
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of SrTiO3 and Al-doped SrTiO3via the deep eutectic solvent route†
Adedoyin N.
Adeyemi
 a,
Amrit
Venkatesh
ab,
Chengcan
Xiao
c,
Zeqiong
Zhao
c,
Ying
Li
a,
Tori
Cox
a,
Dapeng
Jing
bd,
Aaron J.
Rossini
ab,
Frank E.
Osterloh
c and
Julia V.
Zaikina
*a
a,
Amrit
Venkatesh
ab,
Chengcan
Xiao
c,
Zeqiong
Zhao
c,
Ying
Li
a,
Tori
Cox
a,
Dapeng
Jing
bd,
Aaron J.
Rossini
ab,
Frank E.
Osterloh
c and
Julia V.
Zaikina
*a
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: yzaikina@iastate.edu
bAmes Laboratory, US DOE, Iowa State University, Ames, Iowa 50011, USA
cDepartment of Chemistry, University of California at Davis, Davis, California 95616, USA
dMaterials Analysis and Research Laboratory, Iowa State University, Ames, IA 50011, USA
First published on 5th May 2022
Abstract
SrTiO3 and aluminum-doped SrTiO3 are synthesized by calcination of metal salts dissolved in a deep eutectic solvent (DES) without any post-synthesis treatment. The DES used is the eutectic mixture of choline chloride (hydrogen bond acceptor) and malonic acid (hydrogen bond donor). Titanium(IV) oxide bis(2,4-pentanedionate) is utilized as the non-volatile, easy-to-handle, DES-soluble titanium precursor. The ammonia gas evolved during the calcination process provides a reducing atmosphere, resulting in the formation of Ti3+ and oxygen vacancies within the SrTiO3 matrix. According to UV-Vis spectroscopy and X-ray photoelectron spectroscopy, the amount of Ti3+ species and oxygen vacancies (VO) in the synthesized perovskite can be tuned by varying the duration of the calcination process and by adding Al3+ dopants. Solid state 27Al NMR spectroscopy and powder X-ray diffraction confirm the doping of aluminum into the octahedral site of the perovskite structure. Surface photovoltage spectroscopy confirms that Al3+ dopants can eliminate Ti3+ defects in Al-doped SrTiO3. Ultraviolet illumination experiments in water and aqueous methanol show that SrTiO3 and aluminum-doped SrTiO3, after modification with RhxCr2−xO3 or Pt co-catalysts, evolve small amounts of H2 (EQE of 0.0113–0.0173% at 375 nm) with only traces of O2 detected. The lack of photocatalytic activity is attributed to rapid electron-hole recombination in the oxygen vacancy-rich materials and to the lack of crystal facets that could aid charge separation.
Introduction
Deep eutectic solvents (DESs) are analogs of ionic liquids (ILs).1–3 Unlike conventional ILs, which are made up of discrete ions, DESs are made of a hydrogen bond acceptor (usually quaternary ammonium halides) and a hydrogen bond donor, thus DESs possess both hydrogen bonding and ionic interactions. The majority of DESs utilize 2-hydroxyethyltrimethylammonium chloride (choline chloride) as the hydrogen bond acceptor combined with a hydrogen bond donor such as urea, oxalic acid, citric acid, or acetamide, etc.2,4 When these two solids are mixed, they form a low melting eutectic mixture, that is a liquid at room temperature.1 DESs are non-toxic and biodegradable solvents; they have low melting points and are environmentally friendly.5 Owing to the high solubility of selected binary oxide or metal precursors in DESs,3 they are attractive replacements for corrosive inorganic acids as a reaction medium in solution synthesis of materials.3 Several ternary oxides such as spinel-type ferrite nanoparticles,6 zinc and copper vanadates like M2V2O7 and MV2O6 (M = Zn or Cu),7,8 and barium titanate BaTiO39 have been previously synthesized using the DES route. Except for BaTiO3, these ternary oxides were synthesized by dissolving binary metal oxides in DESs, followed by calcination of the resulting solution in air at 500 °C or higher temperatures. BaTiO3 was synthesized from barium acetate and titanium isopropoxide,9 because unlike many other binary oxides, TiO2 is insoluble in any of the studied DESs.3Strontium titanate, SrTiO3 is a functional oxide often used as a dielectric material10–12 and has been recently investigated for thermoelectric applications.13,14 It is also a semiconductor with reported bandgaps in the range of 3.0 eV to 3.88 eV.15–18 Due to the size of the band gap and the position of the valence/conduction band edge, strontium titanate, is a suitable catalyst for overall photocatalytic water splitting.19,20
SrTiO3 exists as an intrinsic non-stoichiometric compound which contains two types of defects: oxygen vacancies and Ti3+ species. When the Ti–O bond breaks, O2 gas is released to yield oxygen vacancies as well as free electrons, which reduce Ti4+ to Ti3+. The doping of SrTiO3 with aliovalent metal cations, specifically a lower valence cation in either of the cation sites (i.e. doping Sr2+ site with a +1 cation or doping Ti4+ site with a +3 cation) can be used to enhance its photocatalytic activity by suppressing the formation of Ti3+, which acts as an electron-hole recombination center.21,22 For example, a strong increase in water splitting activity was observed by doping Al3+ into SrTiO3via solid-state synthesis.22–25
SrTiO3 has been made via several synthetic routes such as solid-state,26–29 sol–gel,30–33 and hydrothermal synthetic methods.34–36 Like with most functional compounds, properties of SrTiO3 can be tuned via different synthetic routes that impact the dopant fraction, crystallinity, crystallite size and morphology. Typically, when reactants are liquids or in solution, this leads to a faster reaction because of the enhanced diffusion.
Here we report the synthesis and characterization of SrTiO3 and Al-doped SrTiO3 made through the environmentally benign choline chloride–malonic acid DES route without any post-synthesis treatment. Unlike previously reported synthesis of titanates, we utilized titanium(IV) oxide bis(2,4-pentanedionate) as a convenient and non-volatile Ti-precursor, which is easily dissolved in the employed DES. Two different heating profiles were employed to obtain SrTiO3 and Al-doped SrTiO3 with different properties. Furthermore, the site to which aluminum is being doped was investigated using 27Al solid-state nuclear magnetic resonance (NMR) spectroscopy. Lastly, the ability of the synthesized materials to function as photocatalysts for the overall water splitting reaction was studied.
Experimental
Materials and methods
Malonic acid CH2(COOH)2 (Alfa Aesar, 99%, crystalline), strontium nitrate Sr(NO3)2 (Alfa Aesar, 99.9%), titanium(IV) oxide bis(2,4-pentanedionate) (CH3COCHCOCH3)2TiO (TOPD) (Alfa Aesar, 99.9%), and aluminum 2,4-pentanedionate (CH3COCHCOCH3)3Al (AlPD) (Alfa Aesar, 99.6% min) were used as received without any further purification. Choline chloride (CH3)3N(Cl)CH2CH2OH (Sigma-Aldrich, 98%) was dried at 120 °C for 10 hours under vacuum prior to use. Fluorine-doped tin oxide (FTO) substrates (MTI corporation) were washed with detergent and deionized water, followed by 15 minutes of sonication in acetone, ethanol and ultra-purified water in series and drying in a 70 °C oven in air overnight.The deep eutectic solvent (DES) was prepared by mixing malonic acid and choline chloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) in a parafilm-covered glass beaker at 85 °C while stirring, until a clear viscous liquid DES was obtained.
:1 molar ratio) in a parafilm-covered glass beaker at 85 °C while stirring, until a clear viscous liquid DES was obtained.
In a typical synthesis of SrTiO3, 0.47 mmol (0.0995 g) of Sr(NO3)2 was dissolved in 35.904 g of DES (density of DES is 1.21 g mL−1) to make a 0.16 M solution, while 0.47 mmol (0.123 g) of TOPD was dissolved in 126.6 g of DES to make 0.0045 M solution. These two solutions were made in separate beakers until a clear solution was obtained in both cases. The resulting solutions were further mixed under vigorous stirring for 3 to 5 hours in a parafilm-covered beaker. About 7 g of the resulting solution was transferred to a 50 mL porcelain crucible and calcined in a Thermolyne Thermofisher muffle furnace. Two different heating profiles were used for sample syntheses.
Al-Doped SrTiO3, e.g. SrTixAl(1−x)O3, x = 0, 0.025, 0.05, 0.1, 0.2, was made using heating profile 1 and 2 by similar synthesis procedures as SrTiO3. In a typical synthesis of SrTi0.9Al0.1O3, three separate solutions were prepared: (1) 0.47 mmol (0.0995 g) of Sr(NO3)2 was dissolved in 35.904 g of DES; (2) 0.423 mmol (0.111 g) of TOPD was dissolved in 113.5 g of DES to make 0.0045 M solution; (3) 0.047 mmol (0.0152 g) of AlPD was dissolved in 12.2 g of DES to make 0.0045 M solution of AlPD in DES. The three solutions were then mixed and calcined as previously described for SrTiO3.
Characterization
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m = 4.1566 Å). The data was analyzed by the Rietveld refinement method using Jana2006 software package.38
:Al particles with a weight ratio of Rh:Cr:SrTiO3 (or SrTiO3:Al) = 1:1:1000, as described previously.44 Typically, 150 mg SrTiO3 or SrTiO3:Al was mixed with appropriate amount of RhCl3 and Cr(NO3)3 solutions containing 0.15 mg Rh and 0.15 mg Cr in a vial in a 70 °C water bath, and water was slowly evaporated under constant stirring. The product was then transferred to a furnace and heated at 350 °C in air for 1 h to produce RhxCr2−xO3 loaded SrTiO3 or SrTiO3:Al in 95% yield. Pt-modified SrTiO3 and SrTi0.9Al0.1O3 (heating profile 1) were made by photo-deposition, using 100 mg SrTiO3 (SrTiO3:Al) particles, a H2PtCl6 stock solution (2 mg Pt mL−1) and 100 mL 20% of aqueous methanol solution with the weight percentage of Pt to SrTiO3 (or SrTiO3:Al) being 2%. The mixture was sonicated for 10 min and bubbled with N2 for 10 min, then connected to the online Gas Chromatography system (SRI 8610c) with a 300W Xe lamp and magnetic stirring. H2 generation was recorded during photo-deposition to pinpoint the end point of this process. A typical photo-deposition lasts for 4–5 hours until the H2 generation rate becomes constant. After photo-deposition, the Pt loaded particles were centrifuged and washed with water for 3 times, re-suspended in 20% volume ratio methanol and made ready for hydrogen evolution experiment.
:Al in 118 mL of 20% aqueous methanol solution. The quartz flask was covered with aluminum foil leaving only a 1.531 cm2 window for incident light from the 375 nm LED. The LED light intensity was measured with a SEL623 Thermopile detector connected to an ILT1400 International Light photometer. Details of EQE calculation are given in the Supporting Information.
m = 4.1566 Å). The data was analyzed by the Rietveld refinement method using Jana2006 software package.38
:Al particles with a weight ratio of Rh:Cr:SrTiO3 (or SrTiO3:Al) = 1:1:1000, as described previously.44 Typically, 150 mg SrTiO3 or SrTiO3:Al was mixed with appropriate amount of RhCl3 and Cr(NO3)3 solutions containing 0.15 mg Rh and 0.15 mg Cr in a vial in a 70 °C water bath, and water was slowly evaporated under constant stirring. The product was then transferred to a furnace and heated at 350 °C in air for 1 h to produce RhxCr2−xO3 loaded SrTiO3 or SrTiO3:Al in 95% yield. Pt-modified SrTiO3 and SrTi0.9Al0.1O3 (heating profile 1) were made by photo-deposition, using 100 mg SrTiO3 (SrTiO3:Al) particles, a H2PtCl6 stock solution (2 mg Pt mL−1) and 100 mL 20% of aqueous methanol solution with the weight percentage of Pt to SrTiO3 (or SrTiO3:Al) being 2%. The mixture was sonicated for 10 min and bubbled with N2 for 10 min, then connected to the online Gas Chromatography system (SRI 8610c) with a 300W Xe lamp and magnetic stirring. H2 generation was recorded during photo-deposition to pinpoint the end point of this process. A typical photo-deposition lasts for 4–5 hours until the H2 generation rate becomes constant. After photo-deposition, the Pt loaded particles were centrifuged and washed with water for 3 times, re-suspended in 20% volume ratio methanol and made ready for hydrogen evolution experiment.
:Al in 118 mL of 20% aqueous methanol solution. The quartz flask was covered with aluminum foil leaving only a 1.531 cm2 window for incident light from the 375 nm LED. The LED light intensity was measured with a SEL623 Thermopile detector connected to an ILT1400 International Light photometer. Details of EQE calculation are given in the Supporting Information.
Results and discussion
The DES employed in this synthesis was choline chloride–malonic acid mixed in a 1:1 molar ratio. TiO2 is not soluble in any studied DES, but titanium isopropoxide has previously been utilized for the synthesis of BaTiO3 using a DES.9 However, both titanium isopropoxide and titanium(IV) chloride are flammable fuming liquids which readily undergoes hydrolysis upon contact with humid air, making them unpleasant precursors to work with. Here we utilized titanium(IV) oxide bis(2,4-pentanedionate)(TOPD)45 as a DES-soluble precursor. TOPD, which is the source of titanium in this synthesis dissolves in the choline chloride–malonic acid DES to give a yellow viscous solution in a parafilm covered beaker. When a laser beam (from a laser pointer) is passed through the yellow viscous solution (Fig. 1(a)), no Tyndall scattering was observed, confirming that that TOPD indeed formed a true solution in DES, not just a dispersion of colloidal particles. TOPD is a more convenient precursor to use since it is a powder (thus easy to weigh) and it does not produce fumes unlike other typically used Ti precursors.
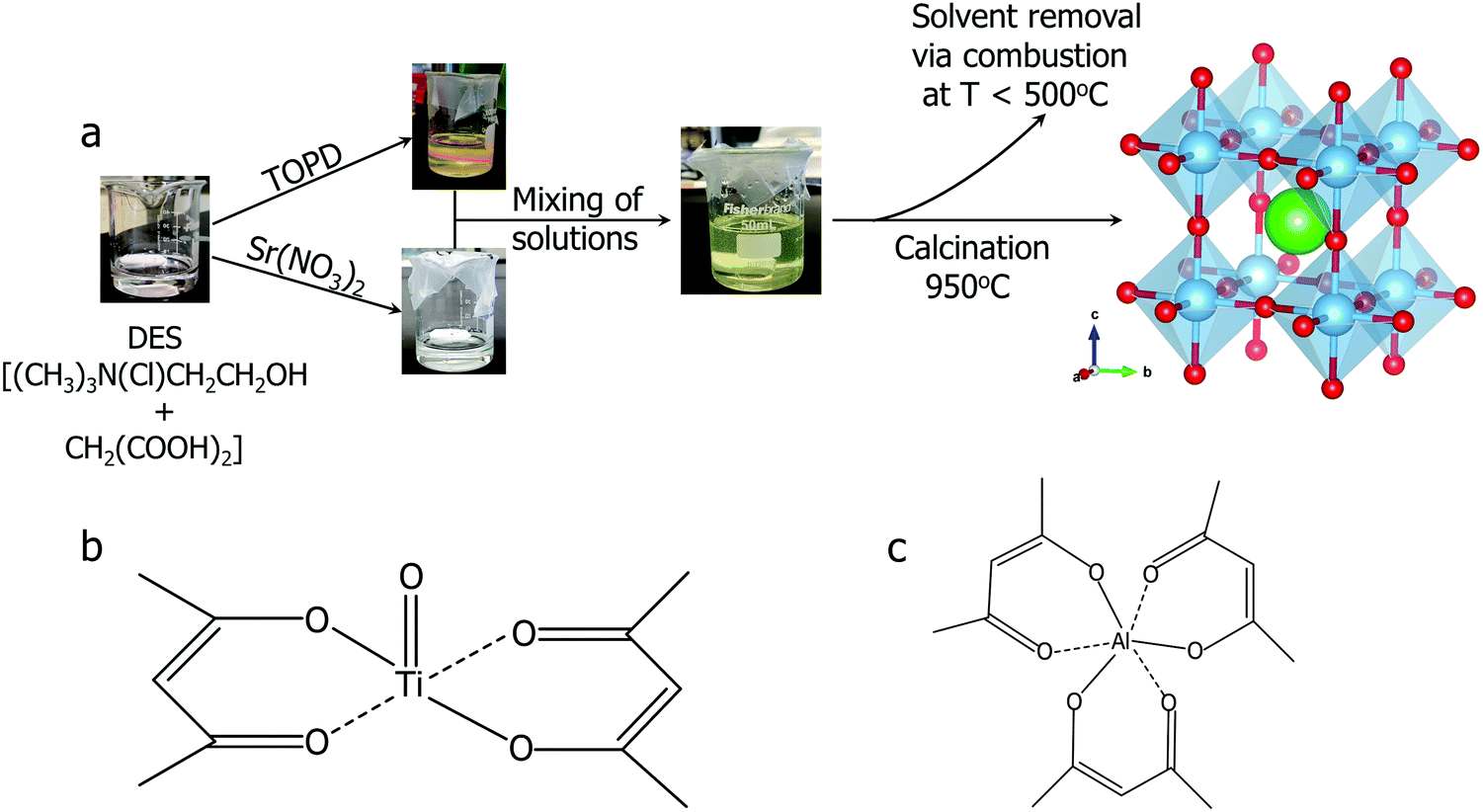 | ||
| Fig. 1 (a) Synthetic route of strontium titanate SrTiO3. Atom color coding used: Sr (green), Ti (blue), O (red); (b) molecular structure of titanium(IV) oxide bis(2,4-pentanedionate) (TOPD); (c) molecular structure of aluminum 2,4-pentanedionate. | ||
The resulting yellow stable viscous solution was transferred into an open porcelain crucible and calcined in air, removing the organic solvent. The organic components are combusted below or at 500 °C. Ammonia, hydrogen chloride, carbon dioxide and water are evolved as gaseous by-products. Powder X-ray diffraction (PXRD) patterns were collected ex situ at different calcination temperatures to monitor the progress of the reaction (Fig. 2). During the calcination of the transparent, yellow viscous solution, the sample remains amorphous up to 300 °C.
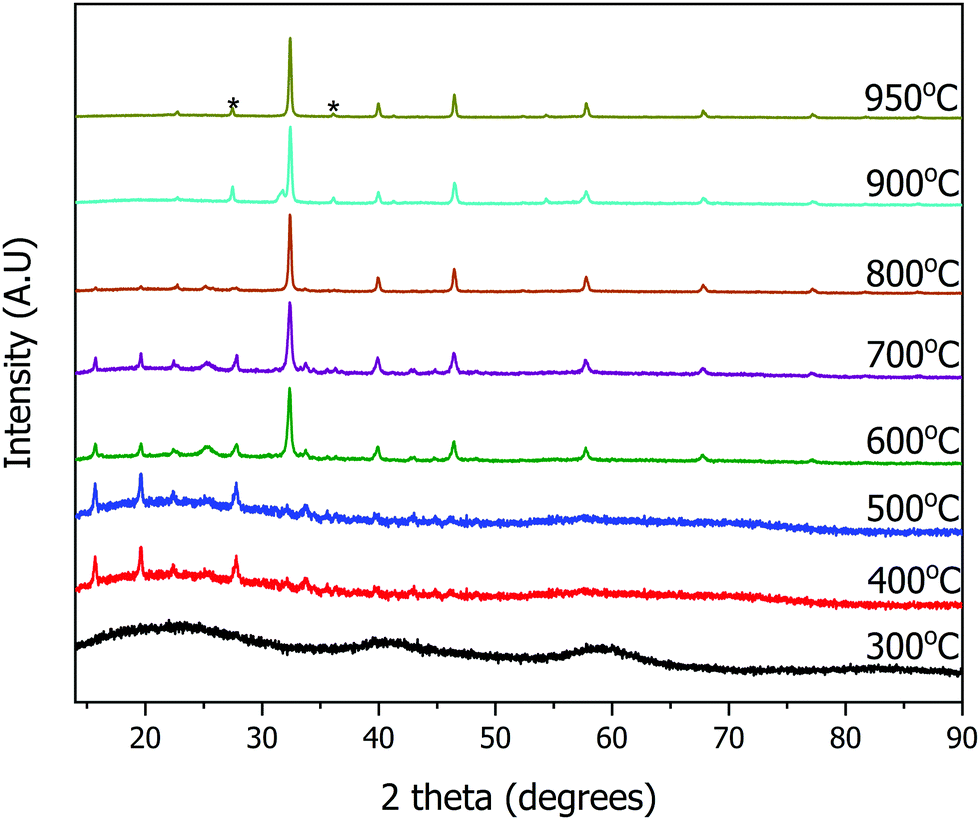 | ||
| Fig. 2 Ex situ powder X-ray diffraction patterns of the calcined sample for the synthesis of SrTiO3 at various temperatures. TiO2 impurity peaks are marked with asterisks (*). | ||
The first set of peaks appearing at 400 °C correspond to two phases: strontium chloride dihydrate (SrCl2·2H2O) and anatase TiO2. Peaks of cubic SrTiO3 (perovskite structure, Pmm) begin to appear at 600 °C with SrCl2·2H2O and anatase TiO2 still present. As the peaks of SrTiO3 become more intense, the intensity of SrCl2·2H2O peaks decrease. At 950 °C the reaction is complete, and SrTiO3 is the primary product with a minor impurity of both anatase and rutile TiO2 (5 wt%), as determined by Rietveld refinement of PXRD data. Overall, SrCl2·2H2O and TiO2 formed first, and further reaction occurs upon an increase in temperature to yield SrTiO3. The result of this stepwise calcination process differs from the previously reported synthesis of BaTiO3,9 since the Bragg's peaks corresponding to SrTiO3 appeared as early as 600 °C whereas peaks corresponding to BaTiO3 were not observed until 850 °C. Additionally, the DES-assisted synthesis of BaTiO3 proceeds through a BaCl2 intermediate while that of SrTiO3 proceeds through a SrCl2·2H2O intermediate. We hypothesize that the difference in the type of the chloride intermediates arises because for BaTiO3 synthesis, precursors were stirred in an open container at 90 °C until water evaporated. In contrast, our synthesis of SrTiO3 used stirring in a parafilm covered beaker which discourages evaporation. Since the little amount of water present in the hygroscopic choline chloride is not allowed to evaporate during mixing, this water may lead to the formation of a hydrated chloride intermediate, SrCl2·2H2O.
Due to the small concentration of metal precursors in the DES, the yellow viscous solution is very dilute. Two heating profiles are employed for the calcination process: heating profile 1 and heating profile 2. Heating profile 2 is similar to that used in the synthesis of BaTiO3,9 it is a rapid calcination and had an overall shorter dwelling time (see experimental details). It was observed that the primary yield of SrTiO3 made from both heating profiles is ∼91%. The secondary yield, however, is different for samples made via different heating profiles. The samples made using heating profile 1 have large clusters, and can be easily transferred from the crucibles, leading to a higher secondary yield of 77% (occasionally, the yield can be as high as 88%). However, samples made by heating profile 2 are mostly sintered to the crucibles. Also, the particles are smaller, unclustered, lightweight, and so are easily lost during the transfer from the crucibles. This leads to a lower secondary yield when heating profile 2 is employed (23%).
The weight percent of impurities present in the synthesized samples was evaluated by Rietveld refinement of PXRD data. Samples made with heating profile 1 contain the cubic SrTiO3 (∼95 wt%) as well as both rutile TiO2 and anatase TiO2 impurity which accounted for 5 wt% of the sample (Fig. 3). The impurity peaks of rutile and anatase TiO2 were absent in the PXRD pattern of SrTiO3 prepared via heating profile 2. We observed that the slower and longer heating profile 1 allowed the formation of TiO2 impurities as opposed to the shorter and faster heating profile 2. As it was further determined by several characterization methods, the primary difference between SrTiO3 samples produced via two heating profiles is the concentration of O vacancies: heating profile 1 resulted in the SrTiO3 that is rich in O vacancies (VO rich), while heating profile 2 leads to SrTiO3 that is poor in O vacancies (VO poor). For the sake of clarity, we refer to the samples prepared via heating profile 1 as “VO rich” and via heating profile 2 as “VO poor” throughout the text.
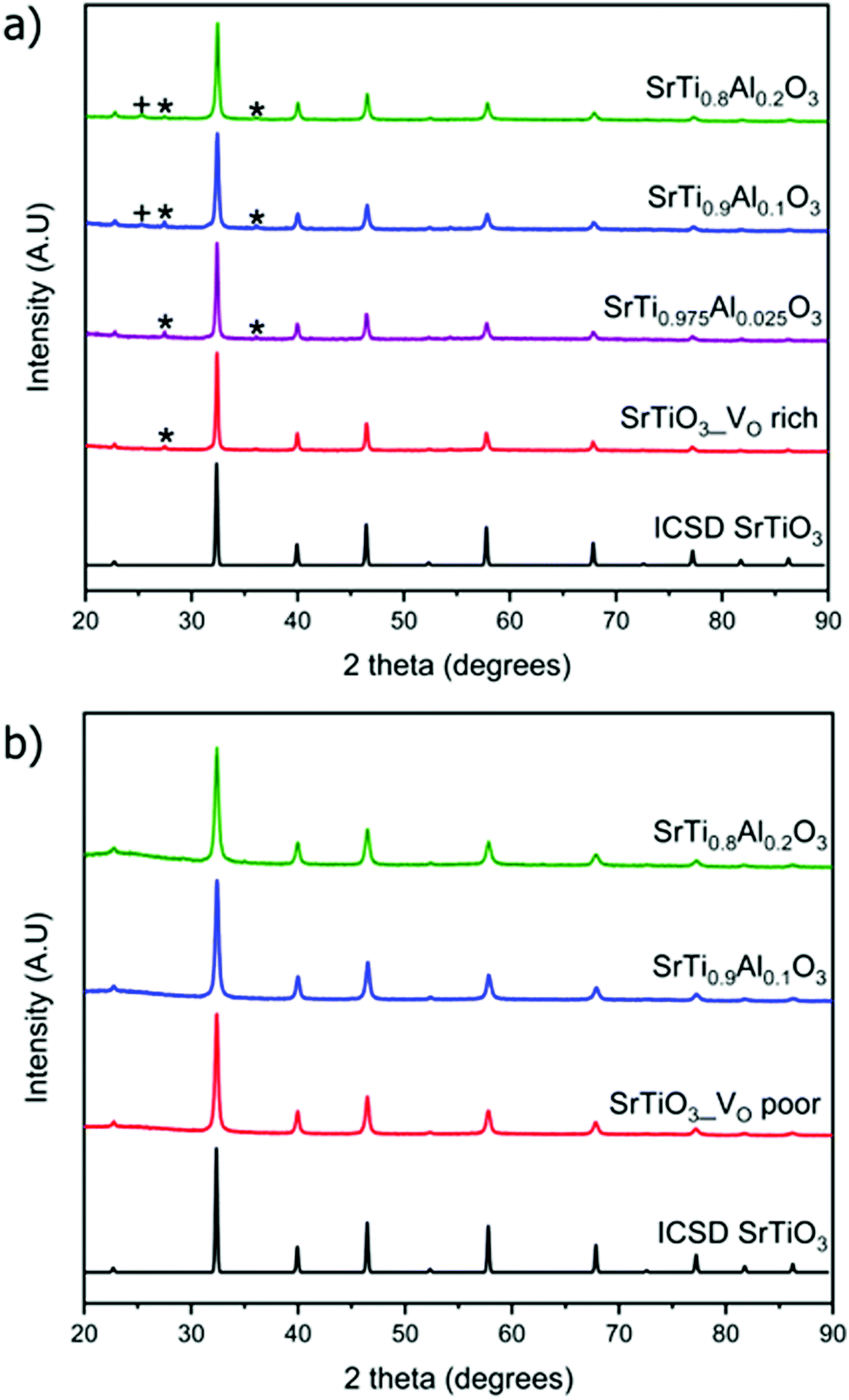 | ||
| Fig. 3 Powder X-ray diffraction patterns of doped and undoped (a) VO rich SrTiO3 made with heating profile 1 and (b) VO poor SrTiO3 made with heating profile 2. Peaks that belong to rutile TiO2 impurity are marked with asterisks (*) while peaks belonging to anatase TiO2 impurity are marked with plus sign (+). ICSD 192314 was used for SrTiO3. | ||
Since the doping of SrTiO3 with aluminum has been reported to increase the rate of photocatalytic activity of SrTiO3,22–25 aluminum-doped samples SrTi1−xAlxO3 were synthesized using the same two heating profiles. The maximum loading fraction of Al used was x = 0.2 because further increase in Al dopant concentration resulted in a SrAlO2 impurity as evident from PXRD data. The doped samples made with heating profile 1 also have the anatase and rutile TiO2 impurities. Similar to undoped SrTiO3, these impurities are absent in the doped samples made with heating profile 2 because the faster and shorter heating profile does not favour the formation of TiO2 impurities (Fig. 3). The replacement of Ti4+ (0.605 Å) in octahedral sites with smaller Al3+ (0.535 Å) is expected to result in the reduction of the cubic unit cell parameters. Rietveld refinement using LaB6 as an internal standard was carried out to determine the change in unit cell upon Al doping (Fig. 4). The unit cell parameter of VO rich SrTi1−xAlxO3 are generally smaller than that VO poor SrTi1−xAlxO3 as shown in Fig. 4. As expected, with the increase of Al content, the unit cell parameter decreases until x = 0.1, i.e., the unit cell parameter of SrTi0.9Al0.1O3 is very similar to that of SrTi0.8Al0.2O3, suggesting that maximal Al dopant concentration is x < 0.2.
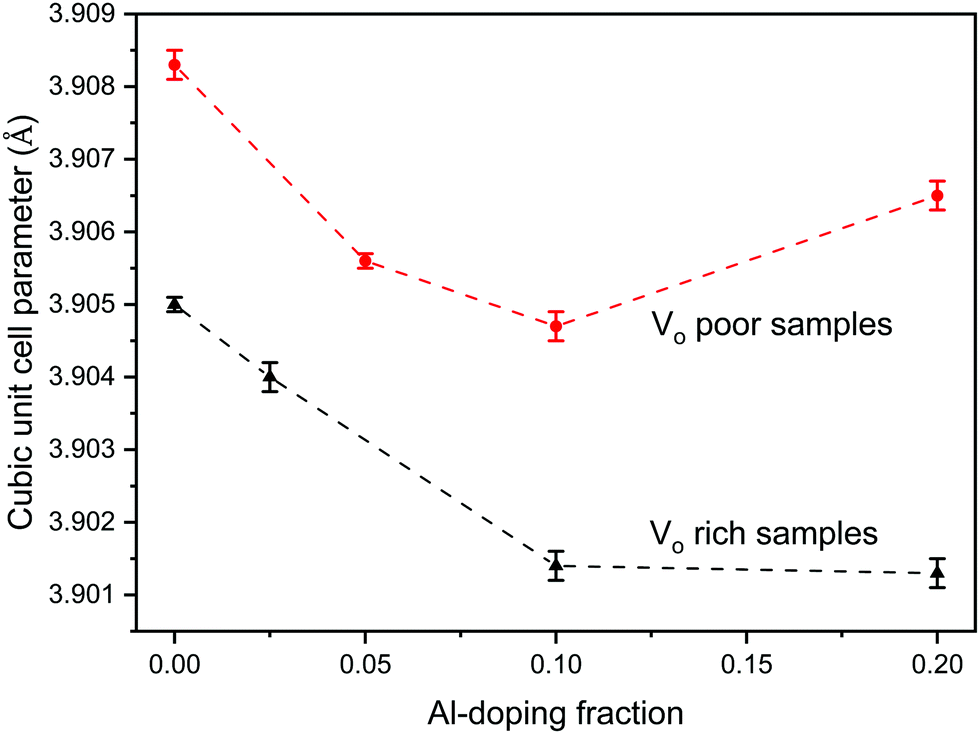 | ||
| Fig. 4 Change in cubic unit cell parameter of the VO rich SrTi1−xAlxO3 made with heating profile 1 (black) and VO poor SrTi1−xAlxO3 made with heating profile 2 (red) as a function of Al doping, x. | ||
The crystallite size of VO rich samples calculated from the Debye Scherrer equation on average decreases with increase in aluminum doping while the crystallite size of VO poor samples remain constant regardless of aluminum doping (Table 1).
Scanning electron microscopy (SEM) images of the samples showed the doped and undoped VO rich SrTiO3 have structured crystallites (in red, Fig. 5(a)), nanoclusters (in yellow, Fig. 5(a)), and flakes (in blue, Fig. 5(a)). On the contrary, the VO poor samples all appeared to be in the form of flakes (or sheets), regardless of the presence or absence of aluminum (Fig. 5 and Fig. S1, ESI†). Energy Dispersive X-ray Spectroscopy (EDXS) of Al-doped sample indicates the homogeneous distribution of Sr, Ti and Al in the samples.
 | ||
| Fig. 5 SEM image of (a) VO rich SrTiO3 made with heating profile 1 and (b) VO poor SrTiO3 made with heating profile 2. | ||
27Al solid-state NMR spectroscopy
Aluminum is expected to replace titanium in the octahedral site of the SrTiO3 perovskite structure based on the similarity of ionic radii. 27Al solid-state NMR spectra of the doped samples were obtained to prove doping of aluminum in a symmetric octahedral site in the perovskite structure (Fig. 6). 27Al is a spin-5/2 quadrupolar nucleus with a natural abundance of 100% and a high sensitivity to the coordination environment and symmetry. We note that solid-state NMR spectroscopy has previously been used to extensively characterize SrTiO3 and related perovskite materials.46–49 The NMR spectra were fitted to three signals: a broad peak with an isotropic shift (δiso) of ca. 83 ppm corresponding to a tetrahedral site, a broad signal with δiso of 16 ppm corresponding to an octahedral site, and a narrow signal with δiso of 9 ppm that is assigned to the doped octahedral site of the perovskite structure. The broad tetrahedral and octahedral signals are characteristic of alumina,50 and were assigned to an amorphous alumina impurity that could not be detected by PXRD.49 The tail of the broad 27Al NMR signals at lower chemical shifts is indicative of a distribution of quadrupolar couplings that is intrinsic to amorphous alumina.50 The sharp signal at 9 ppm represents a 27Al site with low quadrupolar broadening and must therefore correspond to a site of high spherical symmetry; this observation confirms the aluminum doping into the titanium octahedral site in SrTiO3. To ensure quantitative excitation of the various 27Al signals, a small tip angle pulse was used (see experimental).39,40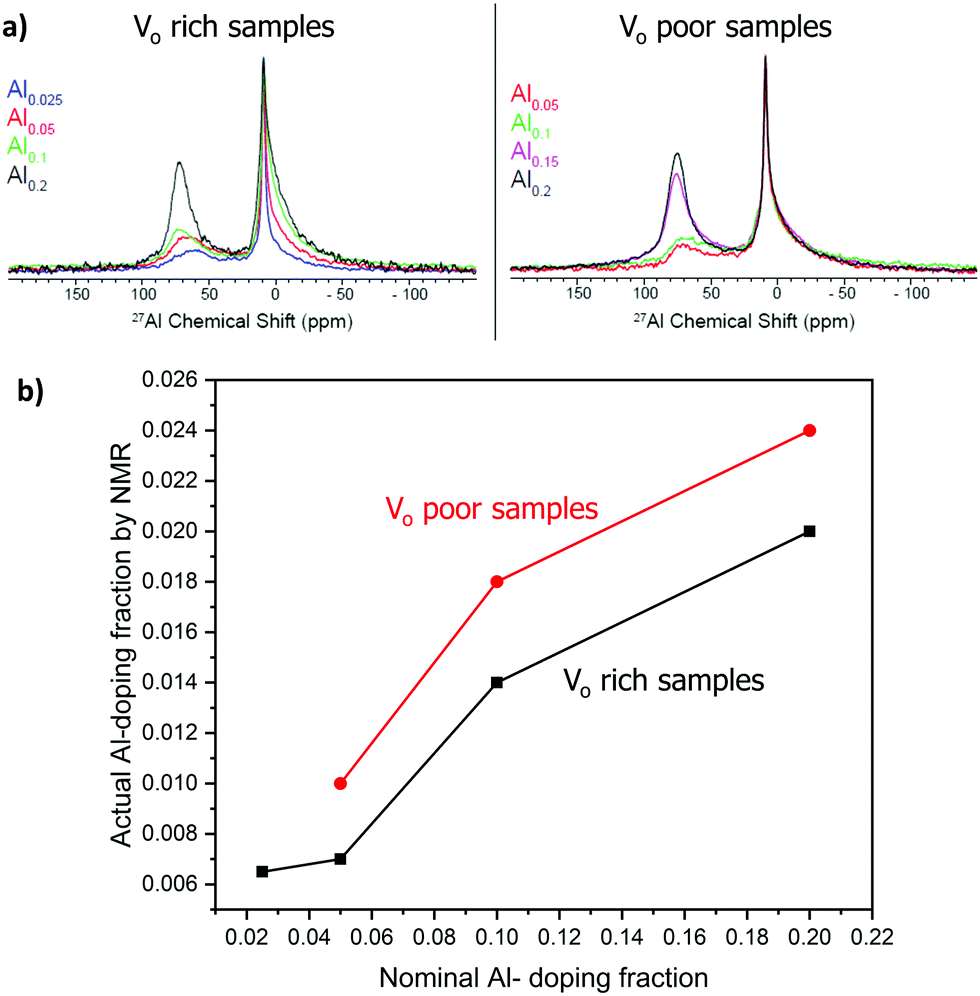 | ||
| Fig. 6 27Al solid-state NMR. (a) Comparison of 1D spectra; signal intensities are normalized with respect the sharp signal at ca. 9 ppm. (b) Plot showing the actual and nominal aluminum doping concentration trend for VO rich samples made with heating 1 (black curve) and VO poor samples made heating 2 (red curve). | ||
Fig. 6(a) compares the 27Al solid-state NMR obtained with different Al doping levels. Note that the intensities are normalized with respect to the sharp 27Al NMR signal at 9 ppm. It is evident that as the Al doping level increases the concentration of the alumina impurity also increases. Next, the spectra were deconvoluted and fit using the Czjzek distribution to model the distribution in quadrupolar couplings of the alumina impurity (Fig. S2, ESI†). The fraction of the octahedral doped 27Al sites in perovskite structure (% doping, Table 2) was then estimated from the absolute integrals of the deconvoluted peaks.
Notably, the actual fraction of aluminum doped into the structure (actual x, Table 2) was lower than the nominal fraction. As the nominal doping fraction increases, so does the actual doping fraction (Fig. 6(b)). However, the percentage of aluminum being doped into the structure decreases as the aluminum doping fraction increases, suggesting that at higher nominal Al fraction more alumina is forming (Table 2).
Diffuse reflectance and bandgap measurement
The diffuse reflectance was measured for all samples and expressed as Tauc plots to calculate the direct band gap (Fig. 7). All the VO rich samples showed an approximate direct band gap of 3.0 eV and all VO poor samples showed an approximate direct band gap of 3.2 eV as shown in Fig. 7 and Fig. S3 (ESI†). The doping of the samples with Al does not significantly affect their bandgaps for either heating profile. Doped and undoped SrTiO3 samples made with heating profiles 1 and 2 showed beige colors but with different intensities. The color of the synthesized samples can be attributed to the presence of oxygen vacancies and high concentration of Ti3+. It has been previously reported that SrTiO3 samples containing a high amount of Ti3+ usually possess color,22 in support of our observation.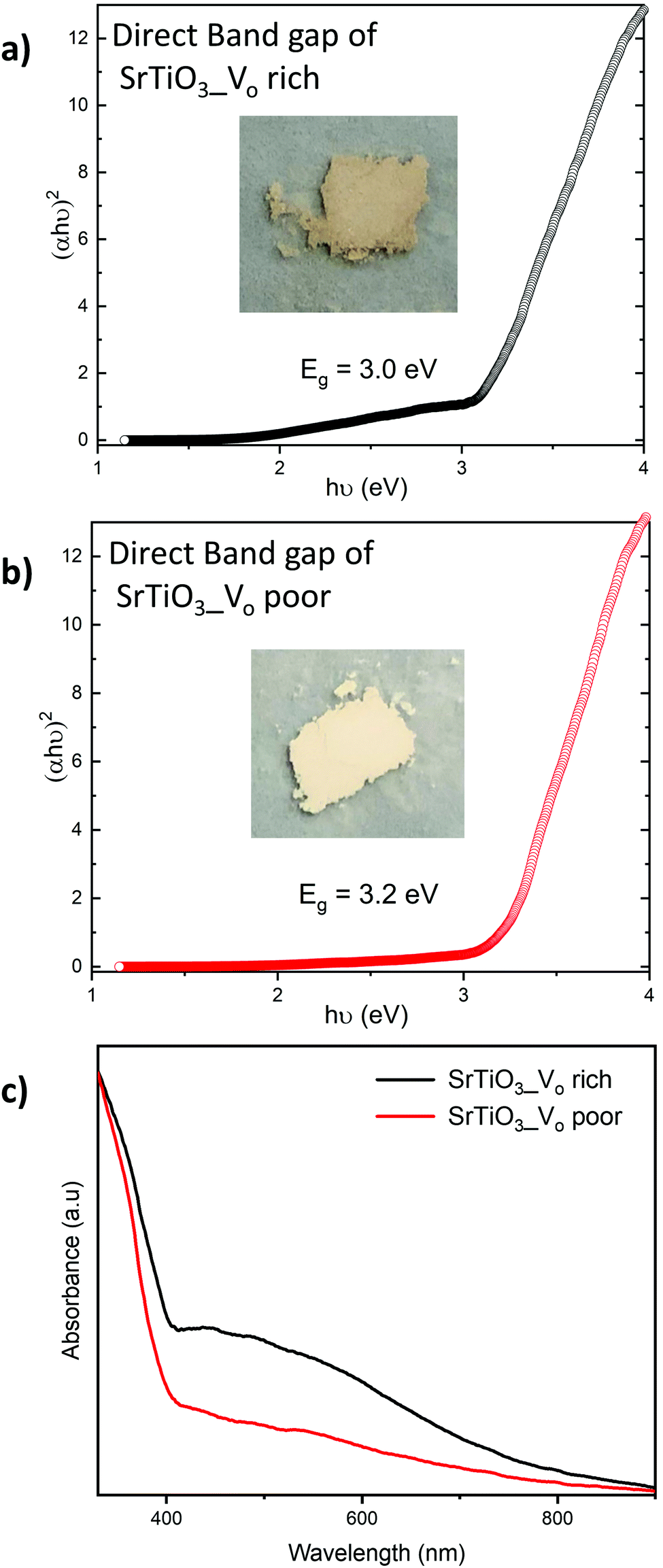 | ||
| Fig. 7 Tauc plots used to determine direct band gaps of (a) VO rich SrTiO3 obtained via heating profile 1 and (b) Vo poor SrTiO3 obtained via heating profile 2. Insets show the images of SrTiO3 samples obtained via respective heating profiles. c) UV-Vis spectra of VO rich SrTiO3 obtained via heating profile 1 (black curve) and Vo poor SrTiO3 obtained via heating profile 2 (red curve). | ||
Both undoped and doped samples made with heating profile 1 were darker beige in color, as shown in Fig. 7(a). A band tail (or an additional absorption edge) was also noticed in their absorption data (Fig. 7(c)). The darker beige color and a band tail suggest that samples made with heating profile 1 have more Ti3+ species and oxygen vacancies (VO rich). This is in agreement with previous reports where the concentration of Ti3+/oxygen vacancies in SrTiO3 was increased by heating the sample in a reducing environment.52,53 Similarly, the decomposition of the DES provides a reducing atmosphere during the synthesis which favors the formation of oxygen vacancies as it was previously shown for zinc and copper vanadates.7,8
Samples made with heating profile 2 (VO poor) showed a lighter beige color and also a slightly wider bandgap (Fig. 7(b)). The absence of a pronounced band tail in the absorbance spectra of samples made with heating profile 2 (Fig. 7(c)) indicates a lower concentration of Ti3+ states.51 Since the same color was observed in undoped sample and doped sample, we presume that the Al3+ dopant did not significantly suppress Ti3+ states.
X-Ray photoelectron spectroscopy
XPS measurements were carried out on the SrTiO3 samples to quantify the amount of reduced Ti species. Fig. 8 shows Ti 2p core level spectra of SrTiO3 made with both heating profiles. The two titanium peaks shown originate from spin–orbit splitting of Ti 2p3/2 and Ti 2p1/2, each peak is a superposition of 3 components corresponding to Ti4+, Ti3+, and Ti2+. Fig. 8(a) shows the titanium species in VO rich SrTiO3. The doublet in red at 458.7 eV (Ti 2p3/2) and 464.4 eV (Ti 2p1/2) are assigned to Ti4+, the doublet in blue at 457.6 eV (Ti 2p3/2) and 463.0 eV (Ti 2p1/2) are assigned to Ti3+, while the doublet in magenta at 455.4 eV and 461.4 eV are assigned to Ti2+. Based on the relative Ti 2p3/2 peak ratios, the VO rich sample has 73.5% of titanium present as Ti3+, 19.2% as Ti4+, and 7.3% as Ti2+. The large quantity of Ti3+ present in oxygen vacancy rich SrTiO3 agrees with the optical properties as discussed above. Fig. 8(b) shows the titanium species in the VO poor SrTiO3. The doublet in red at 458.5 eV (Ti 2p3/2) and 464.2 eV (Ti 2p1/2) are assigned to Ti4+, the doublet in blue at 457.1 eV (Ti 2p3/2) and 462.9 eV (Ti 2p1/2) are assigned to Ti3+, while the doublet in magenta at 455.3 eV and 461.1 eV are assigned to Ti2+. In contrast to the VO rich sample, the VO poor SrTiO3 sample made with heating profile 2 has 72.5% of titanium present as Ti4+, 20.8% as Ti3+ and 6.7% as Ti2+. The values of the binding energy of the titanium species observed in this work are similar to those that have previously been reported.22,54 The presence of Ti3+ and Ti2+ in the VO rich and poor SrTiO3 suggests that the malonic acid–choline chloride DES provides a reducing atmosphere during the calcination of doped or undoped SrTiO3 samples. The prolonged heating of a sample in this reducing atmosphere leads to the increased amount of Ti3+ in the VO rich samples made with heating profile 1. The XPS spectra in the Sr 3d region are similar for SrTiO3 prepared by both heating methods, suggesting that Sr is in +2 oxidation state, irrespective of the heating profile used (Fig. S4, ESI†).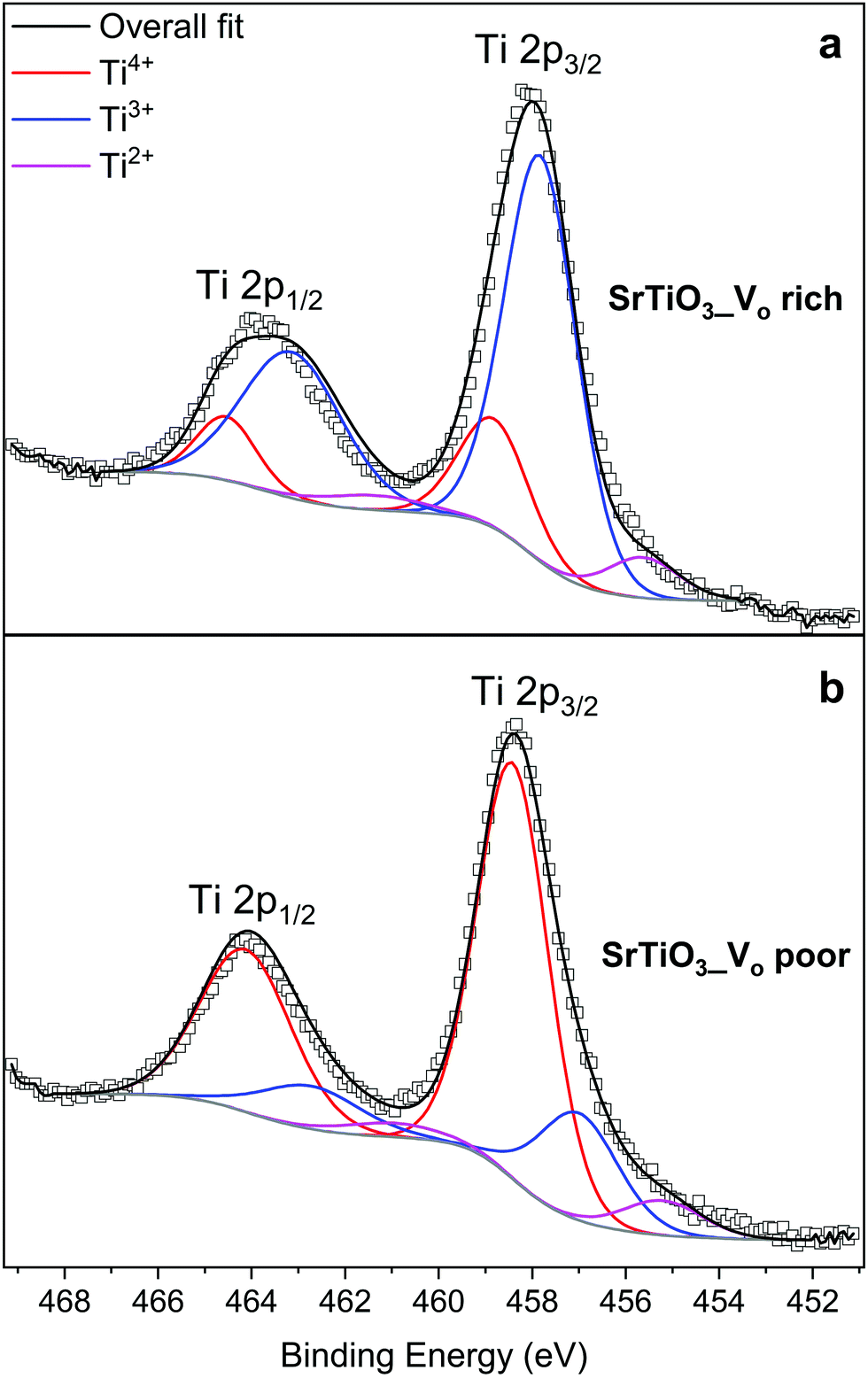 | ||
| Fig. 8 XPS spectra of the Ti 2p region: (a) VO rich SrTiO3 made with heating profile 1 and (b) VO poor SrTiO3 made with heating profile 2. | ||
The O 1s spectra shown in Fig. 9 can also be fitted with 3 peaks. Fig. 9(a) represents the O 1s spectra of VO rich SrTiO3. The peak in red at 529.0 eV22,52 corresponds to the lattice oxygen in the crystal structure of SrTiO3, the blue peak at 531.2 eV52 suggests the oxygen vacancies within the synthesized sample, and the green peak at 532.555 likely corresponds to loosely bound oxygen species at the sample due to atmospheric moisture. The O 1s spectra of VO poor SrTiO3 shown in Fig. 9(b) are shifted to higher binding energies by 0.6 eV. The fitted peak with the red line observed at 529.6 eV is attributed to the lattice oxygen, the fitted peak with the blue line at 531.8 eV corresponds to the oxygen vacancy and the fitted peak in green at 533.1 eV corresponds to the loosely bound oxygen from surface moisture. The amount of oxygen vacancy in the VO rich SrTiO3 sample was deduced from the fitted peaks in blue to be 32.4% while the VO poor SrTiO3 sample had 23.6% oxygen vacancy. The larger quantity of oxygen vacancies in VO rich SrTiO3 made with heating profile 1 can also explain its smaller unit cell parameter (Fig. 4). VO rich SrTiO3 has a unit cell parameter of 3.9050(4) Å which is 0.003 Å smaller than the unit cell parameter of VO poor SrTiO3. The same trend is seen for Al-doped SrTiO3 prepared via two heating profiles with VO rich Al-doped SrTiO3 having overall smaller unit cell parameter compared to VO poor Al-doped SrTiO3 (Fig. 4). It should be noted that an atomic vacancy cannot be directly measured by the XPS, so the peaks assigned to the oxygen vacancy actually correspond to oxygen atoms near the vacant site. The survey spectra (Fig. S5, ESI†) suggest the absence of any other impurity element except the expected surface carbon, albeit the detection limit of XPS is 0.1–1 at%. It should also be noted that XPS is a surface technique and the percent of Ti3+/oxygen vacancies is not representative of the bulk sample.
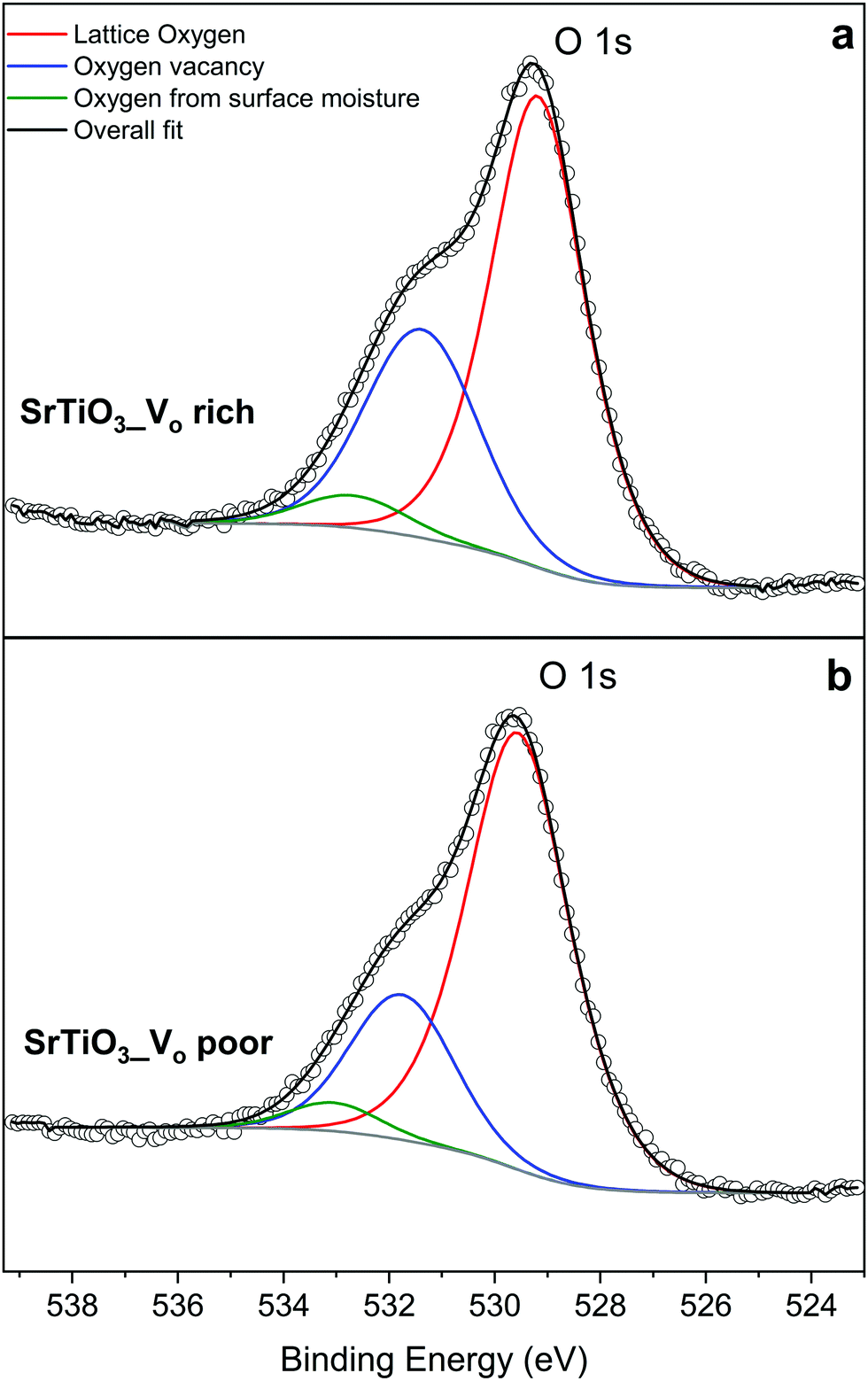 | ||
| Fig. 9 XPS data of the oxygen species in (a) VO rich SrTiO3 made with heating profile 1 and (b) VO poor SrTiO3 made with heating profile 2. | ||
Photocatalytic activity
The photocatalytic activity of SrTiO3 for overall water splitting under UV illumination was investigated after loading a RhxCr2−xO3 proton reduction cocatalyst (for details see experimental section). As can be seen from Fig. 10 and Fig. S6 (ESI†), all samples evolve only small amounts of H2 and only trace amounts of O2 can be detected, likely from residual dissolved oxygen in the reaction mixtures. This suggests that H2 evolution is stoichiometric, not catalytic, and the SrTiO3 samples themselves serve as the electron source for the evolved H2. We find that VO poor SrTiO3 showed a higher H2 evolution rate than that of the VO rich SrTiO3. This suggests that photocatalytic H2 evolution activity and VO/Ti3+ are inversely correlated. The H2 amounts also varied with Al3+ content, although no activity increase or trend could be seen. These findings are in contrast with previous observations of strong water splitting activity with micro- and nanosized Al-doped SrTiO3 made by flux or hydrothermal syntheses.22–25,56 Those samples had mostly cubic morphology and well-defined facets which promoted carrier separation. The necessity for a well-faceted highly crystalline photocatalyst was further emphasized in the recent work by Takata et al.25 It was reported that oxygen evolution reaction preferably occurred on the (110) facets while hydrogen evolution reactions occurred on the (100) facets. As previously reported by Zhao et al.,22 the Ti3+ sites act as recombination sites for photogenerated holes and electrons which leads to a reduction in the photocatalytic activity of SrTiO3. We therefore hypothesize that the absence of water splitting activity in the DES-synthesized materials is a result of both the absence of a faceted morphology, as well as the presence of a substantial amount of Ti3+ recombination sites. This hypothesis is supported by separate H2 evolution experiments on Pt-modified VO rich SrTiO3 and Al–SrTiO3 samples in aqueous methanol (Fig. S7, ESI†). Again, only low H2 amounts are formed with external quantum efficiencies (EQE) of 0.017% and 0.0121% (both at 375 nm) for Pt/SrTiO3 and Pt/SrTi0.9Al0.1O3, respectively. These reactions use methanol as an easy-to oxidize sacrificial electron donor and Pt as an efficient proton reduction co-catalyst. The low EQE values confirm that the low photocatalytic activity is intrinsic to the DES-synthesized materials.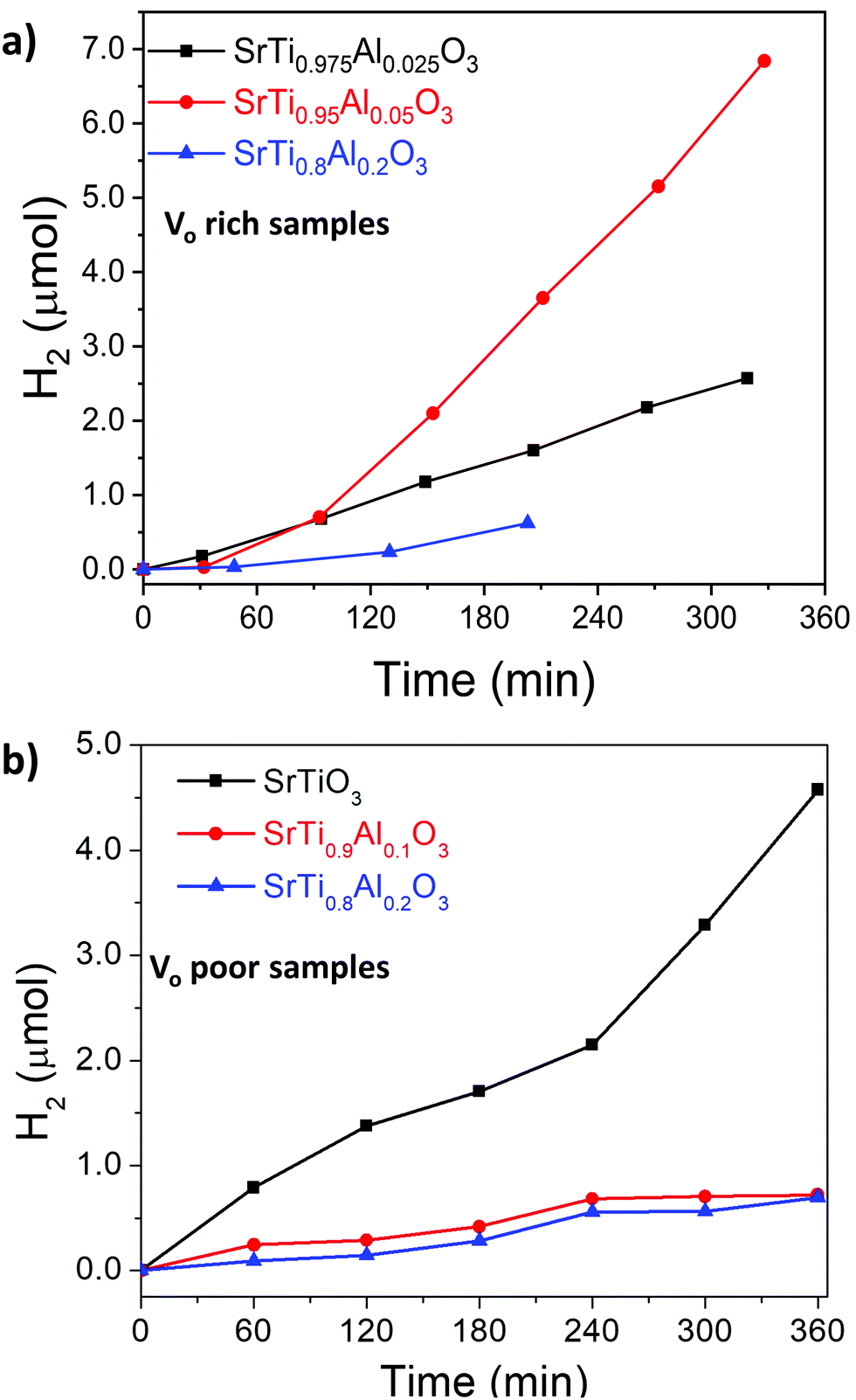 | ||
| Fig. 10 Hydrogen evolution of (a) VO rich samples made with heating profile 1 and (b) VO poor samples made with heating profile 2 from water. Conditions: 100 mW cm−2 UV light from Xe-arc lamp, 100 mg RhxCr2−xO3/SrTiO3 (or RhxCr2−xO3/SrTiO3:Al) powder in 100 mL water. Oxygen was also detected for Vo rich samples made with heating 1 (see also Fig. S6, ESI†), while no oxygen was detected for Vo poor samples made with heating 2. | ||
To study the photochemical charge separation ability of VO rich and poor SrTiO3 and SrTi0.9Al0.1O3, surface photovoltage (SPV) spectroscopy was conducted on particle films on FTO (Fig. 11). Here, a vibrating gold probe measures the contact potential difference (CPD) of the sample. The CPD value in the dark is given by the Fermi level difference between the gold probe and the sample, while the change (ΔCPD) under illumination equals the surface photovoltage.57,58 The SPV spectrum for VO rich Al3+-doped SrTiO3 in Fig. 11(a) shows an SPV onset at 2.63 eV and a maximum SPV signal at 3.6 eV. From the tangent of this major signal, the effective band gap of 3.09 eV can be extracted. This value is in good agreement with the optical bandgap in Fig. 7. Hence, the major photovoltage signal is attributed to band gap excitation of Al3+-doped SrTiO3 and charge transfer as shown in Fig. 12. The spectrum for the VO rich Al3+-free SrTiO3 is also shown in Fig. 11(a). Here, the photovoltage signal starts at 1.8 eV and the effective bandgap appears at 2.63 eV. These low energy features are attributed to excitation of Ti3+ mid-gap states as shown in Fig. 12, and hole trapping in SrTiO3 surface states. Based on the SPV spectroscopy data, the Ti3+ mid-gap state's concentration in VO rich Al3+-doped SrTiO3 is greatly reduced. This confirms that Al3+ doping eliminates Ti3+ states for the VO rich samples obtained via heating profile 1.
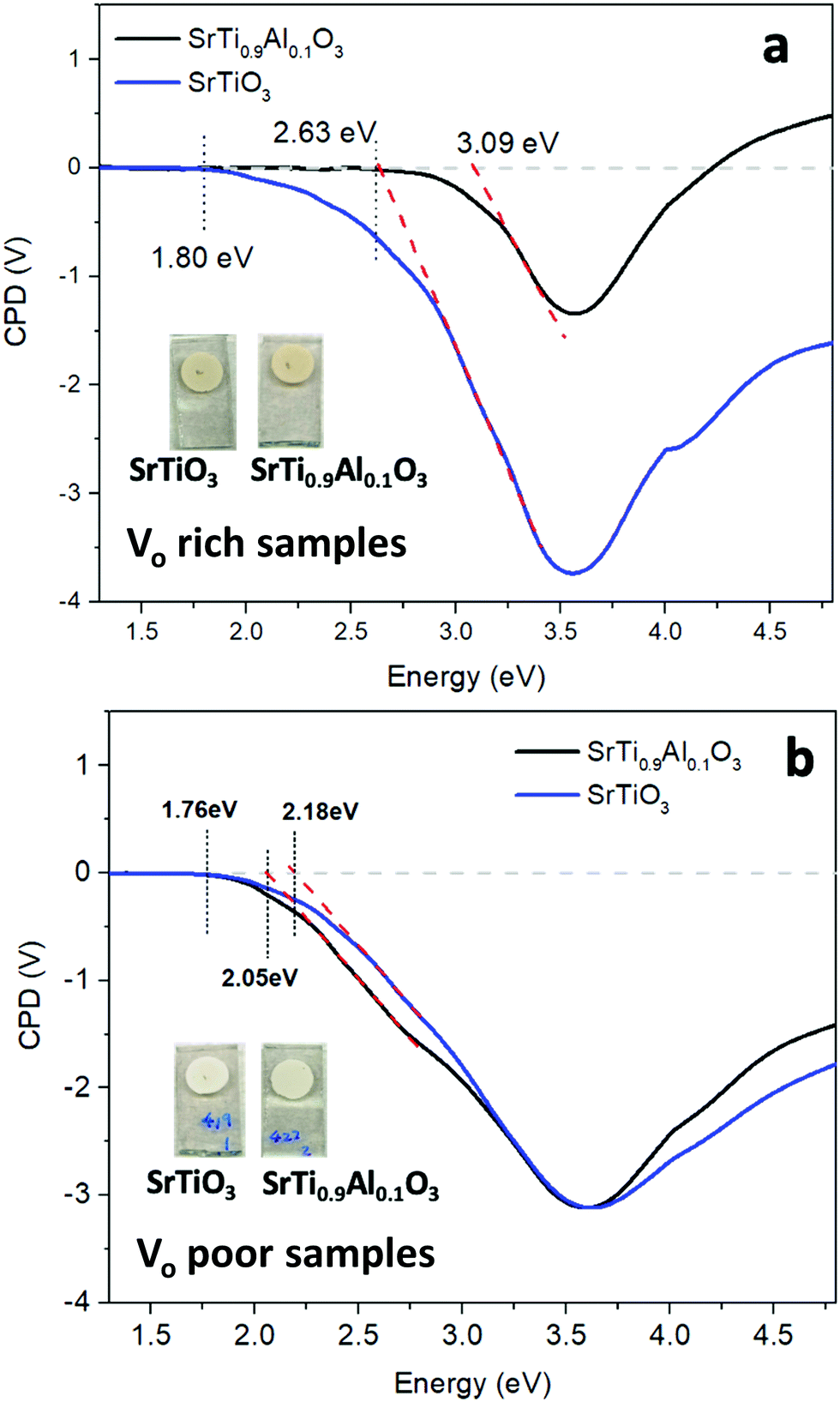 | ||
| Fig. 11 Surface photovoltage (SPV) spectra of SrTiO3 (blue) and SrTi0.9Al0.1O3 (black) (a) VO rich made by heating profile 1 and (b) VO poor made by heating profile 2, respectively. CPD values under illumination are reported relative to the dark. The band-like onset was approximated by linear extrapolation (red dash). The inset pictures show the appearance of the respective films used for SPV measurement. | ||
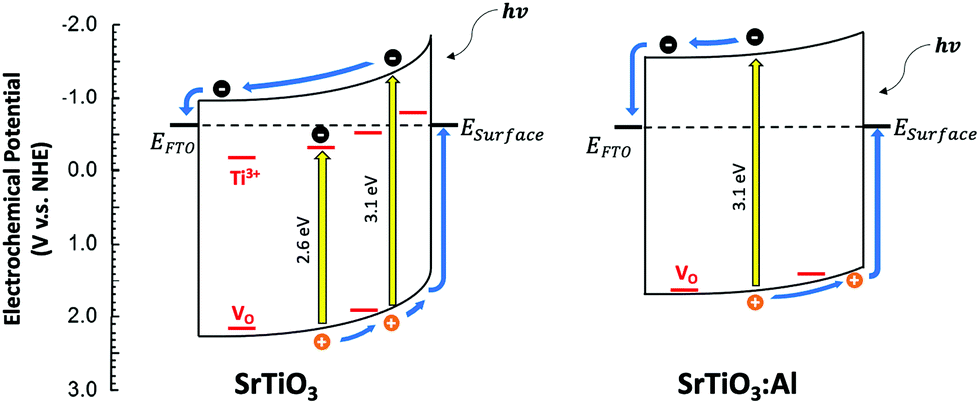 | ||
| Fig. 12 Charge transfer and energetics during the SPV experiment for VO rich SrTiO3 and Al-doped SrTiO3 (heating 1). In SrTiO3:Al, Ti3+ trap states were eliminated by Al3+ doping, leading to a less reducing Fermi level, fewer trapped electrons, and less band bending. The lower band bending in SrTiO3:Al is indicated by the lesser surface photovoltage signal. ESurface denotes the energy of surface states resulting from dangling bonds, reduced ions, or surface adsorbates. Ti3+ and VO lattice defects (shown in red) are responsible for the sub-band gap photovoltage signal. EFTO is the Fermi level of the fluorine doped tin oxide substrate. | ||
SPV spectra for the VO poor samples are shown in Fig. 11(b). Again, the photovoltage maximum is seen at 3.6 eV, but this time the photovoltage onset and the effective band gaps of both samples occur much earlier (2.05–2.18 eV). This suggests that VO poor undoped and Al-doped SrTiO3 have higher mid-bandgap defect concentrations than VO rich samples. Also, this time, Al3+ doping does not eliminate the sub-bandgap signal, suggesting that the defects are not from Ti3+. This suggests that VO poor Al-doped and undoped SrTiO3 contain additional impurities, likely at their surface. As shown in the SEM images (Fig. 5(a), (b) and Fig. S1a, b, ESI†), fast heating profile 2 produces VO poor SrTiO3 with more irregular particle morphology, which may be correlated with structural defects or the unintentional doping of a minute amount of carbon, nitrogen or chlorine arising from trapping of volatiles or incomplete burn-off of the organic eutectic. It should be noted that SPV band gaps are often smaller than optical band gaps because SPV is more sensitive to states in the forbidden region near the band edges. Additionally, the sensitivity of SPV measurement to the impurity elements is higher than that of XPS.59
Conclusions
SrTiO3 and Al-doped SrTiO3 were synthesized using a deep eutectic solvent consisting of a choline chloride and malonic acid mixture; the properties of the synthesized samples were investigated without any post-synthesis treatment. Titanium(IV) oxide bis(2,4-pentanedionate) employed in the synthesis as a titanium precursor is a stable powder which is easy to handle and dissolve in a choline chloride–malonic acid DES. 27Al solid-state NMR showed that Al3+ was indeed doped into the symmetric octahedral Ti4+ site and also revealed the presence of an amorphous alumina impurity which was not detected by PXRD. The unit cell size, oxygen deficiency, Al-doping fraction and bandgap were tuned by variation in heating profile. Samples made with longer dwelling time and slower ramping (heating profile 1) had more of TiO2 impurity and bigger crystallite sizes. Perovskite SrTiO3 prepared via longer heating exhibits higher concentration of oxygen vacancies (VO rich) and Ti3+ species, resulting in darker beige color, smaller bandgap and a smaller unit cell size. Samples made with shorter dwelling times and faster ramping rate (heating profile 2) are characterized by higher purity and smaller crystallite sizes. Strontium titanate prepared via this method has lower concentration of Ti3+/oxygen vacancies (VO poor), resulting in lighter beige color, wider bandgap, and larger cubic unit cell volume. Surface photovoltage spectroscopy confirms that Al3+ dopants can eliminate Ti3+ defects in Al-doped SrTiO3 (heating 1), but that Al3+ doping has no effect on the defects in SrTiO3 (heating 2). The latter defects might be a result of unintentional doping of trace amount of carbon, nitrogen or chlorine from the DES which are below the detection of X-ray photoelectron spectroscopy. The defects and the lack of clear facets are reasons for the lack of photocatalytic activity of the synthesized materials for water splitting or hydrogen evolution from aqueous methanol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for J. V. Z. in the form of start-up funds from Iowa State University is gratefully acknowledged. 27Al solid-state NMR experiments and data analysis (A. V. and A. J. R.) were supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Materials Science and Engineering Division. The Ames Laboratory is operated for the U.S. DOE by Iowa State University under Contract DE-AC02-07CH11358. We thank Dr Kirill Kovnir (Department of Chemistry, Iowa State University and Ames Laboratory) for the access to the PXRD diffractometer; Dr Javier Vela (Department of Chemistry, Iowa State University and Ames Laboratory) for the access to the UV-vis spectrometer; Dr Warren Straszheim (Materials Analysis Research Laboratory, Iowa State University) and Aishwarya Mantravadi (Iowa State University) for the help with SEM/EDS data acquisition. The photocatalytic and the surface photovoltage spectroscopy results are based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0015329.References
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
- Q. H. Zhang, K. D. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- X. Ge, C. D. Gu, X. L. Wang and J. P. Tu, J. Mater. Chem. A, 2017, 5, 8209–8229 RSC.
- A. Soldner, J. Zach, M. Iwanow, T. Gartner, M. Schlosser, A. Pfitzner and B. Konig, Chem. – Eur. J., 2016, 22, 13108–13113 CrossRef PubMed.
- S. Hong, R. M. Doughty, F. E. Osterloh and J. V. Zaikina, J. Mater. Chem. A, 2019, 7, 12303–12316 RSC.
- S. Hong, S. J. Burkhow, R. M. Doughty, Y. Cheng, B. J. Ryan, A. Mantravadi, L. T. Roling, M. G. Panthani, F. E. Osterloh, E. A. Smith and J. V. Zaikina, Chem. Mater., 2021, 33(5), 1667–1682 CrossRef CAS.
- R. Boston, P. Y. Foeller, D. C. Sinclair and I. M. Reaney, Inorg. Chem., 2017, 56, 542–547 CrossRef CAS PubMed.
- R. C. Neville, C. A. Mead and B. Hoeneise, J. Appl. Phys., 1972, 43, 2124 CrossRef CAS.
- A. Tkach, O. Okhay, P. M. Vilarinho and A. L. Kholkin, J. Phys.: Condens. Matter, 2008, 20, 415224 CrossRef.
- J. Qi, M. Cao, J. P. Heath, J. S. Dean, H. Hao, Z. Yao, Z. Yu and H. Liu, J. Mater. Chem. C, 2018, 6, 9130–9139 RSC.
- J. B. Li, J. Wang, J. F. Li, Y. Li, H. Yang, H. Y. Yu, X. B. Ma, X. B. Yaer, L. Liu and L. Miao, J. Mater. Chem. C, 2018, 6, 7594–7603 RSC.
- A. I. Abutaha, S. R.-S. Kumar, A. M. Dehkordi, T. M. Tritt and H. N. Alshareef, J. Mater. Chem. C, 2014, 2, 9712–9719 RSC.
- K. van Benthem, C. Elsasser and R. H. French, J. Appl. Phys., 2001, 90, 6156–6164 CrossRef CAS.
- T. Tomio, H. Miki, H. Tabata, T. Kawai and S. Kawai, J. Appl. Phys., 1994, 76, 5886–5890 CrossRef CAS.
- B. L. Phoon, C. W. Lai, J. C. Juan, P.-L. Show and G.-T. Pan, Int. J. Hydrogen Energy, 2019, 44, 14316–14340 CrossRef CAS.
- A. Frye, R. H. French and D. A. Bonnell, Z. Metallkd., 2003, 94, 226–232 CrossRef CAS.
- J. G. Mavroides, J. A. Kafalas and D. F. Kolesar, Appl. Phys. Lett., 1976, 28, 241–243 CrossRef CAS.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543–544 RSC.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- Z. Q. Zhao, R. V. Goncalves, S. K. Barman, E. J. Willard, E. Byle, R. Perry, Z. K. Wu, M. N. Huda, A. J. Moule and F. E. Osterloh, Energy Environ. Sci., 2019, 12, 1385–1395 RSC.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 RSC.
- Z. Q. Zhao, E. J. Willard, H. Li, Z. K. Wu, R. H.-R. Castro and F. E. Osterloh, J. Mater. Chem. A, 2018, 6, 16170–16176 RSC.
- T. Takata, J. Z. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- J. S. Wang, S. Yin, M. Komatsu, Q. W. Zhang, F. Saito and T. Sato, Appl. Catal., B, 2004, 52, 11–21 CrossRef CAS.
- E. Rocha-Rangel, J. Lopez-Hernandez, J. A. Rodriguez-Garcia, E. N. Armendariz-Mireles, C. A. Calles-Arriaga, W. J. Pech-Rodriguez and J. A. Castillo-Robles, J. Ceram. Process. Res., 2017, 18, 590–593 Search PubMed.
- Y. Z. Fan, Z. Y. Zhou, Y. Chen, W. Huang and X. L. Dong, J. Mater. Chem. C, 2020, 8, 50–57 RSC.
- Y. Yamaguchi, Y. Kanamaru, M. Fukushima, K. Fujimoto and S. Ito, J. Am. Ceram. Soc., 2015, 98, 3054–3061 CrossRef CAS.
- Y. Ma, Z. J. Wu, H. W. Wang, G. Q. Wang, Y. K. Zhang, P. C. Hu, Y. M. Li, D. K. Gao, H. Q. Pu, B. Z. Wang and X. W. Qi, CrystEngComm, 2019, 21, 3982–3992 RSC.
- Z. R. Wang, X. C. Ren, C. W. Leung, S. Q. Shi and P. K.-L. Chan, J. Mater. Chem. C, 2013, 1, 3825–3832 RSC.
- G. Pfaff, J. Mater. Chem., 1993, 3, 721–724 RSC.
- L. H. Hu, C. D. Wang, R. M. Kennedy, L. D. Marks and K. R. Poeppelmeier, Inorg. Chem., 2015, 54, 740–745 CrossRef CAS PubMed.
- L. A. Crosby, B. R. Chen, R. M. Kennedy, J. G. Wen, K. R. Poeppelmeier, M. J. Bedzyk and L. D. Marks, Chem. Mater., 2018, 30, 841–846 CrossRef CAS.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, ACS Nano, 2012, 6, 7420–7426 CrossRef CAS PubMed.
- PDXL: Integrated X-ray powder diffraction software, Version 2.8.1.1, Rigaku, 2018 Search PubMed.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229(5), 345–352 CAS.
- A. Samoson and E. Lippmaa, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 6567–6570 CrossRef CAS.
- E. Lippmaa, A. Samoson and M. Magi, J. Am. Chem. Soc., 1986, 108, 1730–1735 CrossRef CAS.
- J.-B. d'Espinose de la Caillerie, C. Fretigny and D. Massiot, J. Magn. Reson., 2008, 192, 244–251 CrossRef PubMed.
- S. G.-J. van Meerten, W. M.-J. Franssen and A. P.-M. Kentgens, J. Magn. Reson., 2019, 301, 56–66 CrossRef CAS PubMed.
- R. M. Doughty, F. A. Chowdhury, Z. Mi and F. E. Osterloh, J. Chem. Phys., 2020, 153, 144707 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- F. Wang, Q. Xu, Z.-a Tan, D. Qian, Y. Ding, L. Li, S. Li and Y. Li, Org. Electron., 2012, 13, 2429–2435 CrossRef CAS.
- S. F. Dec, M. F. Davis, G. E. Maciel, C. E. Bronnimann, J. J. Fitzgerald and S. S. Han, Inorg. Chem., 1993, 32, 955–959 CrossRef CAS.
- A. Faucher, V. V. Terskikh, E. Ye, G. M. Bernard and R. E. Wasylishen, J. Phys. Chem. A, 2015, 119, 11847–11861 CrossRef CAS PubMed.
- I. Bykov, M. Makarova, V. Trepakov, A. Dejneka, L. Yurchenko, A. Jaeger and L. Jastrabik, Phys. Status Solidi B, 2013, 250, 821–824 CrossRef CAS.
- C. Gervais, D. Veautier, M. E. Smith, F. Babonneau, P. Belleville and C. Sanchez, Solid State Nucl. Magn. Reson., 2004, 26, 147–152 CrossRef CAS PubMed.
- L. Samain, A. Jaworski, M. Eden, D. M. Ladd, D. K. Seo, F. J. Garcia-Garcia and U. Haussermann, J. Solid State Chem., 2014, 217, 1–8 CrossRef CAS.
- C. V. Chandran, C. E.-A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Chem. Soc. Rev., 2019, 48, 134–156 RSC.
- H. Q. Tan, Z. Zhao, W. B. Zhu, E. N. Coker, B. S. Li, M. Zheng, W. X. Yu, H. Y. Fan and Z. C. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 19184–19190 CrossRef CAS PubMed.
- W. L. Zhao, W. Zhao, G. L. Zhu, T. Q. Lin, F. F. Xu and F. Q. Huang, CrystEngComm, 2015, 17, 7528–7534 RSC.
- M. C. Biesinger, L. W.-M. Lau, A. R. Gerson and R. S.-C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W.-M. Lau, A. Gerson and R. S.-C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- M. Klusackova, R. Nebel, K. M. Macounova, M. Klementova and P. Krtil, Electrochim. Acta, 2019, 297, 215–222 CrossRef CAS.
- T. Dittrich and S. Fengler, Surface Photovoltage Analysis of Photoactive Materials, World Scientific Publishing Europe Ltd., London, 2020, p. 287 Search PubMed.
- J. Zhao and F. E. Osterloh, J. Phys. Chem. Lett., 2014, 5, 782–786 CrossRef CAS PubMed.
- J. Lagowski, P. Edelman, A. M. Kontkiewicz, O. Milic, W. Henley, M. Dexter, L. Jastrzebski and A. M. Hoff, Appl. Phys. Lett., 1993, 63, 3043–3045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00404f |
| This journal is © The Royal Society of Chemistry 2022 |