
Synthesis and characterization of a novel pH-responsive drug-releasing nanocomposite hydrogel for skin cancer therapy and wound healing†
Andrea
Gonsalves
a,
Pranjali
Tambe
bc,
Duong
Le
a,
Dheeraj
Thakore
bc,
Aniket S.
Wadajkar
bcde,
Jian
Yang
f,
Kytai T.
Nguyen
bc and
Jyothi U.
Menon
 *ag
*ag
aDepartment of Biomedical and Pharmaceutical Sciences, College of Pharmacy, University of Rhode Island, 7 Greenhouse Road, Kingston, RI 02881, USA. E-mail: jmenon@uri.edu; Fax: +1-401-874-5787; Tel: +1-401-874-4914
bDepartment of Bioengineering, University of Texas at Arlington, Arlington, TX 76019, USA
cDepartment of Biomedical Engineering, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA
dDepartment of Neurosurgery, University of Maryland School of Medicine, Baltimore, MD 21201, USA
eMarlene and Stewart Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD 21201, USA
fDepartment of Biomedical Engineering, Pennsylvania State University, University Park, PA 16802, USA
gDepartment of Chemical Engineering, University of Rhode Island, Kingston, RI 02881, USA
First published on 4th November 2021
Abstract
Local skin cancer recurrence occurs in ∼12% of the patients post-surgery due to persistent growth of residual cancer cells. Wound infection is another significant complication following surgery. We report a novel in situ-forming nanocomposite hydrogel (NCH) containing PLGA-carboxymethyl chitosan nanoparticles (186 nm) for localized pH-responsive skin cancer therapy and wound healing. This injectable hydrogel, comprising of a citric acid-derived polymer backbone, gelled within 5 minutes, and demonstrated excellent swelling (283% of dry weight) and compressive strengths (∼5.34 MPa). Nanoparticle incorporation did not significantly affect hydrogel properties. The NCH effluents were cytocompatible with human dermal fibroblasts at 500 μg ml−1 concentration and demonstrated pH-dependent drug release and promising therapeutic efficacy against A431 and G361 skin cancer cells in vitro. Significant zones of inhibition were observed in S. aureus and E. coli cultures on NCH treatment, confirming its antibacterial properties. Our studies show that the pH-responsive NCH can be potentially used for adjuvant skin cancer treatment and wound healing.
1 Introduction
The incidence of skin cancer continues to rise annually, ranking fourth among new cancer cases in the United States. In 2021, skin cancer accounts for 6% of the nearly 1.9 million newly diagnosed cancer cases, leading to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 540 new deaths.1 Most types of skin cancer, including melanoma with deep invasion, are treated with surgery, immunotherapy, chemotherapy, or radiotherapy. Surgery is regarded as the gold standard in skin cancer treatment in terms of efficacy and cosmetic results.2 However, incomplete surgical excision of localized melanoma and other types of skin cancer can result in recurrence due to persistent growth of residual cancer cells, even though the surgical margins were clear upon histological examination.3 On the other hand, using a large margin of excision can lead to unnecessary morbidity, slow healing, and high cost.4 Although Mohs Micrographic surgery is now considered a better option than standard excision, local recurrences are still possible due to incorrect histological interpretation.5 Postoperative bacterial infection also remains a major challenge during and after surgical resection of skin tumors, leading to increased hospital stays and associated expenses as well as reduced wound healing and skin regeneration.6,7 A 7-year study of surgical site infections in cancer patients reported that Escherichia coli (E. coli) was the most frequently observed microorganism in these areas, followed by Staphylococcus aureus (S. aureus).8 The latter in particular is a known skin pathogen and has also been implicated in the carcinogenesis of squamous cell carcinoma and melanoma.9–11 Therefore, there is a need for a proactive antimicrobial treatment strategy following surgery to minimize the chances of recurrence of skin cancer in the surgical region as well as to facilitate wound healing.
540 new deaths.1 Most types of skin cancer, including melanoma with deep invasion, are treated with surgery, immunotherapy, chemotherapy, or radiotherapy. Surgery is regarded as the gold standard in skin cancer treatment in terms of efficacy and cosmetic results.2 However, incomplete surgical excision of localized melanoma and other types of skin cancer can result in recurrence due to persistent growth of residual cancer cells, even though the surgical margins were clear upon histological examination.3 On the other hand, using a large margin of excision can lead to unnecessary morbidity, slow healing, and high cost.4 Although Mohs Micrographic surgery is now considered a better option than standard excision, local recurrences are still possible due to incorrect histological interpretation.5 Postoperative bacterial infection also remains a major challenge during and after surgical resection of skin tumors, leading to increased hospital stays and associated expenses as well as reduced wound healing and skin regeneration.6,7 A 7-year study of surgical site infections in cancer patients reported that Escherichia coli (E. coli) was the most frequently observed microorganism in these areas, followed by Staphylococcus aureus (S. aureus).8 The latter in particular is a known skin pathogen and has also been implicated in the carcinogenesis of squamous cell carcinoma and melanoma.9–11 Therefore, there is a need for a proactive antimicrobial treatment strategy following surgery to minimize the chances of recurrence of skin cancer in the surgical region as well as to facilitate wound healing.
Biodegradable hydrogels have been studied for drug delivery and wound healing applications as they can be used to deliver bioactive molecules at the wound site to facilitate tissue healing and regeneration following surgery, as well as to provide localized cancer therapy.2–5 Poly(ethylene glycol) (PEG)-based hydrogels are widely used due to their excellent biocompatibility, proven safety, good mechanical properties and high hydrophilicity that maintains a moist environment for faster wound healing.12,13 Although hydrophilicity is essential for maintaining the bioactivity of the encapsulated molecules, high water content can lead to increased permeability of the hydrogel, resulting in rapid or burst release of the drugs. This reduces the efficacy of the hydrogel system, and the subsequent ‘dose dumping’ may cause toxicity to the surrounding tissues.14 Most hydrogels, including PEG-based hydrogels, exhibit a strong burst release within the first two days, followed by total release of the drug within one week or less.12,15 To reduce local toxicity from rapid drug release, drug-loaded nanoparticles (NPs) can be embedded within the hydrogels to form a nanocomposite system for long-term treatment through controlled drug release over a desired time range.
In this project, we hypothesized that the novel in situ-forming injectable and biocompatible PEG-based hydrogel containing pH-responsive carboxymethyl chitosan-based NPs will provide stimuli-responsive and sustained therapy in response to skin cancer recurrence while also providing a hydrated environment for wound healing. Previously, we had developed PEG-diacrylate (PEGDA) hydrogels in which thermo-responsive poly(N-isopropylacrylamide-co-acrylamide) NPs were embedded for sustained and controlled release of proteins for wound healing.16 However, the non-degradability of the PEGDA hydrogels led to the development of a biodegradable poly(ethylene glycol-maleate-citrate) (PEGMC) polymer.17 The soft and elastomeric PEGMC consisting of citric acid, PEG, and maleic acid components, mimics the mechanical properties of a wide range of soft biological tissues. It is biocompatible17,18 and an excellent injectable cell carrier.19 Furthermore, the citric acid offers biocompatibility, and tunable mechanical and degradation properties to the polymer.20 Therefore, in this work, we developed the nanocomposite hydrogel (NCH) system using PEGMC.
The acidic microenvironment of melanoma and most human cancers is an indication of disease progression.21,22 While normal cells tend to have an extracellular pH of 7.4, the acidic tumor microenvironment has a pH between 5.5 to 7.4.23,24 To provide controlled anti-cancer drug release in response to changing pH in the tumor region, a pH-responsive polymer is critical. Therefore, we embedded NPs consisting of poly lactic-co-glycolic acid (PLGA) and the pH-sensitive biodegradable polymer carboxymethyl chitosan (CMC) polymers, hereafter referred to as PC NPs, within the hydrogel. The PC NPs were loaded with the chemotherapeutic drugs doxorubicin (Dox) and 5-fluorouracil (5-FU) in the PLGA core and the CMC shell respectively, for combination therapy. Finally, to test the feasibility of the NCH system for skin cancer treatment and wound healing, we investigated it's in vitro therapeutic efficacy against skin cancer cells and antibacterial activity against bacterial cells like S. aureus and E. coli, which are most commonly linked to wound infections and implant fouling.25,26
To the authors’ knowledge, this is the first nanocomposite hydrogel system being reported for proactive, pH-responsive treatment of local skin cancer recurrence, while also providing a hydrated and anti-bacterial environment to facilitate wound healing. The combination of PEGMC polymeric hydrogels and PC nanoparticles for this application is innovative. The injectability of the nanocomposite system also makes it an attractive platform for localized injection at the site of the surgery. This property makes the formulation appealing for a wide range of drug delivery, tissue engineering and clinical applications where it can take on the shape of irregular wounds, to provide effective and localized treatment.
2 Experimental
2.1 Materials
PEG, citric acid, maleic acid, dichloromethane (DCM), acryloyl chloride (AC), magnesium sulfate (MgSO4), ammonium persulfate (APS), tetramethylethylenediamine (TEMED), and 5-FU were purchased from Sigma Aldrich (St. Louis, MO). Dox-hydrochloride (Tocris Bioscience, Ellisville, MO), PLGA 50:50 (Lakeshore Biomaterials, Birmingham, AL), CMC (AK Scientific Inc., Union City, CA), potassium carbonate (K2CO3) (Alfa Aesar, Ward Hill, MA), and triethylamine (TEA, MP Biomedicals, Solon, OH) were purchased and used as obtained. Dulbecco's Modified Eagle Media (DMEM) and trypsin-ethylenediaminetetraacetic acid (Trypsin-EDTA) were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Adult human dermal fibroblast (HDFs), A431 epidermoid carcinoma cells and G361 human melanoma cells were purchased from ATCC (Rockville, MD).
2.2 Synthesis of pH-responsive PC NPs
Two different batches of PC NPs were prepared by varying the CMC concentration, using a standard double emulsion solvent evaporation technique. A primary emulsion was prepared by dropwise addition of 0.6% w/v Dox-hydrochloride solution to 3% w/v PLGA solution in 3 ml chloroform. Following sonication (20 W, 2 min), the primary emulsion was added dropwise to an aqueous solution of polyvinyl alcohol (PVA) and CMC. For the PC-0.5 NPs, an aqueous solution containing 4.5% w/v PVA, and 0.5% CMC was used. For the PC-2.5 NPs, an aqueous solution containing 2.5% w/v PVA, and 2.5% w/v CMC was used. The final emulsion was stirred overnight to allow chloroform evaporation, following which the NPs were washed, collected, and lyophilized. 5-FU was loaded into the CMC shell of the particles by absorption. Briefly, 5 mg of NPs were incubated with 1 mg of 5-FU in de-ionized water (pH 7.4) and placed on a shaker at 4 °C for 1 day. 5-FU-loaded NPs were then collected by centrifugation and lyophilized for later use.2.3 Characterization of PC NPs
Particle size and zeta potential were measured by dynamic light scattering (DLS, ZetaPALs, Brookhaven Instruments Inc. Holtsville, NY). Incorporation of both PLGA and CMC in the final formulation was confirmed using Fourier transform infrared (FTIR) spectroscopy (Thermo Nicolet 6700 FTIR Spectrometer, Thermo Fisher Scientific, Waltham, MA). To observe the morphology of the NP formulations, transmission electron microscopy (TEM, FEI Tecnai G2 Spirit BioTWIN, Hillsboro, OR) was utilized. For TEM, the particle suspension was air-dried on a 200-mesh formvar-coated copper grid (Electron Microscopy Sciences, Hartfield, PA) and imaged without further staining. To determine drug release kinetics, the NP stock solution (1 mg ml−1) was added to the dialysis bag of 100 kDa molecular weight cut-off (Spectrum Laboratories, Rancho Dominguez, CA), placed in 10 ml phosphate buffered saline (PBS) at pH 7.4 or 5.5 and incubated on a shaker at 37 °C. The two different pH were used to confirm pH-responsive drug release from the CMC coating on the NPs. At predetermined time points, 1 ml of the dialysate was removed for analysis and stored at −20 °C. The dialysate volume was replenished with fresh PBS. Drug release at each time point was measured for fluorescent intensity at λex/λem 470/585 nm for Dox or absorbance intensity at 266 nm for 5-FU using a UV-Vis spectrophotometer (Tecan Infinite M200 plate reader, Männedorf, Switzerland). The NP formulation demonstrating distinct pH-dependent release kinetics was chosen for incorporation within the NCH system.2.4 Synthesis of PEGMC and PEGDA polymers for hydrogel formation
PEGMC was prepared by random polymerization as described previously (Gyawali et al., 2010). Briefly, PEG 200, maleic acid, and citric acid at a molar ratio of 6:4:1 were added to a 100 ml stoppered round bottom flask immersed in a silicon oil bath preheated to 160 °C. As the mixture was dissolved, the stopper was removed, and the temperature was reduced to 140 °C. The stirring speed was reduced by half after each twitch of the stir bar, and the PEGMC obtained was washed using de-ionized water. PEGDA was prepared as described by Durst et al. (Durst et al., 2011). Briefly, 12 g of PEG (3.3 kDa, 6 kDa or 8 kDa) was dissolved in 36 ml of anhydrous DCM and 1.3 ml of TEA was added to it. Separately, 0.61 ml of AC was dissolved in DCM and added dropwise to the reaction. The reaction was stirred continuously under low light and an inert environment with argon purging in an ice bath. The resultant product was washed with 2 M K2CO3 and dried using anhydrous MgSO4. Ethyl ether was then added to precipitate PEGDA that was filtered and vacuum dried. The purified PEGDA was denoted as PEGDA 3.3 kDa, 6 kDa and 8 kDa, depending on the molecular weight (MW) of the PEG used for synthesis.
2.5 Development and optimization of the PEGMC hydrogel
Factorial analysis studies were performed using Design-Ease 8 DoE software (Stat-Ease Inc., Minneapolis, MN) to study the effect of formulation factors on curing time (for injectability) and swelling ratio of the hydrogels. PEGMC concentrations, PEGDA (3.3 kDa, 6 kDa, 8 kDa) crosslinker concentrations and TEMED concentrations (with fixed w/w ratio to APS) were the three factors (independent variables) chosen for the experiments with realistic high- and low-level values for each factor, since previous studies (Xie et al., 2015) have shown that these factors have a significant impact on hydrogel properties. Curing time studies were performed by immediately placing hydrogel precursor solutions into glass tubes with a magnetic bar stirring at 120 rpm. The duration until the first twitch of the stir bar was considered the curing time of the hydrogel. Further, the swelling ratio of the hydrogels with the most promising curing time was determined. The hydrogels (n = 4 per run) were first lyophilized to get the dry weight (Wd). Dry hydrogels were then immersed in 5 ml of de-ionized (DI) water and allowed to swell for 2 hours before measuring the swollen weight (WS). Swelling ratio was calculated using eqn (1) below.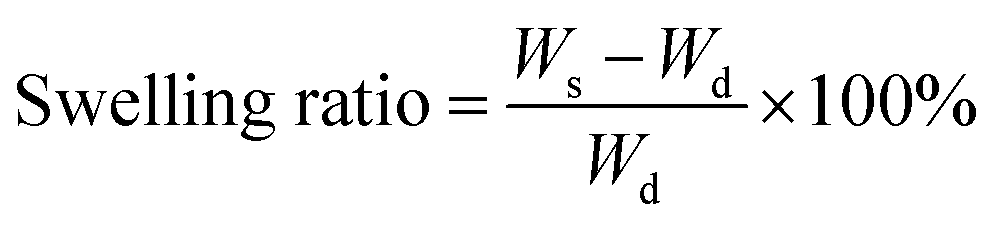 | (1) |
A cross-section of the optimized hydrogel formulation was then imaged using Zeiss Sigma VP field emission scanning electron microscope (FE-SEM). Briefly, a small hydrogel sample was mounted on the holder and flash frozen in the slurry of liquid nitrogen. The frozen sample was then immediately placed in the Gatan Aalto 2500 cryoSEM preparation chamber where the frozen hydrogel was cooled to −130 °C and fractured using a cold-flat edge knife. After fracture, the hydrogel surface was sublimed at −100 °C for 1 minute, cooled back to −130 °C and sputter coated with gold. The prepared samples were kept at −130 °C during imaging with an accelerating voltage of 3 kV, Everhart–Thornley secondary electron detector and a working distance of approximately 9 mm. Images obtained were analyzed using ImageJ to determine pore size distribution. Compressive mechanical tests were also conducted on the hydrogel using an MTS Criterion Model 43 equipped with a 500 N load cell. Cylindrical hydrogel samples of 6 mm diameter and 7 mm thickness were used for the measurement. The load endpoint was set at 50 N with a strain endpoint of 1 mm mm−1 and test speed of 0.02 mm s−1. The results were recorded as mean ± SD. Further, to determine the drug-eluting properties of the hydrogel, Dox was added to the hydrogel precursors prior to hydrogel formation. The formed hydrogel was cut into 4 equal pieces of similar weights, immersed in 1 ml of PBS, and placed at 37 °C on a shaker. At specific time points, the PBS containing the released drug was collected and replaced with an equal volume of fresh PBS. Released Dox was quantified by its fluorescent intensity as described above.
2.6 Synthesis and characterization of the NCH system
For degradation studies, pre-cut hydrogels were lyophilized, and their dry weights were recorded. The hydrogels were then submerged in PBS and incubated over time. At predetermined timepoints, the hydrogels were removed, washed thoroughly in deionized (DI) water, and lyophilized. Degradation was calculated as the change in weight of the hydrogel over a period of time as shown in eqn (2), where W0 represents the initial weight of the specimen and Wt represents its weight at each timepoint.
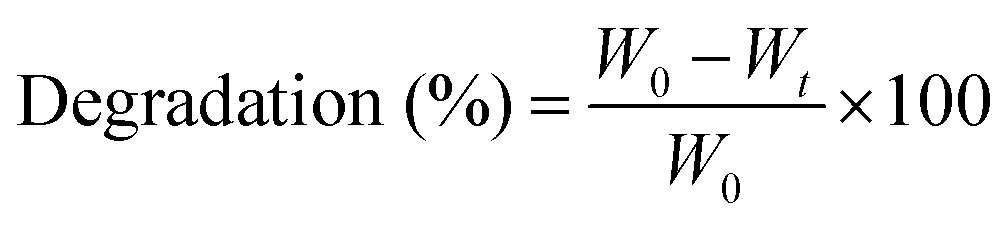 | (2) |
2.7 In vitro cytocompatibility studies
To study the cytocompatibility of PC-2.5 NPs, HDFs were seeded in a 96-well plate at 5000 cells per well seeding density and allowed to attach overnight at 37 °C. NPs at pre-determined concentrations (0, 250, 500, 1000 and 2000 μg ml−1) were incubated with HDFs for 24 hours. At the end of the incubation time, the cells were washed with PBS and MTS assays were performed following the manufacturer's instructions. Cells that were not treated with the NPs were used as control.To study cytocompatibility of the NCH system, the hydrogel was first incubated in DI water at 37 °C for 24 hours and the effluents were collected and lyophilized. Pre-determined concentrations (0, 100, 200, 500, 1000, and 2000 μg ml−1) of the NCH effluents suspended in media were then added to HDFs (seeding density of 5000 cells per well in a 96-well plate). The cells were incubated for 24 hours and MTS assays were performed according to the manufacturer's procedure. Cells that were exposed to only media were used as control.
2.8 In vitro anti-cancer activity evaluation
To study the therapeutic potential of the NCH system, A431 epidermoid carcinoma and G361 melanoma cell lines were seeded (5000 cells per well) in a 96-well plate (n = 5 per group) and allowed to attach for 24 hours. Similar to the cytocompatibility study, the effluents of the NCH (1, 2, and 4 mg ml−1) system containing blank or drug-loaded NPs were collected after 24 hours incubation at 37 °C and pH 5.5. The cancer cells were then treated with the NCH sample effluents for 3 days, where effluent of the NCH with blank NPs was used for comparison. The cells were then washed, and the viability was determined by MTS assays. Cells exposed to only media also served as a control.2.9 In vitro antibacterial activity assessment
Antibacterial properties of the NCH system were determined by placing the hydrogel in agar plates containing freshly cultured S. aureus or E. coli and incubating for 16–18 hours. Ampicillin, a broad-spectrum antibiotic drug was selected to be used as a positive control. For this positive control group, ampicillin was mixed with agar (final concentration = 25 μg ml−1) before plating, and the bacteria was then introduced. The treatment groups included no treatment (control), ampicillin (positive control), PLGA scaffold (negative control), PEGMC hydrogel without NPs, PEGMC hydrogel containing ampicillin (25 μg ml−1 dissolved in PEGMC polymer solution before gelling), and the PEGMC hydrogel with drug-loaded PC-2.5 NPs (no ampicillin). The images were taken at different time points, and zone of inhibition of bacterial growth was determined using ImageJ software.3 Results and discussion
Hydrogels have been widely implemented in drug delivery, wound healing, and tissue engineering over the past several decades due to their unique properties including ease of handling, tunable mechanical strength, high water content, and controllable curing and swelling kinetics.28–30 We had previously developed a hydrogel system consisting of protein-loaded thermo-responsive poly(N-isopropylacrylamide) (PNIPAAm) NPs embedded in a non-degradable PEGDA photo cross-linkable hydrogel.16,31 The limitations of this system lies in its non-degradability and the requirement of UV light irradiation for cross-linking, which can lead to skin-related problems.28,32 We have now improved upon this system and report the development of a biocompatible and partially degradable in situ-forming NCH system consisting of a citric acid-based PEGMC hydrogel and pH-responsive, drug-loaded PC NPs for skin cancer therapy and wound healing applications (Fig. 1).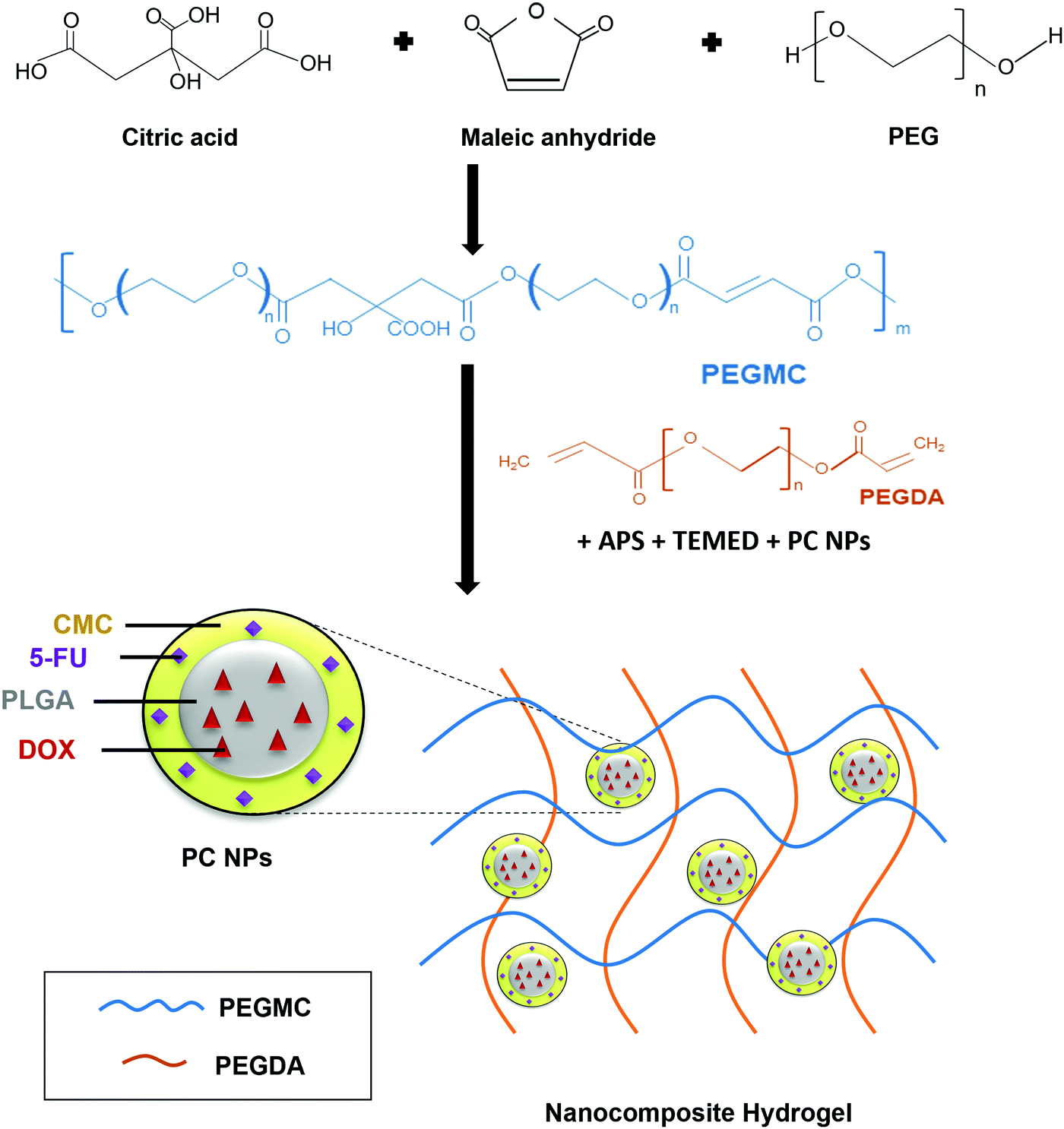 | ||
| Fig. 1 Schematic illustration of the synthesis and use of PEGMC polymer to form the NCH system containing pH-responsive PC NPs. | ||
3.1 Characterization of PC NPs
The PC NPs reported here consisted of a PLGA core and a CMC shell. CMC is a biodegradable and hydrophilic pH-responsive polymer used frequently in drug delivery.33 The core–shell structure of the NP results in a dual-responsive system where the materials used in the core and the shell demonstrate different drug release kinetics.34 Such a NP system is most advantageous for delivering localized combinatorial therapy, especially in cases where one of the drugs needs to be administered first or in larger quantities than the other drug, hence improving the overall efficacy of the treatment.35 Furthermore, the pH-responsive CMC shell offers some control over the DOX release from the PLGA core. The DOX released from PLGA will pass through the pH-responsive CMC layer prior to release, which allows for pH-dependent controlled release.36 We previously reported the development of NPs using a copolymer of PNIPAAm and CMC, which demonstrated greater drug release at pH 6 than at pH 7.4, confirming the pH-responsive nature of CMC.37In the present work, the CMC coating on PLGA NPs serves two purposes – (i) combination therapy via encapsulation of an additional drug (5-FU) in the coating along with the Dox in the PLGA core, and (ii) pH-responsive drug release. In addition, the CMC shell also regulates the release of Dox from the PLGA core into the surrounding environment by providing an additional barrier before release. Thus, there is a synergistic effect of both polymers – PLGA and CMC, on the Dox release kinetics. The PC-0.5 NPs and PC-2.5 NPs had hydrodynamic diameters of 190 ± 8 nm and 186 ± 13 nm, respectively. The particles had homogeneous size distribution as confirmed by the narrow polydispersity index (PDI) of < 0.2. A high zeta potential of −27.9 mV for PC-0.5 NPs and −26.6 mV for PC-2.5 NPs indicates colloidal stability of the NPs (Table S1, ESI†). The high zeta potential values observed for both NPs concur with previous research on CMC NPs, where a highly negative surface charge was observed at pH > 6.8 due to the presence of excess amounts of negatively charged deprotonated carboxyl groups of the CMC.38
TEM images showed a distinct core–shell structure for both NP formulations (Fig. 2A and B). FTIR characterization confirmed the incorporation of both PLGA and CMC in the final NP formulation (Fig. 2C). The peaks corresponding to O–H stretching and NH2 presented in CMC were visible between 3100–3500 cm−1 and at 1519 cm−1 respectively, in the spectra for PC NPs. Symmetric stretching of carboxylic bonds was seen at 1415 cm−1 and peaks related to PLGA, mainly C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, were observed at 1743 cm−1 and 1072 cm−1, respectively.
O and C–O, were observed at 1743 cm−1 and 1072 cm−1, respectively.
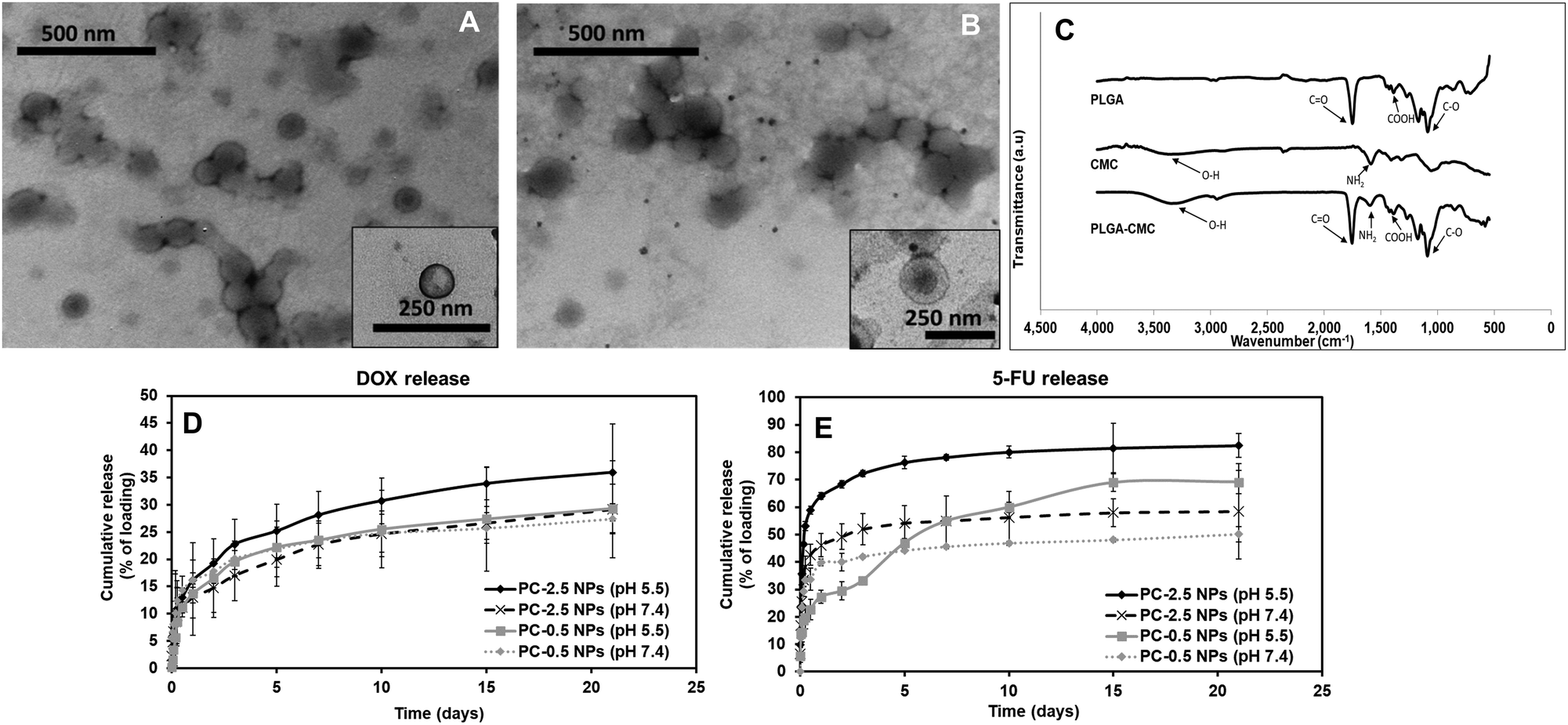 | ||
| Fig. 2 NP characterization. TEM images showing core/shell structures of (A) PC-0.5 and (B) PC-2.5 NPs. (C) FTIR spectra confirming the incorporation of both CMC and PLGA in the PC-2.5 NPs. Release profile of (D) Dox and (E) 5-FU from PC-0.5 and PC-2.5 NPs at pH 5.5 and 7.4. Greater pH-dependent release was observed for PC-2.5 NPs than for PC-0.5 NPs. Lines represent mean values ± SD (n = 4). | ||
Drug release kinetics at different pH were studied for both PC-0.5 and PC-2.5 NPs loaded with Dox in the PLGA core and 5-FU in the CMC shell, to confirm the pH responsive properties of the CMC shell. The Dox from the PLGA core was released in a sustained manner (Fig. 2D) while 5-FU adsorbed in the CMC shell demonstrated pH-responsive release kinetics (Fig. 2E). Therefore, 5-FU – an antimetabolite39 that downregulates ribosomal RNA (rRNA),40 will be delivered as the first line of attack to cancer cells in a pH-dependent manner, while Dox will be released in a sustained manner to kill cancer cells via the DNA-intercalating mechanism.41–43
On day 3, the release of Dox from PC-2.5 NPs was ∼23% of the encapsulated drug at pH 5.5 and 17% in pH 7.4. The release of 5-FU from the NPs was ∼72%, at pH 5.5 and 51% at pH 7.4. After 3 weeks, 36% of the encapsulated Dox was released at pH 5.5 and ∼29% at pH 7.4; meanwhile, 5-FU release was 83% and 58%, respectively. In comparison, the PC-0.5 NPs released 69% and 49% of the encapsulated 5-FU at pH 5.5 and pH 7.4 respectively after 3 weeks, while Dox release from these NPs was ∼29% and ∼27%, respectively, within the same time. These results indicate that the CMC coating on the PC-2.5 NPs demonstrated a superior pH-responsive release behavior compared to the coating on PC-0.5 NPs. This is due to the greater CMC content in the formulation, which had a predominant effect on drug release in response to changing environmental pH. The pH-responsive CMC shell enabled greater release of 5-FU at pH 5.5, which is approximately the acidic pH observed in a cancer microenvironment.24,44 The pH-responsive release behavior concurs with our previous studies using CMC polymer-based NPs.37 The PC-2.5 NPs were therefore chosen for incorporation within the NCH system due to their smaller diameters and greater sensitivity to changes in environmental pH.
3.2 Characterization of blank PEGMC hydrogels
Following characterization and optimization of the NPs, the hydrogel system to be used for embedding the drug-loaded NPs was characterized to ensure that the formulation has optimal properties for application on the skin. A factorial analysis was done to evaluate the effects of three independent formulation factors – PEGMC, PEGDA and TEMED concentrations (w/w ratio of TEMED:APS kept constant) on hydrogel properties (Table S2, ESI†). System outcome parameters, i.e., curing times and swelling ratios, were taken as the dependent variables. PEGDA with different MWs was also used for optimization. The model selection used is tabulated in Table S3 (ESI†). The two-level factorial design usually requires 16 different combinations to be tested. However, with the DoE software, it is possible to design and run a half-factorial experiment using eight combinations that are mirror images of the remaining eight combinations and would therefore generate the same data as a full-factorial design (sixteen runs) (Table S4, ESI†). Therefore, a half-factorial experiment was done.
A gelation time of ∼5 min has been previously found to be optimal for injectable and in situ forming hydrogels for transdermal and other drug delivery applications.45–47 Among the eight formulations tested, six hydrogel formulations, namely 3.3 kDa-H6, 3.3 kDa-H7, 6 kDa-H2, 6 kDa-H5, 8 kDa-H1 and 8 kDa-H5 (denoted with a), demonstrated the desirable curing times for in situ injectable hydrogel applications (Table 1).
| Hydrogel combination | PEGMC (mg ml−1) | PEGDA (mg ml−1) | TEMED (μl ml−1) | Curing time (seconds) | ||
|---|---|---|---|---|---|---|
| PEGDA (3.3 kDa) | PEGDA (6 kDa) | PEGDA (8 kDa) | ||||
| APS:TEMED ratio was kept constant at 8:10 (mg:μl).a Optimal curing time of 3–5 minutes for injectable hydrogels. |
||||||
| H1 | 200 | 50 | 20 | 30 | 60 | 313a |
| H2 | 200 | 100 | 20 | 34 | 240a | 50 |
| H3 | 200 | 50 | 10 | 55 | Did not gel | >500 |
| H4 | 200 | 100 | 10 | 175 | Did not gel | >500 |
| H5 | 100 | 100 | 10 | 96 | 420a | 320a |
| H6 | 100 | 50 | 10 | 270a | >500 | >500 |
| H7 | 100 | 50 | 20 | 300a | 56 | 120 |
| H8 | 100 | 100 | 20 | 15 | 15 | 198 |
As seen in Fig. 3A–C, the PEGDA molecular weight (MW) and concentration, had a significant impact on the curing time, with PEGDA 3.3 kDa showing major variations in curing times with increasing concentrations compared to PEGDA 6 kDa and 8 kDa.
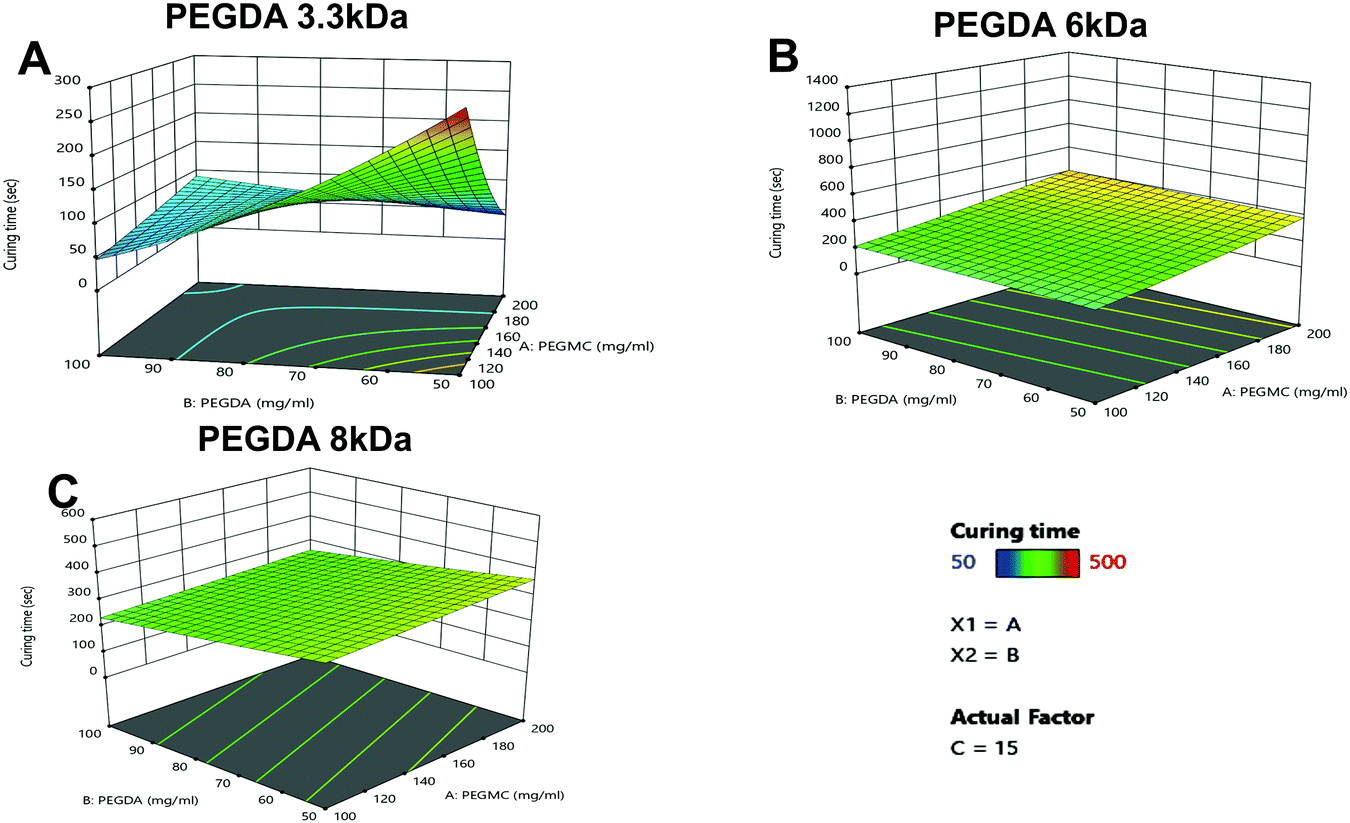 | ||
| Fig. 3 Effects of the formulation factors on PEGMC hydrogel curing time. 3D graphs of curing time (in seconds) with varying concentrations of PEGMC and PEGDA, in terms of the actual factor TEMED (represented as ‘C’), at 15 μl ml−1. Three different MWs of PEGDA, i.e., (A) 3.3 kDa, (B) 6 kDa, (C) 8 kDa, were used. Correlation factors were obtained as R2 = 0.9988, 0.8766 and 0.7665 for PEGDA 3 kDa, 6 kDa and 8 kDa, respectively. Correlation factors were analyzed using square root transformation and either linear or 2FI fit models. | ||
Upon curing, the hydrogels were soft, moist, and non-brittle. The swelling ratio of the six hydrogel formulations with promising curing times was then determined. The MW of PEGDA was found to have a significant effect on swelling ratio of the hydrogels, which concurs with our previously reported results.16 Low MW crosslinkers have short chains, which leads to the generation of highly compact networks with high cross-linking density.48,49 As a result, there will be less void space available within the hydrogel for the diffusion of solutions into and out of the hydrogel. The use of high MW crosslinkers, on the other hand, will allow for greater diffusion of water and/or drugs into and out of the hydrogel. Among the 8 kDa hydrogels, which showed higher swelling compared to other formulations, PEGMC concentration also appeared to play a role in regulating the swelling ratio. Therefore, the 8 kDa-H1 hydrogel was considered the most promising among the tested combinations as it had the highest swelling ratio of about 283% in 2 hours (Fig. 4A). This hydrogel had also undergone complete gelling in 313 seconds or 5.2 minutes, which was confirmed visually (Fig. 4B).
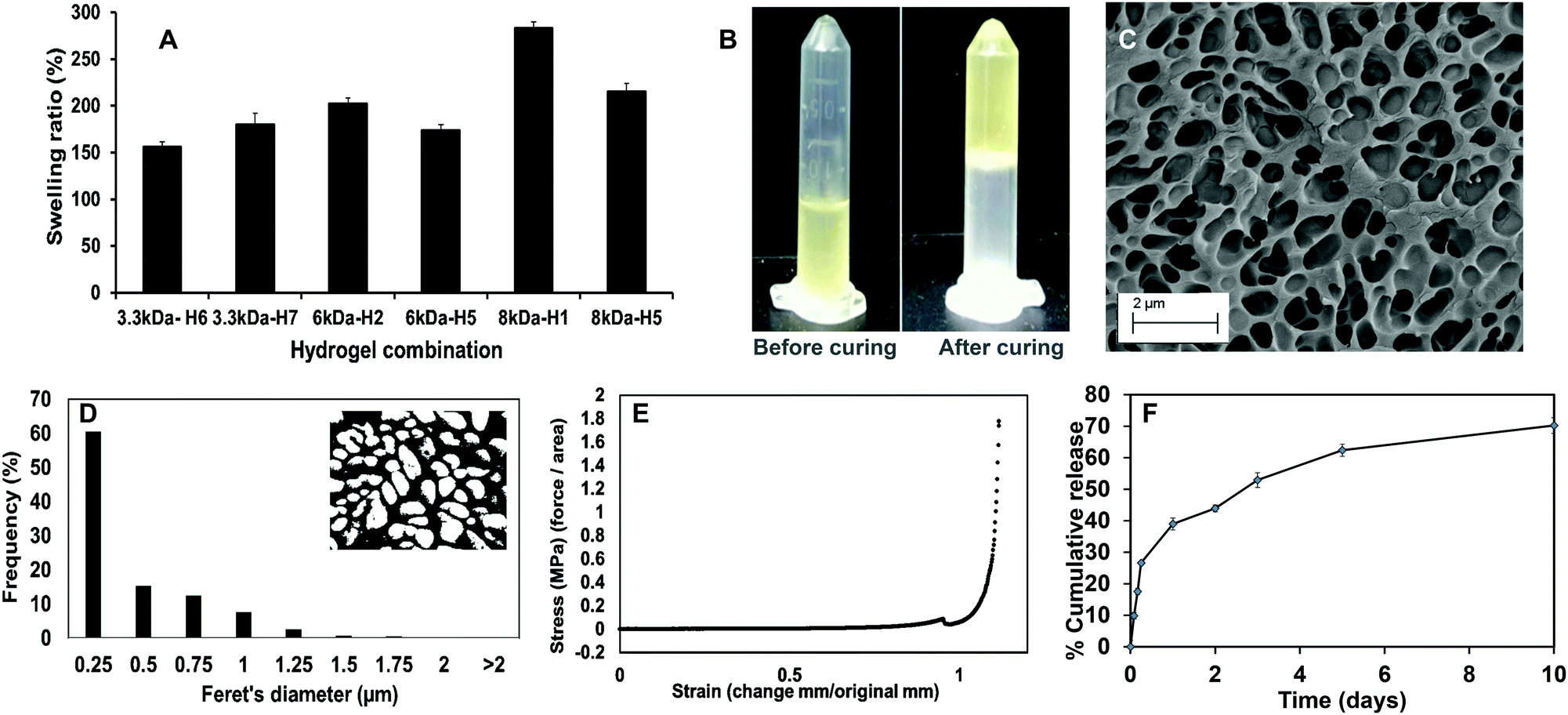 | ||
| Fig. 4 Physicochemical characterization of the blank PEGMC hydrogels. (A) Swelling ratios of the different hydrogel formulations showing greater swelling of 8 kDa-H1 hydrogels (n = 4). (B) Images of the 8 kDa-H1 hydrogels before and after curing, confirming complete gelation. (C) SEM image confirming porous structure of 8 kDa-H1 hydrogels. (D) Feret's diameter of pores within the 8 kDa-H1 hydrogels. (E) Compressive stress–strain curves for the PEGMC hydrogel with the load endpoint set at 50 N. (F) Dox release from the 8 kDa-H1 hydrogels demonstrating ∼70% release in 10 days (n = 4). | ||
The surface morphology of the 8 kDa-H1 hydrogel was also observed using SEM (Fig. 4C). The hydrogel had a highly interconnected porous structure. Using ImageJ analysis, the majority (∼60%) of the pore sizes were found to be in the range of 0.25–0.5 μm (Fig. 4D). This concurs with previous observations of PEGMC hydrogels, where an interconnected porous structure was observed by SEM.19 Interconnected porous structures are desirable in hydrogels to facilitate release of macromolecules (e.g., proteins, DNA) from them.50
The mechanical strength of PEGMC-based and PEGMC-PEGDA hydrogels have already been studied by us in detail.51 Our previously reported study by Xie et al.51 demonstrated that PEGDA concentrations had the largest effect on hydrogel mechanical properties while PEGMC concentrations also positively impacted the mechanical strength of the hydrogel. The mechanical properties of 8 kDa-H1 hydrogels were evaluated through compression tests and an elastic modulus of 5.34 MPa was determined (Fig. 4E).
The release of therapeutics from the 8 kDa-H1 hydrogel was also examined. Dox was released from the 8 kDa-H1 hydrogel in a biphasic manner (Fig. 4F), where a burst release up to ∼40% of the embedded drug took place in 1 day, followed by a sustained release that delivered another 30% of the drug over the next 10 days. Incorporation of the non-degradable PEGDA may have aided in controlling the release of Dox from the hydrogel. Studies by Ahan et al.52 have shown that increasing PEGDA concentrations within hydrogels leads to slower release (75% release from 30 wt% PEGDA gels vs. 50% release from 50 wt% gels within 180 min). The injectability of the hydrogel was also confirmed by extruding the hydrogel solution through a 21G syringe. The hydrogel could be easily extruded through the needle and gelled within 5 min (Fig. S1, ESI†). Based on the data obtained on the curing time, swelling properties, mechanical strength, drug release kinetics and injectability, the 8 kDa-H1 hydrogel was chosen as the optimal formulation for developing the NCH with the PC-2.5 NPs.
3.3 Characterization of the NCH system
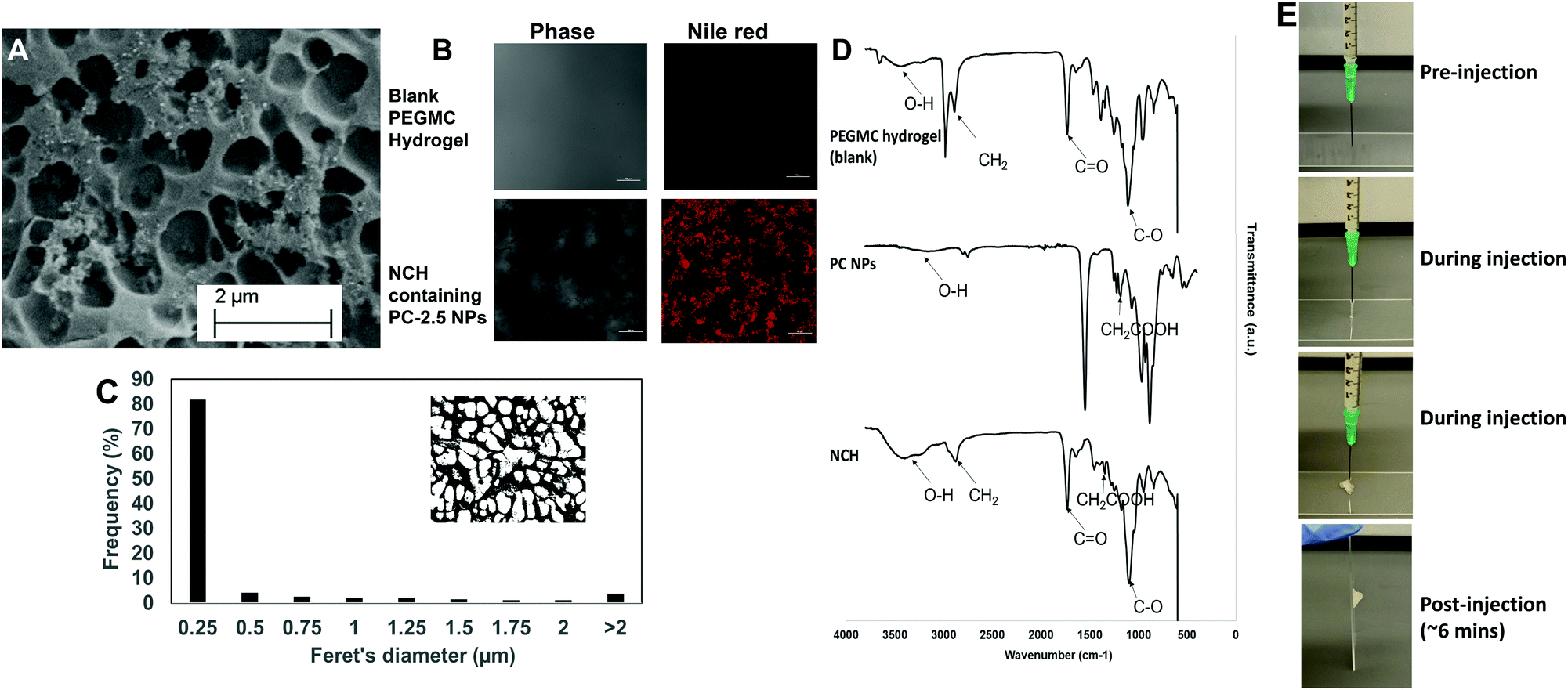 | ||
| Fig. 5 Characterization of PC-2.5 NP incorporation and distribution in the NCH, and NCH injectability. (A) SEM image showing porous structure of the NCH. (B) Confocal images of blank PEGMC hydrogel and NCH containing Nile red-loaded PC-2.5 NPs (scale bar: 100 μm). (C) Feret's diameter of pores within the NCH. (D) FTIR analysis of blank PEGMC hydrogel, PC-2.5 NPs and the NCH system confirming successful incorporation of all components in the final system. (E) Images of NCH injection through a 21-G needle pre-, during and post-injection, confirming injectability of the formulation. | ||
The pore structures of the hydrogel were not affected by incorporation of the NPs, with most pores remaining in the 0.25–0.5 μm range (Fig. 5C). This concurs with our previous findings where incorporation of hydroxyapatite NPs within PEGMC hydrogel did not alter the pore structure, and the NPs were observed in the matrix and not within the pores.19
The curing time of the NCH system remained the same at around 6 minutes, and the hydrogel also maintained its shape after curing. Furthermore, the hydrogel solution containing the NPs could be extruded from a 21G needle to form the NCH, as shown in Fig. 5E. This confirms the injectability of the NCH system.
O) stretching of both the PEGDA and PLGA polymer while the peak at 2800 cm−1 represents the methylene group (−CH2) of PEGDA.54 Symmetric and asymmetric stretching of carboxylic bonds (–COOH) present in PEGMC and PLGA was seen at 1639 cm−1 and 1400 cm−1 respectively indicating the incorporation of the PC NPs within the hydrogel while the peak at 1093 cm−1 represented the C–O group.17,53
Degradation studies were conducted on the NCH system. As shown in (Fig. S2, ESI†), the hydrogel's weight decreased to 54% of its original weight in 3 days, following which it maintained constant weight for the duration of the study (21 days), possibly due to the non-degradable nature of PEGDA.55 This degradation rate was in accordance with a study by Stillman et al. where PEGDA-based hydrogel maintained 50–70% of its weight after 4 weeks’ incubation at a pH range of 5–7.4.56
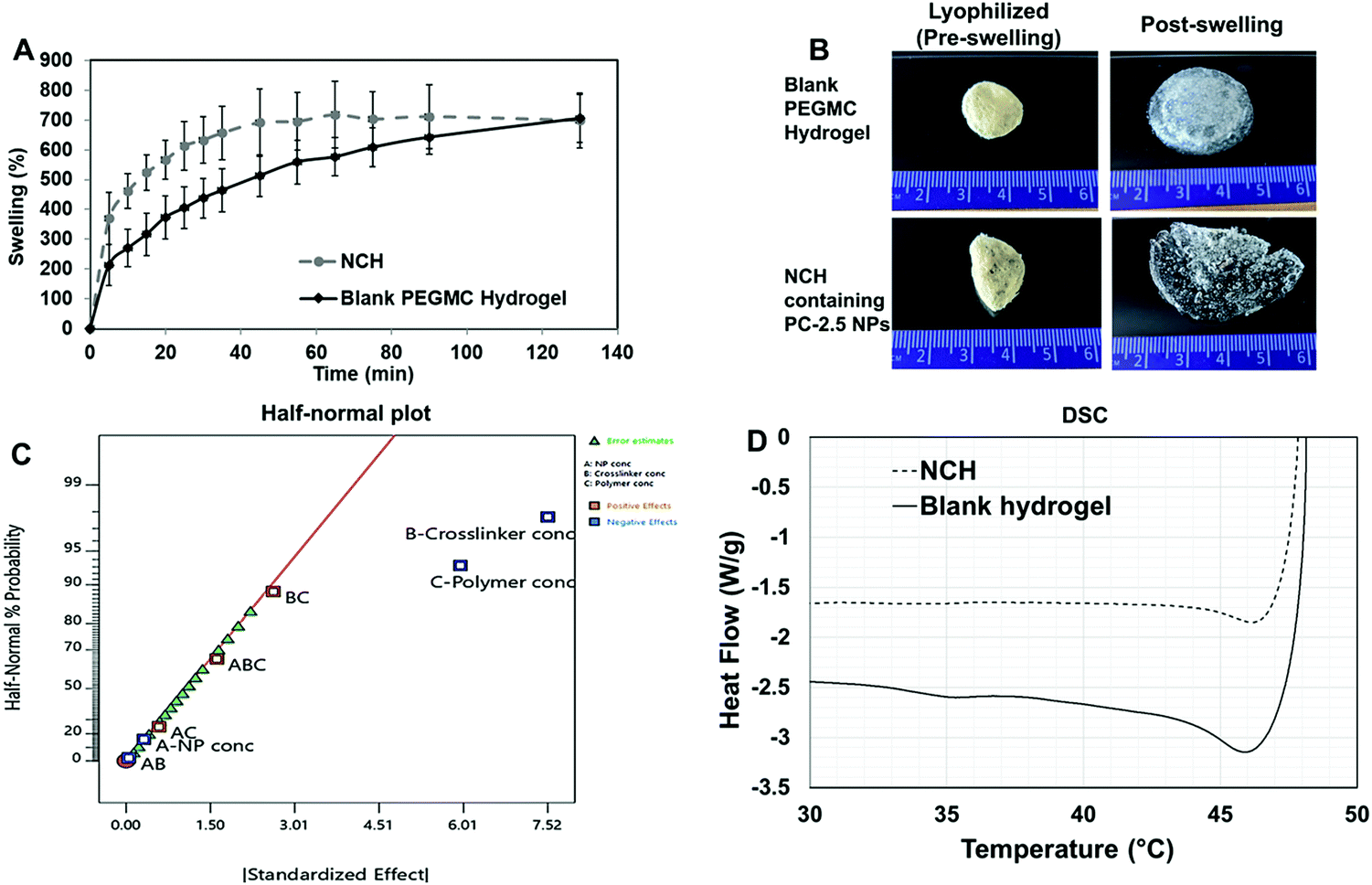 | ||
| Fig. 6 Characterization of the NCH system. (A) Swelling behavior of the blank PEGMC hydrogel and the NCH system showing slightly higher % swelling ratio following incorporation of NPs. (B) Visual observation of the swelling behavior of the lyophilized blank PEGMC hydrogel and the NCH system. (C) Half-normal probability plot for swelling behavior of NCH showing that the swelling is dependent on the concentrations of backbone polymer (PEGMC) and crosslinker (PEGDA). Graphs were analyzed with ANOVA (P = 0.0025) and plotted on Design Expert software (n = 4). (D) DSC curves of the blank PEGMC hydrogel and the NCH system indicating NP incorporation does not affect critical temperatures of phase transition. | ||
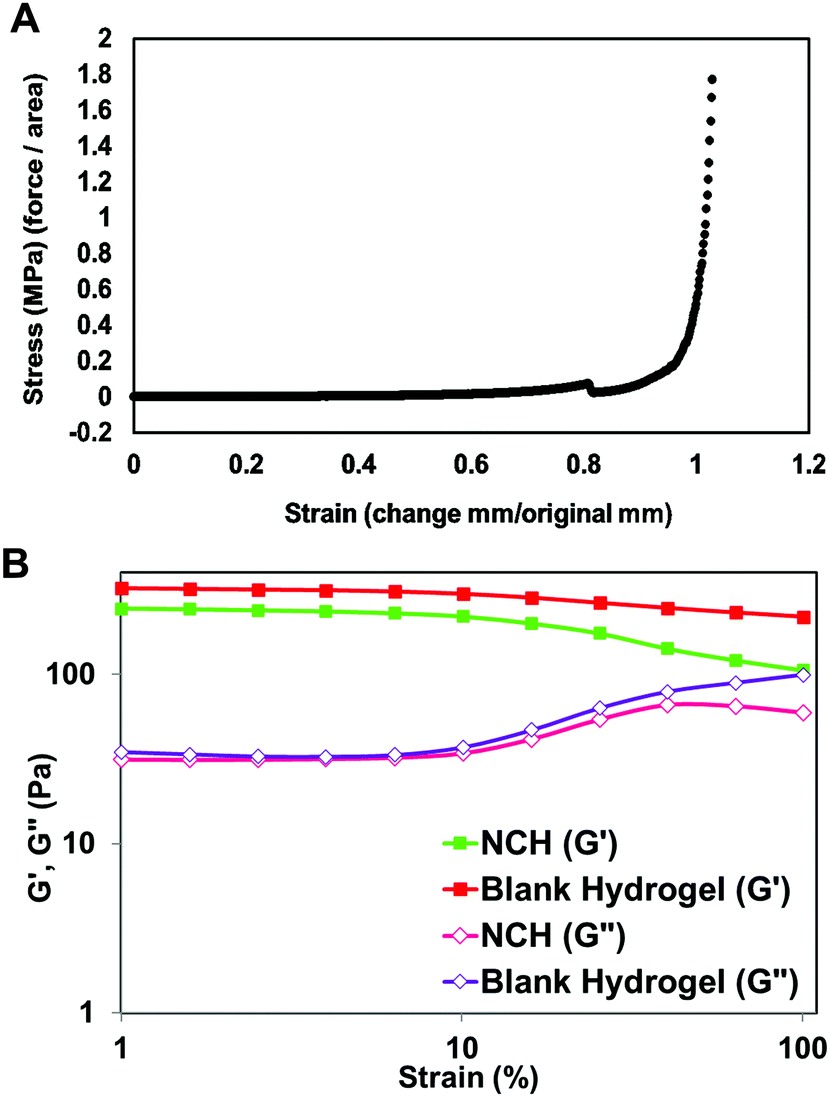 | ||
| Fig. 7 Mechanical properties of the hydrogels. (A) Compressive stress–strain curves for the NCH system with the load endpoint set at 50 N. (B) Storage (G′) and loss (G′′) moduli obtained using dynamic frequency sweep test from 0.1 to 100 rad s−1. | ||
The viscoelastic properties of NCH were then determined using a rheometer by applying an amplitude sweep. The changes in the storage modulus (G′) and loss modulus (G′′) of NCH and blank hydrogels were investigated. The storage modulus corresponds to the elasticity exhibited by the energy stored in the hydrogel while the loss modulus corresponds to the energy depleted in the hydrogel. As seen in Fig. 7B, under the strain of 1–10%, the G′ of both hydrogels was nearly 7 times greater than the G′′ the storage modulus, indicating that the gels were stable and behaved predominantly as a solid upon gelling.64 The results also confirm that the incorporation of PC-2.5 NPs did not significantly impact the viscoelastic properties of the hydrogel.27
3.4 Cytocompatibility studies
The HDFs showed dose-dependent viability when treated with PC-2.5 or PC-0.5 NPs. Both NP types were cytocompatible with >80% viability observed up to a concentration of 2000 μg ml−1 (Fig. 8A). Similarly, about 80% or more of HDFs were viable when treated with effluents from the NCH system at concentrations up to 500 μg ml−1 (Fig. 8B).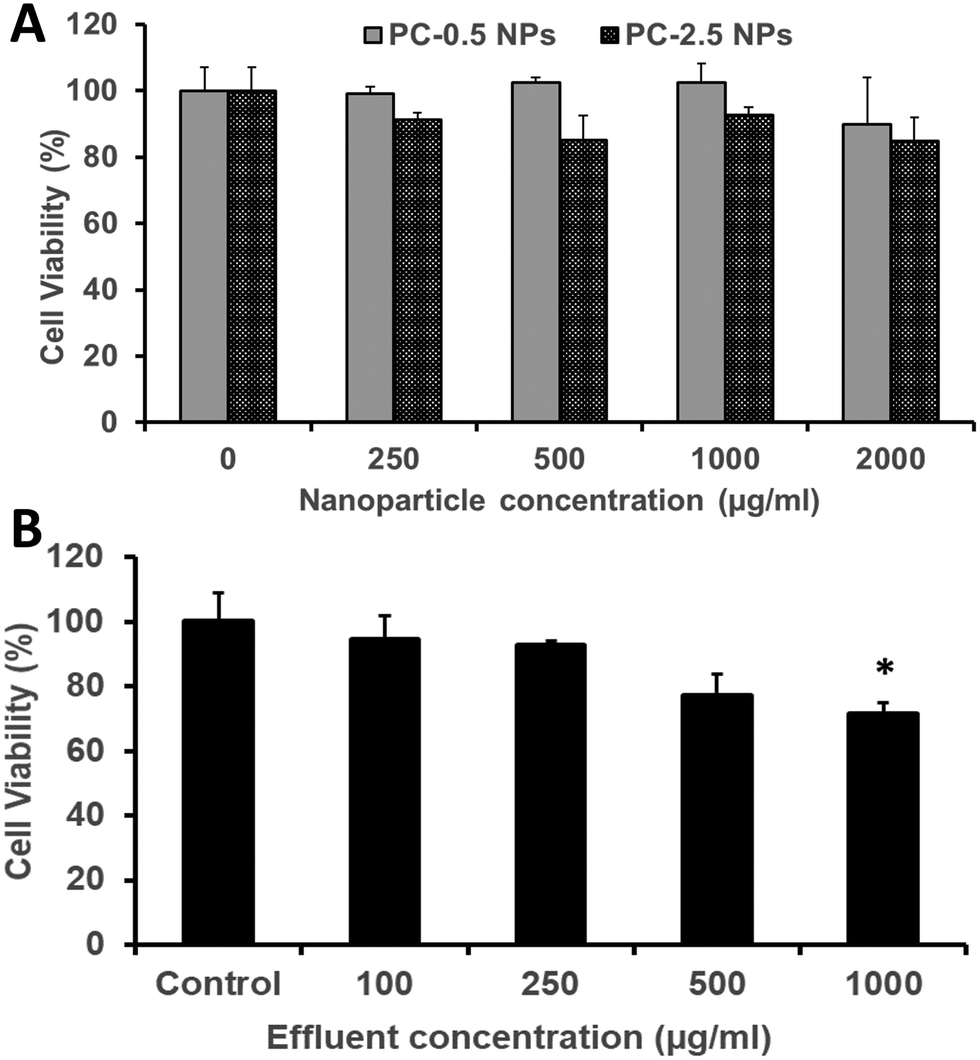 | ||
| Fig. 8 Cytocompatibility studies. Human dermal fibroblasts (HDFs) following treatment with (A) PC NPs and (B) the NCH system containing empty PC-2.5 NPs confirming the cytocompatibility of both systems. Student t-test was performed to analyse statistical difference. *P < 0.05 with respect to control (n = 4). | ||
This concurs with our previously reported results that both PLGA-based NPs65,66 and the hydrogel effluents17,19 are non-toxic against healthy mammalian cells (fibroblasts, smooth muscle cells, osteoblasts). Previous studies on PEGMC hydrogels have also demonstrated minimal inflammation in Sprague-Dawley rats following subcutaneous implantation.17 This indicates that the NCH system is safe to use for wound healing and drug delivery applications, and that its by-products are relatively not toxic.
3.5 Therapeutic efficacy studies
The NCH was then evaluated in terms of its ability to cause cancer cell death in vitro. 1 mg of NCH effluent contained approximately 2.8 μg of Dox and 2.2 μg of 5-FU. When the A431 epidermoid carcinoma cell line was treated with effluents from the NCH system, significant and dose-dependent cancer cell death was observed. About 23% and 50% cell death compared to the untreated control was observed upon treatment with 2 mg ml−1 and 4 mg ml−1 of effluents, respectively (Fig. 9A). In contrast, the NCH effluents were less toxic towards the G361 malignant melanoma cell line. Only about 13% and 23% cell death were observed following treatment of G361 cells with 2 mg ml−1 and 4 mg ml−1 of NCH effluents, respectively (Fig. 9B).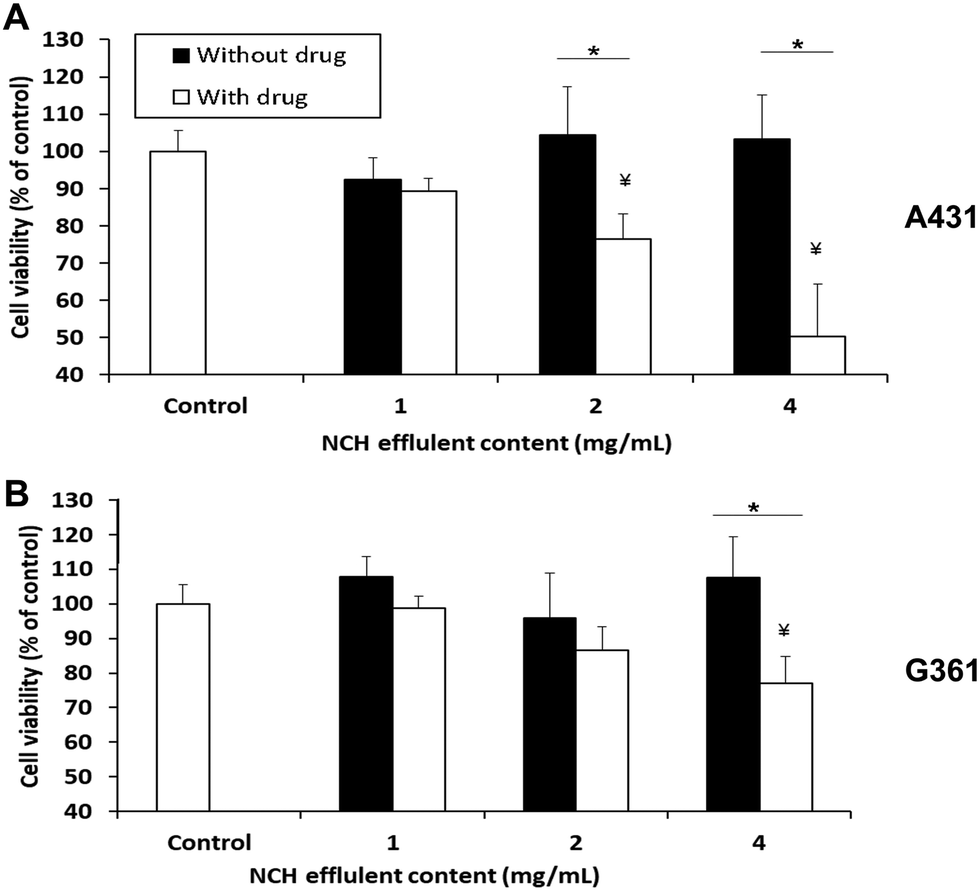 | ||
| Fig. 9 In vitro therapeutic efficacy study. Viability of (A) human epidermal carcinoma A431 and (B) melanoma G361 cell lines following treatment with varying concentrations of NCH effluents for 3 days. NCH hydrogel containing drug-loaded PC-2.5 NPs had a significant effect on cell viability while NCH hydrogel containing blank NPs had no effect on viability. Student t-test was performed to analyse statistical difference. *P < 0.05 with respect to blank NCH; ¥P < 0.05 with respect to control (n = 4). | ||
Our results are in an agreement with the previous studies by Ohori et al.67 who reported that the IC50 value of drugs like 5-FU in A431 cells was much lower than that in G361 cells. Furthermore, the presence of epidermal growth factor receptor (EGFR) on the surface of cells has been reported to increase the sensitivity of the cells to Dox treatment.68 A431 is known to overexpress EGFR while G361 has lower EGFR expression,68–70 which may have resulted in their varied responses to the same Dox concentration. Drugs that are more effective against the highly proliferative and malignant G361 will be incorporated in the NCH system in the future, for further studies. The effluents of the NCH system containing blank NPs had negligible effects on the cell viability of both cancer cell lines.
3.6 Antibacterial activities
In addition to its anti-cancer properties, the NCH also displayed antibacterial properties, which is favorable for post-surgical wound healing. Bacterial wound infections as a result of dermal tissue damage and microbial invasions leads to the delay in the wound healing process.71 The high-water content present in hydrogels prevents tissue dehydration and bacterial infections.72 Additionally, we along with others have previously reported that citric acid (one of the degradation byproducts of PEGMC) possesses antibacterial properties, and so does PEGMC.51,73 Specifically, Su et al. reported that citric acid can easily pass through cell membranes and reduce the pH of the surrounding environment, leading to a decrease in the internal pH of bacteria which will negatively affect their membrane permeability.73 Maleic acid, another degradation byproduct of PEGMC, also has antimicrobial activity and has shown to eradicate bacterial biofilms alone and in combination with citric acid.74 Microorganisms especially belonging to the family of Pseudomonas aeruginosa, Klebsiella pneumonia, S. aureus, and Enterobacter species have the ability to produce biofilms on wounds which increases resistance to antimicrobial agents thus, resulting in wound infection and delaying the wound healing process.75,76 Therefore, in this study, the Gram-positive S. aureus and Gram-negative E. coli were cultured on agar plates. The growth of both E. coli and S. aureus were not inhibited by the untreated agar plate (negative control group) and the PLGA scaffold (negative control group) (Fig. 10A and B). On the other hand, agar plates containing ampicillin (positive control group) strongly inhibited growth of S. aureus, but not of the ampicillin-resistant E. coli.77 The PEGMC hydrogel, PEGMC hydrogel containing 25 μg ml−1 ampicillin, and the NCH system (PEGMC hydrogel containing drug-loaded NPs) resulted in significantly less bacterial growth with a clear zone of inhibition around the hydrogels. The zone of inhibition around the hydrogels was quantified using ImageJ. The PEGMC hydrogel had inherent antibacterial properties. Upon incorporation of drug-loaded NPs, the NCH demonstrated significantly greater antibacterial effects against both E. coli and S. aureus in comparison to the untreated control and the PLGA scaffold (Fig. 10C). The area of the zone of inhibition around the NCH system containing drug-loaded NPs were ∼2.2 cm2 and ∼3.8 cm2 for E. coli and S. aureus, respectively, indicating that the NCH has promising antibacterial properties. Dox can elevate the antibacterial effects, since Dox acts as a DNA intercalating agent and kills bacteria through topoisomerase II poisoning.43,51 Campbell et al. has previously reported that Dox has strong antibacterial effects against Gram-positive bacteria (e.g., S. aureus) while 5-FU can effect both Gram-positive and Gram-negative bacteria.78 Our studies thus confirm the potential of the developed innovative biodegradable and cytocompatible NCH system for use in sustained skin cancer treatment in response to changing pH, while also facilitating wound healing by preventing bacterial infections.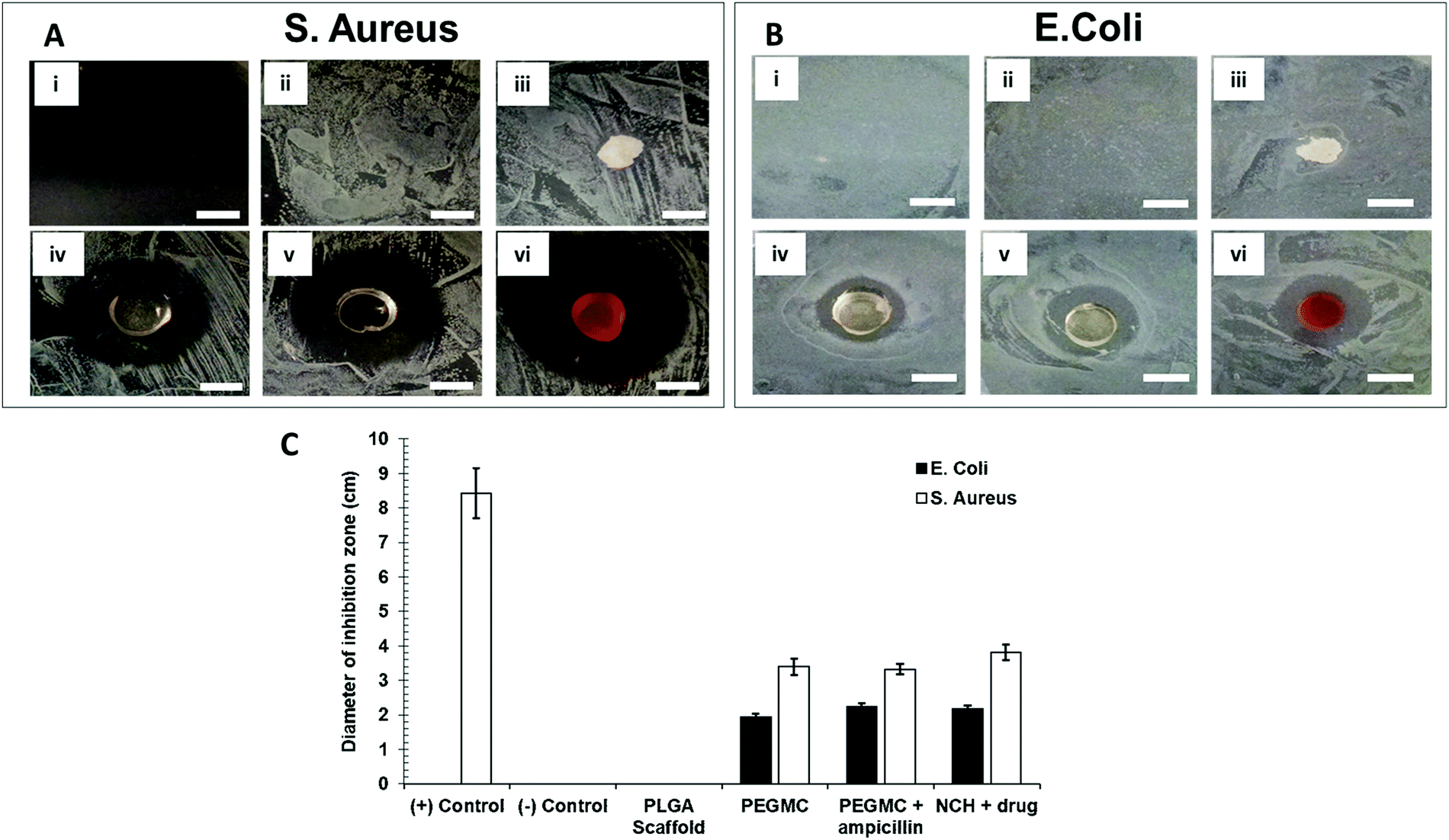 | ||
| Fig. 10 Bacterial zone of inhibition study against S. aureus and E. coli. Representative images of (A) S. aureus and (B) E. coli against treatment groups of (i) ampicillin treatment [(+) control for S. aureus], (ii) no treatment [(−) control], (iii) PLGA scaffold, (iv) PEGMC hydrogel only, (v) PEGMC hydrogel containing ampicillin and (vi) NCH containing drug-loaded NPs (scale = 1 cm). (C) Quantification of the area of zone of inhibition for the different treatment groups. S. aureus and E. coli were cultured to reach OD 0.5 before plating on agar plates (n = 4). | ||
4 Conclusions
In this work, we have built upon our previous research by developing a novel biodegradable and chemically cross-linkable NCH system consisting of the citric acid-based PEGMC polymer, PEGDA and pH-responsive PC-2.5 NPs. The PC-2.5 NPs demonstrated significantly higher release of the encapsulated chemotherapeutic drugs at acidic pH confirming their pH-responsive behavior. The NCH system had a curing time of 6 minutes, which is favorable for use as injectable and in situ forming hydrogels. The PC-2.5 NPs were distributed relatively uniformly within the system. The NCH also demonstrated superior swelling and mechanical properties. It was cytocompatible and can effectively cause cancer cell death, while minimizing infections at the site due to its demonstrated antibacterial properties. However, since the NCH system is not fully biodegradable due to the presence of PEGDA, future studies will focus on identifying suitable alternative crosslinkers that can be used to optimize the system to ensure complete degradability over time. We will also investigate the optimized NCH system loaded with therapeutic reagents in greater detail in vivo in an animal model of melanoma.Author contributions
A. G., P. T. and D. T. designed and carried out the studies. A. G., P. T. and J. U. M. compiled and analysed the data. D. L. and A. S. W. also assisted with data analysis. A. G., D. L., A. S. W. and J. U. M. drafted, reviewed and edited the manuscript. K. T. N., J. Y., and J. U. M. acquired funding for the work and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Rhode Island Institutional Development Award (IDeA) Network of Biomedical Research Excellence from the National Institute of General Medical Sciences of the National Institutes of Health (Grant number P20GM103430), the Rhode Island Foundation Medical Research Grant (#20174376) and the URI College of Pharmacy New faculty start-up funds (J. U. M). This work was also partly supported by NIH R15 HL156076 (K. T. N.), CPRIT RP210206 (K. T. N.), and NIH R01 AR072731 (J. Y.). We would like to thank Dr Irene Andreu for assistance in obtaining the cryo-SEM images, which were acquired at the RI Consortium for Nanoscience and Nanotechnology, a URI College of Engineering core facility partially funded by the National Science Foundation EPSCoR, Cooperative Agreement #OIA-1655221.Notes and references
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, CA: A Cancer J. Clin., 2021, 71, 7–33 Search PubMed.
- L. L. Griffin, F. R. Ali and J. T. Lear, Clin. Med., 2016, 16, 62 CrossRef PubMed.
- P. J. Heenan and M. Ghaznawie, Br. J. Plast. Surg., 1999, 52, 209–213 CrossRef CAS PubMed.
- J. M. Thomas, J. Newton-Bishop, R. A'Hern, G. Coombes, M. Timmons, J. Evans, M. Cook, J. Theaker, M. Fallowfield and T. O'Neill, N. Engl. J. Med., 2004, 350, 757–766 CrossRef CAS PubMed.
- M. Zabielinski, L. Leithauser, T. Godsey and H. M. Gloster Jr, Dermatol. Surg., 2015, 41, 913–916 CrossRef CAS PubMed.
- C. P. Page, J. M. Bohnen, J. R. Fletcher, A. T. McManus, J. S. Solomkin and D. H. Wittmann, Arch. Surg., 1993, 128, 79–88 CrossRef CAS PubMed.
- Y. Xi, J. Ge, M. Wang, M. Chen, W. Niu, W. Cheng, Y. Xue, C. Lin and B. Lei, ACS Nano, 2020, 14, 2904–2916 CrossRef CAS PubMed.
- J. C. Hernaiz-Leonardo, M. F. Golzarri, P. Cornejo-Juárez, P. Volkow, C. Velázquez, M. Ostrosky-Frid and D. Vilar-Compte, Am. J. Infect. Control, 2017, 45, 761–766 CrossRef PubMed.
- N. Madhusudhan, M. R. Pausan, B. Halwachs, M. Durdević, M. Windisch, J. Kehrmann, V. Patra, P. Wolf, P. Boukamp and C. Moissl-Eichinger, Cancers, 2020, 12, 541 CrossRef CAS PubMed.
- J. Kullander, O. Forslund and J. Dillner, Cancer Epidemiol. Prevent. Biomark., 2009, 18, 472–478 CrossRef CAS PubMed.
- B. Nardone, S. Nocadello, D. Massimino, T. Kiguradze, W. Anderson and D. West, J. Am. Acad. Dermatol., 2015, 72(5), AB130 Search PubMed.
- P. S. Hume, C. N. Bowman and K. S. Anseth, Biomaterials, 2011, 32, 6204–6212 CrossRef CAS PubMed.
- J. Liang, Z. Guo, A. Timmerman, D. Grijpma and A. Poot, Biomed. Mater., 2019, 14, 024102 CrossRef CAS PubMed.
- C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- K. J. Rambhia and P. X. Ma, J. Controlled Release, 2015, 219, 119–128 CrossRef CAS PubMed.
- A. Sabnis, A. S. Wadajkar, P. Aswath and K. T. Nguyen, Nanomedicine, 2009, 5, 305–315 CrossRef CAS PubMed.
- D. Gyawali, P. Nair, Y. Zhang, R. T. Tran, C. Zhang, M. Samchukov, M. Makarov, H. K. Kim and J. Yang, Biomaterials, 2010, 31, 9092–9105 CrossRef CAS PubMed.
- R. T. Tran, P. Thevenot, D. Gyawali, J. Chiao, L. Tang and J. Yang, Soft Matter, 2010, 6, 2449–2461 RSC.
- D. Gyawali, P. Nair, H. K. Kim and J. Yang, Biomater. Sci., 2013, 1, 52–64 RSC.
- R. Salihu, S. I. Abd Razak, N. A. Zawawi, M. R. A. Kadir, N. I. Ismail, N. Jusoh, M. R. Mohamad and N. H. M. Nayan, Eur. Polym. J., 2021, 110271 CrossRef CAS.
- R. Martínez-Zaguilán, E. A. Seftor, R. E. Seftor, Y. Chu, R. J. Gillies and M. J. Hendrix, Clin. Exp. Metastasis, 1996, 14, 176–186 CrossRef PubMed.
- Z. Boussadia, J. Lamberti, F. Mattei, E. Pizzi, R. Puglisi, C. Zanetti, L. Pasquini, F. Fratini, L. Fantozzi and F. Felicetti, J. Exp. Clin. Cancer Res., 2018, 37, 245 CrossRef PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- I. Böhme and A. K. Bosserhoff, Pigm. Cell Melanoma Res., 2016, 29, 508–523 CrossRef PubMed.
- A. Sun, X. He, L. Li, T. Li, Q. Liu, X. Zhou, X. Ji, W. Li and Z. Qian, NPG Asia Mater., 2020, 12, 1–11 CrossRef.
- A. Salomé Veiga and J. P. Schneider, Pept. Sci., 2013, 100, 637–644 CrossRef PubMed.
- K. Phogat and S. Bandyopadhyay-Ghosh, Cellulose, 2018, 25, 5821–5830 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed.
- J. Qu, X. Zhao, Y. Liang, Y. Xu, P. X. Ma and B. Guo, Chem. Eng. J., 2019, 362, 548–560 CrossRef CAS.
- A. Sabnis, M. Rahimi, C. Chapman and K. T. Nguyen, J. Biomed. Mater. Res., Part A, 2009, 91, 52–59 CrossRef PubMed.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- J. Huang and X. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 361–370 CrossRef CAS PubMed.
- S. Deshpande, S. Sharma, V. Koul and N. Singh, ACS Omega, 2017, 2, 6455–6463 CrossRef CAS PubMed.
- B. N. H. M. Neerooa, L. Ooi, K. Shameli, N. A. Dahlan, J. M. Islam, J. Pushpamalar and S. Teow, Gels, 2021, 7, 60 CrossRef CAS PubMed.
- S. Khanal, U. Adhikari, N. P. Rijal, S. R. Bhattarai, J. Sankar and N. Bhattarai, J. Funct. Biomater., 2016, 7, 21 CrossRef PubMed.
- J. U. Menon, A. Kuriakose, R. Iyer, E. Hernandez, L. Gandee, S. Zhang, M. Takahashi, Z. Zhang, D. Saha and K. T. Nguyen, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- S. Kalliola, E. Repo, V. Srivastava, F. Zhao, J. P. Heiskanen, J. A. Sirviö, H. Liimatainen and M. Sillanpää, Langmuir, 2018, 34, 2800–2806 CrossRef CAS PubMed.
- M. Toloudi, P. Apostolou and I. Papasotiriou, J. Cancer Ther., 2015, 6, 345 CrossRef.
- D. A. Greenhalgh and J. H. Parish, Br. J. Cancer, 1990, 61, 415–419 CrossRef CAS PubMed.
- B. Jawad, L. Poudel, R. Podgornik, N. F. Steinmetz and W. Ching, Phys. Chem. Chem. Phys., 2019, 21, 3877–3893 RSC.
- N. Chen, C. Wu, C. Chung, Y. Hwu, S. Cheng, C. Mou and L. Lo, PLoS One, 2012, 7, e44947 CrossRef CAS PubMed.
- F. Yang, S. S. Teves, C. J. Kemp and S. Henikoff, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1845, 84–89 CrossRef CAS PubMed.
- E. Boedtkjer and S. F. Pedersen, Annu. Rev. Physiol., 2020, 82, 103–126 CrossRef CAS PubMed.
- L. Nguyen, C. Hsu, H. Ye and Z. F. Cui, Biomed. Mater., 2020, 15, 055005 CrossRef CAS PubMed.
- S. Khan, M. U. Minhas, I. A. Tekko, R. F. Donnelly and R. R. S. Thakur, Drug Delivery Transl. Res., 2019, 9, 764–782 CrossRef CAS PubMed.
- L. S. Mun, M. M. Nadzir, S. R. Chowdhury and M. F. M. Busra, Int. J. Eng. Technol., 2018, 7, 1402–1404 CrossRef CAS.
- M. A. Wisniewska, J. G. Seland and W. Wang, J. Appl. Polym. Sci., 2018, 135, 46695 CrossRef.
- H. Salimi-Kenari, F. Mollaie, E. Dashtimoghadam, M. Imani and B. Nyström, Carbohydr. Polym., 2018, 181, 141–149 CrossRef CAS PubMed.
- M. Kim and C. Cha, Sci. Rep., 2018, 8, 1–12 Search PubMed.
- Z. Xie, N. V. Aphale, T. D. Kadapure, A. S. Wadajkar, S. Orr, D. Gyawali, G. Qian, K. T. Nguyen and J. Yang, J. Biomed. Mater. Res., Part A, 2015, 103, 3907–3918 CrossRef CAS PubMed.
- F. Ayhan and S. Özkan, Drug Delivery, 2007, 14, 433–439 CrossRef CAS PubMed.
- J. Fan, Z. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433–7443 RSC.
- V. Dinh, D. Nguyen, Q. Nguyen, T. Luu, T. H. Y. Pham, T. T. H. Vu, H. Chuang and H. Pham, J. Anal. Methods Chem., 2021, 2021 DOI:10.1155/2021/6613154.
- T. J. Lujan, K. M. Wirtz, C. S. Bahney, S. M. Madey, B. Johnstone and M. Bottlang, Tissue Eng., Part C, 2011, 17, 367–374 CrossRef CAS PubMed.
- Z. Stillman, B. M. Jarai, N. Raman, P. Patel and C. A. Fromen, Polym. Chem., 2020, 11, 568–580 RSC.
- C. Choipang, P. Chuysinuan, O. Suwantong, P. Ekabutr and P. Supaphol, J. Drug Delivery Sci. Technol., 2018, 47, 106–114 CrossRef CAS.
- H. Tang, H. Chen, B. Duan, A. Lu and L. Zhang, J. Mater. Sci., 2014, 49, 2235–2242 CrossRef CAS.
- M. Xiao, J. C. Hu and L. M. Zhang, Adv. Mater. Res., 2014, 1033, 352–356 Search PubMed.
- A. Dabbagh, R. Mahmoodian, B. J. J. Abdullah, H. Abdullah, M. Hamdi and N. H. Abu Kasim, Int. J. Hyperthermia, 2015, 31, 920–929 CrossRef CAS PubMed.
- Y. Kou, S. Wang, J. Luo, K. Sun, J. Zhang, Z. Tan and Q. Shi, J. Chem. Thermodyn., 2019, 128, 259–274 CrossRef CAS.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, Int. J. Mol. Sci., 2014, 15, 3640–3659 CrossRef CAS PubMed.
- A. Kalra, A. Lowe and A. M. Al-Jumaily, J. Mater. Sci. Eng., 2016, 5, 1000254 Search PubMed.
- X. Liu, K. Yang, M. Chang, X. Wang and J. Ren, Carbohydr. Polym., 2020, 240, 116289 CrossRef CAS PubMed.
- R. H. Patel, A. S. Wadajkar, N. L. Patel, V. C. Kavuri, K. T. Nguyen and H. Liu, J. Biomed. Opt., 2012, 17, 046003 CrossRef PubMed.
- J. U. Menon, S. Kona, A. S. Wadajkar, F. Desai, A. Vadla and K. T. Nguyen, J. Biomed. Mater. Res., Part A, 2012, 100, 1998–2005 CrossRef PubMed.
- H. Ohori, H. Yamakoshi, M. Tomizawa, M. Shibuya, Y. Kakudo, A. Takahashi, S. Takahashi, S. Kato, T. Suzuki and C. Ishioka, Mol. Cancer Ther., 2006, 5, 2563–2571 CrossRef CAS PubMed.
- T. T. Kwok and R. M. Sutherland, Int. J. Cancer, 1991, 49, 73–76 CrossRef CAS PubMed.
- J. L. Fiori, T. Zhu, M. P. O'Connell, K. S. Hoek, F. E. Indig, B. P. Frank, C. Morris, S. Kole, J. Hasskamp and G. Elias, Endocrinology, 2009, 150, 2551–2560 CrossRef CAS PubMed.
- B. Boone, K. Jacobs, L. Ferdinande, J. Taildeman, J. Lambert, M. Peeters, M. Bracke, P. Pauwels and L. Brochez, J. Cutan. Pathol., 2011, 38, 492–502 CrossRef PubMed.
- L. Wang, X. Li, T. Sun, Y. Tsou, H. Chen and X. Xu, Macromol. Biosci., 2018, 18, 1700325 CrossRef PubMed.
- B. Tao, C. Lin, Y. Deng, Z. Yuan, X. Shen, M. Chen, Y. He, Z. Peng, Y. Hu and K. Cai, J. Mater. Chem. B, 2019, 7, 2534–2548 RSC.
- L. Su, Z. Xie, Y. Zhang, K. T. Nguyen and J. Yang, Front. Bioeng. Biotechnol., 2014, 2, 23 RSC.
- C. M. Ferrer-Luque, M. T. Arias-Moliz, M. P. González-Rodríguez and P. Baca, J. Endod., 2010, 36, 1673–1675 CrossRef PubMed.
- O. Sarheed, A. Ahmed, D. Shouqair and J. Boateng, Wound Healing-New Insights into Ancient Challenges, ed. V. Alexandrescu, 2016, pp. 373–398 Search PubMed.
- V. Patrulea, G. Borchard and O. Jordan, Pharmaceutics, 2020, 12, 840 CrossRef CAS PubMed.
- M. Li, Q. Liu, Y. Teng, L. Ou, Y. Xi, S. Chen and G. Duan, Infect. Drug Resist., 2019, 12, 2853 CrossRef CAS PubMed.
- O. Campbell, J. Gagnon and J. E. Rubin, Lett. Appl. Microbiol., 2019, 69, 353–357 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01934a |
| This journal is © The Royal Society of Chemistry 2021 |