
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the role of the strong field ligands in [FeFe] hydrogenase: spectroscopic and functional characterization of a semi-synthetic mono-cyanide active site†
Marco
Lorenzi‡
 a,
Joe
Gellett‡
b,
Afridi
Zamader
ac,
Moritz
Senger
d,
Zehui
Duan
b,
Patricia
Rodríguez-Maciá
*b and
Gustav
Berggren
*a
a,
Joe
Gellett‡
b,
Afridi
Zamader
ac,
Moritz
Senger
d,
Zehui
Duan
b,
Patricia
Rodríguez-Maciá
*b and
Gustav
Berggren
*a
aDepartment of Chemistry – Ångström, Molecular Biomimetics, Uppsala University, Lägerhyddsvägen 1, 75120 Uppsala, Sweden. E-mail: Gustav.berggren@kemi.uu.se
bDepartment of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, OX1 3QR, UK. E-mail: patricia.rodriguezmacia@chem.ox.ac.uk
cLaboratoire de Chimie et Biologie des Metaux, iRTSV-LCBM/Biocat, Commissariat à l’Energie Atomique (CEA) Grenoble, 17, Rue des Martyrs, UMR 5249, 38054 Grenoble, Cedex 09, France
dDepartment of Chemistry – Ångström, Physical Chemistry, Uppsala University, Lägerhyddsvägen 1, 75120 Uppsala, Sweden
First published on 11th August 2022
Abstract
Artificial maturation of hydrogenases provides a path towards generating new semi-synthetic enzymes with novel catalytic properties. Here enzymes featuring a synthetic asymmetric mono-cyanide cofactor have been prepared using two different hydrogenase scaffolds. Their structure and reactivity was investigated in order to elucidate the design rationale behind the native di-cyanide cofactor, and by extension the second coordination sphere of the active-site pocket. Surprisingly, the choice of host enzyme was found to have a dramatic impact on reactivity. Moreover, the study shows that synthetic manipulations of the active-site can significantly increase inhibitor tolerance, as compared to native [FeFe] hydrogenase, while retaining the enzyme's native capacity for reversible catalysis.
Introduction
[FeFe] hydrogenases are gas-processing enzymes, catalyzing the interconversion of H+ and H2. They have been extensively studied due to their remarkable efficiencies,1 and often serve as model systems for (redox) catalysis.2–4 Albeit a diverse enzyme family, all [FeFe] hydrogenases feature the so-called H-domain that harbors the active-site, which contains a hexanuclear iron complex: the “H-cluster” (Fig. 1, panel A). The H-cluster can be regarded as a canonical iron–sulfur cluster ([4Fe–4S]H) connected to an organometallic binuclear iron complex ([2Fe]H) via a bridging cysteine. Each iron ion in the [2Fe]H subsite is further coordinated by one terminal CO and CN− ligand while its dimeric structure is enforced by a bridging CO and an aza-dithiolate ligand (–SCH2NHCH2S–, adt).5–8 Thus, the only protein derived ligand of the [2Fe]H subsite is the cysteine that connects it to the [4Fe–4S] cluster. Still, the [2Fe]H subsite is further anchored to the protein matrix through electrostatic interactions involving the cyanide ligands and specific active-site pocket residues (Fig. 1, panel A). The importance of the latter binding is underscored by the observation that those residues are among the few that are strictly conserved within all known subclasses of [FeFe] hydrogenase.9–11 A key structural feature of [2Fe]H is the so-called “rotated structure” of the distal Fe ion (Fed), with a bridging CO ligand and two diatomic ligands in a basal position. This geometry is considered critical for enabling heterolytic cleavage of H2via a frustrated Lewis pair (FLP) mechanism involving the open coordination site placed in an apical position of the Fed ion and the amine group of the adt ligand. Generating structural and functional mimics of the H-cluster and the [2Fe]H subsite has been the focus of intense efforts in the organometallic community.12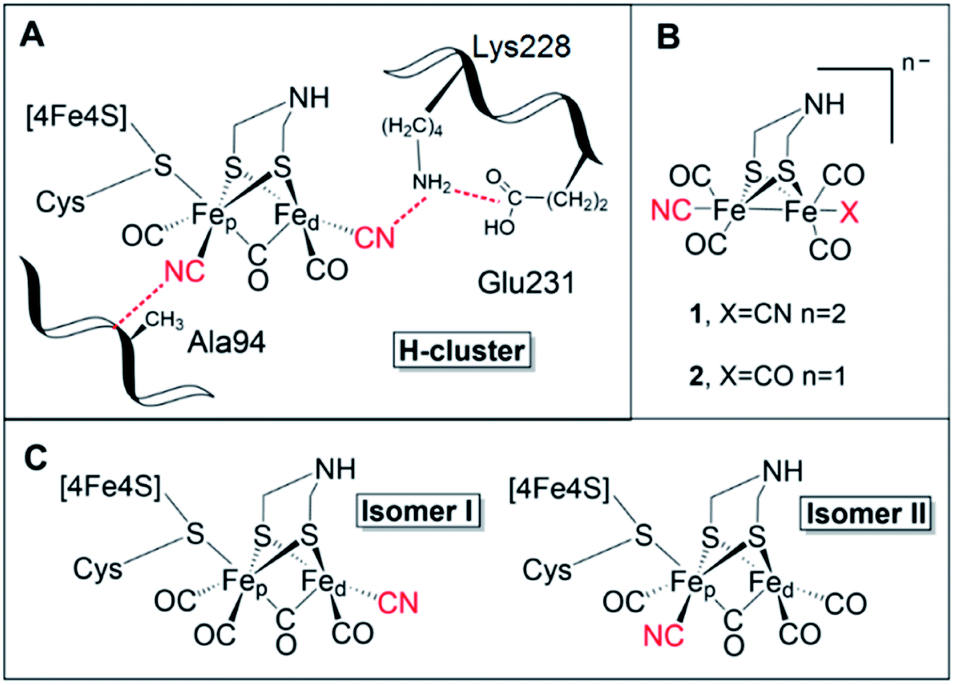 | ||
| Fig. 1 (A) Schematic representation of the active site of [FeFe] hydrogenase, the H-cluster, and selected amino acids (numbering based on CrHydA1). (B) The synthetic mimics used in this study [Fe2(μ-adt)(CO)4(CN)2]2− (1) and [Fe2(μ-adt)(CO)5(CN)]− (2). (C) Possible H-cluster isomer forms following maturation of apo-hydrogenase using complex 2. Cyanide ligands are highlighted in red. Possible rotamers of isomer II are shown in Fig. S9.† | ||
The strong field ligands CO and CN− are highly unusual in biology, but are critical for stabilizing the Fe ions in low oxidation and spin states. The [2Fe]H subsite cycles between Fe(II) and Fe(I) during the catalytic cycle, with the so-called super reduced state representing the most reduced catalytic intermediate. The latter has been described with a Fe(I)Fe(I) valence in the binuclear site and a reduced [4Fe–4S]H+ cluster.13
The biosynthesis and insertion of the [2Fe]H subsite is dependent on a minimum of three [FeFe] hydrogenase-specific maturation enzymes.14 Standard heterologous expression of these enzymes consequently yields inactive apo-[FeFe] hydrogenase, containing the [4Fe–4S]H cluster, but lacking the [2Fe]H subsite.15 However, the maturation machinery can be circumvented via the process of “artificial maturation”. Indeed, apo-[FeFe] hydrogenase treated with the synthetic [2Fe]H mimic [(μ-adt)Fe2(CO)4(CN)2]2− (1) (Fig. 1, panel B) yields an enzyme identical to the one found in nature, and alternative cofactors can be generated via suitable synthetic [2Fe]H pre-cursors.7,16 Artificial maturation has to-date primarily been a tool for elucidating mechanistic aspects of native [FeFe] hydrogenases,17 but it also holds the promise of developing semi-synthetic hydrogenases with new properties operating both in vitro and in vivo.7,18,19
A wide range of semi-synthetic hydrogenases incorporating structurally modified [2Fe]H mimics have been reported, although few have been studied in-depth.20–22 In this context, the mono-cyanide version of complex 1, [(μ-adt)Fe2(CO)5(CN)]− (2) (Fig. 1, panel B), represents a particularly intriguing model to elucidate the role of the biologically unique ligand structure of the H-cluster. Other than complex 1, and its azadiselenate analogue,7,18,21 complex 2 is the only reported [2Fe]H mimic capable of generating a semi-synthetic hydrogenase with significant catalytic activity for H2 conversion, as observed in earlier screening efforts.20 However, as no detailed characterization of the system has been reported to-date, how complex 2 achieves its high activity upon incorporation into the HydA1 hydrogenase scaffold from Chlamydomonas reinhardtii (CrHydA1) remains to be understood.
Apart from its activity when incorporated into CrHydA1, complex 2 is also highly interesting from a molecular design perspective. It not only features a decreased negative charge relative to 1, but the complex also displays intrinsic asymmetry. Consequently, 2 can yield two different isomers of the resulting semi-synthetic H-cluster (Fig. 1, panel C). Each of these two isomers would lack the second-coordination sphere interactions associated with either the proximal (pCN) or distal (dCN) cyanide ligand, which by extension could result in different ligand geometries of the [2Fe]H subsite. Thus, semi-synthetic [FeFe] hydrogenases incorporating 2 allow us to probe both the influence of electron density as well as structural factors on the reactivity of the H-cluster.
Herein, we report the characterization of 2 incorporated into two different [FeFe] hydrogenase scaffolds, i.e. the monomeric HydA1 enzyme from Chlamydomonas reinhardtii (Cr) and the dimeric HydAB from Desulfovibrio desulfuricans (Dd). The active-site pocket of both enzymes is well-conserved. However, the DdHydAB enzyme also contains two additional [4Fe–4S] clusters (so-called F-clusters), which serve as relays to ensure efficient electron transfer between the buried active site and the protein surface.1 Our data shows that the mono-cyanide complex is able to bind the [4Fe–4S]H cluster and strongly favours isomer II in both hydrogenase scaffolds (Fig. 1, panel C). Still, the H-clusters display distinct electronic structures in HydA1 and HydAB. Interestingly, the activity of the resulting semi-synthetic hydrogenase is strongly dependent on the choice of host protein.
Results and discussion
Complex 2 was successfully introduced into the two different [FeFe] hydrogenase enzymes to yield 2-HydA1 and 2-HydAB, respectively. Similar to what is observed during maturation with complex 1, the reaction with 2 proceeded rapidly with HydA1, while incubation for 48 h was required with HydAB. After purification, both of the resulting holo-enzymes displayed distinct FTIR spectra, with sharp H-cluster-like features (Fig. 2 and Table S1†), indicative of the complex being inserted inside the protein scaffold.7 The ‘as prepared’ 2-HydAB FTIR spectrum (Fig. 2, panel A) displays a pure state represented by five narrow bands. Four distinct sharp bands in the 1800–2000 cm−1 region (1835, 1903, 1923 and 1969 cm−1) corresponding to the four CO ligands, and one high-energy band at 2057 cm−1 corresponding to the CN− ligand. We note that the lower frequency CO band (1835 cm−1) appears at higher energy than the bridging CO (μ-CO) in the Hox and Hred states of the ‘native-like’ 1-HydAB (1802 and 1789 cm−1, respectively), potentially reflecting a ‘semi-bridging’ CO ligand as previously proposed for the HredH+ state.13,23 In stark contrast to the ‘native-like’ 1-HydAB, 2-HydAB is not sensitive to light,24 and no change in the IR spectrum was observed after exposing the ‘as prepared’ sample to white-light overnight (data not shown).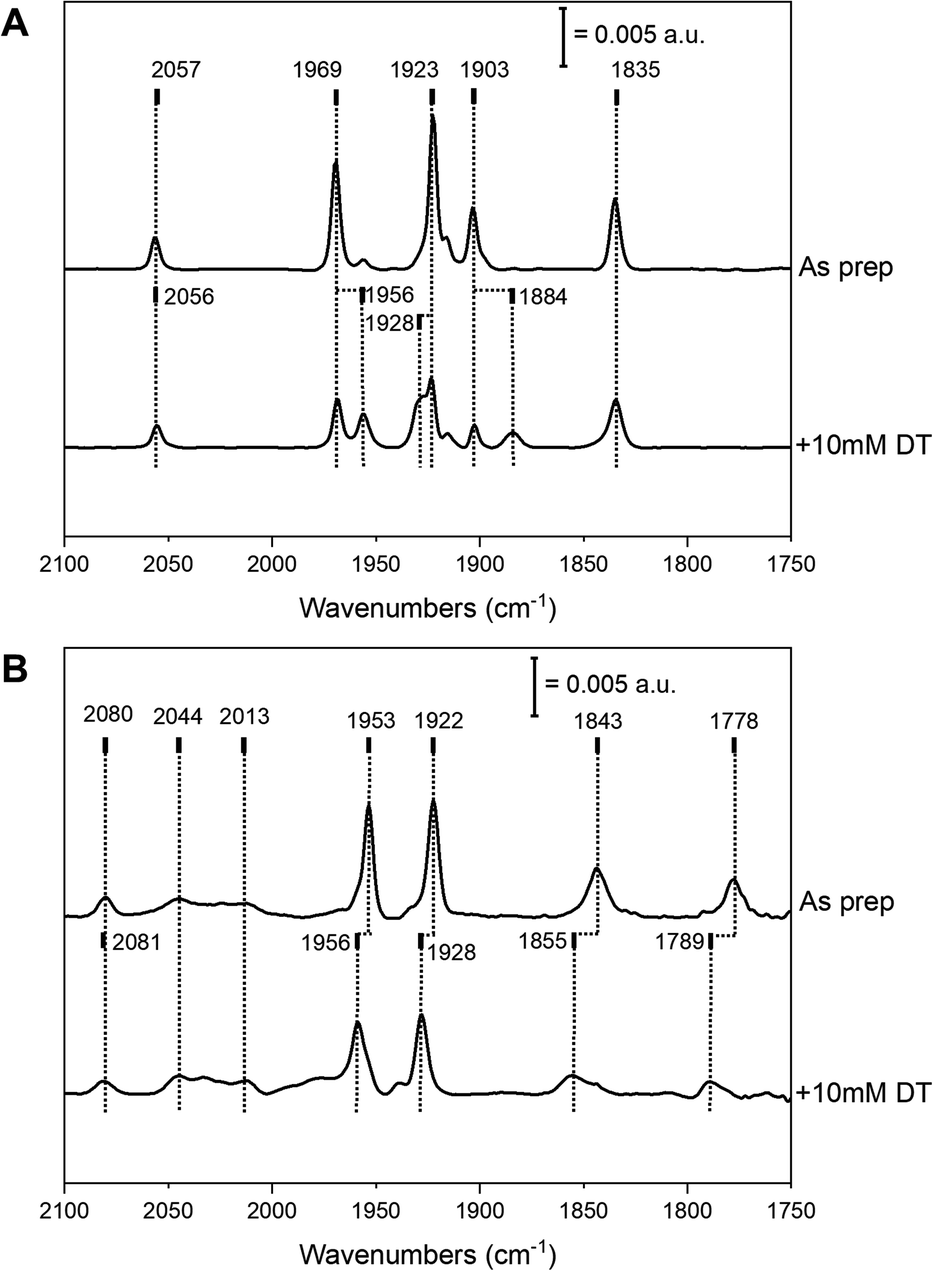 | ||
| Fig. 2 FTIR spectra obtained for 2-HydAB and 2-HydA1. (A): FTIR spectra of 2-HydAB ‘as prepared’ (upper trace) and 10 mM NaDT reduced (bottom trace), recorded in a transmission cell ([2-HydAB] ≈ 1 mM). (B): FTIR spectra of 2-HydA1 ‘as prepared’ after white-light treatment (upper trace) and 10 mM NaDT reduced (bottom trace), recorded on a hydrated film by ATR-FTIR. The most prominent bands are marked by dotted lines and the corresponding frequencies are indicated on top. Spectra collected at room temperature in pH 8 buffer, with a 2 cm−1 spectral resolution. | ||
The FTIR spectrum of 2-HydA1 displayed an overall spectral shape similar to that of 2-HydAB, albeit shifts of up to 60 cm−1 are observed for low and high frequency bands. Additional broader bands were also present in the spectrum, attributable to complex 2 adventitiously binding to the surface of the enzyme (indicated at 2044 and 2013 cm−1 in Fig. 2, panel B). Indeed, these broad features can be reproduced by maturating a metal-free HydA1 protein scaffold, lacking the anchoring [4Fe–4S]H cluster (Fig. S1,† panel A). Moreover, the broad features exhibit light sensitivity, greatly fading upon white light illumination (Fig. S1,† panel B). Conversely, the sharp bands associated with the H-cluster appeared practically insensitive to light also in 2-HydA1. More specifically, the spectrum of light-treated 2-HydA1 is also dominated by one set of five ligand bands (Fig. 2, panel B, as prepared). The same bands were observed in the original report on 2-HydA1,20 although in combination with additional signals. As compared to 2-HydAB, the presence of a broad signal at a low energy (1778 cm−1) is particularly noteworthy. This band is substantially lower in energy that the μ-CO band in ‘native-like’ 1-HydA1. This strongly supports the presence of a μ-CO ligand in 2-HydA1.
In both 2-HydA1 and 2-HydAB, only one distinct CN band is observable, at 2080 and 2057 cm−1 respectively, in contrast to what is observed for apo-[FeFe] hydrogenases matured with complex 1 (a summary of observed band positions and a comparison to 1-HydA1 and 1-HydAB is provided in Table S1†).17,25 In combination with the distinct positions for the CO vibrations, this confirms the mono-cyanide nature of the obtained H-cluster variants in both host proteins. In addition, our data supports the notion that the H-cluster population in the ‘as prepared’ samples is highly homogeneous in both 2-HydA1 and 2-HydAB, suggesting that the active-site pocket strongly favors one possible isomer (Fig. 1, panel C). However, the frequency differences of the bands in the FTIR spectra (e.g. the large shift of the low frequency CO band, 1778 cm−1 and 1835 cm−1 for 2-HydA1 and 2-HydAB respectively) reveals variations in the geometry and potentially oxidation state of the [2Fe]H subsite in the two enzymes.
Addition of the reducing agent sodium dithionite (NaDT, E0′ = − 0.66 V vs. NHE) to 2-HydA1 triggered a hypsochromic (blue-)shift of several bands (Fig. 2, panel B), instead of the expected bathochromic (red-)shifts usually associated with reduction of the H-cluster. The two states observed in the presence and absence of NaDT rationalizes the FTIR spectra originally reported for 2-HydA1, as they appear to reflect a mixture of these two species.202-HydAB displayed more limited reactivity towards NaDT, but partial formation of a new state was discernible from new bands appearing at 1956, 1928 and 1884 cm−1 (Fig. 2, panel A). The magnitude of the shifts implies that they arise from reduction of the [4Fe–4S]H cluster or the F-clusters, rather than the [2Fe]H subsite.26,27 Addition of the stronger reductant, Eu-DTPA (E0′ = −1.09 V vs. NHE) also did not cause complete H-cluster conversion, but resulted in a clearly discernible hypsochromic shift of the high and low frequency bands (Fig. S2†).
Electron paramagnetic resonance (EPR) spectroscopy was employed to obtain additional insight into the structure and oxidation states of the modified H-clusters. Surprisingly, EPR spectra of as-prepared 2-HydA1 (Fig. 3) show a mixture of two different rhombic signals, in contrast with the single set of peaks showing up in FTIR. We attribute this discrepancy to the cryogenic temperatures employed for the EPR measurements. Low temperatures can be expected to freeze different conformations, or rotamers, of the H-cluster that would instead average out in room temperature spectroscopy (Fig. S9†), as observed for related model complexes.28 Distinct effects of applying cryogenic temperatures have also been observed for the native H-cluster.29,30 More specifically, a rotation of the distal iron has previously been proposed to occur upon CO inhibition of the H-cluster,31 and increased structural flexibility is in line with a more facile rotation of the non-cyanide ligated Fe ion, as it would lack the aforementioned stabilizing H-bonding interactions. Moreover, the relatively large anisotropy of the EPR signals and their fast relaxation indicate that both components of the spectrum belong to species with unpaired spin density localized on the [4Fe–4S]H cluster. These results point toward the H-cluster of 2-HydA1 residing in a “super-reduced-like” state (i.e. [4Fe–4S]+-[FeIFeI]H) under conditions that generally yield the oxidized resting state Hox (i.e. [4Fe–4S]2+-[FeIIFeI]H) in native [FeFe]-hydrogenase. Conversely, EPR spectra recorded on as-prepared samples of 2-HydAB did not reveal any discernible signal, further supporting the idea that the enzymes stabilize two different oxidation states of the modified H-cluster. We attribute the EPR silent as-prepared form of 2-HydAB to an HredH+-like oxidation state (i.e. [4Fe–4S]2+-[FeIFeI]H). A di-ferric [FeIIFeII]H subsite could also explain the lack of EPR signal. However, we consider this assignment highly unlikely considering the relatively low frequencies of the carbonyl ligand vibrations. Moreover, considering their relative sigma donating properties, exchanging a CN ligand by a CO ligand will result in stabilization of lower oxidation states.
 | ||
| Fig. 3 EPR spectrum recorded on a 2-HydA1 (150 μM) in 100 mM Tris buffer pH 8 + 200 mM KCl. (a) Experimental data (black) is overlaid with the simulated spectrum (pink dashed line) obtained fitting two rhombic signals. (b) Simulated spectrum of the first rhombic signal (gzyx = 2.085 1.936 1.845). (c) Simulated spectrum of the second rhombic signal (gzyx = 2.105 1.909 1.865). Experimental parameters: frequency: 9.365 GHz, temperature = 10 K; microwave power = 64 μW. | ||
The FTIR data strongly supports the notion of selective formation of one of the two possible isomers, still the question remained open on which was being predominantly formed. To further address this issue, we prepared variants of HydA1 via site-directed mutagenesis, targeting amino acids in the [2Fe]H second and third coordination sphere of the cyanides of the native co-factor (Fig. 1, panel A). In the case of dCN, its environment is closely connected to the maturation channel by which the [2Fe]H precursor is suggested to enter the active-site.15,32 Thus, we decided to prepare the third-coordination sphere E231D variant targeting a glutamate residue involved in a salt-bridge with Lysine228, which in turn is thought to be important to fix the distal CN position. Interestingly, despite repeated efforts, maturation of this mutant with complex 2 could not be achieved. This agrees with previous reports of Lysine228 as the last amino acid in the positively charged maturation channel. Mutations interfering with its positioning unavoidably generate variants defective in cofactor incorporation.32,33 Thus, the A94S variant was also prepared, targeting an alanine residue known to be directly interacting with pCN. The introduction of a polar side chain in this position influences the cyanide stretching mode,33 and causes a distinct red-shift of the only identifiable CN band of 2-HydA1A94S by 20 cm−1 (Fig. S3†). The effect of the A94S mutation can also be observed as a 3–5 cm−1 blue-shift on two CO bands. The comparison is complicated by the appearance of two extra bands in the 2-HydA1A94S FTIR spectrum, at 1959 and 1936 cm−1. As the preparation protocol is identical to that of the wild-type enzyme, they can only be attributed to the introduction of this point-mutation. It is possible that the introduction of a polar residue, which can also act as hydrogen bond donor, could trigger the formation of additional conformations for the mono-CN H-cluster in 2-HydA1A94S. Alternatively, the effect of the mutation could be that of slowing down ligand rotation resulting in an observation analogous to the double signal seen with EPR. Regardless, the distinct shifts observed for the CO bands and in particular the CN band provide compelling evidence for the occurrence of isomer II (Fig. 1, panel C) as the predominant conformation for 2-HydA1. Indeed, the observed shift are strikingly similar to earlier observations with the HydA1A94S variant also for the native cofactor (1-HydA1A94S).33 The formation of isomer II is also indirectly supported by the reactivity observed towards the well-known inhibitors CO and O2. Exposing 2-HydA1 to air resulted in appearance of a small population of a species reminiscent of the Hox–air state observed in TamHydS (Fig. S4†).34 As this state is thought to feature a partially decomposed [2Fe]H subsite with a single Fe ion coordinated by the bridging cysteine, the presence of a band attributable to Fe coordinated CN in this spectrum is consistent with the loss of an all-carbonyl distal Fe ion of isomer II.
In the case of 2-HydAB, the enzyme also exhibits unusual reactivity towards O2, displaying substantially lower O2 sensitivity compared with the native-like 1-HydAB. The IR signature of 2-HydAB was significantly more long-lived than that of 1-HydAB after exposing the two enzymes to air, retaining 45% of the IR signal intensity after 30 min of air exposure for 2-HydAB compared with only 16% after 5 min of exposing 1-HydAB to air (Fig. S5†).
In contrast to the eventual degradation under air, both 2-HydAB and 2-HydA1 appeared completely unreactive towards CO. This is remarkable, as CO is a well-known competitive inhibitor of hydrogenases.35 Especially 1-HydAB has a very high affinity toward CO, and is often isolated in a CO inhibited state (Hox–CO).35,36 However, incubation of as-prepared 2-HydAB under a neat N2 atmosphere for up to 48 h to promote CO release did not result in any significant difference in the FTIR spectrum, strongly arguing against the as-prepared spectrum representing a CO inhibited form of the enzyme. Likewise, exposing as prepared samples of 2-HydA1 and 2-HydAB to 1 atm CO for up to one hour, did not alter the state of the sample, and no extra IR bands could be detected in the IR spectrum (data not shown). Finally, 2-HydAB and 2-HydA1 were treated with one equivalent of KCN to explore the possibility of generating the native di-cyanide cofactor via post-synthetic exchange of a CO ligand. However, after 60 minutes of incubation at pH 8 and room temperature, the FTIR spectra revealed no formation of new species (e.g. no extra CN or CO bands) either for 2-HydAB or 2-HydA1 (data not shown).
Both the band positions of the FTIR spectrum and the characteristics of the EPR signals reveal that the H-cluster of 2-HydA1 and 2-HydAB are clearly distinct from the native H-cluster. Moreover, the decreased anionic charge of complex 2 is expected to result in more positive reduction potentials compared to complex 1.37 We investigated how these changes in redox and structural properties would impact the catalytic behavior of the semi-synthetic mono-cyanide [FeFe] hydrogenases, using a combination of protein film electrochemistry (PFE) and solution activity assays.
As reported by Siebel and coworkers, 2-HydA1 maintains a significant amount of residual H2 evolution activity. In our hands 2-HydA1 show H2 evolution activity values of 223 ± 25 μmol H2 mgprotein−1 min−1, whereas samples of 1-HydA1 display a H2 evolution activity of 370 ± 64 μmol H2 mgprotein−1 min−1. In contrast, 2-HydAB only showed a 1% residual activity for H2 production (84 ± 13 μmol mgprotein−1 min−1) and 0.02% residual activity for H2 oxidation (15.23 ± 0.57 μmol mgprotein−1 min−1).
Similarly, cyclic voltammograms (CVs) recorded with PFE following adsorption of 2-HydAB onto a rotating-disk pyrolytic graphite edge (PGE) electrode, showed only small residual electrocatalytic currents attributable to H+ reduction and H2 oxidation (Fig. S6†). Care should be taken when attributing significance to absolute currents in PFE, as it reflects both intrinsic enzyme activity as well as enzyme coverage on the electrode surface. Still, the CV for 2-HydAB is in line with the data obtained from solution activity assays where only residual activity for both H2 production and H2 oxidation was detected. The inactive nature of 2-HydAB is remarkable, as the native-like 1-HydAB is one of the most active semi-synthetic [FeFe] hydrogenases studied so far, operating reversibly over a wide pH range.26,35,38 Thus, we conclude that in the case of HydAB, the mono-cyanide semi-synthetic enzyme dramatically lowers its activity for H2 conversion.
Conversely, CVs of 2-HydA1 absorbed on a PGE electrode show electrocatalytic current in both directions, displaying the typical behavior of a reversible catalyst, with no overpotential requirement either in the oxidative or in the reductive direction (Fig. 4 and S7†). The Michaelis constant (Km) for H2 was determined to be above 1 atm through chronoamperometric studies under continuously varying H2 concentration (Fig. S8†), as compared to 0.64 ± 0.05 atm reported for 1-HydA1.39 Additionally, the intrinsic catalytic bias of 2-HydA1 was estimated from the ratio between oxidative and reductive currents at 100 mV over-potential. The observed (iox/ired) ratio of ≈1.6 indicates a bias towards H2 oxidation, comparable to what is observed for 1-HydA1 (iox/ired ≈ 1.5).
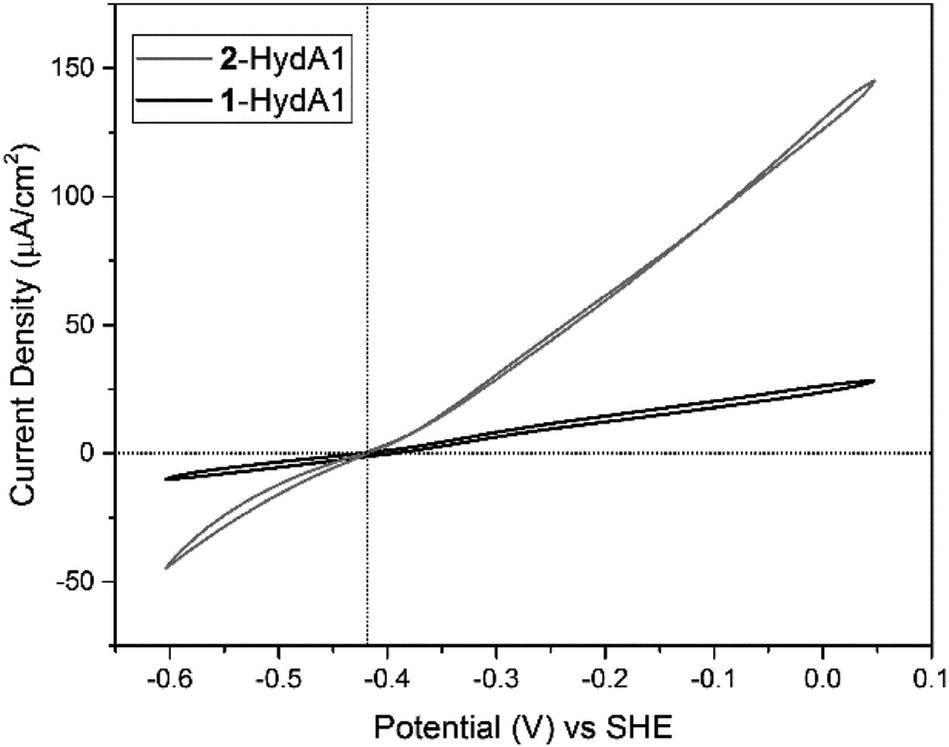 | ||
| Fig. 4 Cyclic voltammograms of 2-HydA1 (black trace) and 1-HydA1 (grey trace) films absorbed on a rotating-disk PGE electrode, measured in pH 7 buffer mix in the absence of halides under 1 atm H2 gas, scan rate: 10 mV s−1, temperature: 30 °C and rotation rate: 3000 rpm. The vertical dotted-line indicates the thermodynamic potential for the 2H+/H2 couple at pH 7 (E0 = −0.413 V, vs. SHE), while the horizontal dotted-line represent the zero current. | ||
Conclusions
In summary, the unsymmetrical H-cluster generated via artificial maturation with the monocyanide complex 2 has been functionally and spectroscopically characterized in-detail. The study employed two different hydrogenase scaffolds, also providing insight into how the protein scaffold tunes the active-site properties. Apart from the complex [(μ-pdt)Fe2(CO)4(CN)2]2− (3, pdt = –SCH2CH2CH2S–), which is analogous to complex 1 but the adt amine is replaced by a methylene group, this is the first time that an H-cluster variant has been prepared and characterized in DdHydAB. In a hydrogenase context, this study sheds new light on the role of both the primary and secondary coordination sphere in structure and reactivity of the H-cluster. The reactivity of 2-HydA1 implies that the presence of a symmetric di-cyanide cofactor in native [FeFe] hydrogenase is not a strict requirement from an electron density perspective. The decreased electron density of the mono-cyanide variant could also at least partially rationalize the lower affinity for binding additional CO ligands, as it lowers the capacity of the [2Fe]H subsite for π back-bonding.In light of these observations, the symmetric di-atomic ligand pattern of the native [2Fe]H subsite is likely an adaption to the biosynthesis of the [2Fe]H subsite, proceeding via dimerization of two Fe(CO)2CN synthons.40,41 In native [FeFe] hydrogenase a high tolerance towards CO can be achieved via changes to the protein scaffold but appears to be connected to a significant decrease in catalytic efficiencies, as exemplified by the putative sensory hydrogenases from Thermoanaerobacter mathranii34,42 and Thermotoga maritima.43 Thus, a mono-cyanide cofactor could provide an evolutionary advantage, as it can combine high activity and CO tolerance. However, it would come with a significant cost, as it would require at least one additional enzyme in the maturation machinery complementing HydG to produce a suitable monomeric Fe-ion precursor.
With regards to the second coordination sphere, K228 (CrHydA1 numbering) is an extremely well conserved residue, and its interaction with the dCN ligand is thought to play a key role in stabilizing the rotated structure of the distal iron, and preventing the formation of dead-end intermediates.44 However, it has been challenging to probe directly, as H-cluster assembly is prevented in K228 variants.45 Despite the inactivity of 2-HydAB, the results for 2-HydA1 shows that the distal CN ligand (dCN), and by extension its H-bonding interactions, does not represent a critical structural feature for catalysis. Still, we note that K228 might be needed for decreasing the electron donation of dCN in the native di-cyanide cofactor.
Additionally, the K228-dCN interaction evidently contributes to the stabilization of the specific rotamer constituting the rotated structure (Fig. 1 and S9†), which holds the open coordination site in an apical position. This notion is supported by the observation of two separate H-cluster species in low temperature EPR measurements of 2-HydA1. In parallel, the significant loss of activity in 2-HydAB can be attributed to the stabilization of an inactive rotamer in the absence of the dCN-K228 interaction (Fig. S9,† rotamer I or II). This is in line with the exceptional stability of the CO-inhibited state Hox–CO in native DdHydAB.35 The high affinity of DdHydAB towards CO is not well understood yet. However, it implies a strong tendency to stabilize a CO ligand in the apical position of the distal Fe ion. We hypothesize that in 2-HydAB, lacking the H-bonding interactions of the dCN ligand enforcing the “rotated structure”, one of the CO ligands of the distal Fe ion is likely to be stabilized in the apical position. This would result in a rotamer where the vacant site is not at the apical position, thus losing the critical FLP interaction with the adt-nitrogen, rationalizing the catalytically inactive nature 2-HydAB (Fig. S9,† rotamers I or II). Such a structural arrangement is likely to be an additional factor contributing to the enhanced O2 and CO tolerance of 2-HydAB, as it would decrease inhibitor accessibility to the [2Fe]H subsite. In short, formation of the key rotated structure must be triggered primarily by other interactions in the active site pocket or protein surrounding, and then subsequently stabilized by K228.
From a molecular design perspective, the 2-HydA1 system allowed us to probe the effect of the H-cluster on the overall catalytic performance in a direct way: exchange of a CN for CO causes a slight decrease in rate for H2 production and an increase in Km, but comes with significant improvements in inhibition tolerance. Furthermore, it does not introduce an over-potential for catalysis in any direction. In a broader context, the reactivity observed for 2-HydA1 provides an example of how rate and over-potential for catalysis are decoupled parameters in (redox) catalysis.2,3 Finally, this study shows that it is possible to rely on the protein scaffold to introduce asymmetric mimics in the hydrogenase active site in a selective fashion, paving the way for highly elaborate catalyst design. Moreover, the observed differences in catalytic activity of 2-HydAB and 2-HydA1 highlights the importance of exploring different hydrogenase scaffolds when constructing semi-synthetic hydrogenases.
Author contributions
Conceptualization: M. L., G. B. and P. R.-M.; methodology: M. L., J. G., P. R.-M.; investigation: M. L., J. G., A. Z., M. S. Z. D.; writing – original draft: M. L., G. B. and P. R.-M.; writing – review & editing: all authors; supervision: G. B. and P. R.-M.; funding acquisition: G. B. and P. R.-M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Research Council (ERC, to GB, Contract No. 714102) as well as the European Union's Horizon 2020 research and innovation program (Marie Skłodowska Curie Grant No. 897555 to MS) are gratefully acknowledged for funding. PR-M thanks the University of Oxford for a Glasstone Research Fellowship and Linacre College Oxford for a Junior Research Fellowship. The University of Oxford Santander/Oxford Travel Awards is gratefully acknowledged for supporting JG's research stay at the University of Uppsala. The authors would also like to express their gratitude to Dr James Birrell for critical reading of the manuscript.Notes and references
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS.
- V. Fourmond, N. Plumeré and C. Léger, Nat. Rev. Chem., 2021, 5, 348–360 CrossRef CAS.
- V. Fourmond, E. S. Wiedner, W. J. Shaw and C. Léger, J. Am. Chem. Soc., 2019, 141, 11269–11285 CrossRef CAS PubMed.
- D. W. Mulder, J. W. Peters and S. Raugei, Chem. Commun., 2021, 57, 713–720 RSC.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- A. Silakov, E. J. Reijerse, S. P. J. Albracht, E. C. Hatchikian and W. Lubitz, J. Am. Chem. Soc., 2007, 129, 11447–11458 CrossRef CAS PubMed.
- L. S. Mészáros, H. Land, H. J. Redman and G. Berggren, Curr. Opin. Green Sustainable Chem., 2021, 32, 100521 CrossRef.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- M. Winkler, J. Esselborn and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 974–985 CrossRef CAS PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, Coord. Chem. Rev., 2021, 449, 214191 CrossRef CAS.
- R. D. Britt, G. Rao and L. Tao, Chem. Sci., 2020, 11, 10313–10323 RSC.
- D. W. Mulder, E. S. Boyd, R. Sarma, R. K. Lange, J. A. Endrizzi, J. B. Broderick and J. W. Peters, Nature, 2010, 465, 248–251 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- N. Khanna, C. Esmieu, L. S. Meszaros, P. Lindblad and G. Berggren, Energy Environ. Sci., 2017, 10, 1563–1567 RSC.
- L. S. Meszaros, B. Nemeth, C. Esmieu, P. Ceccaldi and G. Berggren, Angew. Chem., Int. Ed., 2018, 57, 2596–2599 CrossRef CAS PubMed.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- L. Kertess, F. Wittkamp, C. Sommer, J. Esselborn, O. Rudiger, E. J. Reijerse, E. Hofmann, W. Lubitz, M. Winkler, T. Happe and U. P. Apfel, Dalton Trans., 2017, 46, 16947–16958 RSC.
- C. Sommer, C. P. Richers, W. Lubitz, T. B. Rauchfuss and E. J. Reijerse, Angew. Chem., Int. Ed., 2018, 57, 5429–5432 CrossRef CAS PubMed.
- Y. Nicolet, A. L. de Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- M. Sensi, C. Baffert, V. Fourmond, L. de Gioia, L. Bertini and C. Léger, Sustainable Energy Fuels, 2021, 5, 4248–4260 RSC.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Biochemistry, 2009, 48, 7780–7786 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, K. Pawlak, O. Rüdiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- W. Wang, M. J. Nilges, T. B. Rauchfuss and M. Stein, J. Am. Chem. Soc., 2013, 135, 3633–3639 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- S. T. Stripp, S. Mebs and M. Haumann, Inorg. Chem., 2020, 59, 16474–16488 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, F. Wittkamp, U.-P. Apfel, J. Heberle, M. Haumann and S. T. Stripp, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8454–8459 CrossRef CAS PubMed.
- O. Lampret, J. Esselborn, R. Haas, A. Rutz, R. L. Booth, L. Kertess, F. Wittkamp, C. F. Megarity, F. A. Armstrong, M. Winkler and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15802–15810 CrossRef CAS PubMed.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U.-P. Apfel, E. J. Reijerse, W. Lubitz, O. Rüdiger, T. Happe and M. Winkler, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS PubMed.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Németh, N. Polidori, L. S. Mészáros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
- B. R. Glick, W. G. Martin and S. M. Martin, Can. J. Microbiol., 1980, 26, 1214–1223 CrossRef CAS PubMed.
- V. Fourmond, C. Baffert, K. Sybirna, S. Dementin, A. Abou-Hamdan, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Chem. Commun., 2013, 49, 6840–6842 RSC.
- Y. Zhang, L. Tao, T. J. Woods, R. D. Britt and T. B. Rauchfuss, J. Am. Chem. Soc., 2022, 144, 1534–1538 CrossRef CAS PubMed.
- J. M. Kuchenreuther, W. K. Myers, D. L. M. Suess, T. A. Stich, V. Pelmenschikov, S. A. Shiigi, S. P. Cramer, J. R. Swartz, R. D. Britt and S. J. George, Science, 2014, 343, 424–427 CrossRef CAS.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS PubMed.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, Chem. Sci., 2014, 5, 215–221 RSC.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including experimental details, additional FTIR spectra and PFE data. See https://doi.org/10.1039/d2sc02271k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |