamline at PSI, for access to their facilities.
References
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740 CrossRef CAS.
- S. Mitchell and J. Pérez-Ramírez, Nat. Rev. Mater., 2021, 6, 969 CrossRef.
- L. Liu and A. Corma, Nat. Rev. Mater., 2020, 6, 244 CrossRef.
- H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Nat. Commun., 2020, 11, 5884 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2021, 33, e2003075 CrossRef.
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W. C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9, 2353 CrossRef PubMed.
- E. Vorobyeva, E. Fako, Z. Chen, S. M. Collins, D. Johnstone, P. A. Midgley, R. Hauert, O. V. Safonova, G. Vilé, N. López, S. Mitchell and J. Pérez-Ramírez, Angew. Chem., 2019, 131, 8816 CrossRef.
- D. Faust Akl, A. Ruiz-Ferrando, E. Fako, R. Hauert, O. Safonova, S. Mitchell, N. López and J. Pérez-Ramírez, ChemCatChem, 2021, 13, 3247 CrossRef CAS.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, e1905622 CrossRef.
- S. Dai, J. P. Chou, K. W. Wang, Y. Y. Hsu, A. Hu, X. Pan and T. Y. Chen, Nat. Commun., 2019, 10, 440 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397 CrossRef CAS.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231 CrossRef CAS.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M. X. Chen, E. Jin, J. Yang, H. W. Liang, Y. Zhao, X. Feng, A. Narita and K. Müllen, Nat. Commun., 2020, 11, 5892 CrossRef CAS.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. T. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887 CrossRef CAS PubMed.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425 CrossRef CAS PubMed.
- J. Wu, L. Xiong, B. Zhao, M. Liu and L. Huang, Small Methods, 2019, 4, 1900540 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981 CrossRef CAS PubMed.
- K. Morgan, A. Goguet and C. Hardacre, ACS Catal., 2015, 5, 3430 CrossRef CAS.
- S. Feng, X. Song, Y. Liu, X. Lin, L. Yan, S. Liu, W. Dong, X. Yang, Z. Jiang and Y. Ding, Nat. Commun., 2019, 10, 5281 CrossRef PubMed.
- S. Feng, P. Hemberger, A. Bodi, X. Song, T. Yu, Z. Jiang, Y. Liu and Y. Ding, J. Catal., 2020, 382, 347 CrossRef CAS.
- A. Krapp, K. K. Pandey and G. Frenking, J. Am. Chem. Soc., 2007, 129, 7596 CrossRef CAS.
- T. Wang, J. Catal., 1981, 70, 187 CrossRef CAS.
- S. K. Kaiser, R. Lin, F. Krumeich, O. V. Safonova and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2019, 58, 12297 CrossRef CAS PubMed.
- R. Lin, S. K. Kaiser, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2018, 8, 1114 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072 CrossRef CAS.
- O. Muller, M. Nachtegaal, J. Just, D. Lutzenkirchen-Hecht and R. Frahm, J. Synchrotron Radiat., 2016, 23, 260 CrossRef.
- P. M. Abdala, O. V. Safonova, G. Wiker, W. van Beek, H. Emerich, J. A. van Bokhoven, J. Sa, J. Szlachetko and M. Nachtegaal, Chimia, 2012, 66, 699 CrossRef CAS PubMed.
- A. H. Clark, J. Imbao, R. Frahm and M. Nachtegaal, J. Synchrotron Radiat., 2020, 27, 551 CrossRef.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322 CrossRef CAS.
- E. Gallegos-Suarez, A. Guerrero-Ruiz, I. Rodriguez-Ramos and A. Arcoya, Chem. Eng. J., 2015, 262, 326 CrossRef CAS.
- J. L. Gómez de la Fuente, M. V. Martínez-Huerta, S. Rojas, P. Hernández-Fernández, P. Terreros, J. L. G. Fierro and M. A. Peña, Appl. Catal., B, 2009, 88, 505 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS.
-
J. R. Pilbrow, Transition Ion Electron Paramagnetic Resonance, Clarendon Press, Oxford, 1990 Search PubMed.
- Y. Pu, J. Zhang, L. Yu, Y. Jin and W. Li, Appl. Catal., A, 2014, 488, 28 CrossRef CAS.
- B. Wang, Y. Yue, S. Wang, Z. Chen, L. Yu, S. Shao, G. Lan, Z. Pan, J. Zhao and X. Li, Chem. Commun., 2020, 56, 10722 RSC.
- X. Duan, X. Tian, J. Ke, Y. Yin, J. Zheng, J. Chen, Z. Cao, Z. Xie and Y. Yuan, Chem. Sci., 2016, 7, 3181 RSC.
- H. Hillebrecht, P. J. Schmidt, H. W. Rotter, G. Thiele, P. Zönnchen, H. Bengel, H. J. Cantow, S. N. Magonov and M. H. Whangbo, J. Alloys Compd., 1997, 246, 70 CrossRef CAS.
- E. Solari, S. Antonijevic, S. Gauthier, R. Scopelliti and K. Severin, Eur. J. Inorg. Chem., 2007, 2007, 367 CrossRef.
- P. J. Carl and S. C. Larsen, J. Catal., 2000, 196, 352 CrossRef CAS.
- B.-Z. Wan and J. H. Lunsford, Inorg. Chim. Acta, 1982, 65, L29 CrossRef CAS.
- S. K. Kaiser, I. Surin, A. Amoros-Perez, S. Büchele, F. Krumeich, A. H. Clark, M. C. Román-Martínez, M. A. Lillo-Ródenas and J. Pérez-Ramírez, Nat. Commun., 2021, 12, 4016 CrossRef CAS.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta09238c |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Selina K.
Kaiser
a,
Selina K.
Kaiser