
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular conductors from bis(ethylenedithio)tetrathiafulvalene with tris(oxalato)gallate and tris(oxalato)iridate†
Toby J.
Blundell
 a,
Alexander L.
Morritt
a,
Elizabeth K.
Rusbridge
a,
Luke
Quibell
a,
Jakob
Oakes
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b,
Shusaku
Imajo
c,
Tomofumi
Kadoya
d,
Jun-ichi
Yamada
d,
Simon J.
Coles
e,
Jeppe
Christensen
e and
Lee
Martin
*a
a,
Alexander L.
Morritt
a,
Elizabeth K.
Rusbridge
a,
Luke
Quibell
a,
Jakob
Oakes
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b,
Shusaku
Imajo
c,
Tomofumi
Kadoya
d,
Jun-ichi
Yamada
d,
Simon J.
Coles
e,
Jeppe
Christensen
e and
Lee
Martin
*a
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Clifton, Nottingham NG11 8NS, UK. E-mail: lee.martin@ntu.ac.uk
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
cThe Institute for Solid State Physics, The University of Tokyo, Kashiwa, Chiba 277-8581, Japan
dGraduate School of Material Science, University of Hyogo, Kamigori-cho, Ako-gun, Hyogo, 678-1297, Japan
eSchool of Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 13th May 2022
Abstract
We present the synthesis, crystal structures and conducting properties of six new BEDT-TTF radical-cation salts with the tris(oxalato)gallate or -iridate anion. The use of halobenzenes gives 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 β′′ metallic or superconducting salts with tris(oxalato)gallate – fluorobenzene (PhF) or chlorobenzene (PhCl) as a guest molecule gives a metal, whilst the larger bromobenzene (PhBr) or iodobenzene (PhI) gives a superconductor. A 4:1 pseudo-κ semiconducting salt is obtained with benzonitrile as the guest molecule for tris(oxalato)iridate, and a novel 5:1 metal–insulator phase with tris(oxalato)iridate is also reported.
:1 β′′ metallic or superconducting salts with tris(oxalato)gallate – fluorobenzene (PhF) or chlorobenzene (PhCl) as a guest molecule gives a metal, whilst the larger bromobenzene (PhBr) or iodobenzene (PhI) gives a superconductor. A 4:1 pseudo-κ semiconducting salt is obtained with benzonitrile as the guest molecule for tris(oxalato)iridate, and a novel 5:1 metal–insulator phase with tris(oxalato)iridate is also reported.
Introduction
The organic donor molecule BEDT-TTF has produced the largest number of molecular superconductors, with the highest superconducting Tc being 11.6 K at ambient pressure in κ-(BEDT-TTF)2Cu[N(CN)2]Br,1 and 14.2 K under pressure in β′-(BEDT-TTF)2ICl2.2Multifunctionality has been achieved in BEDT-TTF salts with the use of various transition-metal complexes with particular focus on the tris(oxalato)metallate anion to combine conductivity in the same lattice with paramagnetism,3 anti-ferromagnetism,4 ferromagnetism,5 proton conductivity,6 or chirality.7 The first radical-cation salt in this family, β′′-(BEDT-TTF)4[(A)Fe(C2O4)3]·PhCN3 (PhCN = benzonitrile, A = initially reported as H2O and later as H3O+), was synthesised by the group of Professor Peter Day at The Royal Institution of Great Britain in 1995.3 This salt has a superconducting Tc of 8.5 K which is still the highest superconducting Tc for the BEDT-TTF-tris(oxalato)metallate salts to date.8 A polymorph, pseudo-κ-(BEDT-TTF)4[(A)Fe(C2O4)3]·PhCN (A = K+ and NH4+),3 differing only in the spatial arrangement of the enantiomers of Fe(C2O4)3, was also reported. This initial discovery opened pathways to produce a family of materials which now numbers well over a hundred examples with the majority of the structures being 4:1 salts of the β′′ or pseudo-κ type.8
The packing of the BEDT-TTF donor layers determines the conducting behaviour and this packing can be subtly changed through modifications of the neighbouring anion layers. The anion layers form a hexagonal network of tris(oxalato)metallate with a counter cation which produces honeycomb cavities capable of accommodating a guest molecule of the solvent used for electrocrystallisation. Changing the metal ion of Fe(C2O4)33− from iron (Al3+,9 Co3+,9 Cr3+,10 Ga3+,11 Mn3+,12 Rh3+,13 or Ru3+9,14) or changing the counter cation (A = H3O+, K+, NH4+)3 has a small effect on the donor packing arrangement. However, changing the size and shape of the included solvent guest molecule can have a marked effect on the donor packing and thus the conducting properties.
Guest molecules similar in size to benzonitrile produce isostructural 4:1 β′′ salts, whilst bulkier guest molecules protrude on one side of the anion layer to produce two different faces for the anion layer which leads to a different BEDT-TTF packing type on either side of the anion layer (e.g. α + β′′ with PhCH2CN15 or PhCH(OH)CH3,7 or α + pseudo-κ with 1,2-Br2Ph16). With smaller guest molecules (CH3NO2, CH3CN, and CH2Cl2)17 a 3:1 semiconducting salt is obtained. In some salts 18-crown-6 ether, which is added to aid solubility of the ammonium or potassium salt of the anion, is included in the lattice to produce a 2:1 superconducting salt, β′′-(BEDT-TTF)2[(H2O)(NH4)2M(C2O4)3]·18-crown-6 (M = Cr3+ or Rh3+)18 or a 2:1 proton conductor/metal β′′-(BEDT-TTF)4[(A)M(C2O4)3]2·[(A)218-crown-6](H2O)5 (M = Cr3+ or Ga3+, A = H3O+ or NH4+).6
A variety of other unusual donor and anion packings have also been obtained with Fe(C2O4)33−,19 as is also the case when replacing the M3+ ion with M4+ with dianionic Ge(C2O4)32−.20
This paper reports the syntheses, structures and properties of six new additions to this family of BEDT-TTF salts with tris(oxalato)metallates of gallium and iridium. Four tris(oxalato)gallate salts are 4:1 salts β′′-(BEDT-TTF)4[(H3O)Ga(C2O4)3]·halobenzene – fluorobenzene (PhF) or chlorobenzene (PhCl) as a guest molecule gives a metal, whilst the larger bromobenzene (PhBr) or iodobenzene (PhI) gives a superconductor. A semiconducting 4:1 salt pseudo-κ-(BEDT-TTF)4[(H3O)Ir(C2O4)3]·benzonitrile (PhCN) is reported. The synthesis, structure and band calculations of a new 5:1 metal–insulator phase, β′′-(BEDT-TTF)5Ir(C2O4)3·ethanol, are also reported.
Results and discussion
4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 β′′ salts with tris(oxalato)gallate
:1 β′′ salts with tris(oxalato)gallate
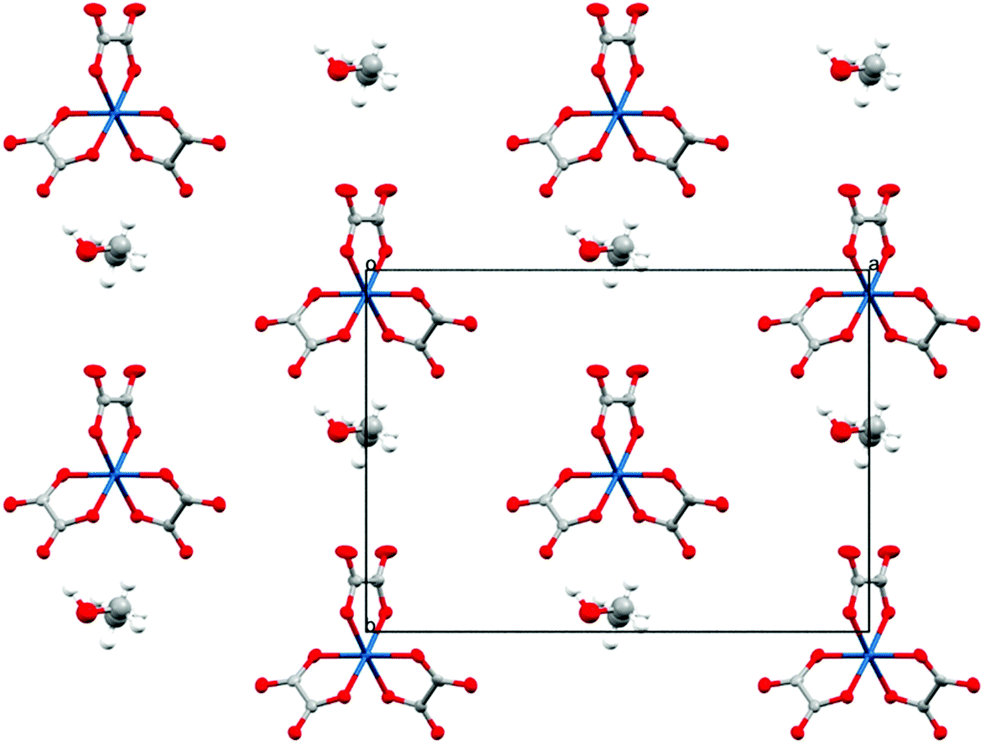
Isostructural salts 1–4 are new additions to the β′′ family of BEDT-TTF radical-cation salts with the tris(oxalato)gallate anion. All four salts have the formula β′′-(BEDT-TTF)4[(H3O)Ga(C2O4)3]·guest (guest = PhF fluorobenzene 1, PhCl chlorobenzene 2, PhBr bromobenzene 3, and PhI iodobenzene 4). Salts 1–4 all crystallise in the monoclinic space group C2/c with an asymmetric unit consisting of two crystallographically independent BEDT-TTF molecules, half a Ga(C2O4)33− anion, half a H3O+ counter cation, and half a guest molecule.
Fig. 1 shows the layered structure of these salts which consists of alternating donor layers of BEDT-TTF and anion layers of M(C2O4)33−/guest/H3O+. The two crystallographically independent BEDT-TTF molecules adopt a β′′ packing motif in stacks along the a/b crystallographic axis (Fig. 2). The two independent BEDT-TTF molecules follow an AABBAA sequence within each stack. There are a number of side-to-side S⋯S interactions between donor stacks which are below the sum of the van der Waals radii (Table 1). Using the method of Guionneau et al.21 to estimate the charge on a BEDT-TTF molecule from its C–S and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths we estimate charges of approximately +0.5 for all BEDT-TTFs (Table 2) as expected for these 4:1 β′′ salts where the anion layer has a 2-charge [(H3O)Ga(C2O4)3]2−.
C bond lengths we estimate charges of approximately +0.5 for all BEDT-TTFs (Table 2) as expected for these 4:1 β′′ salts where the anion layer has a 2-charge [(H3O)Ga(C2O4)3]2−.
 | ||
| Fig. 1 Structure of 4 viewed down the a axis. Salts 1–4 are isostructural. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
 | ||
| Fig. 2 Packing arrangement of the donor layer for 4. Salts 1–4 are isostructural. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
| Contact (Å) | Ga-PhF (1) | Ga-PhCl (2) | Ga-PhBr (3) | Ga-PhI (4) |
|---|---|---|---|---|
| S1⋯S7 | 3.3817(19) | 3.417(2) | 3.4206(10) | 3.4381(10) |
| S3⋯S7 | 3.5225(18) | 3.517(2) | 3.5041(10) | 3.4915(10) |
| S2⋯S9 | 3.3397(19) | 3.329(2) | 3.3147(10) | 3.297(4) |
| S2⋯S11 | 3.3811(18) | 3.3792(19) | 3.3695(10) | 3.368(4) |
| S6⋯S15 | 3.528(2) | 3.533(2) | 3.5180(10) | 3.5123(10) |
| S8⋯S15 | 3.612(2) | 3.594(2) | 3.5612(11) | 3.5404(12) |
| S8⋯S10 | 3.600(2) | 3.600(2) | 3.5793(11) | 3.5753(11) |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Salt | Donor | a | b | C | D | δ | Q |
| Ga-PhF | A | 1.355 | 1.73725 | 1.7455 | 1.347 | 0.781 | +0.52 |
| 1 | B | 1.360 | 1.73525 | 1.746 | 1.3475 | 0.774 | +0.57 |
| Ga-PhCl | A | 1.360 | 1.744 | 1.747 | 1.3505 | 0.781 | +0.52 |
| 2 | B | 1.357 | 1.741 | 1.745 | 1.352 | 0.777 | +0.54 |
| Ga-PhBr | A | 1.358 | 1.738 | 1.748 | 1.346 | 0.782 | +0.51 |
| 3 | B | 1.370 | 1.735 | 1.749 | 1.341 | 0.773 | +0.58 |
| Ga-PhI | A | 1.360 | 1.7345 | 1.7425 | 1.3455 | 0.772 | +0.59 |
| 4 | B | 1.359 | 1.73525 | 1.744 | 1.346 | 0.774 | +0.57 |
The anion layer is a honeycomb created using M(C2O4)33− anions and H3O+ cations with a guest solvent molecule in the hexagonal cavities (Fig. 3). Each anion layer consists of a single M(C2O4)33− enantiomer with adjacent layers being the opposite enantiomer to give a repeating ΔΛΔΛΔΛ pattern and a racemic lattice. The dimensions of the hexagonal cavities for salts 1–4 are defined in Fig. 4 and the values given in Table 3. It has previously been observed for rhodium13 and iron22 salts that increasing the size of the guest molecule from fluorobenzene to chlorobenzene to bromobenzene leads to an increase in the height and a decrease in the width of the cavity. In gallium salts we observe the same trend going from fluorobenzene to bromobenzene to iodobenzene, however the chlorobenzene salt has a longer height than bromobenzene and a narrower width than fluorobenzene.
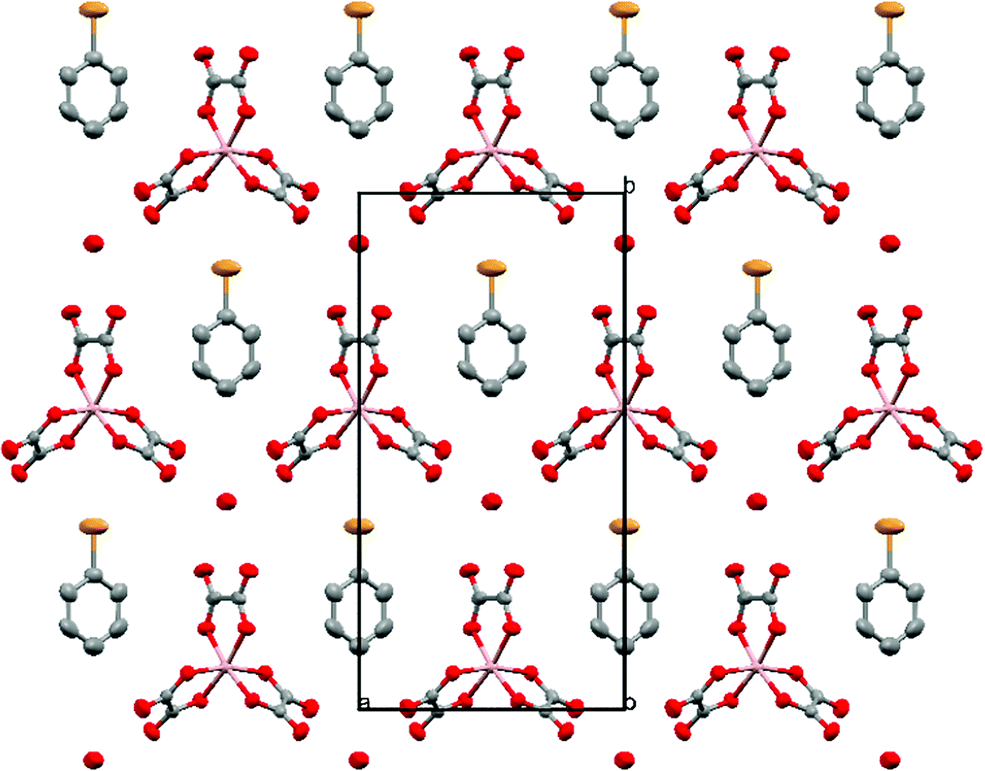 | ||
| Fig. 3 Packing arrangement of the anion layer for 4. Salts 1–4 are isostructural. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
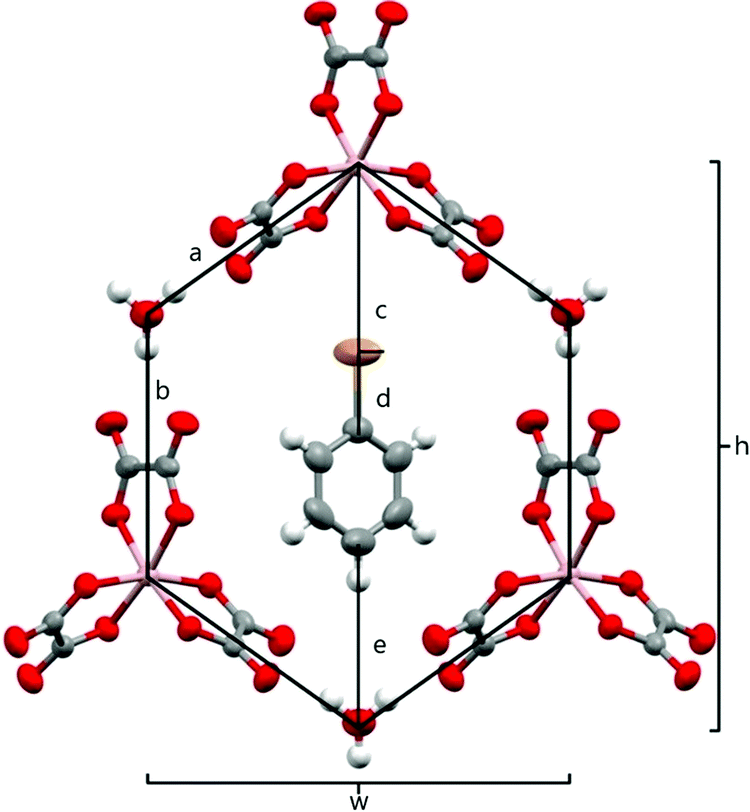 | ||
| Fig. 4 Honeycomb cavity in the anion layer for 4 with measurement parameters labelled. Salts 1–4 are isostructural. | ||
| Salt temp. | Ga-PhF (1) 294 K | Ga-PhCl (2) 293 K | Ga-PhBr (3) 293 K | Ga-PhI (4) 294 K | Ga-PhCN (5) 293 K |
|---|---|---|---|---|---|
| Distances (Å) | |||||
| a | 6.259(4) | 6.282(4) | 6.279(2) | 6.298(2) | 6.288(4) |
| b | 6.378(7) | 6.395(7) | 6.360(4) | 6.349(4) | 6.254(7) |
| c | 4.802(14) | 4.614(3) | 4.5533(7) | 4.5571(6) | c + d = 5.168(9) |
| d | 1.311(18) | 1.740(9) | 1.889(5) | 2.078(4) | 0.950 |
| e | 4.674(17) | 4.497(14) | 4.405(7) | 4.294(7) | 5.375(14) |
| h | 13.520(7) | 13.597(7) | 13.579(4) | 13.658(4) | 13.352(7) |
| w | 10.2801(4) | 10.2964(7) | 10.2760(2) | 10.2593(2) | 10.38141(15) |
| O4-cation | 2.960(6) | 3.005(6) | 3.022(4) | 3.073(3) | 2.930(7) |
| O6-cation | 2.866(5) | 2.851 (5) | 2.839(3) | 2.828(3) | 2.899(5) |
| O1-cation | 3.011(8) | 3.013(8) | 2.994(4) | 2.986(4) | 2.881(8) |
| Angles (°) | |||||
| δ | 33.4(3) | 32.7(2) | 33.5(2) | 34.60(16) | 25.4(2) |
The size, shape and orientation of the guest solvent molecule within the hexagonal cavity influence the order–disorder of the terminal ethylene groups of adjacent BEDT-TTF molecules which affects the transport properties or destabilises the superconducting transition.8 The BEDT-TTF molecules in all of the salts 1–4 contain disordered ethylene groups (C1, C2, C9, and C10) irrespective of the guest solvent molecule present. In the case of 1 (Ga PhF) we also observe disorder in the position of fluoride on the guest molecule (Fig. 5). The fluoride occupies both the standard position, pointing directly at the metal atom along the c axis, as well as a roughly 16% occupancy at each meta position to the standard position. This results in a disordered component where the fluoride protrudes out of the cavity. Presumably, this is possible due to the small size of fluoride compared to the other substituents on the phenyl of the guest molecules, allowing this orientation to be favourable without interrupting the crystal packing.
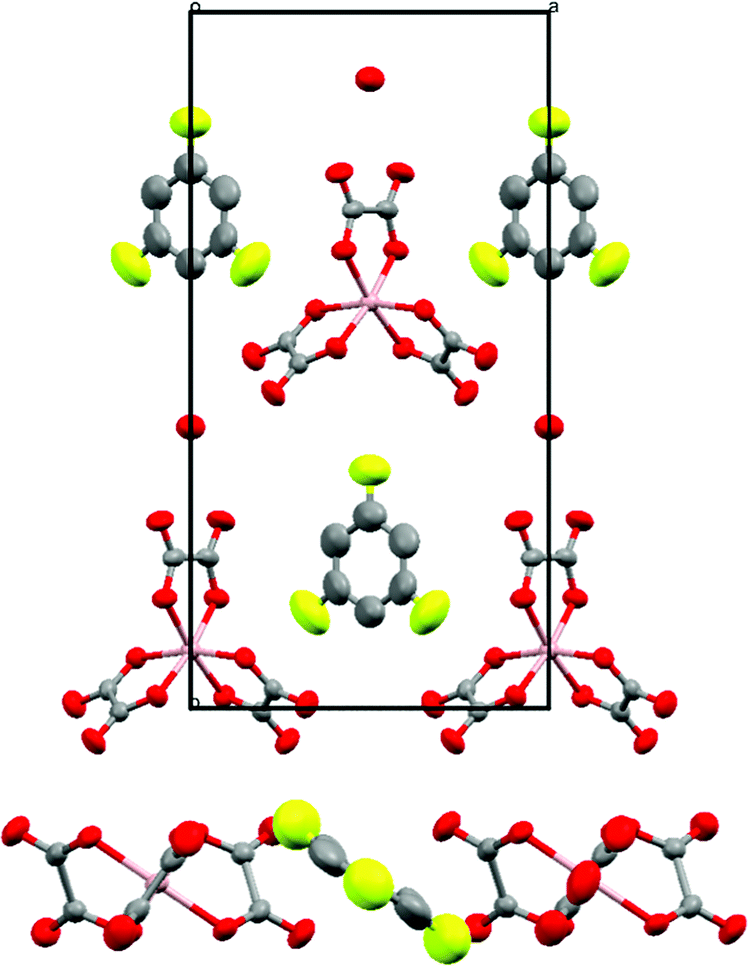 | ||
| Fig. 5 Hexagonal cavity of 1 containing fluorobenzene showing the orientation of the disordered fluorine atoms. The major component (∼68%) points directly at the gallium atom whereas the minor component (∼16% each side) protrudes out of the cavity. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
The only previously reported β′′ superconducting salts with the tris(oxalato)gallate anion incorporated the guest molecules pyridine (Tc = 1.5 K) or nitrobenzene (Tc = 7.5 K).11 Guest molecules 2-chloropyridine or 2-bromopyridine show metallic behaviour down to 0.5 K.23
A salt synthesised from potassium tris(oxalato)gallate with bromobenzene has previously been reported (β′′-(BEDT-TTF)4[K0.33(H3O)0.67Ga(C2O4)3]·PhBr)11 and showed no superconductivity down to 0.5 K. Salt 3 is the isostructural salt starting from ammonium tris(oxalato)gallate with bromobenzene and shows superconductivity with a Tc of 3.0 K. When using ammonium tris(oxalato)gallate instead of potassium only the H3O+ counter cation is included in the lattice (β′′-(BEDT-TTF)4[(H3O)Ga(C2O4)3]·bromobenzene). The non-superconducting salt with K0.33(H3O)0.67 has a b axis of 19.860(2) Å whilst the H3O+ salt (3) has a longer b of 19.9923(14) Å (both at 293 K).
Analysis of the crystal structures of the β′′-(BEDT-TTF)4[(A)M(C2O4)3]·solvent salts by Imajo et al.24 has revealed a correlation between the cell parameters and the conducting properties. Changing the size and shape of the included guest molecule has a marked effect on the cell parameters, especially the length of the b axis, with which the superconducting Tc can be correlated. The halogen atom of the guest molecule is directed along the b axis in these salts so we expect an increase of the b axis as the guest molecule is increased in size from PhF up to PhI. In these Ga salts the b axis for the PhCl salt 2 is longer than that of the PhBr salt 3 (Table 4‡). Salts 1 and 2 with smaller PhF and PhCl, respectively, do not show superconductivity (Fig. 6). Salts 3 and 4 with larger PhBr and PhI show superconducting Tcs of 3.0 K and 2.4 K respectively.
| Ga-PhF (1) | Ga-PhCl (2) | Ga-PhBr (3) | Ga-PhI (4) | |
|---|---|---|---|---|
| Formula | C52H40FO13GaS32 | C52H40ClO13GaS32 | C52H40BrO13GaS32 | C52H40IO13GaS32 |
| Fw [g mol−1] | 1987.52 | 2003.93 | 2048.39 | 2095.38 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c | C2/c |
| Z | 4 | 4 | 4 | 4 |
| T (K) | 294(2) | 293(2) | 293(2) | 294(2) |
| a [Å] | 10.2801(4) | 10.2964(7) | 10.2880(3) | 10.2593(2) |
| b [Å] | 19.8975(10) | 19.9923(14) | 19.9599(6) | 20.0074(4) |
| c [Å] | 35.2455(13) | 35.423(3) | 35.5191(8) | 35.5849(7) |
| α [°] | 90 | 90 | 90 | 90 |
| β [°] | 92.680(3) | 93.080(3) | 93.392(2) | 93.7398(18) |
| γ [°] | 90 | 90 | 90 | 90 |
| Volume [Å3] | 7201.5(5) | 7281.3(9) | 7280.98(16) | 7288.67(14) |
| Density [g cm−3] | 1.832 | 1.828 | 1.871 | 1.911 |
| μ [mm−1] | 1.373 | 1.392 | 1.905 | 1.778 |
| R 1 | 0.0709 | 0.0745 | 0.0573 | 0.0413 |
| wR [all data] | 0.1479 | 0.1447 | 0.1254 | 0.0934 |
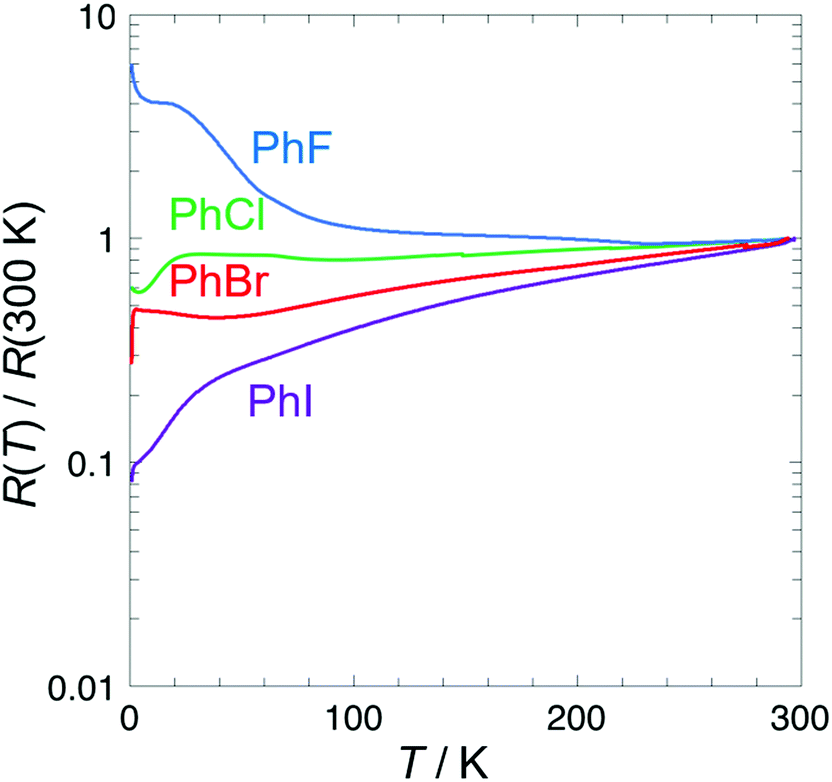 | ||
| Fig. 6 Electrical resistivity for salts 1–4. | ||
4:1 Pseudo-κ salt with tris(oxalato)iridate
A large number of guest molecules have been included in this family of salts but the pseudo-κ phase is only obtained when the guest is benzonitrile (or a mixture of benzonitrile with a 2nd guest – nitrobenzene or dichlorobenzene).
Here we report the results of crystal growth of BEDT-TTF with tris(oxalato)iridate with benzonitrile as a guest which gives only the semiconducting pseudo-κ phase (BEDT-TTF)4[(H3O)Ir(C2O4)3]·PhCN and no crystals of β′′. The best crystals were obtained when using the crystal growth method which includes 1,2,4-trichlorobenzene.25 Using the method of Peter Day's group that was used to prepare the β′′ Fe3 and Cr10 phases in the original salts also gave only pseudo-κ crystals with Ir.
The structure of pseudo-κ-(BEDT-TTF)4[(H3O)Ir(C2O4)3]·PhCN (5) is isostructural with the previously reported salts in the family, crystallising in the orthorhombic space group Pbcn with two crystallographically independent BEDT-TTF donors, half a tris(oxalato)iridate anion, half a benzonitrile guest molecule, and half an H3O+ counter cation.
Fig. 7 shows the alternating donor and anion layers. Fig. 8 shows the pseudo-κ packing motif of the BEDT-TTFs in the donor layer. Short S⋯S contacts are given in Table 5. (BEDT-TTF+)2 dimers are surrounded by six BEDT-TTF0 monomers which are responsible for the semiconducting behaviour observed in these salts (Table 6).
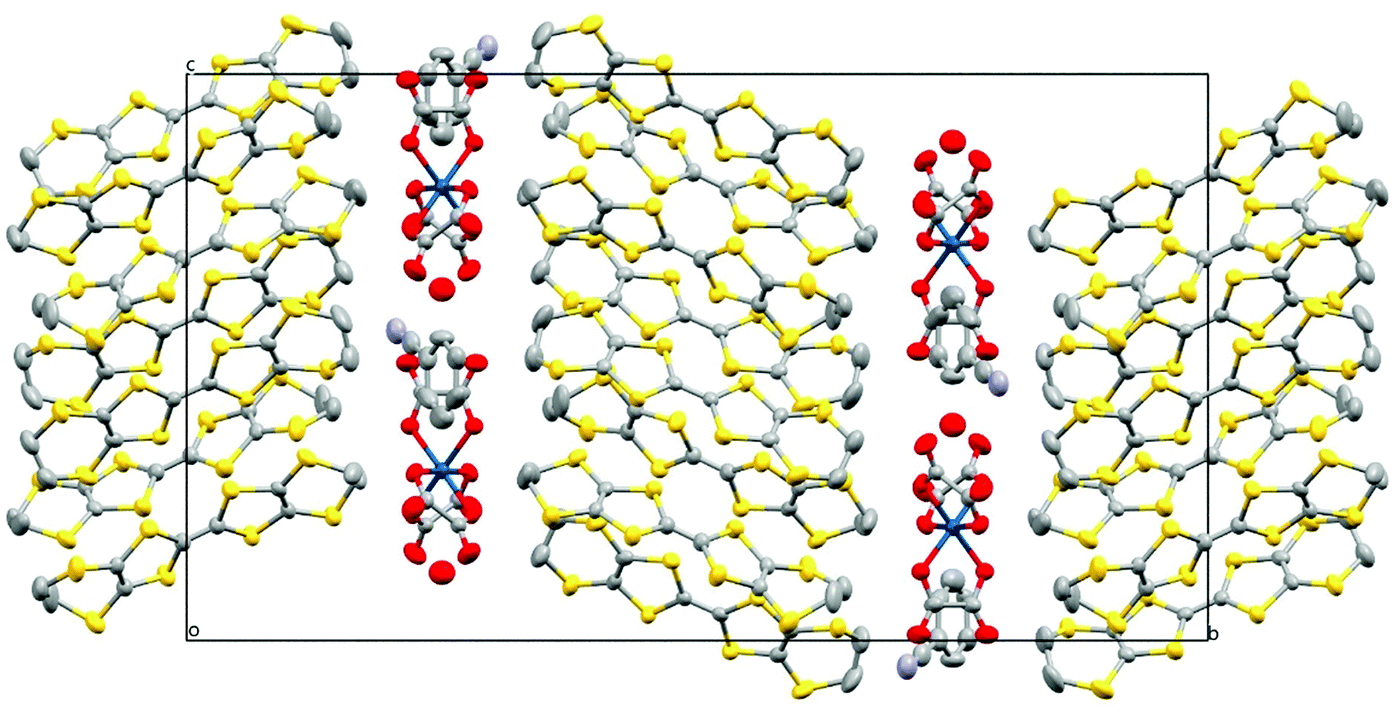 | ||
| Fig. 7 Structure of 5 viewed down the a axis. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
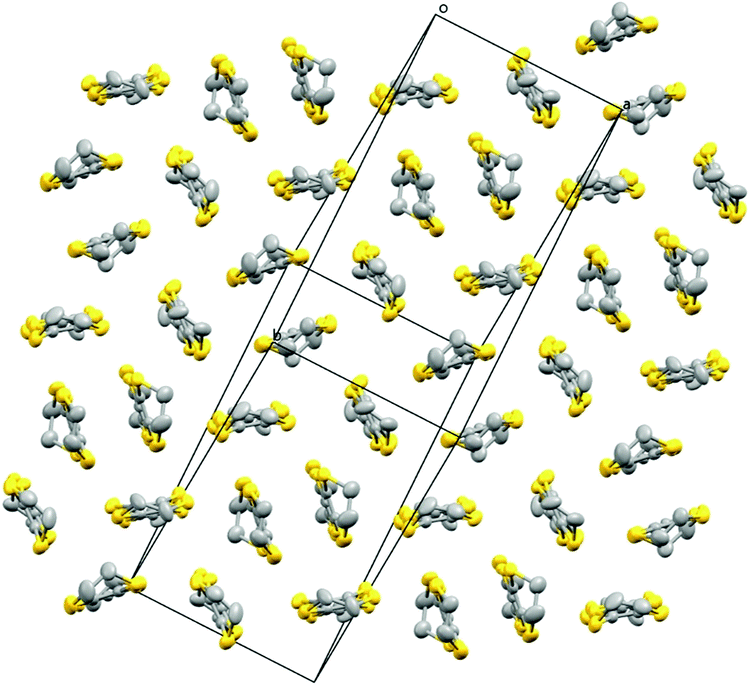 | ||
| Fig. 8 Packing arrangement of the donor layer for 5. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
| Contact (Å) | Ir-PhCN (5) |
|---|---|
| S1⋯S14 | 3.2190(17) |
| S1⋯S16 | 3.4418(19) |
| S7⋯S10 | 3.4384(18) |
| S2⋯S15 | 3.5170(19) |
| S8⋯S9 | 3.5214(18) |
| S2⋯S11 | 3.4333(17) |
| S6⋯S15 | 3.5106(18) |
| S11⋯S14 | 3.4460(17) |
| S12⋯S13 | 3.4768(17) |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Salt 5 | Donor | a | b | c | d | δ | Q |
| A | 1.381 | 1.725 | 1.743 | 1.3505 | 0.736 | +0.85 | |
| B | 1.343 | 1.756 | 1.758 | 1.340 | 0.831 | +0.15 | |
The anion layer (Fig. 9) is a honeycomb arrangement of Ir(C2O4)33− and H3O+. The benzonitrile molecule sits in the honeycomb's hexagonal cavity with the nitrile group disordered over two positions pointing towards H3O+ cations (Fig. 10 and Table 3). The hexagonal cavity is therefore wider than in the β′′ phase (Fig. 4 and Table 3). Every anion layer in the pseudo-κ phase is built up of alternating rows of the Δ and Λ enantiomers of the tris(oxalato)iridate. The β′′ phase differs in the dispersion of enantiomers in having an entire anion layer consisting of a single enantiomer whilst the adjacent anion layers consist of the opposite enantiomer. Both result in a net overall racemic lattice.3,10
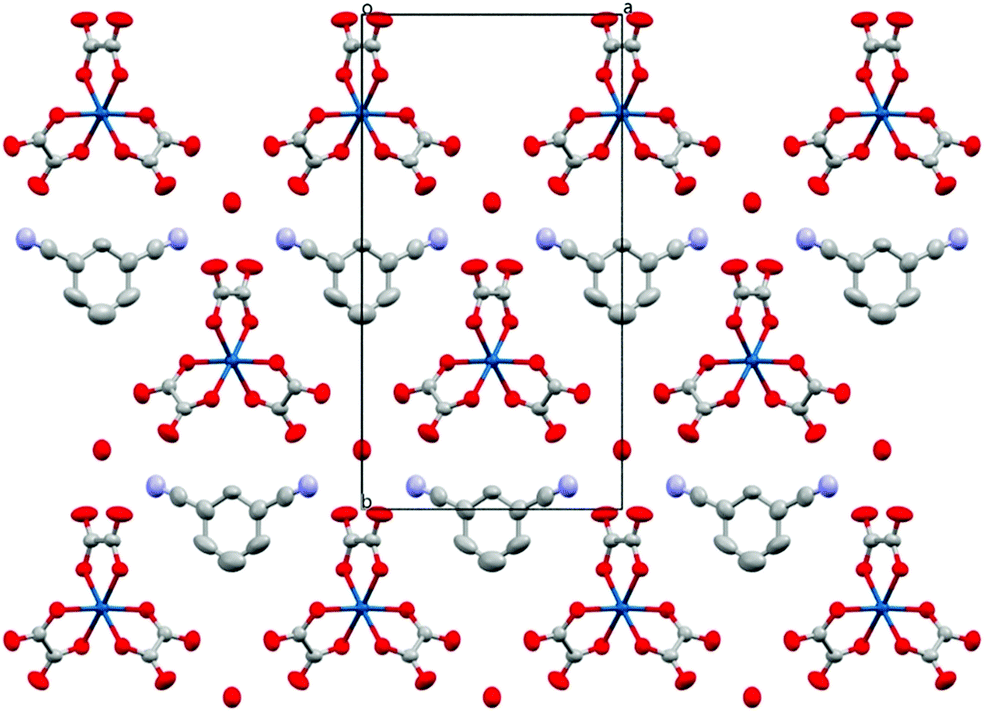 | ||
| Fig. 9 Packing arrangement of the anion layer for 5 viewed down the c axis. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
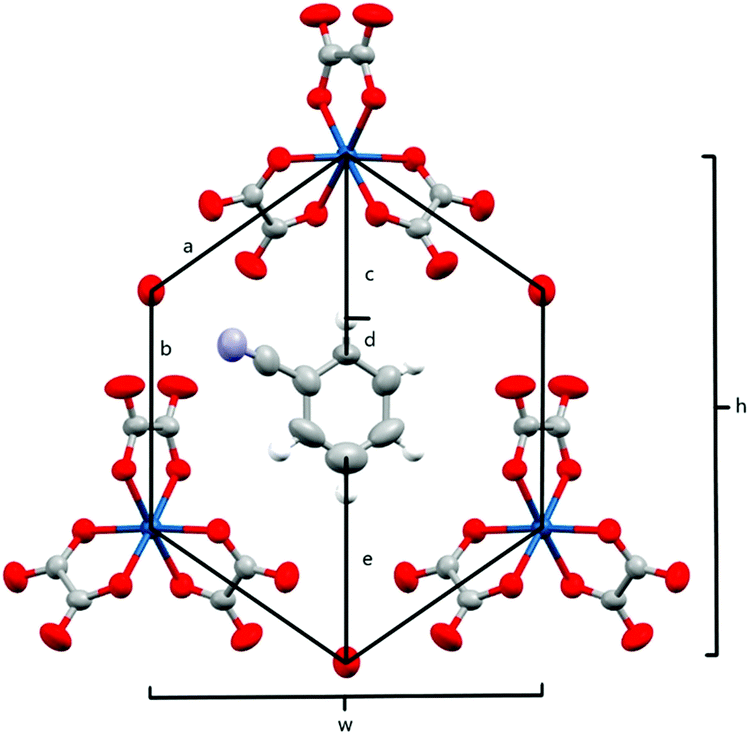 | ||
| Fig. 10 Honeycomb cavity in the anion layer for 5 with measurement parameters labelled. | ||
Previously reported pseudo-κ-(BEDT-TTF)4[(A)M3+(C2O4)3]·PhCN salts are all semiconductors with an Ea between 140 and 245 meV.8 Salt 5 also shows semiconducting behaviour (Fig. 11) with an Ea of 116 meV.
 | ||
| Fig. 11 Electrical resistivity for salt 5. | ||
5 : 1 salt with tris(oxalato)iridate
:1 salt, β′′-(BEDT-TTF)2[(H2O)(NH4)2Ir(C2O4)3]·18-crown-6, which was the first example in this family to contain a 5d tris(oxalato)metallate anion.18 A 4:1 BEDT-TTF salt has previously been reported with 5d bis(oxalato)platinate, Pt(C2O4)22−, which shows metallic behaviour down to 60 K.26
When using benzonitrile with tris(oxalato)iridate in salt 5 we have also used halobenzenes with tris(oxalato)iridate but no crystals could be obtained, in contrast to the relative ease with which they can be obtained when using the 3d and 4d tris(oxalato)metallates.
Crystals of a 5:1 salt (6) have been obtained using acetophenone as the electrocrystallisation solvent with either the ammonium or potassium salt of tris(oxalato)iridate (Fig. 12). Two 5:1 salts in the BEDT-TTF-tris(oxalato)metallate family have previously been reported and both are semiconductors. Instead of the hexagonal anion layer packing arrangement found in the 2:1,18 3:1,17 and 4:13,8 salts the tris(oxalato)ferrate anions are arranged in a parallelogram. (BEDT-TTF)5[Fe(C2O4)3]·(H2O)2·CH2Cl227 has an anion layer containing only a single enantiomer of tris(oxalato)ferrate with the adjacent anion layers being the opposite enantiomer. The parallelogram contains a CH2Cl2 and two H2O molecules. α′′-(BEDT-TTF)5[Ga(C2O4)3]·(H2O)3.4·(EtOH)0.623 differs with every anion layer containing a 50:50 mixture of each tris(oxalate)gallate enantiomer and a parallelogram containing disordered ethanol and water molecules in a void on one side.
 | ||
| Fig. 12 Structure of 6 viewed down the b axis. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
Salt (6) differs from these previously reported 5:1 salts in having a β′′ donor packing arrangement (Fig. 13) with a diamond-shaped packing of tris(oxalato)iridate in the anion layers with no included water molecules (Fig. 14). Salt 6 crystallises in the monoclinic space group C2/c, with two and a half crystallographically independent BEDT-TTF molecules, half an Ir(C2O4)33− anion, and a disordered ethanol molecule. The crystal structure is built up of alternating layers of BEDT-TTF in a β′′ packing motif and layers of Ir(C2O4)33−/ethanol (Fig. 12).
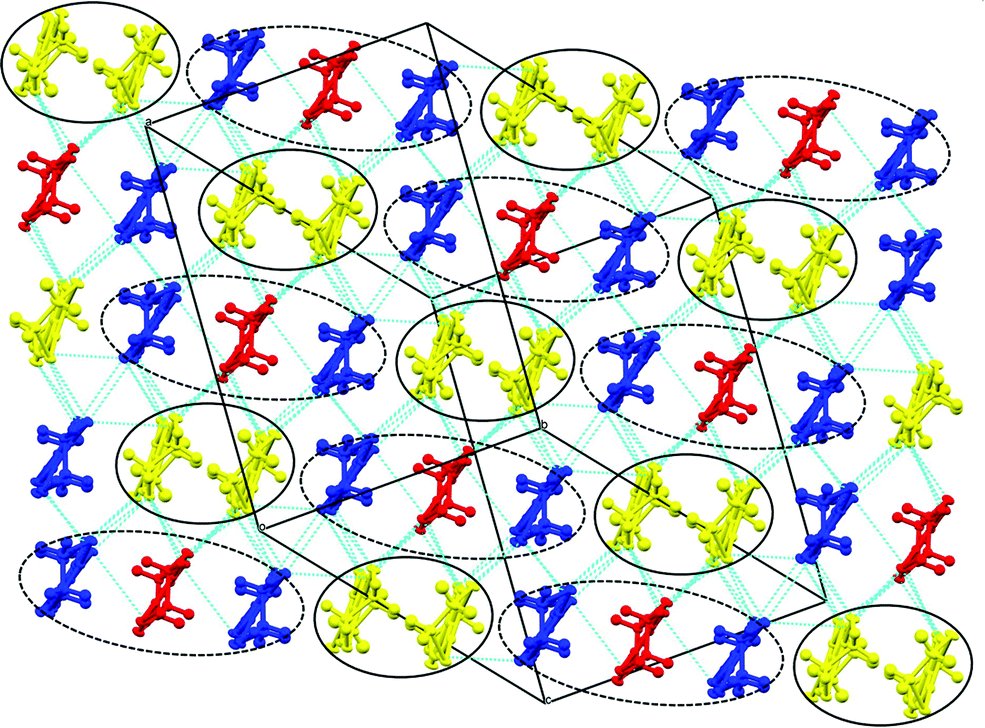 | ||
| Fig. 13 Packing arrangement of the donor layer for 6. Donor A = yellow, B = blue, C = red. | ||
 | ||
| Fig. 14 Packing arrangement of the anion layer for 6 viewed down the c axis. | ||
Each anion layer contains only a single enantiomer of Ir(C2O4)33−, with the adjacent anion layers containing only the opposite enantiomer to give an overall racemic lattice (Fig. 14). The tris(oxalato)iridate anions adopt a diamond shaped arrangement with 11.332 Å between every iridium atom, and a disordered ethanol molecule within the cavity between anions.
The two and half crystallographically independent BEDT-TTF molecules, where A, B and C are coloured yellow, blue and red, respectively, in Fig. 13 and 16, follow an –AABCBAABCB– pattern within each stack. There are a number of side-to-side S⋯S interactions between donor stacks which are below the sum of the van der Waals radii (Table 7) but there are none face-to-face within a stack. The BEDT-TTF donor molecules denoted A (Fig. 13, 15 and 16) form a dimer which is twisted by only 2.6° with respect to the trimer formed using donors B and C. In Fig. 13, we show solid line ellipsoids on the A–A dimers and dashed line ellipsoids on the B–C–B trimers. As mentioned in Fig. 15a, the intermolecular interactions for A–A dimers and B–C–B trimers are relatively strong and weak, respectively. Using the method of Guionneau et al.21 to estimate the charge on a BEDT-TTF molecule from its C–S and CC bond lengths we estimate a charge on each donor (Table 8). This gives the expected 3+ overall charge for five donors balancing the charge of the anion [Ir(C2O4)3]3−. Salt 6 is metallic down to 50 K below which there is a transition to insulating behaviour for the cooling process (Fig. 17). The metal–insulator transition temperature in the heating process is the same as that of the cooling process, but a weak-semiconducting or semi-metallic behaviour is observed from 50 to 225 K. Above 225 K metallic behaviour is observed up to room temperature. Band calculations are performed at 100, 150 and 290 K. The results are shown in Fig. 15. The Fermi surfaces at 290 and 150 K (Fig. 15e and d) are simply cylindrical, suggesting that at each temperature the salt is a stable 2D metal. However, Table 8 suggests a tendency for charge disproportionation. Normalized charges (Qn) suggest that donor A is almost monocationic and C is almost neutral. Therefore, the A–A dimer is dicationic, meaning that spin dimers are formed and hence the dimers do not play any role in the electrical conductivity. C is neutral, meaning that the valence orbital is fully occupied, suggesting that C also has no conducting carriers. The valence orbitals of only B are partially occupied and interact with each other along the a + b direction (≈//side-by-side direction). Actually, the Fermi surface in Fig. 15d has a wide flat region along the a′*-axis in the primitive lattice (≈//side-by-side direction), indicating that this direction is the most conductive. In contrast, at 150 K the A–A dimer and C molecule become monocationic as an electron moves from the A–A dimer to C. At this temperature the cooling curve has no anomaly but the heating curve has an anomaly at 225 K, where the phase transition appears to occur. The resultant B–C–B trimer is dicationic therefore there is a spin dimer on each trimer. Therefore, only the partially occupied monocationic A–A dimer is conductive. However, as shown in Fig. 13, the A–A dimers are isolated by the dicationic B–C–B trimers, suggesting that the charge disproportionate state at 150 K seems to be less conductive than that at 290 K, which was realized on the heating resistivity curve. Although the charge disproportionate states were realized at both temperatures, the salt shows low resistivity in this temperature range. A conductive charge-ordered state is also observed in θ-(BEDT-TTF)2CsCo(NCS)4.28 In addition, there is an incorporated EtOH molecule in the unit cell. It is heavily disordered at 290 K and ordered over two positions at 150 K. The incorporated EtOH molecule is located close to the C BEDT-TTF molecule. So, we guess that the dipole moment of the ordered EtOH may play a significant role in changing the charge of C from neutral at 290 K to cationic at 150 K. As mentioned above, the Fermi surfaces at 290 and 150 K are cylindrical, which suggests that both are stable metals. However, each band dispersion has a gap between the third and fourth lines from the bottom and the gap at 150 K is wider than that at 290 K. We do not know the exact effect of the gap but usually such a gap causes localization of electrons. In addition, the charge of C of 0.79 at 150 K is not so close to 1.0. We suggest that the value is close to 1.0 just below the transition temperature because Table 8 indicates no charge disproportionation at 100 K, namely the charges of A, B and C are ≈ 0.6 which is equal to the BEDT-TTF formula charge of 6. The lower the temperature the weaker the charge disproportionation, which is somewhat strange, but it was recently reported in a salt.29 This suggests that the salt at 100 K is more conductive than that at 150 or 290 K. The band dispersions at 100 K have no gap as shown in Fig. 15d, which again suggests no localization. The Fermi surface at 100 K opens along the b′*-axis (≈//stacking direction). This indicates that the salt is quasi 1D-like below 100 K. These results suggest that a phase transition or crossover occurs between 100 and 150 K. Fig. 17 indicates no anomaly on the cooling curve in this temperature range but a broad peak around 130 K on the heating curve. This might be the temperature at which the phase transition or crossover occurs. We have no structural information below the MI transition. But the discussion above suggests that the salt is not in any charge-ordered states at low temperature. Therefore, a charge-density wave along the side-by-side direction is the most suitable explanation, although it is only speculation.
| Contact (Å) | Ir-EtOH (6) 100 K | Ir-EtOH (6) 150 K | Ir-EtOH (6) 298 K |
|---|---|---|---|
| a 1 − x, −y, 1 − z. b 1.5 − x, 0.5 − y, 1 − z. c 1 − x, 1 − y, 1 − z. d 1.5 − x, 1.5 − y, 1 − z. | |||
| S1⋯S3a | 3.4725(11) | 3.4745(11) | 3.521(6) |
| S1⋯S11a | 3.4159(12) | 3.4273(11) | 3.469(5) |
| S1⋯S12a | 3.4122(11) | 3.4285(11) | 3.490(5) |
| S3⋯S9a | 3.5057(11) | 3.5167(11) | 3.558(5) |
| S4⋯S9a | 3.5775(11) | 3.5893(11) | 3.634(5) |
| S5⋯S16b | 3.5337(13) | 3.5400(12) | 3.607(7) |
| S6⋯S8b | 3.5810(11) | 3.5854(11) | 3.615(5) |
| S6⋯S16b | 3.4319(12) | 3.4442(12) | 3.526(6) |
| S8⋯S13b | 3.5046(10) | 3.5138(10) | 3.560(6) |
| S9⋯S17c | 3.3585(10) | 3.3701(10) | 3.417(5) |
| S9⋯S18c | 3.4240(12) | 3.4356(12) | 3.495(5) |
| S13⋯S18 | 3.5533(12) | 3.5661(11) | 3.581(5) |
| S14⋯S18 | 3.3542(11) | 3.3648(11) | 3.413(5) |
| S16⋯S19d | 3.3860(11) | 3.4022(11) | 3.457(5) |
| S16⋯S20d | 3.3683(11) | 3.3792(11) | 3.431(5) |
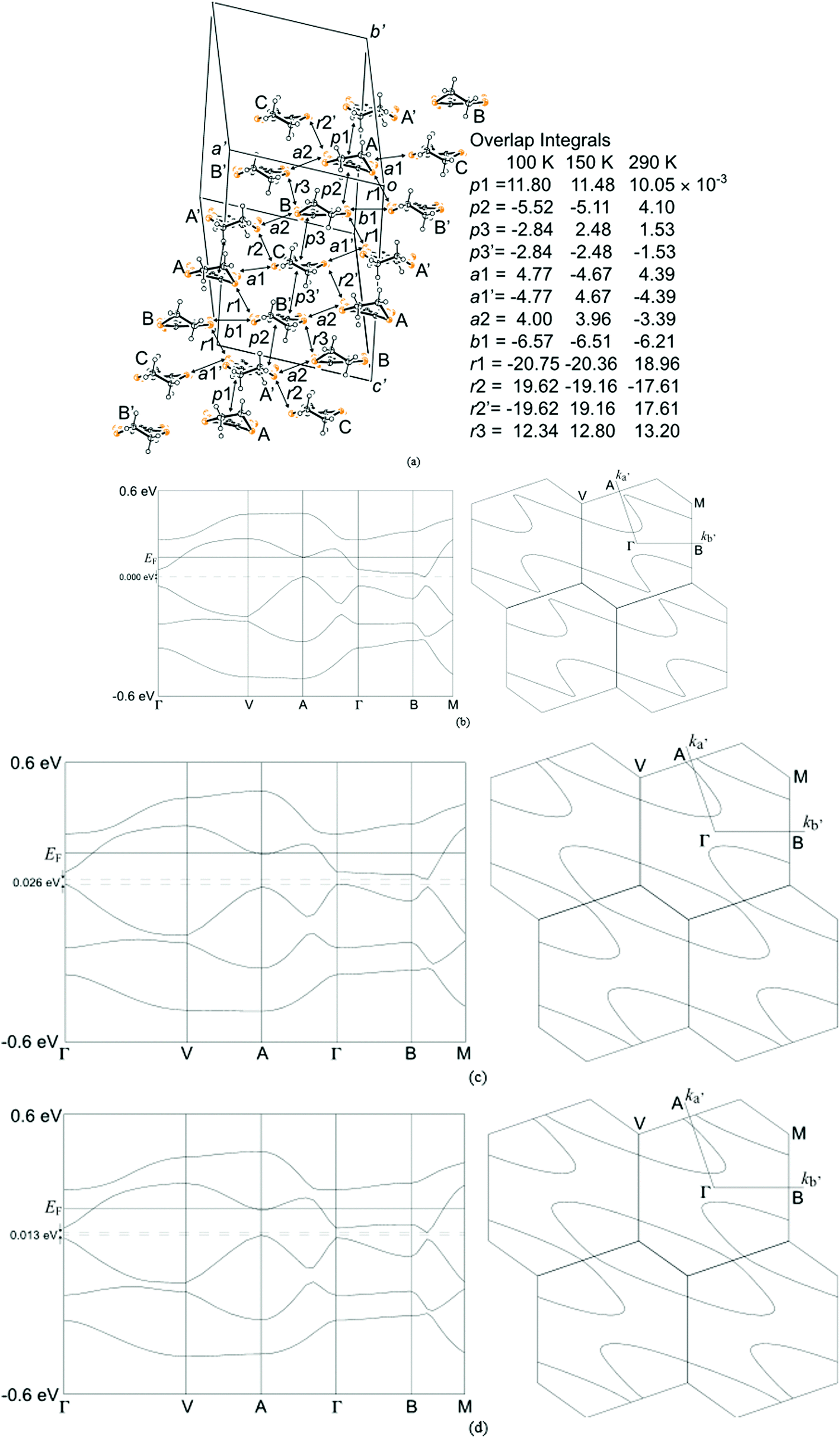 | ||
| Fig. 15 (a) Donor arrangement at 100 K and overlap integrals, band dispersions and Fermi surfaces of salt 6 at 100 (b), 150 (c) and 290 K (d)*1. *1 Because of the software's restriction*1 the band calculation was performed after a unit cell transformation from the C-centered lattice (x, y, z) to a primitive lattice (x′, y′, z′) = (−0.5x − 0.5y, −0.5x + 0.5y, −z). The resulting cell parameters are as follows; a′ = 11.307, 11.332 and 11.434, b′ = 11.307, 11.332 and 11.434, c′ = 33.641, 33.606 and 33.698 Å, α′ = 93.31, 93.36 and 93.65, β′ = 93.31, 93.36 and 93.65, γ′ = 71.28, 71.14 and 70.82°, for 100, 150 and 290 K, respectively, where a* is parallel to the side-by-side direction and b* is close to and parallel to the stacking direction. *1 T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627–633. | ||
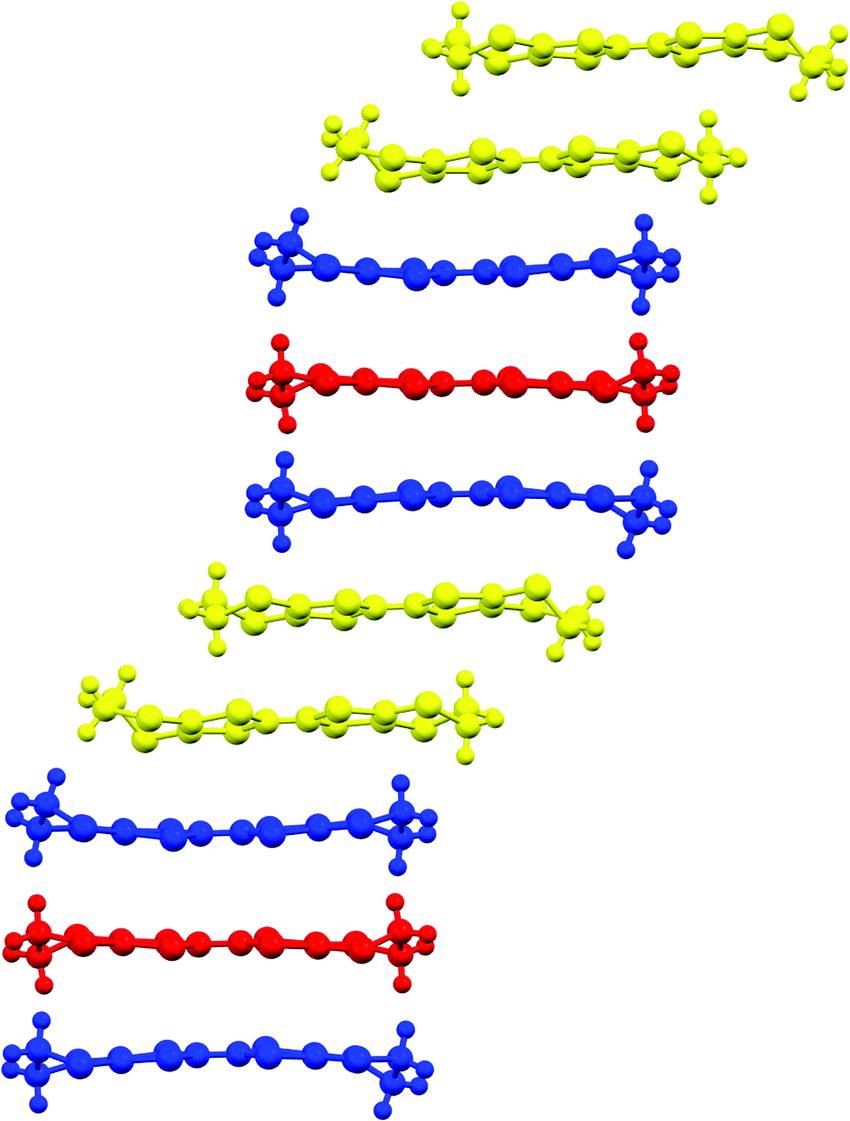 | ||
| Fig. 16 Packing arrangement of the donor layer for 6. A = yellow, B = blue, C = red. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Salt 6 | Donor | a | b | c | D | δ | Q | Q n | Q m | |
| a Normalised by the total charge. b Normalised charges of dimers and trimers. | ||||||||||
| 100 K | A | 1.375 | 1.736 | 1.750 | 1.352 | 0.759 | +0.68 | +0.60 | A–A dimer | +1.20 |
| B | 1.371 | 1.737 | 1.750 | 1.353 | 0.763 | +0.65 | +0.58 | B–C–B trimer | +1.81 | |
| C | 1.376 | 1.734 | 1.753 | 1.358 | 0.753 | +0.73 | +0.65 | |||
| 150 K | A | 1.362 | 1.737 | 1.751 | 1.347 | 0.779 | +0.53 | +0.57 | A–A dimer | +1.14 |
| B | 1.360 | 1.738 | 1.750 | 1.343 | 0.785 | +0.49 | +0.53 | B–C–B trimer | +1.85 | |
| C | 1.375 | 1.732 | 1.752 | 1.357 | 0.752 | +0.73 | +0.79 | |||
| 290 K | A | 1.374 | 1.730 | 1.756 | 1.324 | 0.788 | +0.47 | +0.90 | A–A dimer | +1.80 |
| B | 1.347 | 1.735 | 1.747 | 1.323 | 0.812 | +0.29 | +0.55 | B–C–B trimer | +1.20 | |
| C | 1.312 | 1.747 | 1.751 | 1.329 | 0.857 | +0.05 | +0.10 | |||
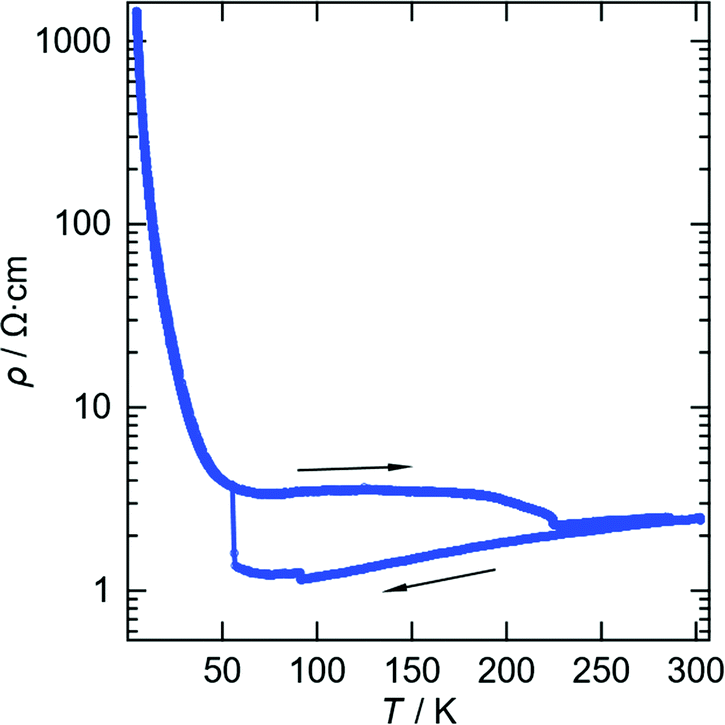 | ||
| Fig. 17 Electrical resistivity for salt 6. | ||
Conclusions
We report six new additions to the family of salts of BEDT-TTF with tris(oxalato)metallate anions. Four new tris(oxalato)gallate salts are 4:1 salts β′′-(BEDT-TTF)4[(H3O)Ga(C2O4)3]·halobenzene with fluorobenzene or chlorobenzene as a guest molecule giving metallic behaviour, whilst the larger bromobenzene or iodobenzene shows superconductivity. Two new tris(oxalato)iridate salts are a semiconducting 4:1 salt pseudo-κ-(BEDT-TTF)4[(H3O)Ir(C2O4)3]·benzonitrile and a 5:1 metal–insulator β′′-(BEDT-TTF)5Ir(C2O4)3·ethanol.
Experimental
Starting materials
Fluorobenzene, chlorobenzene, bromobenzene, iodobenzene, 1,2,4-trichlorobenzene, ethanol and 18-crown-6 were purchased from Sigma-Aldrich and used as received. BEDT-TTF was purchased from TCI and recrystallised from chloroform. Ammonium tris(oxalato)iridate,30 potassium tris(oxalato)iridate,30 and ammonium tris(oxalato)gallate31 were prepared by literature methods and recrystallised several times from water.Synthesis of radical-cation salts
Radical-cation salts 1–6 were synthesized by dissolving 100 mg of ammonium tris(oxalato)gallate or -iridate with 250 mg 18-crown-6 ether in 10 mL 1,2,4-trichlorobenzene: 2 mL ethanol with 10 mL fluorobenzene (1), chlorobenzene (2), bromobenzene (3), iodobenzene, (4), benzonitrile (5), or acetophenone (6). This was added to a H-shaped electrochemical cell containing 10 mg BEDT-TTF in the anode compartment and a current of 1.0 μA was used for up to a month to obtain a black block (β′′ 1–4), black diamonds (pseudo-κ 5), or black prisms (6).Electrical resistivity measurements
Temperature dependent electrical resistivity measurements were performed using four contacts on the single crystals of 1–4 along the out-of-plane direction in the range of 0.8–300 K. Measurements were performed using two contacts on the single crystals of 5 in the range of 240–300 K. Measurements were performed using four contacts on the single crystals of 6 in the range of 4–300 K.X-ray crystallography
Data for 1, 3–5 and for 6 (at 150 K and 290 K) were collected on a Rigaku Oxford Diffraction Xcalibur System equipped with a Sapphire detector.Data for 2 were collected on a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with VHF Varimax optics (70 μm focus).
Data for 6 (at 100 K) were collected on a Rigaku AFC11 quarter chi goniometer equipped with a Rigaku Hypix 6000 detector mounted at the window of a 007 HF copper rotating anode generator with Varimax optics (300 μm focus).
Author contributions
Synthesis, L. M., A. L. M., E. K. R., L. Q., J. O.; X-ray crystallography, T. J. B., H. A., Y. N., S. J. C., J. C.; conductivity measurements and band calculations S. I., H. A., Y. N., J.-I. Y., T. K.; writing—original draft preparation, L. M., T. J. B., H. A.; project administration, L. M.; funding acquisition, L. M; supervision, L. M., T. J. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. and T. B. would like to thank the Leverhulme Trust for financial support (RPG-2019-242). L. M. and E. K. R. would like to thank NTU for a PhD studentship. We thank EPSRC for funding the National Crystallography Service.Notes and references
- A. M. Kini, U. Geiser, H. H. Wang, K. D. Carlson, J. M. Williams, W. K. Kwok, K. G. Vandervoort, J. E. Thompson and D. L. Stupka, Inorg. Chem., 1990, 29, 2555–2557 CrossRef CAS.
- H. Taniguchi, M. Miyashita, K. Uchiyama, K. Satoh, N. Mori, H. Okamoto, K. Miyagawa, K. Kanoda, M. Hedo and Y. Uwatoko, J. Phys. Soc. Jpn., 2003, 72, 468–471 CrossRef CAS.
- A. W. Graham, M. Kurmoo and P. Day, J. Chem. Soc., Chem. Commun., 1995, 2061–2062 RSC; M. Kurmoo, A. W. Graham, P. Day, S. J. Coles, M. B. Hursthouse, J. L. Caulfield, J. Singleton, F. L. Pratt and W. Hayes, J. Am. Chem. Soc., 1995, 117, 12209–12217 CrossRef CAS.
- B. Zhang, Y. Zhang and D. Zhu, Chem. Commun., 2012, 48, 197–199 RSC.
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS PubMed; A. Alberola, E. Coronado, J. R. Galán-Mascarós, C. Giménez-Saiz and C. J. Gómez-García, J. Am. Chem. Soc., 2003, 125, 10774–10775 CrossRef PubMed.
- A. Akutsu-Sato, H. Akutsu, S. S. Turner, P. Day, M. R. Probert, J. A. K. Howard, T. Akutagawa, S. Takeda, T. Nakamura and T. Mori, Angew. Chem., Int. Ed., 2005, 44, 291–295 CrossRef PubMed.
- L. Martin, P. Day, H. Akutsu, J. Yamada, S. Nakatsuji, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, P. McMillan and S. Firth, CrystEngComm, 2007, 9, 865–867 RSC.
- S. Benmansour and C. J. Gómez-García, Magnetochemistry, 2021, 7, 93 CrossRef CAS; L. Martin, Coord. Chem. Rev., 2018, 376, 277–291 CrossRef.
- T. J. Blundell, M. Brannan, J. Mburu-Newman, H. Akutsu, Y. Nakazawa, S. Imajo and L. Martin, Magnetochemistry, 2021, 7, 90 CrossRef CAS; L. Martin, S. S. Turner, P. Day, P. Guionneau, J. K. Howard, D. E. Hibbs, M. E. Light, M. B. Hursthouse, M. Uruichi and K. Yakushi, Inorg. Chem., 2001, 40, 1363–1371 CrossRef PubMed.
- L. Martin, S. S. Turner, P. Day, F. E. Mabbs and E. J. L. McInnes, Chem. Commun., 1997, 1367–1368 RSC.
- H. Akutsu, A. Akutsu-Sato, S. S. Turner, D. Le Pevelen, P. Day, V. Laukhin, A. Klehe, J. Singleton, D. A. Tocher, M. R. Probert and J. A. K. Howard, J. Am. Chem. Soc., 2002, 124, 12430–12431 CrossRef CAS PubMed; H. Akutsu, A. Akutsu-Sato, S. S. Turner, P. Day, D. A. Tocher, M. R. Probert, J. A. K. Howard, D. Le Pevelen, A.-K. Klehe, J. Singleton and V. Laukhin, Synth. Met., 2003, 137, 1239–1240 CrossRef; T. G. Prokhorova, L. I. Buravov, E. B. Yagubskii, L. V. Zorina, S. V. Simonov, R. P. Shibaeva and V. N. Zverev, Eur. J. Inorg. Chem., 2014, 3933–3940 CrossRef.
- S. Benmansour, Y. Sánchez-Máñez and C. J. Gómez-García, Magnetochemistry, 2017, 3, 7 CrossRef.
- L. Martin, A. L. Morritt, J. R. Lopez, Y. Nakazawa, H. Akutsu, S. Imajo, Y. Ihara, B. Zhang, Y. Zhang and Y. Guo, Dalton Trans., 2017, 46, 9542–9548 RSC.
- T. G. Prokhorova, L. V. Zorina, S. V. Simonov, V. N. Zverev, E. Canadell, R. P. Shibaeva and E. B. Yagubskii, CrystEngComm, 2013, 15, 7048–7055 RSC.
- H. Akutsu, A. Akutsu-Sato, S. S. Turner, P. Day, E. Canadell, S. Firth, R. J. H. Clark, J. Yamada and S. Nakatsuji, Chem. Commun., 2004, 18–19 RSC.
- L. V. Zorina, S. S. Khasanov, S. V. Simonov, R. P. Shibaeva, V. N. Zverev, E. Canadell, T. G. Prokhorova and E. B. Yagubskii, CrystEngComm, 2011, 13, 2430–2438 RSC.
- L. Martin, P. Day, P. N. Horton, S. Nakatsuji, J. Yamada and H. Akutsu, J. Mater. Chem., 2010, 20, 2738–2742 RSC; L. Martin, H. Akutsu, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2015, 17, 2783–2790 RSC; L. Martin, H. Akutsu, P. N. Horton, M. B. Hursthouse, R. W. Harrington and W. Clegg, Eur. J. Inorg. Chem., 2015, 1865–1870 CrossRef CAS; L. Martin, P. Day, S. Nakatsuji, J. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369–1372 RSC.
- A. L. Morritt, J. R. Lopez, T. J. Blundell, E. Canadell, H. Akutsu, Y. Nakazawa, S. Imajo and L. Martin, Inorg. Chem., 2019, 58, 10656–10664 CrossRef CAS PubMed; L. Martin, J. R. Lopez, H. Akutsu, Y. Nakazawa and S. Imajo, Inorg. Chem., 2017, 56(22), 14045–14052 CrossRef PubMed; L. Martin, A. L. Morritt, J. R. Lopez, H. Akutsu, Y. Nakazawa, S. Imajo and Y. Ihara, Inorg. Chem., 2017, 56(2), 717–720 CrossRef PubMed.
- L. Martin, P. Day, W. Clegg, R. W. Harrington, P. N. Horton, A. Bingham, M. B. Hursthouse, P. McMillan and S. Firth, J. Mater. Chem., 2007, 31, 3324–3329 RSC; L. Martin, P. Day, S. A. Barnett, D. A. Tocher, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2008, 10, 192–196 RSC.
- L. Martin, P. Day, S.-I. Nakatsuji, J.-I. Yamada, H. Akutsu, P. N. Horton and M. B. Hursthouse, Bull. Chem. Soc. Jpn., 2010, 83(4), 419–423 CrossRef CAS; J. Lopez, H. Akutsu and L. Martin, Synth. Met., 2015, 209, 188–191 CrossRef; L. Martin, S. S. Turner, P. Day, P. Guionneau, J. A. K. Howard, M. Uruichi and K. Yakushi, J. Mater. Chem., 1999, 9, 2731–2736 RSC.
- P. Guionneau, C. J. Kepert, G. Bravic, D. Chasseau, M. R. Truter, M. Kurmoo and P. Day, Synth. Met., 1997, 86, 1973–1974 CrossRef CAS.
- E. Coronado, S. Curreli, C. Giménez-Saiz and C. J. Gómez-García, Inorg. Chem., 2012, 51, 1111–1126 CrossRef CAS PubMed.
- T. G. Prokhorova, E. B. Yagubskii, L. V. Zorina, S. V. Simonov, V. N. Zverev, R. P. Shibaeva and L. I. Buravov, Crystals, 2018, 8, 92 CrossRef.
- S. Imajo, H. Akutsu, A. Akutsu-Sato, A. L. Morritt, L. Martin and Y. Nakazawa, Phys. Rev. Res., 2019, 1, 033184 CrossRef CAS.
- T. G. Prokhorova, L. I. Buravov, E. B. Yagubskii, L. V. Zorina, S. S. Khasanov, S. V. Simonov, R. P. Shibaeva, A. V. Korobenko and V. N. Zverev, CrystEngComm, 2011, 13, 537–545 RSC.
- S. Gärtner, I. Heinen and D. Schweitzer, Synth. Met., 1989, 31, 199–213 CrossRef.
- B. Zhang, Y. Zhang, F. Liu and Y. Guo, CrystEngComm, 2009, 11, 2523–2528 RSC.
- M. Watanabe, Y. Nogami, K. Oshima, H. Mori and S. Tanaka, J. Phys. Soc. Jpn., 1999, 68, 2654–2663 CrossRef CAS.
- H. Akutsu, A. Kohno, S. S. Turner, S. Yamashita and Y. Nakazawa, Mater. Adv., 2020, 1, 3171–3175 RSC.
- M. Delephine, Bull. Soc. Chim. Fr., 1917, 21(4), 157 Search PubMed; H. G. Kruszyna, I. Bodek, L. K. Libby and R. M. Milburn, Inorg. Chem., 1974, 13(2), 434–438 CrossRef CAS.
- N. Bulc, L. Golic and J. Siftar, Acta Crystallogr., 1984, 40, 1829 CAS; P. Neogi and N. K. J. Dutt, J. Indian Chem. Soc., 1938, 15, 83–85 Search PubMed.
Footnotes |
| † CCDC 2163082–2163089 contains supplementary X-ray crystallographic data for 1–6, respectively. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ma00384h |
| ‡ Crystal data 1: C52H40O13S32F1Ga1, M = 1987.52, black block, a = 10.2801(4), b = 19.8975(10), c = 35.2455(13) Å, β = 92.680(3)°, U = 7201.5(5) Å3, T = 294(2) K, space group C2/c, Z = 4, μ = 1.373 mm−1, reflections collected = 19058, independent reflections = 6022, R1 = 0.0725, wR2 = 0.1454 [F2 > 2σ(F2)], R1 = 0.0891, wR2 = 0.1542 (all data). Crystal data 2: C52H40O13S32Cl1Ga1, M = 2003.93, black block, a = 10.2964(7), b = 19.9923(14), c = 35.423(3) Å, β = 93.080(3)°, U = 7281.3(9) Å3, T = 293(2) K, space group C2/c, Z = 4, μ = 1.392 mm−1, reflections collected = 21117, independent reflections = 9327, R1 = 0.0745, wR2 = 0.1117 [F2 > 2σ(F2)], R1 = 0.1483, wR2 = 0.1447 (all data). Crystal data 3: C52H40O13S32Br1Ga1, M = 2048.39, black block, a = 10.2880(3), b = 19.9599(6), c = 35.5191(8) Å, β = 93.392(2)°, U = 7280.98(16) Å3, T = 293(2) K, space group C2/c, Z = 4, μ = 1.901 mm−1, reflections collected = 39889, independent reflections = 8997, R1 = 0.0425, wR2 = 0.0992 [F2 > 2σ(F2)], R1 = 0.0537, wR2 = 0.1057 (all data). Crystal data 4: C52H40O13S32I1Ga1, M = 2095.38, black block, a = 10.2593(2), b = 20.0074(4), c = 35.5849(7) Å, β = 93.7398(18)°, U = 7288.7(2) Å3, T = 294(2) K, space group C2/c, Z = 4, μ = 1.778 mm−1, reflections collected = 35658, independent reflections = 7838, R1 = 0.0347, wR2 = 0.0801 [F2 > 2σ(F2)], R1 = 0.0385, wR2 = 0.0819 (all data). Crystal data 5: C53H39O13S32N1Ir1, M = 2115.99, black block, a = 10.38140(14), b = 19.6057(3), c = 35.5790(5) Å, U = 7241.56(18) Å3, T = 150.00(10) K, space group Pbcn, Z = 4, μ = 12.724 mm−1, reflections collected = 30911, independent reflections = 6344, R1 = 0.0454, wR2 = 0.1171 [F2 > 2σ(F2)], R1 = 0.0503, wR2 = 0.1200 (all data). Crystal data 6 at 100 K: C58H46IrO13S40, M = 2425.56, black prism, a = 18.37840(10), b = 13.17680(10), c = 33.6413(3) Å, β = 94.0750(10)°, U = 8126.27(11) Å3, T = 100(2) K, space group C2/c, Z = 4, μ = 13.310 mm−1, reflections collected = 38584, independent reflections = 7628, R1 = 0.0314, wR2 = 0.0820 [F2 > 2σ(F2)], R1 = 0.0322, wR2 = 0.0824 (all data). Crystal data 6 at 150 K: C58H46IrO13S40, M = 2425.56, black prism, a = 18.4349(2), b = 13.18390(10), c = 33.6063(2) Å, β = 94.1340(10)°, U = 8146.55(12) Å3, T = 150.01(10) K, space group C2/c, Z = 4, μ = 13.276 mm−1, reflections collected = 15759, independent reflections = 7803, R1 = 0.0350, wR2 = 0.0876 [F2 > 2σ(F2)], R1 = 0.0383, wR2 = 0.0899 (all data). Crystal data 6 at 290 K: C56H40IrO12S40 + solvent, M = 2379.50, black prism, a = 18.6386(15), b = 13.2500(8), c = 33.689(2) Å, β = 94.474(6)°, U = 8294.5(10) Å3, T = 290.03(10) K, space group C2/c, Z = 4, μ = 13.016 mm−1, reflections collected = 7329, independent reflections = 5332, R1 = 0.0964, wR2 = 0.2453 [F2 > 2σ(F2)], R1 = 0.1199, wR2 = 0.2645 (all data). |
| This journal is © The Royal Society of Chemistry 2022 |