
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual electrochemical and chemical control in atom transfer radical polymerization with copper electrodes†
Francesco
De Bon
a,
Francesca
Lorandi
 bc,
Jorge F. J.
Coelho
a,
Armenio C.
Serra
a,
Krzysztof
Matyjaszewski
c and
Abdirisak A.
Isse
*b
bc,
Jorge F. J.
Coelho
a,
Armenio C.
Serra
a,
Krzysztof
Matyjaszewski
c and
Abdirisak A.
Isse
*b
aCentre for Mechanical Engineering Materials and Processes (CEMMPRE), Department of Chemical Engineering, University of Coimbra, Rua Sílvio Lima, Pólo II, 3030-790 Coimbra, Portugal
bDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, I-35131, Padova, Italy. E-mail: abdirisak.ahmedisse@unipd.it
cDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Ave, 15213, Pittsburgh, PA, USA
First published on 5th May 2022
Abstract
In Atom Transfer Radical Polymerization (ATRP), Cu0 acts as a supplemental activator and reducing agent (SARA ATRP) by activating alkyl halides and (re)generating the CuI activator through a comproportionation reaction, respectively. Cu0 is also an unexplored, exciting metal that can act as a cathode in electrochemically mediated ATRP (eATRP). Contrary to conventional inert electrodes, a Cu cathode can trigger a dual catalyst regeneration, simultaneously driven by electrochemistry and comproportionation, if a free ligand is present in solution. The dual regeneration explored herein allowed for introducing the concept of pulsed galvanostatic electrolysis (PGE) in eATRP. During a PGE, the process alternates between a period of constant current electrolysis and a period with no applied current in which polymerization continues via SARA ATRP. The introduction of no electrolysis periods without compromising the overall polymerization rate and control is very attractive, if large current densities are needed. Moreover, it permits a drastic charge saving, which is of unique value for a future scale-up, as electrochemistry coupled to SARA ATRP saves energy, and shortens the equipment usage.
Introduction
Atom transfer radical polymerization (ATRP) is among the most explored reversible deactivation radical polymerizations (RDRPs), due to its compatibility with several monomers, the use of inexpensive reactants at T <100 °C, and the possibility to be performed either in bulk or in monomer/solvent mixtures, under homogeneous or heterogeneous conditions.1,2 The most widely used ATRP catalysts are Cu complexes with polydentate nitrogen-based ligands (L).3,4 The polymerization is triggered by the reaction of [CuIL]+ with an alkyl halide initiator (RX, X = Br, Cl), whereby a propagating radical is formed together with the oxidized copper complex, [X–CuIIL]+. The radical adds only few monomer units before it is quenched by [X–CuIIL]+ to a halogen-capped dormant species (Pn–X) and the starting CuI complex (Scheme 1). Conventional ATRP employs a high amount of catalyst, which must be removed from the polymer through expensive and cumbersome methods. To reduce the catalyst to part per million (ppm) levels without affecting the polymerization control, new ATRP methods have been developed,1 including electrochemically mediated ATRP (eATRP) and supplemental activator and reducing agent (SARA) ATRP (Scheme 1).5,6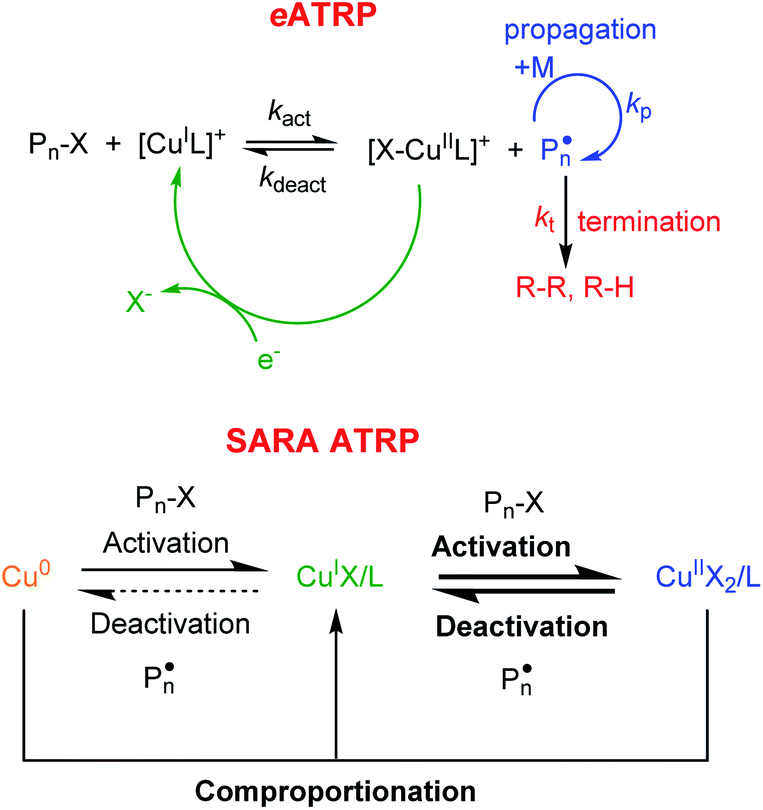 | ||
| Scheme 1 Mechanism of copper-catalyzed eATRP and SARA ATRP. In SARA ATRP bold lines indicate the main reaction routes. | ||
SARA ATRP exploits the comproportionation reaction between CuII species and Cu0 in the presence of free L to (re)generate CuI species (Scheme 1). SARA ATRP has been used for several monomers, such as (meth)acrylates and vinyl chloride.7–19 The method allows temporal control of polymerization as the reaction can be stopped and re-started by lifting and re-immersing a Cu wire in the polymerization mixture.20eATRP permits: (i) the (re)generation of the CuI activator with no by-products, (ii) fine tuning of the reaction rate, and (iii) temporal control of polymerization through the applied potential (or current). The polymerization starts upon generation of the activator [CuIL]+ by applying an appropriate potential (Eapp) or cathodic current (Iapp) to reduce CuII to CuI at an electrode surface. Cycling Eapp or Iapp between suitable values allows for stopping and restarting the polymerization.21 In addition, eATRP can be stopped by completely excluding the electrochemical stimulus.22 The (re)generation of the catalyst in the activator form can occur at the surface of non-noble metal cathodes such as stainless steel SS304, NiCr alloy, Ti or glassy carbon.23,24 Aluminum is most commonly used as a sacrificial anode in a single compartment cell.25 For industrial applications, noble metal cathodes with large area are too expensive, therefore non-noble metal alternatives were successfully tested and implemented.26,27
Remarkably, Cu was never tested as an electrode in eATRP, despite the abundance and relatively low cost of the metal. In principle, a Cu cathode can provide electrons to trigger the polymerization, as demonstrated for other non-noble metals. However, Cu0 is also an activator of alkyl halide dormant species and more importantly, in the presence of free ligand, the comproportionation reaction between Cu0 and CuII species can re-generate [CuIL]+ (as in SARA ATRP). The use of Cu cathodes can therefore imply that a dual regeneration of [CuIL]+ is at place, via both comproportionation and electrochemical reduction. In the first part of this study, Cu was employed as a cathode for a model eATRP system. This allowed evaluating the relative contributions of SARA mechanism and electrochemical reduction, exploring their potential synergy or opposition.
In addition, Cu can replace Al as a sacrificial anode. In principle, Cu ions released in solution following the anodic oxidation reaction are reduced again at the cathode without affecting the polymerization. However, if the ligand (L) is in excess, Cu complexes can be eventually formed and participate in the polymerization mechanism, thus the contribution of SARA ATRP cannot be neglected. Yet, when using a Cu anode, eventual side reactions can lead to contamination and more difficult purification of the final polymer, making this setup impractical for a pilot/industrial plant. Nevertheless, in eATRP with Cu as a both cathode and sacrificial anode, Cu ions can be removed by a facile and clean electrodeposition onto the Cu cathode. The combination of Cu anode and cathode was attempted on a selected model system in the second part of this work. However, the cost-benefit equilibrium tends to favor Al over Cu, as Al is less expensive than Cu, and three electrons are needed to release one Al3+ ion in solution, while one or two electrons are needed to release a Cu ion. Therefore, the combination of Al anode and Cu cathode was also studied. The different electrochemical setups employed herein are shown in Fig. 1.
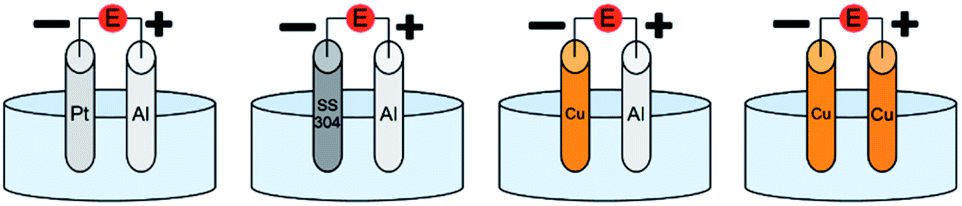 | ||
| Fig. 1 Schematic representation of a general undivided cell with Pt, SS304 and Cu cathodes combined with an Al or Cu sacrificial anode. Reference electrode was omitted for clarity. | ||
The electrochemical control over polymerizations offers a still unexplored degree of freedom. One can modulate the waveform of the electrochemical stimulus, switching from a conventional continuous galvanostatic electrolysis (CGE) to a pulsed galvanostatic electrolysis (PGE) by introducing a duty cycle. Inspired by an electrochemical switch developed for the eATRP of styrene,22 we attempted an on-off keying, which closely resembles a pulsed wave. This is a non-sinusoidal periodic waveform in which the amplitude alternates at a steady frequency between a minimum and a maximum value, which are held for the same duration. The ratio of the high period to the total period of a pulsed wave is called duty cycle. A perfect pulsed wave has a 50% duty cycle.
From a practical point-of-view, the use of galvanostatic electrolysis in eATRP is more appealing than a potentiostatic mode, as it requires a simpler and less expensive equipment. However, to maintain good control over the polymerization, galvanostatic eATRP is generally conducted by applying a sequence of Iapp values that mimics the current profile recorded during a similar potentiostatic eATRP. Thus, the development of a galvanostatic eATRP where only one current value is applied in an intermittent manner, through a PGE, preserving the reaction control would greatly simplify the operations. In such approach, the use of a Cu cathode offers additional advantages. In a typical eATRP on inert cathodes, if the electrochemical stimulus is stopped, [CuIL]+ reacts with dormant halogen-capped polymer chains until complete consumption resulting in total conversion of [CuIL]+ to [Br–CuIIL]+, then the reaction stops. The time needed to stop the polymerization under conventional conditions can be employed as the PGE duty cycle, and its value depends on the specific system. However, with a Cu cathode, the polymerization does not stop when the cell is switched OFF, but rather proceeds via SARA ATRP. Therefore, in the last part of the study we investigated the application of a PGE, where Iapp at a Cu cathode is cycled between a certain value and zero. This toggling procedure is extremely attractive if large current densities are needed and if, for some reasons, the electrochemical equipment does not entirely fulfil the current output requisites, or if the reaction must be suddenly shut down. In this way, the SARA mechanism can sustain the ATRP.
The model system employed in the studies comprised [CuIIMe6TREN]2+ (Me6TREN = tris[2-dimethylaminoethyl]amine) as a catalyst, ethyl 2-bromoisobutyrate (EBiB) as initiator and 50 vol% butyl acrylate (nBA) in dimethylformamide (DMF) (Fig. 2). The detailed investigation on the model system enabled to extend the PGE approach to other polymerization systems with different solvents, monomers, catalysts, and initiators. When employing an Al sacrificial anode in DMF, Al3+ interferes with Cu/L complexes, therefore excess L was used to simultaneously quench the Al3+ ions and trigger the SARA process.28
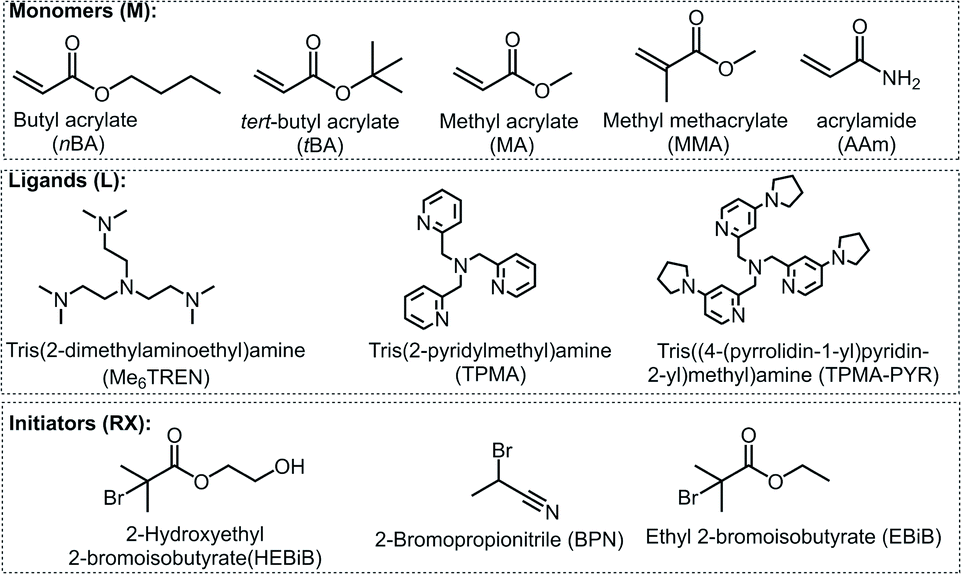 | ||
| Fig. 2 Chemical structures of monomers, ligands, and initiators used in this work. | ||
Results and discussion
eATRP with a Cu cathode
Prior to performing polymerizations, the electrochemical behavior of 10−3 M [Br–CuIIMe6TREN]+ was investigated by cyclic voltammetry (CV) in DMF + 50 vol% nBA (Fig. S1a in the ESI†). The standard reduction potential of the catalyst was determined as E1/2 = (Epc + Epa)/2 = −0.286 V vs. saturated calomel electrode (SCE), where Epc and Epa correspond to the cathodic and anodic peak potentials, respectively. The CV signal was modified upon introduction of 10−2 M EBiB with a remarkable enhancement of the cathodic peak (Fig. S1b†), proving the catalytic behavior of the Cu complex.Potentiostatic eATRP of nBA in DMF was initially conducted with a conventional Pt/Pt electrode pair (Table 1, entry 1), where the Pt counter electrode (CE) was placed in a separated compartment. Throughout the paper we will use the notation cathode/anode (e.g., Pt/Pt, Cu/Pt, Cu/Al, etc.) to denote the setup of the electrodes. The polymerization was conducted under potentiostatic conditions at Eapp = E1/2 − 0.06 V; at this Eapp value, the reaction reached high conversion (>90%) in a relatively short time. P(nBA)–Br had Đ < 1.20 and expected molecular weight (MW), in line with previous reports.23,25 Then, an activated Cu wire was used as working electrode (WE), while a Pt foil was maintained as CE in a separate compartment. The Cu/Pt pair was employed to focus on the Cu WE without interference from an Al or Cu sacrificial anode. First, an eATRP was performed with no free L (Table 1, entry 2), so that the Cu WE could only act as an inert cathode, merely providing electrons. A well-controlled polymerization was obtained, albeit slower than the corresponding eATRP with a Pt/Pt setup (Table 1, entry 1). This was likely due to the lower surface area of the Cu wire relative to the Pt mesh (geometrical area: Pt mesh ≈ 6 cm2, Cu wire ≈ 4.41 cm2), as the rate of electrochemical reduction of CuII species in eATRP is proportional to the electrode surface area. Vis-NIR spectra and CV of the Cu/Pt eATRP solution before and after polymerization confirmed that the Cu cathode acts only as an electron source and virtually no Cu ions are released into the solution (Fig. S2a and S3a†).
| Entry | Cathode | Anode | E app − E1/2 (V) |
|
C 0L,free (mM) | Q (C) | t (h) | Conversionb (%) | k p,app (h−1) | M GPCn (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Other conditions: nBA/EBiB = 349/1; CnBA = 3.49 M in DMF + 0.1 M Et4NBF4 + 10−3 M Et4NBr, T = 45 °C; activated Cu wire: l = 14 cm; stirring rate = 700 rpm. b Calculated from 1H-NMR in CDCl3 using DMF as internal standard. c Apparent propagation rate constants calculated as the slopes of ln([M]0/[M]) vs. t plots. d Calculated from THF GPC with narrow PMMA standards at T = 30 °C. e Calculated from 1H-NMR: Mthn = Conv. × DP × MnBA + MEBiB. f Đ = Mw/Mn. g The polymerization nearly stopped after 5 min and monomer conversion and polymer properties (Mn, Đ) remained practically unchanged. h SARA ATRP using a Cu wire identical to the one used as eATRP cathode. | ||||||||||||
| 1 | Pt | Pt | −0.06 | 1 | 2 | 2.6 | 1.5 | 92 | 1.40 | 36.4 | 41.6 | 1.16 |
| 2 | Cu | Pt | −0.06 | 1 | — | 1.83 | 2 | 78 | 0.85 | 39.3 | 35.0 | 1.18 |
| 3 | Cu | Pt | −0.18 | — | 1 | 1.49 | 3 | 92 | 0.92 | 24.8 | 41.1 | 1.35 |
| 4 | Cu | Pt | −0.06 | — | 1 | 1.64 | 3 | 90 | 0.90 | 30.6 | 40.3 | 1.32 |
| 5 | Cu | Pt | 0.06 | — | 1 | 2.17 | 3 | 53 | 0.33 | 18.4 | 23.7 | 1.28 |
| 6 | Cu | Pt | 0.18 | — | 1 | 2.04 | 3 | 61 | 0.39 | 20.5 | 27.4 | 1.17 |
| 7 | Cu | Pt | 0.30 | — | 1 | 4.02 | 3 | 18g | —g | 8.1 | 8.1 | 1.17 |
| 8 | (Cu)h | — | — | — | 1 | — | 3 | 94 | 1.06 | 34.7 | 42.0 | 1.27 |
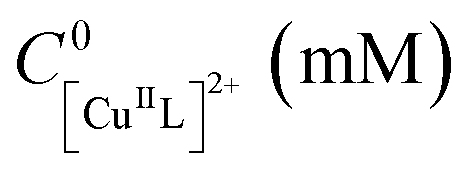
Then, a set of experiments was conducted with the Cu/Pt setup, but without a CuII salt in the initial polymerization mixture. Instead, free L was present at the beginning, so that SARA ATRP could occur in the system. Different values of Eapp were employed (ranging from −0.18 V to +0.3 V relative to E1/2 of the catalyst) to explore the potential synergistic, as well as any adverse effects between eATRP and SARA ATRP involving the same Cu surface (Table 1, entries 3–7). When Eapp = E1/2 − 0.06 was used, fast and controlled polymerization was observed, reaching 90% conversion within 3 h. Shifting Eapp to E1/2 − 0.18 V slightly worsened the outcome, likely due to the interference of an organocupric intermediate that can also be reduced at such negative potential values.29 It should be noted that at Eapp = E1/2 − 0.06, the control over the polymerization was worse than in a similar polymerization with CuII initially present in solution (compare entries 2 and 4 in Table 1), indicating that the presence of CuII at the early stages is crucial for the control. On the other hand, when Eapp = E1/2 + 0.06 V and E1/2 + 0.18 V were used (Table 1, entries 5 and 6), the polymerization remained controlled but strongly slowed down after reaching 50–60% monomer conversion (30–60 min). When Eapp = E1/2 + 0.3 V was used, the polymerization stopped after few minutes (Table 1, entry 7).
Interestingly, the polymerization rate during the first 30 min was only slightly affected by the Eapp value (Fig. S4†), and a similar rate was measured for a SARA ATRP performed with an identical setup (except that no electrochemical potential was applied; Table 1, entry 8). Notably, at E1/2 − 0.06 V the Cu/Pt eATRP exhibited a slightly faster polymerization than the SARA ATRP within the first 30 min, suggesting that the electrochemical reduction and the SARA mechanism acted in concert (additional discussion is provided in section S6 of the ESI). At E1/2 − 0.18 V the polymerization was slightly slower than SARA ATRP, despite the very negative Eapp which should lead to a much higher concentration of CuI. The lower rate could be due to side reactions that occur at this very negative potential.29 At Eapp > E1/2, oxidation of CuI is more favorable than reduction of CuII but the polymerization proceeded with a moderate rate in the initial stage, suggesting that the SARA ATRP mechanism was dominant. However, after the first 30 min the polymerization slowed down considerably. When a very positive potential (E1/2 + 0.30 V) was applied, the reaction stopped within the first few minutes (Table 1, entry 7). Due to the high activity of the catalyst and rapid monomer propagation, 18% conversion was still observed within the first ∼5 min of reaction, although a more effective stop at low or negligible conversion is expected for less active systems. Nevertheless, this result indicates that the application of a significantly more positive potential than E1/2 to a Cu wire is a viable strategy to halt a SARA ATRP on demand.
The dual contribution of SARA and electrochemical reduction can also be appreciated by observing the trends in polymer dispersity at the beginning of the polymerizations (Fig. S5†). In fact, Đ values during the first 15 minutes of polymerization increased as Eapp was shifted to more negative potentials, i.e. by relatively decreasing the amount of CuII deactivator present in solution, as Đ is inversely proportional to the concentration of ATRP deactivator.30 The dispersity of low MW P(nBA)–Br made by SARA ATRP fell in between the Đ values found for eATRPs at Eapp > E1/2 and Eapp < E1/2.
eATRP with a Cu cathode and a sacrificial anode
To study the effect of a sacrificial anode, model polymerizations were initially performed using Pt/Al and SS304/Al electrode pairs (Table 2, entries 1 and 2). The Pt/Al system showed identical polymerization rate and control as the Pt/Pt system employing a divided cell (Table 1, entry 1). On the other hand, a slower but well-controlled polymerization was obtained with SS304 cathode, likely due to the smaller surface area.| Entry | Cathode | Anode | E app − E1/2 (V) |
|
C 0L,free (mM) | Q (C) | m CE (mg) | t (h) | Conversionc (%) | k p,app (h−1) | M GPCn (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Other conditions: nBA/EBiB = 349/1; CnBA = 3.49 M in DMF + 0.1 M Et4NBF4, 10−3 M Et4NBr, T = 45 °C; activated Cu wire: l = 14 cm; stirring rate = 700 rpm. b Estimated mass of CE consumed during electrolysis (see ESI, Section S5). c Calculated from 1H-NMR in CDCl3 using DMF as internal standard. d Apparent propagation rate constants calculated as the slopes of ln([M]0/[M]) vs. t plots. e Calculated from THF GPC with narrow PMMA standards at T = 30 °C. f Calculated from 1H-NMR: Mthn = Conv. × DP × MnBA + MEBiB. g Đ = Mw/Mn. | |||||||||||||
| 1 | Pt | Al | −0.06 | 1 | 2 | 2.9 | 0.27 | 2 | 90 | 1.35 | 38.5 | 40.4 | 1.13 |
| 2 | SS304 | Al | −0.16 | 1 | 2 | 5.9 | 0.55 | 3 | 79 | 0.48 | 32.5 | 36.9 | 1.11 |
| 3 | Pt | Cu | −0.06 | 1 | — | 20.2 | 6.6 | 2 | 64 | 0.59 | 34.0 | 28.8 | 1.22 |
| 4 | Cu | Cu | −0.06 | 1 | — | 20.9 | 6.7 | 3 | 83 | 0.66 | 46.1 | 37.4 | 1.17 |
| 5 | Cu | Al | −0.06 | 1 | 2 | 1.66 | 0.15 | 2 | 86 | 1.15 | 36.6 | 38.6 | 1.10 |
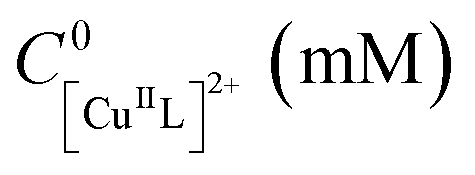
Then, the possibility of employing Cu as a sacrificial anode was explored. eATRPs were performed at Eapp = E1/2 − 0.06 V, with CuII initially present in solution without free ligand. The Pt/Cu system enabled to reach 64% conversion in 2 h (Table 2, entry 3). Cu0 started depositing on the WE surface few minutes after the electrolysis was started. During a potentiostatic eATRP, in a divided or undivided cell, the recorded current vs. time plot (i.e. chronoamperometry) typically shows a current decrease over time during the initial polymerization stage, after which a small, nearly constant value is maintained (see e.g. Fig. S7†).23,24 Unexpectedly, however, the Pt/Cu system showed a rapid enhancement in current, which then remained almost constant at |I| ≈ 3 mA for the entire duration of the process (Fig. S8†), leading to the passage of a very high charge in the system (>10 times higher than the theoretical charge, Qth, value of 1.5 C). A similar result was obtained when the experiment was repeated using a Cu/Cu setup (Table 2, entry 4), although the recorded current was lower, |I| ≈ 2 mA (Fig. S9†).
The use of a sacrificial Cu anode causes CuII ions to be released into solution, which are then reduced and deposited at the cathode surface. The extent of Cu anode consumption and of Cu deposition at the cathode can be estimated as described in the ESI (Section S5†). Calculated amounts of Cu “detached” from the anode (mCE) are listed in Table 2. Note that only a small fraction of sacrificial Cu anode is consumed in a typical eATRP experiment and >300 h of polymerization are required within this setup to “dissolve” a substantial portion of the anode (more details in ESI, Section S5†). Considering that ∼2 g of P(nBA) are produced in 1 h, it is possible to make ∼1 kg of polymer before the Cu anode must be replaced.
Nevertheless, released CuII ions can perturb the eATRP equilibrium and contaminate the polymer. Therefore, the Cu/Al electrode pair was tested (Table 2, entry 5 and Fig. S10†). This is an all-non-noble setup, which is easily scalable, with the advantage of the sacrificial Al anode. Since the Al3+ ions released in solution can interact with Me6TREN,23,28 a 3-fold excess of L was employed. Therefore, the contribution of SARA ATRP cannot be neglected with this setup. The polymerization reached 86% conversion in 2 h and P(nBA)–Br exhibited excellent dispersity, Đ = 1.10; also, a more typical current vs. time plot was observed, thus the charge consumption was much smaller than with a Cu anode. The polymerization rate was comparable to the case of a Pt/Al setup (Table 2, entry 1), despite the 27% lower surface area of the Cu wire relative to the Pt mesh, which would result in a slower polymerization in a pure eATRP system (see ESI, Section S6†). Therefore, the observed comparable rates suggest that SARA and eATRP worked synergistically, enhancing the rate of the process. A conventional SARA ATRP performed under similar conditions gave similar outcomes (Table S1, entry 1 and Fig. S11†).
The observed constant current value with Cu as anode suggested the possibility of performing galvanostatic eATRP by applying only one Iapp value rather than a sequence of decreasing current values, using different cathode/anode combinations: Pt/Al, Cu/Al, and Cu/Cu. Galvanostatic eATRP should be the preferred choice for large volume reactions due to the simpler and less expensive setup, and it has previously been successfully used.31–34 To test galvanostatic electrolysis on a Cu cathode, a series of experiments was performed (Table 3) under continuous galvanostatic electrolysis (CGE). Initially, CGEs were attempted with a Cu/Cu pair and a single Iapp value (Table 3, entries 1–4). When |Iapp| = 1 mA was applied at the cathode, the polymerization was well-controlled and produced P(nBA)–Br with excellent dispersity, Đ = 1.11 (Table 3, entry 1). Raising |Iapp| to 2 mA afforded similar results but slightly higher polymer dispersity, Đ = 1.18. Further enhancing |Iapp| to 3 mA did not affect the results (Table 3, entry 3), probably because the process was already diffusion controlled. The initial concentration of [Br–CuIIMe6TREN]+ was then decreased from 10−3 M to 10−4 M (Table 3, entry 4). As expected, the polymerization was slower and the polymer dispersity increased (Đ = 1.39), but control was acceptable. This experiment was then repeated without CuII salt in the initial mixture, to exploit the possibility of generating the Cu catalyst from the sacrificial Cu anode. Thus, the initial solution contained only 10−4 M Me6TREN and Br− ions. Nevertheless, the polymerization was faster and better controlled (Đ = 1.30) than the one with initial 10−4 CuII complex (Table 3, entries 4 vs. 5, and Fig. S12†). Thus, the contribution of the SARA mechanism to the eATRP process increased the polymerization rate, as already discussed for the Pt/Al vs. Pt/Cu and Cu/Pt systems with potentiostatic electrolysis. Moreover, the combination of SARA and eATRP seems to improve the polymerization control when the loading of the ligand is low. A regular SARA ATRP with only 10−4 M Me6TREN and Br− ions (Table 3, entry 6) exhibited very poor conversion and control. Therefore, the synergy between eATRP and SARA ATRP can be applied to run polymerizations with very limited loadings of reagents.
| Entry | Anode |
|
C 0L,free (mM) | |Iapp| (mA) | t (h) | Q (C) | m CE (mg) | Conversionc (%) | k p,app (h−1) | M GPCn (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Other conditions: nBA/EBiB = 349/1; CnBA = 3.49 M in DMF + 0.1 M Et4NBF4, 10−3 M Et4NBr (except for entries 5 and 6: 10−4 M Et4NBr), T = 45 °C; WE = activated Cu wire; all wires used as WE or CE had 1 mm diameter and l = 14 cm; stirring = 700 rpm. b Estimated mass of CE consumed during electrolysis (see ESI, Section S5). c Calculated from 1H-NMR in CDCl3 using DMF as internal standard. d Apparent propagation rate constants calculated as the slopes of ln([M]0/[M]) vs. t plots. e Calculated from THF GPC with narrow PMMA standards at T = 30 °C. f Calculated from 1H-NMR: Mthn = Conv. × DP × MnBA + MEBiB. g Đ = Mw/Mn. h SARA ATRP. i The polymerization nearly stopped after 1 h. | ||||||||||||
| 1 | Cu | 1 | — | 1 | 3 | 10.8 | 3.6 | 86 | 0.91 | 37.8 | 35.1 | 1.11 |
| 2 | Cu | 1 | — | 2 | 2 | 14.4 | 4.7 | 81 | 1.01 | 41.9 | 36.4 | 1.18 |
| 3 | Cu | 1 | — | 3 | 2 | 21.6 | 7.1 | 82 | 1.01 | 46.5 | 36.8 | 1.18 |
| 4 | Cu | 0.1 | — | 2 | 3.5 | 25.2 | 8.3 | 73 | 0.39 | 33.2 | 32.9 | 1.39 |
| 5 | Cu | — | 0.1 | 2 | 3 | 21.6 | 7.1 | 84 | 0.70 | 35.4 | 37.6 | 1.30 |
| 6h | — | — | 0.1 | — | 1 | — | — | 24i | 0.26i | 5.5 | 11.2 | 2.49 |
| 7 | Al | 1 | 2 | 0.227 | 2 | 1.66 | 0.15 | 86 | 1.19 | 40.6 | 37.9 | 1.11 |
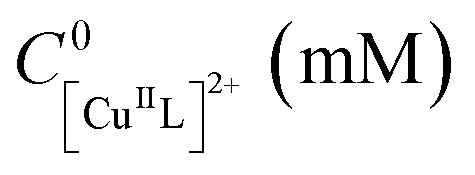
The rather high |Iapp| values used in these reactions resulted in high charge consumption and thus large quantities of CuII ions released from the anode and deposited as Cu0 on the cathode surface. The Cu surfaces of both anode and cathode were analyzed by scanning electron microscopy to determine morphological changes during polymerization at |Iapp| = 1 mA (Fig. S13 and S14†). The surface of the Cu anode showed some signs of corrosion, while electrodeposited Cu particles were observed at the surface of the Cu cathode. In addition, the solutions turned green due to the formation of soluble Cu species and/or dispersed Cu nanoparticles, as in potentiostatic eATRPs with a Cu anode.
Since the Cu/Cu pair is impractical and potentially disadvantageous on an industrial scale because of the heavy contamination of the mixture, a Cu/Al pair was preferred, with a slight excess of Me6TREN (Table 3, entry 7, Fig. S15 and S16†). With this system, the CGE was performed with a much lower applied current, |Iapp| = 227 µA, which was calculated as the average current (Iaverage = Q/t) in a similar eATRP under potentiostatic conditions (Table 2, entry 5, Q = 1.66 C). The polymerization reached 86% conversion within 2 h under well-controlled conditions yielding P(nBA)–Br with a narrow molecular weight distribution (Đ = 1.11). Two other control eATRPs using a Pt/Al pair of electrodes (Table S1,† entries 2 and 3, with Q = 0.83 C and Q = 1.66 C, respectively) showed that P(nBA)–Br can be obtained with excellent dispersity by CGE with a single current value also on a conventional Pt cathode.
From continuous electrolysis to pulsed electrolysis
Since the introduction of eATRP in 2011, almost all electrochemically mediated polymerizations have been performed with continuous electrolysis, mostly by a potentiostatic mode. In order to implement pulsed galvanostatic electrolysis (PGE) in eATRP, the electrolysis duty cycle was first determined, by measuring the time required to consume all [CuIL]+ upon removal of the electrochemical stimulus during a potentiostatic electrolysis with a Pt/Al setup.22eATRP of nBA was carried out at Eapp = E1/2 − 0.06 V with CuII/L ratio = 1/3 for 30 min. Then the current was switched off and monomer conversion was measured every 5 min for 15 min, followed by one last analysis at 60 min (Fig. 3). The polymerization stopped completely within <15 min after switching off the electrolysis. The polymerization halt was not instantaneous upon removal of the electrochemical stimulus, as a certain amount of [CuIL]+ was present in the system, which could still activate polymer chains until being completely converted to CuII species. This behavior agrees with the “imperfect” temporal control previously observed in photoATRP systems.35,36 When the same procedure was repeated on a Cu cathode, the polymerization did not stop, but proceeded as SARA ATRP, reaching 89% conversion after a total of 120 min and affording P(nBA)–Br with Mn = 42.3 kDa and Đ = 1.10 (Fig. S17 and S18†).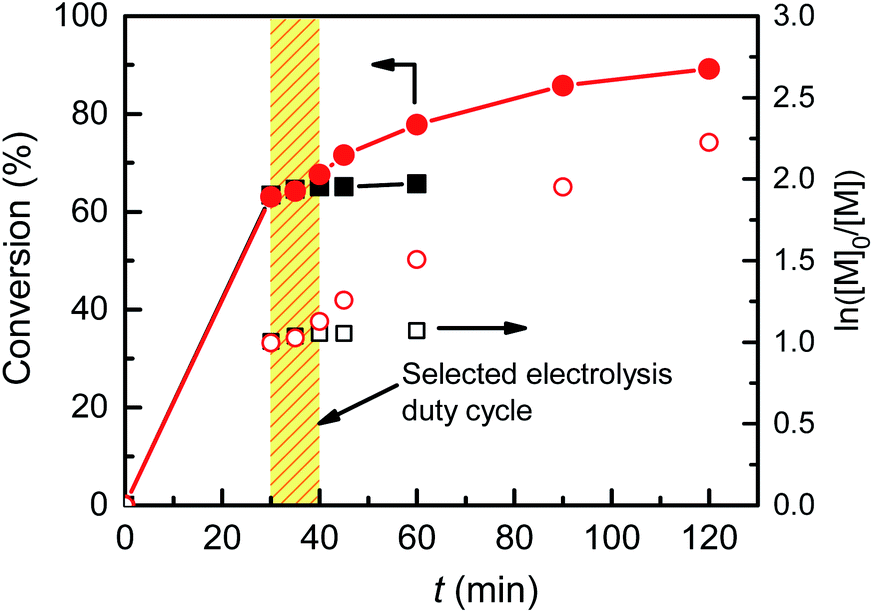 | ||
| Fig. 3 Determination of the duty cycle by following the evolution of conversion and ln([M]0/[M]) vs. t for eATRP of 50 vol% nBA in DMF+ 0.1 M Et4NBF4 at T = 45 °C, performed in an undivided cell with a Pt (squares) or a Cu (circles) cathode at Eapp = E1/2 − 0.06 V and a sacrificial Al anode. Electrolysis was switched off on both electrodes after 30 minutes. Conditions: nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.1/0.3/0.1, CnBA = 3.49 M. | ||
Then, a PGE was attempted by selecting the electrolysis parameters to allow the same charge to pass in the system over the same total reaction time. The CGE reported in Table 3, entry 7, was repeated under PGE mode, with a fixed duty cycle of 10 min (Fig. 3). Under these conditions, the electrolysis was ON for 60 min and OFF for 60 min. Thus, the applied current was doubled (|Iapp| = 454 µA, Table 4, entry 1, Fig. S19 and S20†) to obtain the same total charge of 1.66 C. The polymerization was still well-controlled (Đ = 1.11); however, the conversion reached a lower value of 71% after 2 h.
| Entry |
|
C 0L,free (mM) | |Iapp| (mA) | t (h) | Q (C) | m CE (mg) | Conversionc (%) | k p,app (h−1) | M GPCn (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Other conditions: nBA/EBiB = 349/1 (except for entry 3), CnBA = 3.49 M in DMF + 0.1 M Et4NBF4, 10−3 M Et4NBr (except for entry 5: 10−4 M Et4NBr), T = 45 °C; during PGE, the duty cycle was 10 min; WE = activated Cu wire, l = 14 cm, CE = Al wire, l = 14 cm; all wires had 1 mm diameter; stirring = 700 rpm. b Estimated mass of CE consumed during electrolysis (see Section S5 of ESI). c Calculated from 1H-NMR in CDCl3 using DMF as internal standard. d Apparent propagation rate constants calculated as the slopes of ln([M]0/[M]) vs. t plots. e Calculated from THF GPC with narrow PMMA standards at T = 30 °C or with TriSEC calibration using PS standards (only entry 3) at 30 °C. f Calculated from 1H-NMR: Mthn = Conv. × DP × MnBA + MEBiB. g Đ = Mw/Mn. h DPT = CnBA/CEBiB = 1745. | |||||||||||
| 1 | 1 | 2 | 0.454 | 2 | 1.66 | 0.15 | 71 | 0.70 | 32.6 | 31.9 | 1.11 |
| 2 | 1 | 2 | 0.227 | 2 | 0.83 | 0.08 | 89 | 1.36 | 40.8 | 40.9 | 1.11 |
| 3h | 1 | 2 | 0.227 | 1.5 | 0.83 | 0.08 | 69 | 0.91 | 119.2 | 153.8 | 1.19 |
| 4 | — | 3 | 0.227 | 2 | 0.83 | 0.08 | 92 | 1.10 | 39.9 | 39.5 | 1.13 |
| 5 | 0.1 | 2.9 | 0.227 | 2 | 0.83 | 0.08 | 89 | 1.12 | 39.5 | 39.6 | 1.10 |
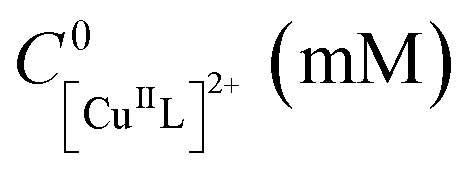
Despite being not necessary from a practical point of view, a 3-electrode setup was employed in these galvanostatic experiments to gain some insight on the process. This allowed monitoring the potential of the Cu cathode, EWE, vs. the reference electrode. The recorded potential was lower than the expected value (∼E1/2 − 0.06 V), particularly in the later stage of the reaction (Fig. S19†). This negative drift of potential over time could cause over-reduction of CuII species to Cu0, ultimately leading to a premature stop of the process. Therefore, the charge was cut by 50% (Q = 0.83 C), by pulsing |Iapp| = 227 µA every 10 min (Table 4, entry 2, Fig. S21 and S22†). Pleasingly, the conversion improved to 89%, producing P(nBA)–Br of very low dispersity (Đ = 1.11). The charge cut, drastically decreased the energy required to drive the polymerization, making these conditions industrially appealing for future scaled-up reactions.
Another attempt was made by targeting a 5-fold higher target degree of polymerization (DPT = 1745) than the one hitherto used in all experiments. Thus, the initiator loading was decreased from 10−2 to 2 × 10−3 M (Table 4, entry 3). After 1.5 h, P(nBA)–Br with MGPCn = 119.2 kDa and Đ = 1.19 was obtained (Fig. S23†). The polymerization was stopped after that time due to the high viscosity, which hampered effective stirring (see Fig. S24†).
Then, the reaction with DPT = 349 was repeated with no CuII initially present in solution but with the same quantity of Me6TREN (Table 4, entry 4). The advantage of this system is that it avoids the addition of a metal salt at the beginning by exploiting the SARA mechanism (see discussion in the ESI, Section S6†). The polymerization reached a high conversion of 92%, producing P(nBA)–Br with Đ = 1.13. A disadvantage of this type of setup could be the corrosion of the anode, so replacement should be provided after a certain number of reactions. However, the Al3+ release is rather small. Calculated amounts of Al released from the anode (mAl) are given in Table 4. Note that only a small fraction of the sacrificial Al anode is consumed during polymerization and 5752 h (240 days) will be needed within this setup to dissolve a substantial portion of the anode (more details in the ESI, Section S5†). Considering that ∼6.14 g of P(nBA) is produced in 2 h, it should be possible to produce 17.8 kg of polymer before the Al anode must be replaced.
To evaluate the amount of CuII ions released in solution, mainly by SARA ATRP during the periods of no applied current, CV and Vis-NIR spectra were recorded before and after the polymerization (Fig. S2c and S3c†). These analyses showed a modest release of CuII ions, with a final concentration of [Br–CuIIL]+ of about 10−3 M. Therefore, the polymer is not strongly contaminated by Cu ions.
One final attempt was carried out with only 10−4 M of initial CuII and Et4NBr, maintaining the usual concentration of Me6TREN. The polymerization reached 90% conversion with still very low Đ and perfect agreement between MGPCn and Mthn (Table 4, entry 5). Examples of kinetic analysis of this set of polymerizations and obtained polymer features are shown in Fig. 4, whereas potential profiles of the WE and GPC traces are reported in Fig. S24–S29.†
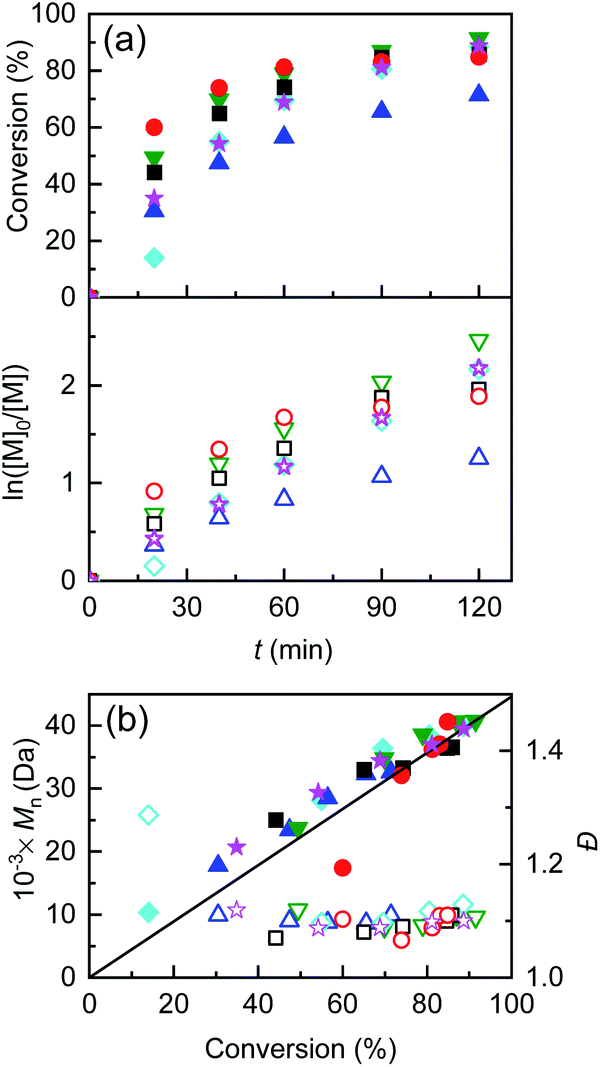 | ||
Fig. 4 (a) Kinetic plots and (b) evolution of Mn and Đ vs. conversion for eATRP of nBA in DMF + 0.1 M Et4NBF4, T = 45 °C, performed using a Cu/Al electrode pair under various conditions. ( ) Potentiostatic electrolysis at Eapp = E1/2 − 0.06 V (Table 2, entry 5); ( ) Potentiostatic electrolysis at Eapp = E1/2 − 0.06 V (Table 2, entry 5); ( ) CGE, Q = 1.66 C (Table 3, entry 7); ( ) CGE, Q = 1.66 C (Table 3, entry 7); ( ) PGE, Q = 1.66 C (Table 4, entry 1), ( ) PGE, Q = 1.66 C (Table 4, entry 1), ( ) PGE, Q = 0.83 C (Table 4, entry 2); general conditions: nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.1/0.3/0.1, CnBA = 3.49 M. ( ) PGE, Q = 0.83 C (Table 4, entry 2); general conditions: nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.1/0.3/0.1, CnBA = 3.49 M. ( ) PGE, Q = 0.83 C nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0/0.3/0.1, CnBA = 3.49 M (Table 4, entry 4); ( ) PGE, Q = 0.83 C nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0/0.3/0.1, CnBA = 3.49 M (Table 4, entry 4); ( ) PGE, Q = 0.83 C, nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.01/0.3/0.1, CnBA = 3.49 M (Table 4, entry 5). The straight line in (b) stands for the theoretical molecular weights. ) PGE, Q = 0.83 C, nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.01/0.3/0.1, CnBA = 3.49 M (Table 4, entry 5). The straight line in (b) stands for the theoretical molecular weights. | ||
Chain extension
The well-controlled polymerizations obtained under PGE suggested that living systems were still achieved. To demonstrate the preserved livingness, P(nBA)–Br produced as in Table 4, entry 2, was extended using tert-butyl acrylate (tBA), without removing unreacted nBA and isolating the macroinitiator. The first block was built by PGE eATRP of nBA on a Cu/Al electrode pair in 1 h, followed by quenching the reaction by applying Eapp = E1/2 + 0.30 V. The electrolysis was continued after the injection of degassed tBA, again by PGE for one additional hour (Fig. S30†), producing the block copolymer P(nBA)-b-P(nBA-stat-tBA)–Br of MGPCn = 40.6 kDa and Đ = 1.06 (Fig. 5), showing excellent end-group fidelity. The second segment of the block copolymer is statistical, considering the similar reactivity of nBA and tBA.37 NMR spectra of the P(nBA)–Br macroinitiator and the obtained copolymer are reported in Fig. S31 and S32.†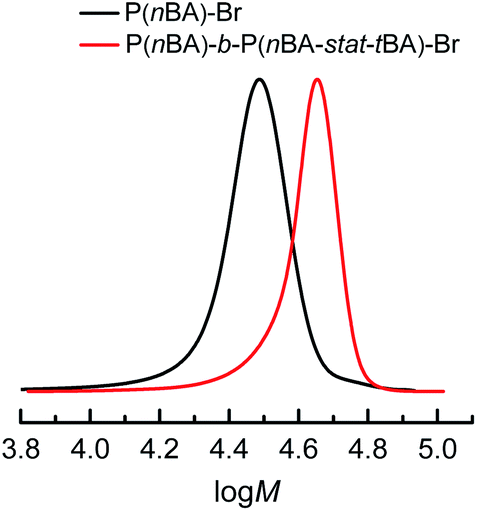 | ||
| Fig. 5 Molecular weight distribution of P(nBA)288–Br macroinitiator (Mn = 37.0 kDa and Đ = 1.11, conv.BA = 83%) and P(nBA)288-b-P(nBA40-stat-tBA56)–Br copolymer (Mn = 50.6 kDa and Đ = 1.06, conv.tBA = 25%, conv.BA = 94%) produced by PGE eATRP of nBA and nBA + tBA at T = 45 °C in DMF + 0.1 M Et4NBF4. tBA was added after 1 h of nBA polymerization under the following conditions: nBA/EBiB/Cu(OTf)2/Me6TREN/Et4NBr = 349/1/0.1/0.3/0.1, CnBA = 3.49 M. | ||
Extension to other monomers, catalysts, and solvents
To further expand the scope of eATRP with Cu electrodes, various monomers, solvents, catalysts, and initiators were investigated (Table 5). First, polymerization of methyl acrylate in dimethyl sulfoxide (DMSO) at T = 40 °C was carried out. The most active ATRP catalyst to-date was used, [CuIITPMA-PYR]2+ (TPMA-PYR = tris(4-pyrrolidinopyridyl-2-methyl)amine).38 Well-controlled polymerizations were obtained via either SARA ATRP or eATRP (with a potentiostatic electrolysis at Eapp = E1/2 = −0.371 V vs. SCE) with the Cu/Al electrode pair, giving PMA–Br with low dispersity (Đ ∼ 1.1, Table 5, entries 1 and 2). The eATRP with the Cu cathode was 1.4-fold faster than the SARA ATRP (Fig. S33†), which can be attributed to the synergy of the two CuI regeneration mechanisms.| Entry | M | ATRP mode | E app (V) | Ligand | Solvent | t (h) | Q (C) | Conv.b (%) | k p,app (h−1) | M GPCn (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: entries 1 and 2: MA/EBiB/CuBr2/TPMA-PYR = 552/1/0.03/0.09, DPT = 552, CMA = 5.52 M in DMSO + 0.1 M Et4NBF4, T = 40 °C; entries 3 and 4: MMA/BPN/CuCl2/TPMA/Bu4NCl = 467/1/0.1/0.3/5, DPT = 467, CMMA = 4.67 M, T = 50 °C; entries 5 and 6: AAm/HEBiB/CuBr2/Me6TREN/NaBr = 141/0.2/0.1/0.4/10, DPT = 705. CAAm = 1.41 M, T = 0 °C. WE = activated Cu wire, l = 14 cm, CE = Al wire, l = 14 cm; all wires had 1 mm diameter. Stirring = 700 rpm. b Calculated from 1H-NMR in CDCl3 or D2O using 2 vol% DMF as internal standard. c Apparent propagation rate constants calculated as the slopes of ln([M]0/[M]) vs. t plots. d Calculated from THF GPC with narrow PMMA standards at T = 30 °C (PMA, PMMA) or aqueous GPC with narrow PEO standards at T = 35 °C (PAAm). e Calculated from 1H-NMR: Mthn = Conv. × DP × MM + MRX. f Đ = Mw/Mn. g E pc = cathodic peak potential. | ||||||||||||
| 1 | MA | SARA | — | TPMA-PYR | DMSO | 2 | — | 62 | 0.55 | 35.6 | 29.1 | 1.09 |
| 2 | MA | eATRP | E 1/2 | TPMA-PYR | DMSO | 2 | 0.19 | 73 | 0.77 | 33.5 | 34.3 | 1.10 |
| 3 | MMA | SARA | — | TPMA | EtOH | 2 | — | 61 | 0.54 | 31.6 | 36.7 | 1.86 |
| 4 | MMA | eATRP | E 1/2 − 0.06 | TPMA | EtOH | 3 | 2.70 | 76 | 0.46 | 32.6 | 35.7 | 1.26 |
| 5 | AAm | SARA | — | Me6TREN | H2O | 2 | — | 49 | 0.28 | 12.7 | 25.0 | 1.40 |
| 6 | AAm | eATRP | E pc | Me6TREN | H2O | 1.5 | 0.31 | 90 | 1.69 | 35.9 | 45.4 | 1.28 |
Then, methyl methacrylate (MMA) was used, which, unlike BA or MA, requires a rather slow regeneration because it is characterized by much higher KATRP. For example, controlled polymerizations were obtained with weak ligands such as bpy or PMDETA.39–43 MMA also requires very active initiators such as ethyl 2-bromophenylacetate or 2-bromopropionitrile (BPN) to balance the propagating radical reactivity and avoid the penultimate effect.39,44,45 We attempted SARA ATRP of MMA in ethanol initiated by BPN at 50 °C and catalyzed by [CuIITPMA]2+ (TPMA = tris(2-pyridilmethyl)amine), utilizing catalytic halogen exchange (cHE, with 0.05 M Bu4NCl) to suppress the mismatch of reactivity.39,46–50 This SARA ATRP (Table 5, entry 3) was unsuccessful, even with cHE, and resulted in a poorly controlled PMMA–Cl, with a multimodal MW distribution (Mn = 31.6 kDa, Đ = 1.86). However, using the same conditions but superimposing electrochemical control with a potentiostatic electrolysis at Eapp = E1/2 + 0.06 V (E1/2 = −0.714 V vs. ferrocenium/ferrocene), the polymerization greatly improved (Table 5, entry 4). Indeed, electrochemistry forces the distribution at the electrode surface in favor of the [ClCuIITPMA]+ deactivator, producing a well-controlled PMMA–Cl with Mn = 32.6 kDa and Đ = 1.26 (Fig. 6).
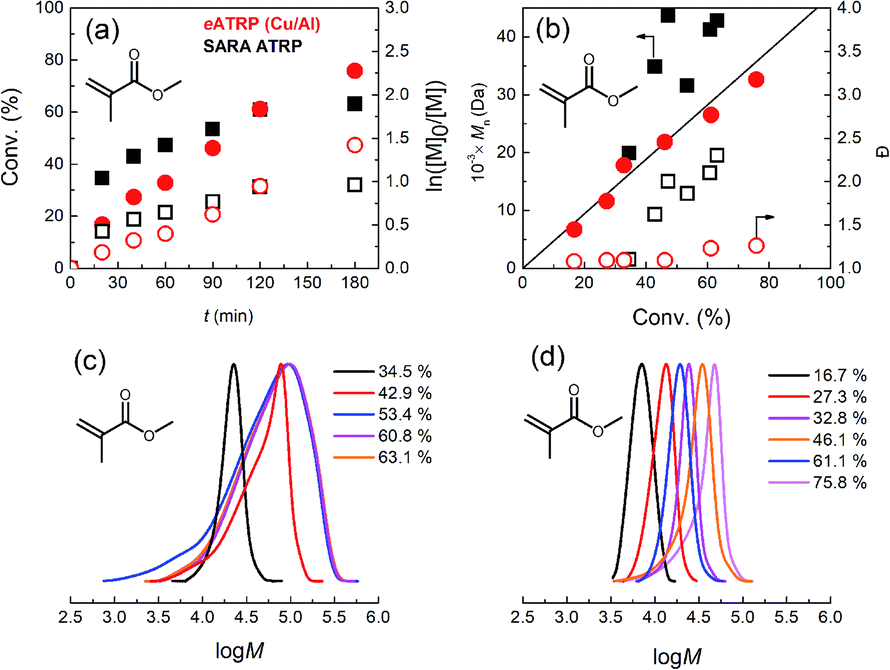 | ||
| Fig. 6 (a) Kinetic plots, (b) evolution of Mn and Đ vs. conv. during SARA ATRP (squares) or eATRP on a Cu/Al pair (circles) of MMA in EtOH + 0.05 M Bu4NCl, T = 50 °C. (c and d) Normalized evolution of molecular weight distribution of PMMA–Cl produced by SARA ATRP (c) or eATRP (d). Conditions: MMA/BPN/CuCl2/TPMA/Bu4NCl = 467/1/0.1/0.3/5, CMMA = 4.67 M. Full and empty symbols refer to the left and right ordinates, respectively. The black straight line represents Mthn in (b). | ||
Finally, eATRP and SARA ATRP of acrylamide (AAm) in H2O were attempted. Water is the most commonly used solvent for acrylamide.51,52 The temperature was set at T = 0 °C, to avoid known side reactions. eATRP at the Cu cathode was carried out via potentiostatic electrolysis at Eapp = Epc = −0.540 V vs. SCE, using 2-hydroxyethyl α-bromoisobutyrate (HEBiB) as initiator and [CuIIMe6TREN]2+ as catalyst under nearly diffusion-controlled conditions. The polymerization was faster and better controlled under similar conditions than using SARA ATRP (Table 5, entries 5, 6). Monomer conversion reached 90% in 90 min in the eATRP with Cu/Al setup, yielding PAAm–Br with Đ = 1.28 (Fig. 7).
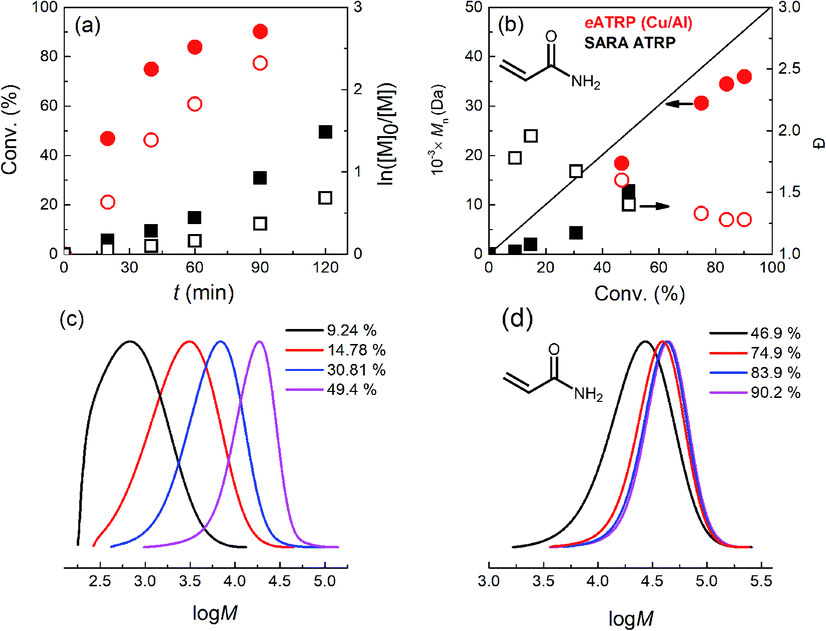 | ||
| Fig. 7 (a) Kinetic plots, (b) evolution of Mn and Đ vs. conv. during SARA ATRP (squares) or eATRP with a Cu/Al pair (circles) of AAm in H2O + 0.1 M NaBr, T = 0 °C. (c and d) Normalized evolution of molecular weight distribution of PAAm–Br produced by SARA ATRP (c) or eATRP (d). Conditions: AAm/HEBiB/CuBr2/Me6TREN/NaBr = 141/0.2/0.1/0.4/10, CAAm = 1.41 M. Full and empty symbols refer to the left and right ordinates, respectively. The black straight line represents Mthn in (b). | ||
Conclusions
This work investigated the use of Cu as an electrode material in electrochemically mediated ATRP in organic solvents and water, with various monomers, catalysts, and initiators, including the most active ATRP catalyst known to-date. Depending on the conditions, the polymerization can proceed via classical eATRP or with some contribution from SARA mechanism, where the CuI activator is (re)generated via both comproportionation and electrochemical reduction of CuII species. Moreover, the electrochemical setup can be used to stop a SARA ATRP on demand, or to obtain well-controlled polymerizations under conditions where SARA ATRP alone is not effective.By employing Cu electrodes, a galvanostatic approach is hence possible, via either a continuous or pulsed manner. The pulsed galvanostatic electrolysis mode takes advantage of the SARA ATRP mechanism that drives the polymerization during the periods when the electrolysis is switched off. Consequently, it is possible to decrease the charge passed into the system, thus lowering the energy consumption, without altering the polymer properties. P(nBA)–Br was obtained with low dispersity, even at high DPT or without any initially added copper salt. Other well-defined polymers (PMA–Br, PAAm–Br and PMMA–Cl) were prepared via eATRP with a Cu cathode, demonstrating the flexibility of this setup.
Metallic Cu is much less expensive than Pt or glassy carbon electrodes. In addition, to reduce process cost, bulk Cu can be replaced by electrodes made of a thin layer of Cu (electro)deposited on less expensive, non-noble metals. The setup proposed is highly suitable for the scale-up of eATRP and for future eATRP studies, in view of a more widespread use of eATRP and electrochemistry in general, as a potent and versatile tool for controlled radical polymerizations.
Data availability
Additional data and detailed experimental details are available in the ESI.†Author contributions
Conceptualization: F. L., F. D. B., A. A. I., K. M.; experiments: F. D. B., F. L.; investigation, formal analysis, writing – original draft: F. D. B., F. L.; writing – review and editing: all authors; funding acquisition: A. A. I., K. M., F. D. B., A. C. S. and J. F. J. C.; validation: all authors; supervision: F. L., F. D. B., A. A. I., K. M.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
F. D. B. thanks FCT – Fundação para a Ciência e a Tecnologia (Portuguese Foundation for Science and Technology) for financial support through the grants POLYCEL (POCI-01-0145-FEDER-029742) and POLYELECTRON (PTDC/EQU-EQU/2686/2020). Support from the U.S. National Science Foundation (NSF, grant CHE 2000391) is gratefully acknowledged. A. A. I. acknowledges the University of Padova for financial support and Dr Andrea Basagni for SEM images. Part of the 1H-NMR data were collected at the UC-NMR facility which is supported in part by FEDER – European Regional Development Fund through the COMPETE Programme (Operational Programme for Competitiveness) and by National Funds through FCT – Fundação para a Ciência e a Tecnologia (Portuguese Foundation for Science and Technology) through Grants REEQ/481/QUI/2006, RECI/QEQ-QFI/0168/2012, CENTRO-07-CT62-FEDER-002012, and Rede Nacional de Ressonancia Magnetica Nuclear (RNRMN). This research was partially sponsored by FEDER funds through the program COMPETE – Programa Operacional Factores de Competitividade – and by national funds through FCT – Fundação para a Ciência e a Tecnologia, under the project UIDB/00285/2020.Notes and references
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- K. Schroder, R. T. Mathers, J. Buback, D. Konkolewicz, A. J. D. Magenau and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1037–1040 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- D. Konkolewicz, Y. Wang, M. J. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- A. S. R. Oliveira, P. V. Mendonça, A. C. Serra and J. F. J. Coelho, J. Polym. Sci., 2019, 58, 145–153 CrossRef.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4409 RSC.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- J. P. Mendes, J. R. Góis, J. R. C. Costa, P. Maximiano, A. C. Serra, T. Guliashvili and J. F. J. Coelho, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1322–1328 CrossRef CAS.
- K. F. Augustine, T. G. Ribelli, M. Fantin, P. Krys, Y. Cong and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3048–3057 CrossRef CAS.
- C. M. R. Abreu, L. Fu, S. Carmali, A. C. Serra, K. Matyjaszewski and J. F. J. Coelho, Polym. Chem., 2017, 8, 375–387 RSC.
- J. P. Mendes, P. V. Mendonca, P. Maximiano, C. M. R. Abreu, T. Guliashvili, A. C. Serra and J. F. J. Coelho, RSC Adv., 2016, 6, 9598–9603 RSC.
- P. Krys, Y. Wang, K. Matyjaszewski and S. Harrisson, Macromolecules, 2016, 49, 2977–2984 CrossRef CAS.
- P. Chmielarz, P. Krys, S. Park and K. Matyjaszewski, Polymer, 2015, 71, 143–147 CrossRef CAS.
- J. P. Mendes, F. Branco, C. M. Abreu, P. V. Mendonca, A. V. Popov, T. Guliashvili, A. C. Serra and J. F. Coelho, ACS Macro Lett., 2014, 3, 544–547 CrossRef CAS PubMed.
- D. Konkolewicz, P. Krys, J. R. Góis, P. V. Mendonça, M. Zhong, Y. Wang, A. Gennaro, A. A. Isse, M. Fantin and K. Matyjaszewski, Macromolecules, 2014, 47, 560–570 CrossRef CAS.
- C. M. R. Abreu, A. C. Serra, A. V. Popov, K. Matyjaszewski, T. Guliashvili and J. F. J. Coelho, Polym. Chem., 2013, 4, 5629–5636 RSC.
- S. Dadashi-Silab and K. Matyjaszewski, Macromolecules, 2018, 51, 4250–4258 CrossRef CAS.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- F. De Bon, G. M. Carlan, E. Tognella and A. A. Isse, Processes, 2021, 9, 1327 CrossRef CAS.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2016, 7, 5357–5365 RSC.
- M. Fantin, F. Lorandi, A. A. Isse and A. Gennaro, Macromol. Rapid Commun., 2016, 37, 1318–1322 CrossRef CAS PubMed.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS PubMed.
- F. De Bon, D. C. M. Ribeiro, C. M. R. Abreu, R. A. C. Rebelo, A. A. Isse, A. C. Serra, A. Gennaro, K. Matyjaszewski and J. F. J. Coelho, Polym. Chem., 2020, 11, 6745–6762 RSC.
- F. De Bon, A. A. Isse and A. Gennaro, Electrochim. Acta, 2019, 304, 505–512 CrossRef CAS.
- J. Luo, C. Durante, A. Gennaro and A. A. Isse, Electrochim. Acta, 2021, 388, 138589 CrossRef CAS.
- M. Fantin, F. Lorandi, T. G. Ribelli, G. Szczepaniak, A. E. Enciso, C. Fliedel, L. Thevenin, A. A. Isse, R. Poli and K. Matyjaszewski, Macromolecules, 2019, 52, 4079–4090 CrossRef CAS.
- F. Lorandi and K. Matyjaszewski, Isr. J. Chem., 2020, 60, 108–123 CrossRef CAS.
- M. Fantin, P. Chmielarz, Y. Wang, F. Lorandi, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2017, 50, 3726–3732 CrossRef CAS PubMed.
- I. Zaborniak and P. Chmielarz, Polym. Adv. Technol., 2020, 31, 2806–2815 CrossRef CAS.
- P. Chmielarz and A. Sobkowiak, J. Polym. Res., 2017, 24, 77 CrossRef.
- B. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- S. Dadashi-Silab, I.-H. Lee, A. Anastasaki, F. Lorandi, B. Narupai, N. D. Dolinski, M. L. Allegrezza, M. Fantin, D. Konkolewicz, C. J. Hawker and K. Matyjaszewski, Macromolecules, 2020, 53, 5280–5288 CrossRef CAS.
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I. H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy, S. Shanmugam, V. Kottisch, B. P. Fors, C. Boyer, G. M. Miyake, K. Matyjaszewski, D. M. Haddleton, J. R. de Alaniz, A. Anastasaki and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268–273 CrossRef CAS PubMed.
- W. Wang, Z. Liu, Z. Guo, J. Zhang, C. Li, S. Qiu, X. Lei and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 50812–50822 CrossRef CAS PubMed.
- A. E. Enciso, F. Lorandi, A. Mehmood, M. Fantin, G. Szczepaniak, B. G. Janesko and K. Matyjaszewski, Angew. Chem., Int. Ed., 2020, 59, 14910–14920 CrossRef CAS PubMed.
- F. De Bon, A. A. Isse and A. Gennaro, ChemElectroChem, 2019, 6, 4257–4265 CrossRef CAS.
- G. Wang, M. Lu and H. Wu, Polymer, 2012, 53, 1093–1097 CrossRef CAS.
- T. J. Aitchison, M. Ginic-Markovic, S. Clarke and S. Valiyaveettil, Macromol. Chem. Phys., 2012, 213, 79–86 CrossRef CAS.
- P. V. Mendonça, A. C. Serra, J. F. J. Coelho, A. V. Popov and T. Guliashvili, Eur. Polym. J., 2011, 47, 1460–1466 CrossRef.
- H. Ma, X. Wan, X. Chen and Q.-F. Zhou, Polymer, 2003, 44, 5311–5316 CrossRef CAS.
- C. Y. Lin, M. L. Coote, A. Petit, P. Richard, R. Poli and K. Matyjaszewski, Macromolecules, 2007, 40, 5985–5994 CrossRef CAS.
- A. K. Nanda and K. Matyjaszewski, Macromolecules, 2003, 36, 8222–8224 CrossRef CAS.
- F. De Bon, C. M. R. Abreu, A. C. Serra, A. Gennaro, J. F. J. Coelho and A. A. Isse, Macromol. Rapid Commun., 2020, 42, 2000532 CrossRef PubMed.
- E. Trevisanello, F. De Bon, G. Daniel, F. Lorandi, C. Durante, A. A. Isse and A. Gennaro, Electrochim. Acta, 2018, 285, 344–354 CrossRef CAS.
- F. De Bon, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2018, 9, 646–655 RSC.
- Y. Wang and K. Matyjaszewski, Macromol. Rapid Commun., 2020, 41, e2000264 CrossRef PubMed.
- C. H. Peng, J. Kong, F. Seeliger and K. Matyjaszewski, Macromolecules, 2011, 44, 7546–7557 CrossRef CAS.
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef CAS.
- P. Chmielarz, S. Park, A. Simakova and K. Matyjaszewski, Polymer, 2015, 60, 302–307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01982e |
| This journal is © The Royal Society of Chemistry 2022 |