
Computational screening of single-atom alloys TM@Ru(0001) for enhanced electrochemical nitrogen reduction reaction†
Gurpreet
Kour
,
Xin
Mao
and
Aijun
Du
 *
*
Centre for Materials Science and School of Chemistry and Physics, Queensland University of Technology, Gardens Point Campus, QLD 4001, Brisbane. E-mail: aijun.du@qut.edu.au
First published on 13th January 2022
Abstract
Searching for highly efficient, active, and stable electrocatalysts for the nitrogen reduction reaction is vital to supersede the energy-intensive Haber–Bosch process. The electrocatalytic nitrogen reduction reaction (NRR) is one of the most promising strategies to synthesize value-added ammonia (NH3) under mild conditions with low energy utilization and less greenhouse gas emission. However, the lack of effective electrocatalysts remains the major hurdle for its practical applications. Ruthenium is generally considered a promising electrocatalyst for the electrochemical NRR. However, it exhibits a high overpotential corresponding to the potential determining step in the reduction pathway. Herein, using density functional theory, we systematically investigated the potential of a series of transition metal-doped Ru-based TM@Ru(0001) (TM = Sc–Zn, Y–Cd) single-atom alloys to evaluate their NRR activity. Among all the studied catalysts, it was found that the V doped Ru(0001) SAA exhibited a reduced kinetic barrier of about 1.14 eV as compared to that of the pure Ru(0001) corresponding to the potential determining step (PDS). In addition, it showed a significantly low negative limiting potential of −0.15 V for the PDS along with thermodynamical stability and high selectivity over the competing hydrogen evolution reaction (HER). The Climbing-Image Nudged Elastic Band (CI-NEB) method was employed to calculate the barrier height between various hydrogenation steps of the reduction reaction followed by the calculation of turnover frequency (TOF) for V@Ru(0001) using a microkinetic modelling approach. The TOF for V@Ru(0001) was found to be 4.24 × 10−3 per s per site at 100 bar and 700 K, which is far better than that of the pure Ru(0001) surface. This report provides a new design strategy to improve the catalytic performance of Ru(0001) for effective NRR.
1. Introduction
Conversion of atmospheric nitrogen (N2) to ammonia (NH3), also known as nitrogen fixation, is one of the most significant processes of industries. NH3 is not only a green energy carrier but also constitutes an important chemical in manufacturing dyes, explosives, medicaments, fertilizers, etc.1 Industrial production of NH3 primarily relies on the well-known Haber–Bosch process that requires very high temperature (300–500 °C) and high pressure (150–200 atm).2 However, this process accounts for 2% of the world's annual total energy consumption and liberates enormous amounts of greenhouse gases into the atmosphere. Thus, it is crucial to develop an alternative approach for sustainable and economical ammonia synthesis under mild conditions of temperature and pressure.Electrocatalytic nitrogen fixation using electrocatalysts has attracted the most intense investigative attention from the research world, benefitting from the ambient operating conditions and the utilization of electricity produced from renewable energy like wind energy, solar energy, etc.3–5 Moreover, electrochemical catalysis can be easily controlled by adjusting the reaction's temperature and potential to get the desired product. However, electrochemical methods are largely hampered by the high overpotential and low selectivity due to sluggish kinetics and the competitive HER.6 To circumvent these issues, it is highly recommended to develop active and stable electrocatalysts to enhance NRR activity while suppressing the accompanying HER. In the recent past, extensive research has been undertaken to explore the catalytic activities of potential NRR electrocatalysts including noble metals,7–9 transition-metal based materials10–15 and non-metallic materials.16–18 Transition metal-based nitrides, as well as sulphides, were also predicted to be promising for nitrogen fixation.19–22 Moreover, efforts are also being undertaken to further optimize their NRR activity by introducing dopants/vacancies,14,23–30 and modifying the catalyst's morphology/size.31–35
Single-atom alloys (SAAs) have recently gained momentum towards the design of extremely selective and active catalysts for the production of desired products.36–40 Substantial work has been done in the area of SAAs particularly on selective hydrogenation41–43 and H2 activation.44 Recently, it was found that an alloy of Pd supported by Cu(111) displayed an excellent activity towards the dissociation of H2.45 Tierney et al. later found that such alloys also promoted the spillover of H atoms across the surface of the catalyst.46 Further, Long et al. reported a CuPd SAA as a highly selective photocatalyst for converting CO2 to CH4. The paired Cu–Pd atoms with tuneable d-band centres enhanced the activity and selectivity of the catalyst. Although SAAs have been shown to catalyze C–O coupling, NO reduction, CO oxidation, hydrogenations, and dehydrogenations,39 not much has been done in the field of electrocatalytic reduction of nitrogen.47 Inspired by this strategy, we proposed SAAs based on Ru(0001) to boost their catalytic activity towards nitrogen fixation.
Ruthenium (Ru)-based catalysts have been extensively studied, both computationally and experimentally, for the NRR and have been found to exhibit good performance.9,48–58 Skúlason et al. in 2011 studied the free energy profile of flat Ru(0001) for the nitrogen reduction reaction.59 They reported a free energy change of 1.08 eV for the potential determining step (PDS). However, in another study by Tayyebi et al., the free energy change for the PDS was found to be 0.80 eV, suggesting poor activity of flat Ru(0001) for efficient NRR.53 In our work, we systematically investigated the feasibility of SAAs based on TM@Ru(0001) for efficient NRR by examining their stability, activity and selectivity. We performed a number of systematic DFT calculations to investigate the structural stability, adsorption of N2, minimum energy pathway, free energy change for the PDS, and kinetic barrier for the hydrogenation steps. Out of 20 candidates, we identified Ti@Ru(0001) and V@Ru(0001) as potential candidates based on a series of results obtained from the adsorption energy of the N2 molecule and the free energy change corresponding to the PDS. Among these, V@Ru(0001) presents enhanced electrocatalytic properties with a very low negative limiting potential of about −0.15 V vs. SHE along with an activation barrier of 1.14 eV. Indeed, many researchers have found that vanadium as a dopant could increase the activity of the substrate for effective NRR.60–63 Furthermore, microkinetic modelling was used to calculate the turnover frequency for the V@Ru(0001) catalyst which was found to be 4.24 × 10−3 per s per site for 25% N2 partial pressure at 100 bar pressure and 700 K temperature, which is excellent as compared to that of the pure Ru(0001). In addition to this, the selectivity and stability of these selected electrocatalysts were studied along with the competitive HER. These results may provide essential guidance for the development of SAA electrocatalysts for ammonia synthesis under mild conditions of temperature and pressure based on ruthenium.
2. Computational details
In this work, a close-packed hcp(0001) surface was used to represent the flat surface of a Ru atom. The Ru(0001) substrate was modelled by a four-layer slab in which the bottom two layers were fixed, and the top two layers were allowed to relax along with the adsorbed moiety to enhance the calculation speed. A vacuum space of 15 Å was employed to decouple the interactions between two adjacent supercells.All the structure optimizations and the electronic properties calculations were undertaken using the Vienna Ab initio Simulation Package (VASP)64–67 code based on the spin-polarized density functional theory (DFT) approach. The electron correlation functional was considered through the generalized gradient approximation (GGA)68 in the form of Perdew–Burke–Ernzerhof (PBE). The projector augmented wave (PAW) method69 was applied to generate the pseudopotentials. The kinetic cut-off energy was set to be 500 eV, while all the self-consistent optimizations were done with thresholds of 1 × 10−5 and 0.001 eV Å−1 for energy and force convergence, respectively. In all the calculations, van der Waals interactions were addressed using the DFT+D3 method.70 An implicit solvation model with water as implemented in VASPsol was used to consider the solvent effect in the reaction.71 The charge populations were studied using the Bader charge analysis method.72 The structural stability of the catalyst was evaluated using ab initio molecular dynamics (AIMD) simulations.73 The time step was set to be 1.0 fs, and simulations were conducted at 300 K.
The NRR process was calculated based on the computational hydrogen electrode (CHE) model. The reference potential was set to be that of the standard hydrogen electrode (SHE). The Gibbs free energy change was calculated using the following equation
| ΔG = ΔEDFT + ΔEZPE − TΔS |
| UL = −ΔGmax/e |
The climbing-image nudged elastic band (CI-NEB) method74 was used to calculate the energy barrier for the transition state. Eight images were added in between the initial and final positions for CI-NEB calculations. The intermediate images were optimized until the forces converged to 0.03 eV Å−1. In micro-kinetic modelling, a quasi-equilibrium approximation was used to study the NRR on V@Ru(0001). The TOF was calculated as the reaction rate for the potential determining step, while all other hydrogenation steps were considered as a fast equilibrium process. All the details for the calculation of TOF are given in the ESI.† Furthermore, to analyze the chemical bonding between the TM and the adsorbate, we used projected crystal orbital Hamilton population75,76 (pCOHP) analysis as implemented in the LOBSTER code.77
3. Results and discussion
The optimized geometry of TM@Ru(0001) SAAs (for example, V@Ru(0001)) is as shown in Fig. 1. One of the atoms from the top layer of bulk Ru(0001) was replaced with a TM (Sc–Zn, Y–Cd), which is expected to enhance the catalytic performance of the pure Ru(0001) due to charge redistribution. A number of different doping sites on the top layer of Ru(0001) were studied, and all the sites were found to be equally favourable and were of the same energy. All the studied TM@Ru(0001) complexes have similar geometry to the one shown in Fig. 1.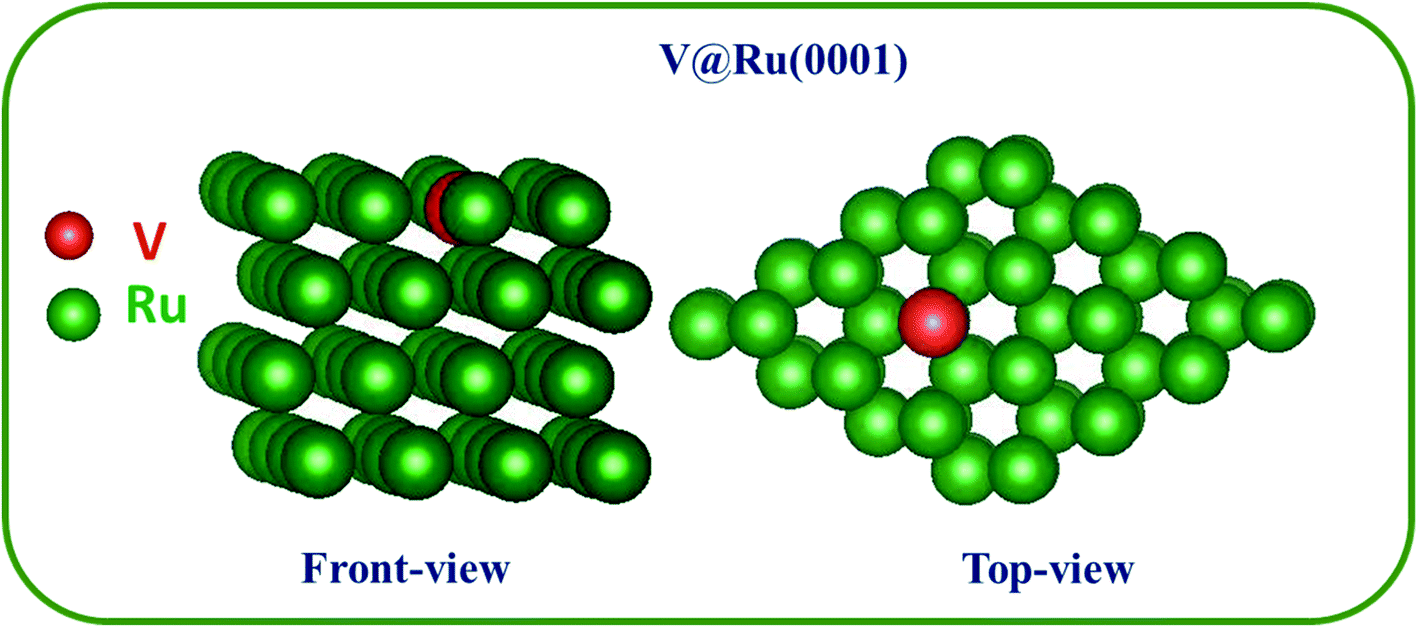 | ||
| Fig. 1 Optimized structure for V@Ru(0001). Both the front view and top view have been provided. The green and red spheres denote ruthenium (Ru) and vanadium (V) atoms, respectively. | ||
3.1 Screening of TMs supported on Ru(0001) as NRR catalysts
The NRR is a very complicated process that can take place through various mechanisms like distal, alternating, enzymatic and consecutive mechanisms, as shown in Fig. 2. Calculating all the intermediates in different mechanisms would cost lots of computational resources; thus, it is very important to frame a strategy to identify promising candidates. In the case of the NRR, the first step involves the N2 adsorption on the catalyst surface. It has been found that in most of the cases, the protonation of the N2 molecule to form *N2H or the last step in which the *NH2 intermediate upon hydrogenation forms *NH3 constitutes the PDS.59,78–81 The weak interaction of N2 with the catalyst results in high energy consumption due to the cleavage of the inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. On the other hand, in the case of the strong binding of N2 to the substrate, the last hydrogenation step becomes difficult. Thus, an efficient NRR catalyst should satisfy certain criteria such as (1) ΔGN2 should be lower than 0 eV to ensure effective adsorption of N2 on the surface of the catalyst, (2) ΔG for the first hydrogenation step, ΔG (N2–NNH), usually corresponding to a rate-determining step should be smaller than 0.50 eV, which is close to the calculated barrier for one of the best electrocatalysts4,78 and (3) high selectivity of the NRR over the HER.
N bond. On the other hand, in the case of the strong binding of N2 to the substrate, the last hydrogenation step becomes difficult. Thus, an efficient NRR catalyst should satisfy certain criteria such as (1) ΔGN2 should be lower than 0 eV to ensure effective adsorption of N2 on the surface of the catalyst, (2) ΔG for the first hydrogenation step, ΔG (N2–NNH), usually corresponding to a rate-determining step should be smaller than 0.50 eV, which is close to the calculated barrier for one of the best electrocatalysts4,78 and (3) high selectivity of the NRR over the HER.
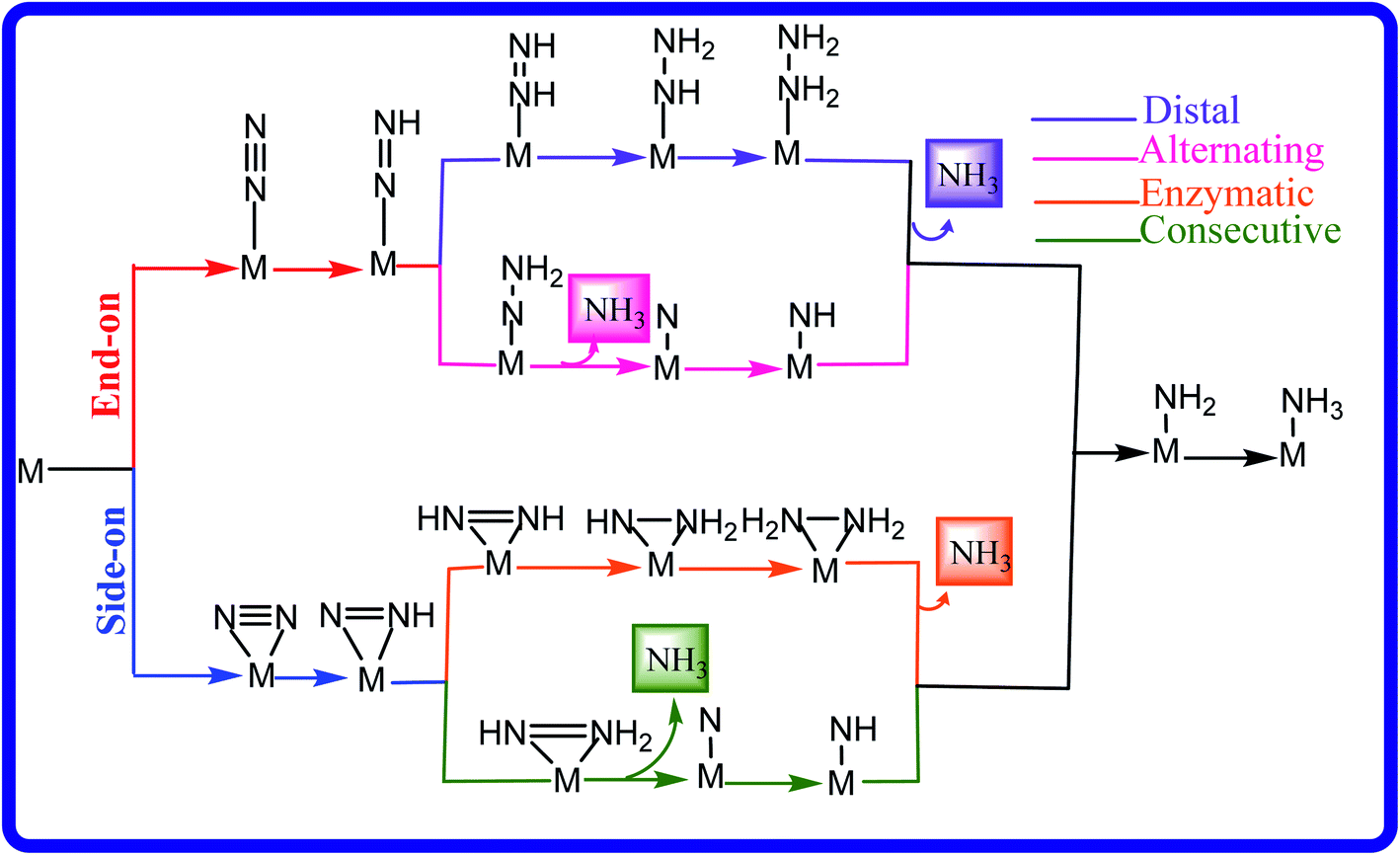 | ||
| Fig. 2 Systematic illustration of the different mechanisms encountered during the NRR. | ||
Based on criterion 1, we screened a number of TM@Ru(0001) (TM = Sc–Zn, Y–Cd) by computing ΔG of N2 adsorption (Fig. 3(a)). It is clear that Ti, V, Cr, Nb and Tc (green area of Fig. 3(a)) based SAAs exhibited a suitable Gibbs free change for N2 adsorption, while some of the SAAs have a positive free energy change, indicating weak adsorption, and the rest of them have a very high negative free energy change. Rh@Ru(0001) is also considered for further analysis as the free energy change for N2 adsorption is quite close to 0 eV. Following criterion 2 (Fig. 3(b)), the catalytic pathway for Ti@Ru(0001) for side-on adsorption and V@Ru(0001) for both side-on and end-on adsorption is considered for further investigations. The HER is an unwanted competitive reaction with respect to the NRR, and often lowers the catalytic performance of the electrocatalyst. To analyze the reaction selectivity, the adsorption energies of H+ and N2 (values are given in Table S1†) are calculated for each of the catalysts via V@Ru(0001) and Ti@Ru(0001) and are found to be highly negative (<1 eV). Strong adsorption would lead to the difficult desorption of H2 from the catalyst surface, making the competitive HER highly suppressed. It was found that the H* species is preferably adsorbed on the neighbouring Ru atom, leaving the active site vacant for the adsorption of intermediates during the reduction process. The NRR intermediates can be hydrogenated by the adsorbed H* species. In addition, the kinetic barrier for hydrogen formation on V@Ru(0001) and Ti@Ru(0001) was calculated using the CI-NEB method and was found to be 1.70 eV and 1.09 eV, respectively. The calculated kinetic barrier for H2 formation is higher than the barrier obtained for ammonia formation (1.14 eV), indicating that the HER is suppressed from the kinetic aspect. Thus, based on criterion 3, V@Ru(0001) and Ti@Ru(0001) exhibit high selectivity towards the NRR.
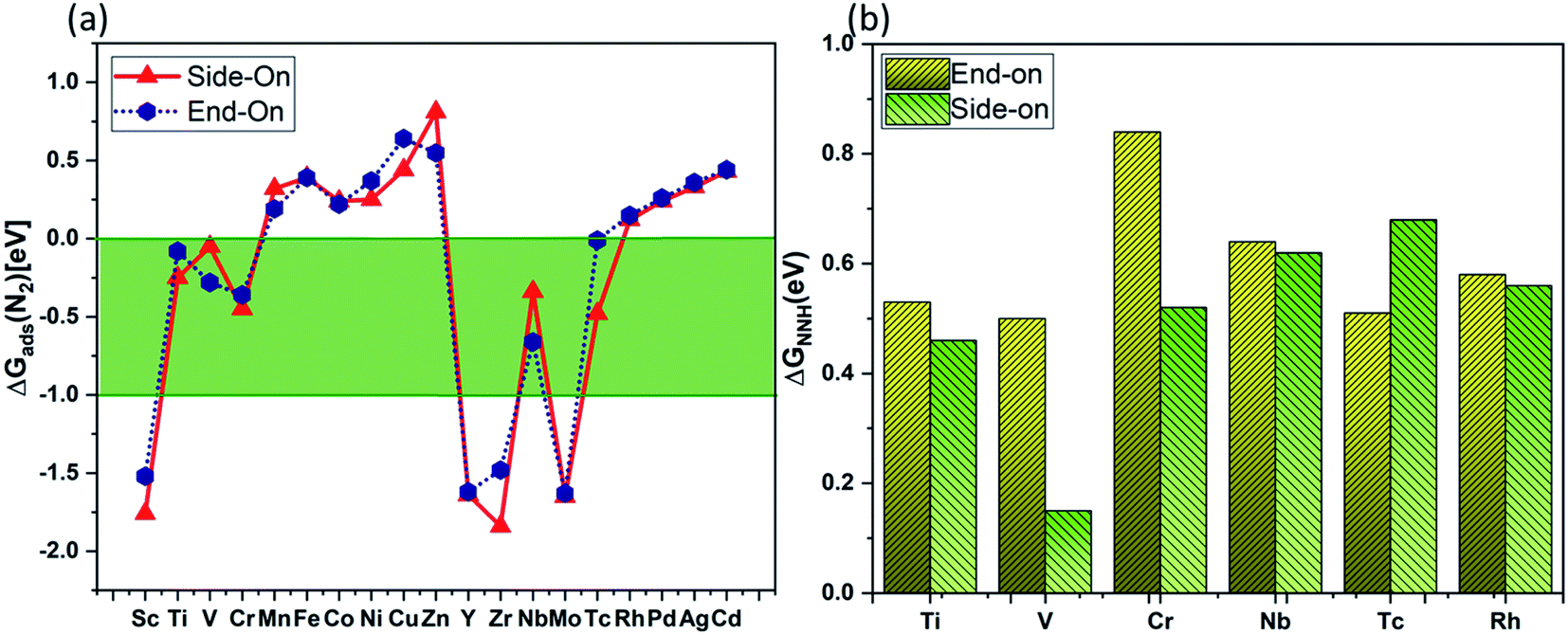 | ||
| Fig. 3 Screening results of the TM@Ru(0001) system for the NRR process using the (a) free energy change of *N2 adsorption and (b) the first hydrogenation step of N2 reduction ΔGNNH. The green shaded portion in (a) corresponds to the acceptable values for the effective adsorption of N2 on the catalyst's surface. | ||
3.2 N2 adsorption on TM@Ru(0001)
Effective adsorption of N2 on the surface of the catalyst is of paramount importance for an efficient nitrogen reduction process. It is well known that TMs with empty d-orbitals can strongly interact with the adsorbed N2 molecule through the formation of a N–metal bond.5,82 The exceptional performance of TM based catalysts can be attributed to the coexistence of their vacant and filled d-orbitals. The empty d-orbital of the TM can accept electron density from the filled s-favourable orbital of the N2 molecule, while the filled orbital of the TM can donate the electron density to the empty π* antibonding orbital of the N2 molecule (Fig. 4a). The “acceptance–back donation” of electrons between the TM and N2 molecule plays a significant role in the activation of the N2 molecule.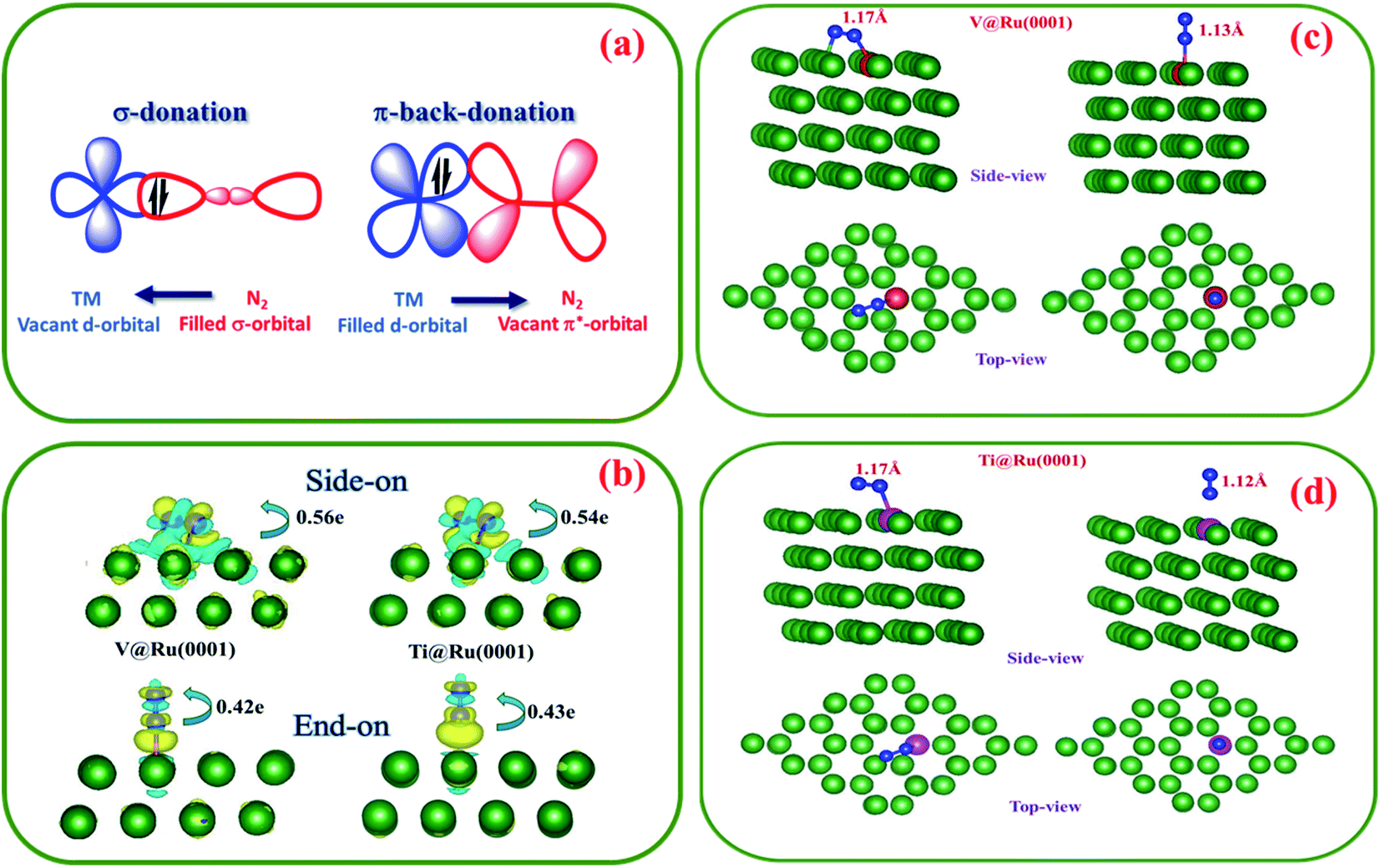 | ||
| Fig. 4 (a) Schematic demonstration of N2 bonding to a transition metal (TM). (b) The charge density difference of N2 adsorbed on V@Ru(0001) and Ti@Ru(0001) in side-on and end-on configurations with the isosurface value set to 0.001 e Å−1; the yellow and cyan depict positive and negative charges, respectively. (c and d) The side-on and end-on binding of N2 on V@Ru(0001) and Ti@Ru(0001), respectively. | ||
The adsorption of N2 on the substrate takes place via two common configurations, side-on and end-on, as shown in Fig. 4c and d. In the case of the end-on configuration, only one nitrogen atom binds to the active site, while in the case of the side-on geometry, both the nitrogen atoms of the N2 molecule form bonds with the active centre. For Ti@Ru(0001), N2 adsorption prefers the side-on configuration while for the V based complex, the end-on configuration is preferred.
In the case of V@Ru(0001), the N2 bond undergoes elongation from 1.098 Å (isolated state) to 1.17 Å and 1.13 Å for the side-on and end-on configurations, respectively. The Ti@Ru(0001) catalyst also displayed similar values for the NN elongation. Furthermore, based on the Bader charge difference analysis (Fig. 4b), it was found that 0.56e/0.42e and 0.54e/0.43e charges were transferred from the V and Ti based catalysts to the adsorbed N2 molecule for side-on/end-on configurations, respectively. All of the above analysis shows that the NN bond becomes activated upon adsorption on the surface of the catalyst.
Furthermore, the adsorbed N2 molecule on the catalyst was further analyzed by using the projected density of states (pDOS) and projected crystal orbital Hamilton population (pCOHP). The pDOS for N2 adsorption on both pure ruthenium and V@Ru(0001) is depicted in Fig. 5(a) and (b). It is clear that in the case of V@Ru(0001), the π* orbitals of the adsorbed nitrogen are partially filled, while the antibonding orbitals lie far above the Fermi level in the case of Ru(0001), revealing the active adsorption of N2 on V@Ru(0001). Further, Fig. 5(c) shows the pCOHP analysis in which the bonding contributions are shown on the right and the antibonding interactions are on the left side of the diagram. As predicted by the above figure, the occupied antibonding orbitals of N2 lie near the Fermi level as the bonding states, indicating the adsorption of a N2 molecule on the surface, with the unoccupied π* orbitals above the Fermi level. The integrated COHP (ICOHP) values were calculated by calculating the energy integral up to the highest occupied level. A higher negative value of ICOHP corresponds to an effective binding of N2 on the catalyst. In the case of V@Ru(0001), the ICOHP for end-on adsorption of N2 is −10.91 eV, pointing towards the strong adsorption of N2, which agrees well with the N2 adsorption data for the end-on configuration. Thus, COHP analysis also supports the active adsorption of N2 on the considered materials. The energy difference between 4d (Ru)/2p (N) and 3d (V)/2p (N) in the case of Ru(0001) and V@Ru(0001), respectively, may be responsible for better N2 adsorption on the surface of SAAs, thus showing an increment in their activity for the NRR over pure Ru.
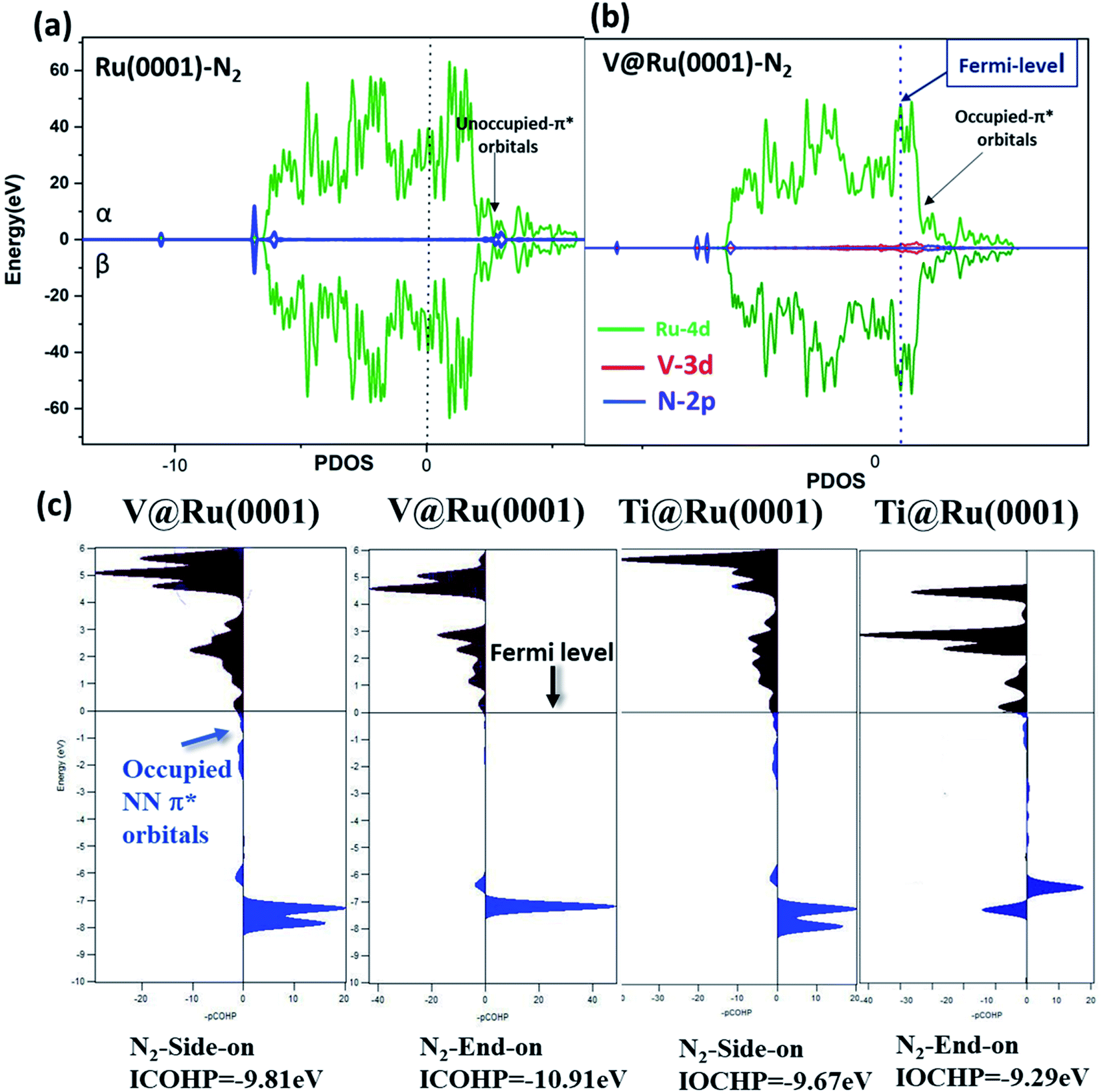 | ||
| Fig. 5 Partial density of states (PDOS) for (a) nitrogen adsorbed on pure Ru(0001), (b) V@Ru(0001) (c) projected crystal orbital Hamilton population (pCOHP) for N2 adsorption on V@Ru(0001) and Ti@Ru(0001) in side-on and end-on configurations. | ||
3.3 Stability of TM@Ru(0001)
The stability of TM@Ru(0001) SAAs was investigated using first-principles calculation. It is essential to have strong binding between the TM and the substrate in order to prevent its agglomeration. Thus, we calculated the binding energy Eb between the transition metal and Ru(0001) to analyze its structural stability using the equation Eb = ETM@Ru(0001) − ETM − ERu(0001), where ETM@Ru(0001), ETM and ERu(0001) are the energies of the TM@Ru(0001), TM and Ru(0001), respectively. It was found that the binding energies of a single V and Ti on Ru(0001) were −8.19 eV and −9.31 eV, respectively. Further, the thermodynamical stability of V@Ru(0001) and Ti@Ru(0001) was examined by performing AIMD simulations at 300 K (Fig. S2†). The structures of the catalyst were well maintained, with no notable geometrical buckling even at high temperatures. The negative binding energy and strong thermal stability indicate that Ru(0001) can provide a good anchoring site for the transition metal.3.4 Electrocatalytic conversion of N2 to NH3 with TM@Ru(0001) (TM = V, Ti) catalysts
Fig. 6 and 7 show the free-energy diagrams for the reduction of N2 to NH3 through consecutive and enzymatic reaction pathways along with the structures of various reactive intermediates encountered along the reaction pathway, respectively. The cartesian coordinates for all the formed intermediates during the reduction process are given in Table S2.† Both these pathways involve six elemental steps of proton-coupled electron transfer to the adsorbed N2, with the liberation of two NH3 molecules. The adsorbed N2 in the side-on configuration reacts with one pair of H+/e− to form *N–*NH with an uphill free energy of only 0.15 eV. In the second step, the reactive intermediate, *N–*NH, after reaction with one H+/e− pair can either form *N–*NH2 through a consecutive mechanism with a free energy change of −0.49 eV or can form *NH–*NH following an enzymatic pathway with ΔG of −0.15 eV. In the case of the consecutive reaction pathway, the H+/e− pair attacks *N–*NH2 and forms a *N intermediate along with the release of an NH3 molecule with a free energy change of −1.47 eV. The formed reactive intermediate after further hydrogenation steps ultimately releases another ammonia molecule accompanied by the liberation of the catalyst. However, in the case of the enzymatic mechanism, *NH–*NH upon reaction with a proton–electron pair forms *NH–*NH2 with ΔG of −0.40 eV, which on further reaction produces *NH2–NH2 with a free energy change of 0.30 eV. This step corresponds to the potential determining step for the enzymatic pathway. The proton–electron pair attacks *NH2–NH2 (ΔG = −1.51 eV) and produces one ammonia molecule along with an *NH2 reactive species which on further reaction produces another ammonia molecule.
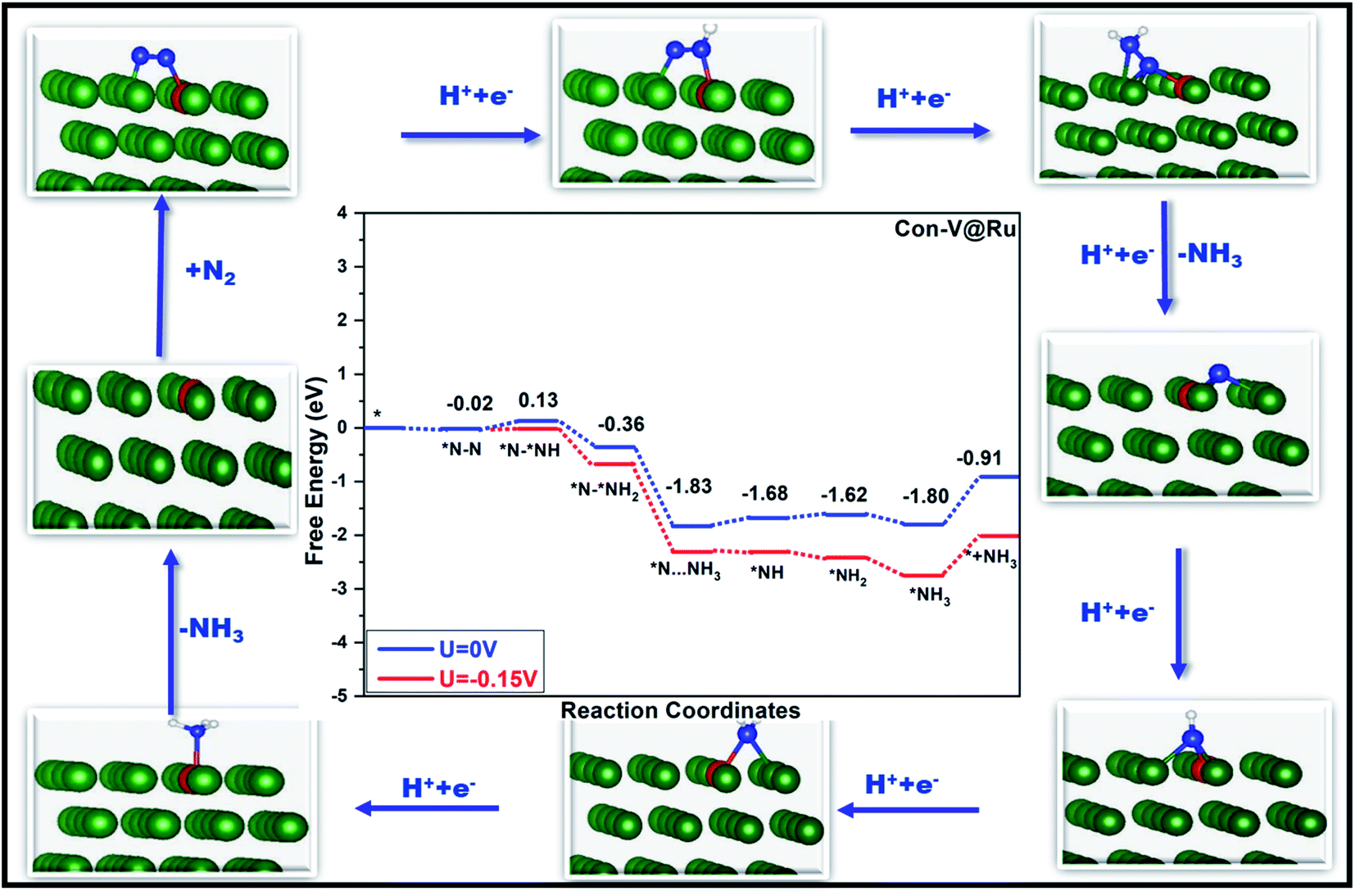 | ||
| Fig. 6 Free-energy diagram for N2 reduction via a consecutive mechanism at different applied potentials along with the corresponding structures of the intermediates involved in the reduction process. Green, red, blue and white balls represent Ru, V, N and H atoms, respectively. | ||
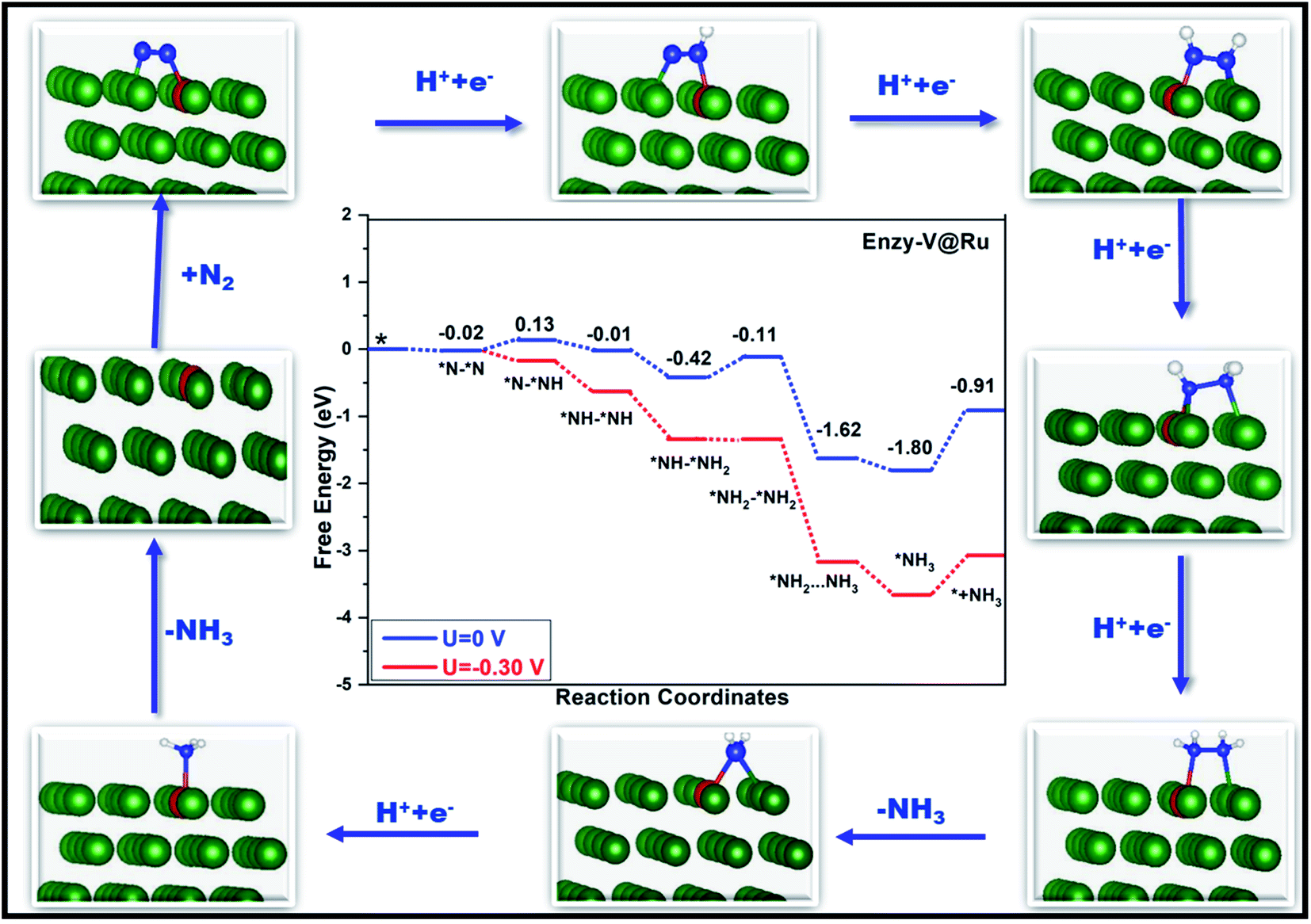 | ||
| Fig. 7 Free-energy diagram for N2 reduction via an enzymatic mechanism at different applied potentials along with the corresponding structures of the intermediates involved in the reduction process. | ||
In the case of the consecutive pathway, only the first and the fourth hydrogenation steps were found to be uphill in energy. During the whole reduction process through the consecutive pathway, the potential limiting step is the hydrogenation of *N2 to *N–*NH with an ultralow limiting potential of only −0.15 V, which is quite low as compared to those of various TM based catalyst for the NRR reported so far.52,83–85 Similar to the consecutive pathway, the enzymatic pathway also encountered two positive free energy changes during the hydrogenation steps. However, in the latter case, the limiting potential was found to be −0.30 V corresponding to the conversion of *NH–NH2 to an *NH2–NH2 intermediate. The rapid desorption of the formed NH3 from the surface of the catalyst is important for the regeneration of the active sites which is in turn good for the catalyst's long durability. The desorption of the produced ammonia is a fast process with a free energy change value of only 0.89 eV, indicating its excellent performance as a catalyst.
In addition to consecutive and enzymatic mechanisms, alternating and distal pathways were also explored for the NRR process. The free energy diagram for each of these pathways with the optimized structures of the reaction intermediates is shown in Fig. S3 and S4,† respectively. In case of the alternating pathway, the H+/e− pair attacks the two nitrogens alternatively to generate two molecules of NH3. However, in the case of the distal mechanism, a H+/e− pair first attacks the remote nitrogen to convert it into NH3 and then the remaining N atom reacts with another H+/e− pair to generate another molecule of NH3. From Fig. S3,† it is clear that the ΔG of N2 adsorption in the end-on mode is negative (ΔG = −0.28 eV), indicating the spontaneity of the reaction. However, the protonation of *N2 is non-spontaneous with ΔG of 0.50 eV, and this step corresponds to the potential-limiting step for the distal mechanism. In the case of the alternating mechanism, the hydrogenation of *N–NH into *NH–NH is the rate-limiting step with ΔG of 0.89 eV, which is quite high compared to all other pathways.
In addition to the associative pathway, the dissociative pathway was also investigated. The kinetic barrier for the scission of the N2 bond was calculated using the CI-NEB method and was found to be 1.90 eV which is very high to be achievable under moderate conditions of temperature and pressure.
Thus, for the V@Ru(0001) catalyst, out of the four studied pathways, consecutive and enzymatic mechanisms are the preferred ones showing excellent catalytic activity with record low negative values of limiting potential of −0.15 V and −0.30 V vs. SHE, respectively, for the conversion of N2 to NH3. Alternating and distal mechanisms, however, correspond to high negative values of limiting potential.
Lastly, in the case of the V@Ru(0001) catalyst, we calculated the solvation effects on the free energy values for the various hydrogenated steps in the case of the consecutive pathway. An implicit solvent model using water as a solvent was employed, and the calculated free energy diagram for V@Ru(0001) is shown in Fig. S5.† From the figure, it is clear that there is not much effect on the values of the free energy even with the solvent effects. The rate-determining step corresponding to the conversion of *N2 to *NNH is 0.14 eV, which is very close to the value obtained for calculations without the solvent effects. Therefore, no solvation treatment was undertaken for the rest of the calculations.
N from 1.098 Å to 1.17 Å. This step is accompanied by the highest value of ΔG = 0.47 eV during the whole reduction process in both the mechanisms and thus corresponds to the rate-determining step. All the protonation steps except the formation of *NH2–NH2 (ΔG = 0.28 eV) in the enzymatic pathway and the generation of *NH (ΔG = 0.05 eV) in the consecutive pathway are spontaneous. The last two steps of proton-coupled electron transfer in both mechanisms are common and are thermodynamically favourable. In the case of the Ti@Ru(0001) catalyst, the desorption of the formed *NH3 molecule in the last step shows a slight uphill with a ΔG of 0.51 eV. Thus, the Ti@Ru(0001) catalyst displays good performance with a limiting potential of −0.47 V vs. SHE following the enzymatic pathway as well as the consecutive mechanism.
Furthermore, to obtain deeper insight into the mechanism for both V and Ti-based catalysts, the CI-NEB method was used to calculate the kinetic barrier for each of the steps of the consecutive and enzymatic pathways. The calculated values for the kinetic barrier for each of the hydrogenation steps for the consecutive and enzymatic pathways for the NRR using V@Ru(0001) are given in Tables S3 and S4,† respectively. In the case of the consecutive pathway, the kinetic barrier for the potential determining step is only 1.14 eV which is quite low as compared to that of the pure Ru(0001) surface (1.85 eV).86 However, for the enzymatic pathway, the energy barrier for the rate-determining step for the conversion of *NH–NH2 to *NH2–NH2 is 1.51 eV. The activation barrier for the rate-determining step for the consecutive and enzymatic pathways for the Ti@Ru(0001) catalyst is 1.48 eV (Tables S5 and S6†). The calculated activation barrier for H2 formation is 1.70 eV which is very high as compared to that for the hydrogenation of adsorbed nitrogen to form *NNH, and thus the competitive HER process would have a minimal effect on the activity of the V@Ru(0001) for nitrogen reduction.
Thus, based on the above calculations, V@Ru(0001) exhibited improved catalytic activity with a record low negative value of limiting potential of −0.15 V vs. SHE for the conversion of N2 to NH3 following the consecutive pathway along with a kinetic barrier of only 1.14 eV for the rate determining step.
3.5 Micro-kinetic modelling
Turnover frequency (TOF) is a measure of the instantaneous efficiency of the catalyst. Thus, microkinetic simulations were employed to evaluate the TOF for the NRR using V@Ru(0001) as a function of temperature and pressure under the quasi-equilibrium approximation.31 The temperature for this reaction was in the range of 500–1000 K while the pressure was in the range of 1–100 bar.The contour plot (Fig. 8(a)) gives the variation of TOF with the change of temperature and pressure. It is evident from Fig. 8(b) and (c) that TOF rises with the rise of temperature and pressure, respectively. The rising temperature help in overcoming the kinetic barrier of the rate-determining step while the increasing pressure can bring more reactant molecules together to react and produce ammonia. The calculated value for the TOF for the formation of NH3 from N2 and H2 using V@Ru(0001) is 4.25 × 10−3 per site per s at 100 bar pressure and 700 K temperature, which is very close to the reported value by Mao et al.86 The obtained value of TOF for the NRR has increased as compared to that of the pure Ru(0001) catalyst (7.04 × 10−5 per site per s). Thus, it can be concluded that SAAs exhibit excellent performance as compared to the pure Ru(0001).
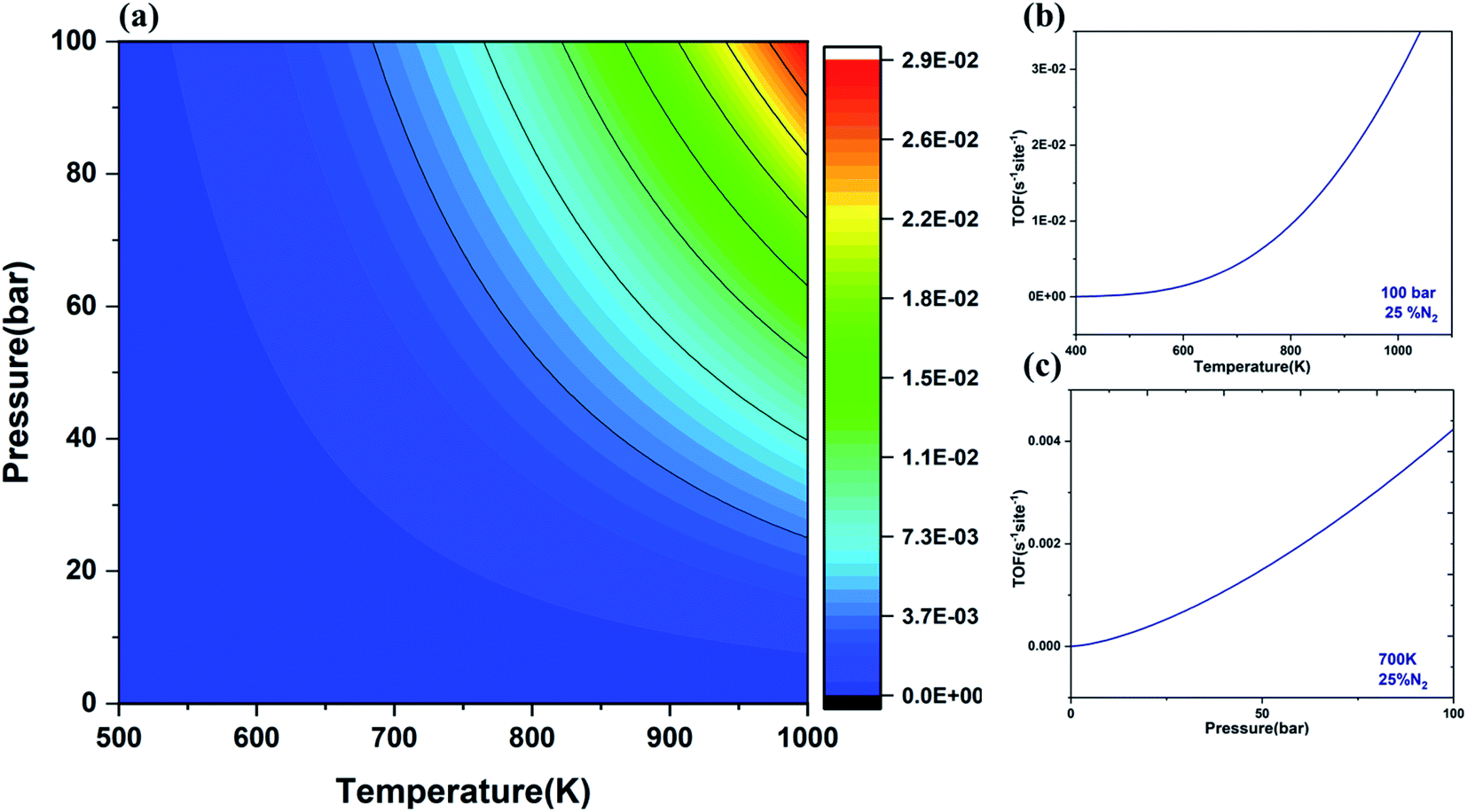 | ||
| Fig. 8 (a) Contour plot depicting the turnover frequency (TOF) for the formation of NH3 over the V@Ru(0001) catalyst at various temperatures and pressures with 25% and 75% partial pressures of nitrogen and hydrogen, respectively; (b) graph depicting the variation of temperature with TOF at 100 bar pressure; (c) graph showing the variation of pressure with TOF at 700 K temperature for the V@Ru(0001) catalyst. | ||
4. Conclusion
In conclusion, we have undertaken high throughput screening of catalysts based on DFT methods to explore the catalytic performance of 20 single-atom alloys based on transition metal doped Ru(0001) for the NRR. All the 3d and 4d elements (Sc–Zn and Y–Cd) were used as doped atoms on the top layer of Ru(0001), one of the best materials considered so far for the NRR.49,87,88 In our calculations, it was found that among all the studied catalysts in this work, the V doped Ru(0001) SAA exhibited enhanced NRR activity with a very low negative limiting potential of only −0.15 V vs. SHE along with a reduced kinetic barrier of about 1.14 eV corresponding to the rate determining step, following a consecutive pathway for nitrogen reduction. Apart from V@Ru(0001), Ti@Ru(0001) also showed good catalytic performance with a limiting potential of −0.47 V vs. SHE for the production of NH3via enzymatic and consecutive pathways. Both these materials exhibited good thermodynamical stability, selectivity and catalytic activity for the NRR process. Alloying of Ru with a transition metal helped in tuning the binding energy of the adsorbed nitrogen on the surface of the catalyst, thereby increasing its catalytic activity. SAAs seem to be favourable for decreasing the energy barrier by enhancing the binding strength of nitrogen. This work could possibly encourage more experimental and computational studies towards the design of highly efficient single atom alloys for the electrochemical conversion of N2 to NH3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the generous grants of high-performance computing resources provided by Queensland University of Technology, the NCI National Facility, and the Pawsey Supercomputing Centre through the National Computational Merit Allocation Scheme supported by the Australian Government and the Government of Western Australia. A. D. greatly appreciates the financial support by the Australian Research Council under the Discovery Project (DP170103598, DP210100721, and DP210100331).References
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Nitrogen Cycle Electrocatalysis, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. K. Nørskov, Exploring the limits: a low-pressure, low-temperature Haber–Bosch process, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- K. C. MacLeod and P. L. Holland, Recent developments in the homogeneous reduction of dinitrogen by molybdenum and iron, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- G.-F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L.-X. Ding and H. Wang, Advances in Electrocatalytic N2 Reduction—Strategies to Tackle the Selectivity Challenge, Small Methods, 2019, 3, 1800337 CrossRef.
- M. Nazemi and M. A. El-Sayed, Electrochemical Synthesis of Ammonia from N2 and H2O under Ambient Conditions Using Pore-Size-Controlled Hollow Gold Nanocatalysts with Tunable Plasmonic Properties, J. Phys. Chem. Lett., 2018, 9, 5160–5166 CrossRef CAS PubMed.
- H. Xie, Q. Geng, X. Zhu, Y. Luo, L. Chang, X. Niu, X. Shi, A. M. Asiri, S. Gao, Z. Wang and X. Sun, PdP2 nanoparticles–reduced graphene oxide for electrocatalytic N2 conversion to NH3 under ambient conditions, J. Mater. Chem. A, 2019, 7, 24760–24764 RSC.
- R. Zhao, C. Liu, X. Zhang, X. Zhu, P. Wei, L. Ji, Y. Guo, S. Gao, Y. Luo, Z. Wang and X. Sun, An ultrasmall Ru2P nanoparticles–reduced graphene oxide hybrid: an efficient electrocatalyst for NH3 synthesis under ambient conditions, J. Mater. Chem. A, 2020, 8, 77–81 RSC.
- W. Gu, Y. Guo, Q. Li, Y. Tian and K. Chu, Lithium Iron Oxide (LiFeO2) for Electroreduction of Dinitrogen to Ammonia, ACS Appl. Mater. Interfaces, 2020, 12, 37258–37264 CrossRef CAS PubMed.
- Y. Liu, Y. Luo, Q. Li, J. Wang and K. Chu, Bimetallic MnMoO4 with dual-active-centers for highly efficient electrochemical N2 fixation, Chem. Commun., 2020, 56, 10227–10230 RSC.
- H. Nan, Y. Liu, Q. Li, P. Shen and K. Chu, A Janus antimony sulfide catalyst for highly selective N2 electroreduction, Chem. Commun., 2020, 56, 10345–10348 RSC.
- P. Shen, Y. Liu, Q. Li and K. Chu, FeVO4 porous nanorods for electrochemical nitrogen reduction: contribution of the Fe2C–V2C dimer as a dual electron-donation center, Chem. Commun., 2020, 56, 10505–10508 RSC.
- T. Wu, H. Zhao, X. Zhu, Z. Xing, Q. Liu, T. Liu, S. Gao, S. Lu, G. Chen, A. M. Asiri, Y. Zhang and X. Sun, Identifying the Origin of Ti3+ Activity toward Enhanced Electrocatalytic N2 Reduction over TiO2 Nanoparticles Modulated by Mixed-Valent Copper, Adv. Mater., 2020, 32, 2000299 CrossRef CAS PubMed.
- T. Xu, D. Ma, C. Li, Q. Liu, S. Lu, A. M. Asiri, C. Yang and X. Sun, Ambient electrochemical NH3 synthesis from N2 and water enabled by ZrO2 nanoparticles, Chem. Commun., 2020, 56, 3673–3676 RSC.
- Y. Tian, D. Xu, K. Chu, Z. Wei and W. Liu, Metal-free N, S co-doped graphene for efficient and durable nitrogen reduction reaction, J. Mater. Sci., 2019, 54, 1–10 CrossRef.
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Sulfur-doped graphene for efficient electrocatalytic N2-to-NH3 fixation, Chem. Commun., 2019, 55, 3371–3374 RSC.
- K. Chu, Q.-q. Li, Y.-p. Liu, J. Wang and Y.-h. Cheng, Filling the Nitrogen Vacancies with Sulphur Dopants in Graphitic C3N4 for Efficient and Robust Electrocatalytic Nitrogen Reduction, Appl. Catal., B, 2020, 267, 118693 CrossRef CAS.
- Y. Abghoui, S. B. Sigtryggsson and E. Skúlason, Biomimetic Nitrogen Fixation Catalyzed by Transition Metal Sulfide Surfaces in an Electrolytic Cell, ChemSusChem, 2019, 12, 4265–4273 CrossRef CAS PubMed.
- M. Gudmundsson, V. Ellingsson, E. Skúlason and Y. Abghoui, Optimizing Nitrogen Reduction Reaction on Nitrides: A Computational Study on Crystallographic Orientation, Top. Catal., 2021, 1–10 Search PubMed.
- H. Li, Y. Liu, K. Chen, J. T. Margraf, Y. Li and K. Reuter, Subgroup Discovery Points to the Prominent Role of Charge Transfer in Breaking Nitrogen Scaling Relations at Single-Atom Catalysts on VS2, ACS Catal., 2021, 11, 7906–7914 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Electrochemical ammonia synthesis: mechanistic understanding and catalyst design, Chem, 2021, 7, 1708–1754 CAS.
- Y.-p. Liu, Y.-b. Li, H. Zhang and K. Chu, Boosted Electrocatalytic N2 Reduction on Fluorine-Doped SnO2 Mesoporous Nanosheets, Inorg. Chem., 2019, 58, 10424–10431 CrossRef CAS PubMed.
- T. Wang, L. Xia, J.-J. Yang, H. Wang, W.-H. Fang, H. Chen, D. Tang, A. M. Asiri, Y. Luo, G. Cui and X. Sun, Electrocatalytic N2-to-NH3 conversion using oxygen-doped graphene: experimental and theoretical studies, Chem. Commun., 2019, 55, 7502–7505 RSC.
- X.-h. Wang, J. Wang, Y.-b. Li and K. Chu, Nitrogen-Doped NiO Nanosheet Array for Boosted Electrocatalytic N2 Reduction, ChemCatChem, 2019, 11, 4529–4536 CrossRef CAS.
- K. Chu, Y.-p. Liu, Y.-h. Cheng and Q.-q. Li, Synergistic boron-dopants and boron-induced oxygen vacancies in MnO2 nanosheets to promote electrocatalytic nitrogen reduction, J. Mater. Chem. A, 2020, 8, 5200–5208 RSC.
- K. Chu, J. Wang, Y.-p. Liu, Q.-q. Li and Y.-l. Guo, Mo-doped SnS2 with enriched S-vacancies for highly efficient electrocatalytic N2 reduction: the critical role of the Mo–Sn–Sn trimer, J. Mater. Chem. A, 2020, 8, 7117–7124 RSC.
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, Activating VS2 basal planes for enhanced NRR electrocatalysis: the synergistic role of S-vacancies and B dopants, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC.
- Y.-b. Li, Y.-p. Liu, J. Wang, Y.-l. Guo and K. Chu, Plasma-engineered NiO nanosheets with enriched oxygen vacancies for enhanced electrocatalytic nitrogen fixation, Inorg. Chem. Front., 2020, 7, 455–463 RSC.
- C. Fu, Y. Li and H. Wei, Double boron atom-doped graphdiynes as efficient metal-free electrocatalysts for nitrogen reduction into ammonia: a first-principles study, Phys. Chem. Chem. Phys., 2021, 23, 17683–17692 RSC.
- K. Chu, Y.-p. Liu, Y.-b. Li, H. Zhang and Y. Tian, Efficient electrocatalytic N2 reduction on CoO quantum dots, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC.
- K. Chu, Y.-p. Liu, J. Wang and H. Zhang, NiO Nanodots on Graphene for Efficient Electrochemical N2 Reduction to NH3, ACS Appl. Energy Mater., 2019, 2, 2288–2295 CrossRef CAS.
- Y.-p. Liu, Y.-b. Li, D.-j. Huang, H. Zhang and K. Chu, ZnO Quantum Dots Coupled with Graphene toward Electrocatalytic N2 Reduction: Experimental and DFT Investigations, Chem.–Eur. J., 2019, 25, 11933–11939 CrossRef CAS PubMed.
- K. Chu, Y.-h. Cheng, Q.-q. Li, Y.-p. Liu and Y. Tian, Fe-doping induced morphological changes, oxygen vacancies and Ce3+–Ce3+ pairs in CeO2 for promoting electrocatalytic nitrogen fixation, J. Mater. Chem. A, 2020, 8, 5865–5873 RSC.
- K. Chu, H. Nan, Q. Li, Y. Guo, Y. Tian and W. Liu, Amorphous MoS3 enriched with sulfur vacancies for efficient electrocatalytic nitrogen reduction, J. Energy Chem., 2021, 53, 132–138 CrossRef.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. Sykes, Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Pt/Cu single-atom alloys as coke-resistant catalysts for efficient C–H activation, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed.
- J. Liu, M. B. Uhlman, M. M. Montemore, A. Trimpalis, G. Giannakakis, J. Shan, S. Cao, R. T. Hannagan, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Integrated Catalysis-Surface Science-Theory Approach to Understand Selectivity in the Hydrogenation of 1-Hexyne to 1-Hexene on PdAu Single-Atom Alloy Catalysts, ACS Catal., 2019, 9, 8757–8765 CrossRef CAS.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Single-Atom Alloy Catalysis, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- R. Réocreux, P. L. Kress, R. T. Hannagan, V. Çınar, M. Stamatakis and E. C. H. Sykes, Controlling Hydrocarbon (De)Hydrogenation Pathways with Bifunctional PtCu Single-Atom Alloys, J. Phys. Chem. Lett., 2020, 11, 8751–8757 CrossRef PubMed.
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Selective hydrogenation of 1,3-butadiene on platinum–copper alloys at the single-atom limit, Nat. Commun., 2015, 6, 8550 CrossRef PubMed.
- F. R. Lucci, M. T. Darby, M. F. G. Mattera, C. J. Ivimey, A. J. Therrien, A. Michaelides, M. Stamatakis and E. C. H. Sykes, Controlling Hydrogen Activation, Spillover, and Desorption with Pd–Au Single-Atom Alloys, J. Phys. Chem. Lett., 2016, 7, 480–485 CrossRef CAS PubMed.
- M. D. Marcinkowski, J. Liu, C. J. Murphy, M. L. Liriano, N. A. Wasio, F. R. Lucci, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Selective Formic Acid Dehydrogenation on Pt–Cu Single-Atom Alloys, ACS Catal., 2017, 7, 413–420 CrossRef CAS.
- F. R. Lucci, M. D. Marcinkowski, T. J. Lawton and E. C. H. Sykes, H2 Activation and Spillover on Catalytically Relevant Pt–Cu Single Atom Alloys, J. Phys. Chem. C, 2015, 119, 24351–24357 CrossRef CAS.
- Q. Fu and Y. Luo, Active Sites of Pd-Doped Flat and Stepped Cu(111) Surfaces for H2 Dissociation in Heterogeneous Catalytic Hydrogenation, ACS Catal., 2013, 3, 1245–1252 CrossRef CAS.
- H. L. Tierney, A. E. Baber, J. R. Kitchin and E. C. Sykes, Hydrogen dissociation and spillover on individual isolated palladium atoms, Phys. Rev. Lett., 2009, 103, 246102 CrossRef PubMed.
- G. Zheng, Y. Li, X. Qian, G. Yao, Z. Tian, X. Zhang and L. Chen, High-Throughput Screening of a Single-Atom Alloy for Electroreduction of Dinitrogen to Ammonia, ACS Appl. Mater. Interfaces, 2021, 13, 16336–16344 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Electride support boosts nitrogen dissociation over ruthenium catalyst and shifts the bottleneck in ammonia synthesis, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- S. Back and Y. Jung, On the mechanism of electrochemical ammonia synthesis on the Ru catalyst, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- Y. Cao, Y. Gao, H. Zhou, X. Chen, H. Hu, S. Deng, X. Zhong, G. Zhuang and J. Wang, Highly Efficient Ammonia Synthesis Electrocatalyst: Single Ru Atom on Naturally Nanoporous Carbon Materials, Adv. Theory Simul., 2018, 1, 1800018 CrossRef.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Achieving a Record-High Yield Rate of 120.9 for N2 Electrochemical Reduction over Ru Single-Atom Catalysts, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, Conversion of dinitrogen to ammonia on Ru atoms supported on boron sheets: a DFT study, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC.
- E. Tayyebi, Y. Abghoui and E. Skúlason, Elucidating the Mechanism of Electrochemical N2 Reduction at the Ru(0001) Electrode, ACS Catal., 2019, 9, 11137–11145 CrossRef CAS.
- G.-R. Xu, M. Batmunkh, S. Donne, H. Jin, J.-X. Jiang, Y. Chen and T. Ma, Ruthenium(III) polyethyleneimine complexes for bifunctional ammonia production and biomass upgrading, J. Mater. Chem. A, 2019, 7, 25433–25440 RSC.
- Y. Yao, H. Wang, X.-z. Yuan, H. Li and M. Shao, Electrochemical Nitrogen Reduction Reaction on Ruthenium, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- L. Lin, L. Gao, K. Xie, R. Jiang and S. Lin, Ru–polyoxometalate as a single-atom electrocatalyst for N2 reduction to NH3 with high selectivity at applied voltage: a perspective from DFT studies, Phys. Chem. Chem. Phys., 2020, 22, 7234–7240 RSC.
- A. Liu, M. Gao, Y. Gao, X. Ren, Y. Yang, Q. Yang, Y. Li, L. Gao, X. Liang and T. Ma, DFT study of Ru/graphene as high-performance electrocatalyst for NRR, Inorg. Chem. Commun., 2020, 120, 108169 CrossRef.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, Q. Yang, W. Guan, L. Gao, X. Liang and T. Ma, A two-dimensional Ru@MXene catalyst for highly selective ambient electrocatalytic nitrogen reduction, Nanoscale, 2020, 12, 10933–10938 RSC.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- M. Zhao, C. Guo, L. Gao, X. Kuang, H. Yang, X. Ma, C. Liu, X. Liu, X. Sun and Q. Wei, Vanadium-doped NiS2 porous nanospheres with high selectivity and stability for the electroreduction of nitrogen to ammonia, Inorg. Chem. Front., 2021, 8, 3266–3272 RSC.
- T. Wu, W. Kong, Y. Zhang, Z. Xing, J. Zhao, T. Wang, X. Shi, Y. Luo and X. Sun, Greatly Enhanced Electrocatalytic N2 Reduction on TiO2via V Doping, Small Methods, 2019, 3, 1900356 CrossRef CAS.
- L. Niu, D. Wang, K. Xu, W. Hao, L. An, Z. Kang and Z. Sun, Tuning the Performance of Nitrogen Reduction Reaction by Balancing the Reactivity of N2 and the Desorption of NH3, Nano Research, 2021 Search PubMed.
- X. Liang, X. Deng, C. Guo and C.-M. L. Wu, Activity origin and design principles for atomic vanadium anchoring on phosphorene monolayer for nitrogen reduction reaction, Nano Res., 2020, 13, 2925–2932 CrossRef CAS.
- G. Kresse, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, A Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- E. Paquet and H. L. Viktor, Computational Methods for Ab Initio Molecular Dynamics, Adv. Chem., 2018, 2018, 9839641 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Bloechl, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: a tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, The Challenge of Electrochemical Ammonia Synthesis: A New Perspective on the Role of Nitrogen Scaling Relations, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- L. Li, X. Wang, H. Guo, G. Yao, H. Yu, Z. Tian, B. Li and L. Chen, Theoretical Screening of Single Transition Metal Atoms Embedded in MXene Defects as Superior Electrocatalyst of Nitrogen Reduction Reaction, Small Methods, 2019, 3, 1900337 CrossRef CAS.
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, A General Two-Step Strategy–Based High-Throughput Screening of Single Atom Catalysts for Nitrogen Fixation, Small Methods, 2019, 3, 1800376 CrossRef.
- G. Zheng, L. Li, S. Hao, X. Zhang, Z. Tian and L. Chen, Double Atom Catalysts: Heteronuclear Transition Metal Dimer Anchored on Nitrogen-Doped Graphene as Superior Electrocatalyst for Nitrogen Reduction Reaction, Adv. Theory Simul., 2020, 3, 2000190 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, Metal-Free Single Atom Catalyst for N2 Fixation Driven by Visible Light, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- X. Chen, X. Zhao, Z. Kong, W.-J. Ong and N. Li, Unravelling the electrochemical mechanisms for nitrogen fixation on single transition metal atoms embedded in defective graphitic carbon nitride, J. Mater. Chem. A, 2018, 6, 21941–21948 RSC.
- X. Zhang, A. Chen, Z. Zhang and Z. Zhou, Double-atom catalysts: transition metal dimer-anchored C 2N monolayers as N2 fixation electrocatalysts, J. Mater. Chem. A, 2018, 6, 18599–18604 RSC.
- Q. Cui, G. Qin, W. Wang, K. R. Geethalakshmi, A. Du and Q. Sun, Mo-based 2D MOF as a highly efficient electrocatalyst for reduction of N2 to NH3: a density functional theory study, J. Mater. Chem. A, 2019, 7, 14510–14518 RSC.
- X. Mao, Z. Gu, C. Yan and A. Du, Unlocking the potential of ruthenium catalysts for nitrogen fixation with subsurface oxygen, J. Mater. Chem. A, 2021, 9, 6575–6582 RSC.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Role of Steps in N2 Activation on Ru(0001), Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- Á. Logadóttir and J. K. Nørskov, Ammonia synthesis over a Ru(0001) surface studied by density functional calculations, J. Catal., 2003, 220, 273–279 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08246a |
| This journal is © The Royal Society of Chemistry 2022 |