
Getting control of hydrogel networks with cross-linkable monomers†
Pamela G.
Cohn
 *a,
Sahar
Qavi
b,
Jasmine
Cubuk
a,
Mihir
Jani
a,
Mohamed Lamine
Megdad
a,
Dhvani
Shah
a,
Cara
Cattafi
a,
Panchatapa
Baul
a,
Shanthi
Rajaraman
a and
Reza
Foudazi
*bc
*a,
Sahar
Qavi
b,
Jasmine
Cubuk
a,
Mihir
Jani
a,
Mohamed Lamine
Megdad
a,
Dhvani
Shah
a,
Cara
Cattafi
a,
Panchatapa
Baul
a,
Shanthi
Rajaraman
a and
Reza
Foudazi
*bc
aChemistry Program, Stockton University, Galloway, NJ 08205, USA. E-mail: Pamela.Cohn@stockton.edu
bDepartment of Chemical and Materials Engineering, New Mexico State University, Las Cruces, NM 88003, USA
cSchool of Chemical, Biological and Materials Engineering, University of Oklahoma, Norman, OK 73019, USA. E-mail: rfoudazi@ou.edu
First published on 23rd September 2021
Abstract
The structure of a hydrogel network determines its ability to dissipate stress upon deformation, as well as its ability to swell in water. By designing systems with cross-linkable thiol groups in the monomers, radical thiol–ene chemistry was used to form controlled networks for acrylamide monomers. The use of radical thiol–ene chemistry effectively suppressed homo-polymerization of the bis(acrylamide) monomer and resulted in networks of alternating thiol and acrylamide monomers. Additionally, if the stoichiometry between the monomers is controlled, the network should approach that of ideality. In the case of bis(acrylamide) monomers, the incorporation of hydrogen-bond donors into the network creates a single network hydrogel with the benefits of high strength and ductility from the simultaneous incorporation of chemical and physical cross-links. Additionally, this strategy suppresses the formation of homo-polymerization in the acrylamide monomer to achieve an alternating network, which is supported with NMR characterization of base-digested fragments. For three different monomer compositions, the resulting gels had high compressive strength (up to 40 MPa) and tunable mechanical properties. The high mechanical strength of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, thiol:ene gel composition is due to the uniform distribution of cross-links, which creates defect-free networks for efficient stress transfer. The present one-pot synthetic strategy toward controlled gel networks affords monomer versatility and synthetic ease, as well as the potential for mechanically robust materials.
:1, thiol:ene gel composition is due to the uniform distribution of cross-links, which creates defect-free networks for efficient stress transfer. The present one-pot synthetic strategy toward controlled gel networks affords monomer versatility and synthetic ease, as well as the potential for mechanically robust materials.
Introduction
Hydrogels are crosslinked polymeric networks that swell in water, and they have use in a variety of applications such as tissue engineering, contact lenses, prostheses, and drug delivery.1–4 Current efforts aim to design gels that are ductile and/or strong for the development of artificial tissues like muscle or cartilage replacement materials. Most hydrogels are not mechanically strong or tough, and this is likely due to inefficient stress dissipation mechanisms within the material. During commonly used one-pot methods for polymerization and crosslinking to form gel networks, the crosslinks are randomized. This phenomenon leads to non-ideal gel networks with mechanical defects and does not allow for tunability of the mechanical properties.Within the last decade, several strategies have emerged for achieving hydrogels with high strength and/or toughness.5–9 Most of these strategies incorporate some variant of the sacrificial bond concept, wherein two different types of covalent and/or noncovalent bonds are incorporated into a gel network to produce a material that has high strength and/or ductility, and high fracture energy. This behavior has been attributed to the breaking of the sacrificial bonds at low stress before breaking the main-chain network at higher levels of applied stress. The types of sacrificial bonds employed depend on the type of gel network. The first tough hydrogels employed two mechanically dissimilar polymers that were entangled to form a double-network gel.10 At low levels of stress, the covalent bonds within the first polymer would break to relieve stress within the material (and no crack propagation resulted), and then at higher levels of stress, the second polymer would rupture and the material would fracture.10 Hybrid double network gels have been synthesized by Sun and coworkers that incorporated two types of crosslinked polymers: one ionically-crosslinked alginate and a covalently-crosslinked polyacrylamide.5 When stress was applied to the material, the ionic crosslinks would break first and leave the covalently crosslinked polyacrylamide chains intact. In this manner, cracks would not form until high levels of stress were applied.
With the goal of achieving a controlled and mechanically tunable gel network with efficient stress transfer, our research focused on incorporating the sacrificial bond concept into a single network material that possesses both covalent and noncovalent crosslinks. These crosslinks are based on two different monomers (one with hydrogen bond donors and the other with an excess of cross-linkable thiol groups) polymerized with thiol–ene chemistry to assemble the main chain polymers and crosslinks. This approach has been demonstrated with copper-mediated azide–alkyne cycloaddition (another click reaction) to assemble controlled gels for drug delivery,11 and more recently for tissue engineering.12 However, there are limited reports of using radical thiol–ene polymerization with acrylamide as a co-monomer to assemble a controlled gel network,13 and no reports of thermally initiated thiol–ene polymerizations with acrylamide as a co-monomer. The strategy would allow for greater control over the crosslinks in the network that minimize mechanical defects, as well as imparting other desirable properties like stimuli responsiveness in a controlled gel network. The precise placement of covalent and noncovalent crosslinks from the acrylamide monomer should allow for efficient stress transfer mechanisms within the network and results in hydrogels with tunable mechanical properties. A similar strategy has been employed with diacrylate and dimethacrylate monomers.14
Results and discussion
Synthesis
By controlling the stoichiometry between monomers, copolymers with a controllable amount of free thiol groups will form via radical thiol–ene polymerization. It was hypothesized that if the thiol–ene radical polymerization used to assemble the main chain of the copolymer was faster than homo-polymerization of monomers, the positions of the cross-linkable thiol groups would be fixed at precise positions to form a gel with controlled structure. The different gel compositions were named according to the stoichiometry of the functional groups in each monomer. The gel composition labeled 1:1 (thiol:ene) groups was predicted to produce an ideal network between pentaerythritol tetrakis(3-mercaptopropionate) (PTK) and N,N-methylene bis(acrylamide) (MBA) monomers (according to Fig. 1). The 1:1 (thiol:ene) network contains uniformly spaced chemical cross-links (blue lines) and physical cross-links from hydrogen bonding groups (green lines). The gel compositions labeled 2:1 and 4:1 (thiol:ene) were predicted to have non-ideal networks and less control over the network compared to the 1:1 composition. When the network contains an excess of free thiol groups in the main chain of the polymer, it can achieve high strength after cross-linking to form disulfide bonds or chain entanglements of the PTK units in a gel network. Such a material can lead to self-healing phenomena and stimuli responsiveness.
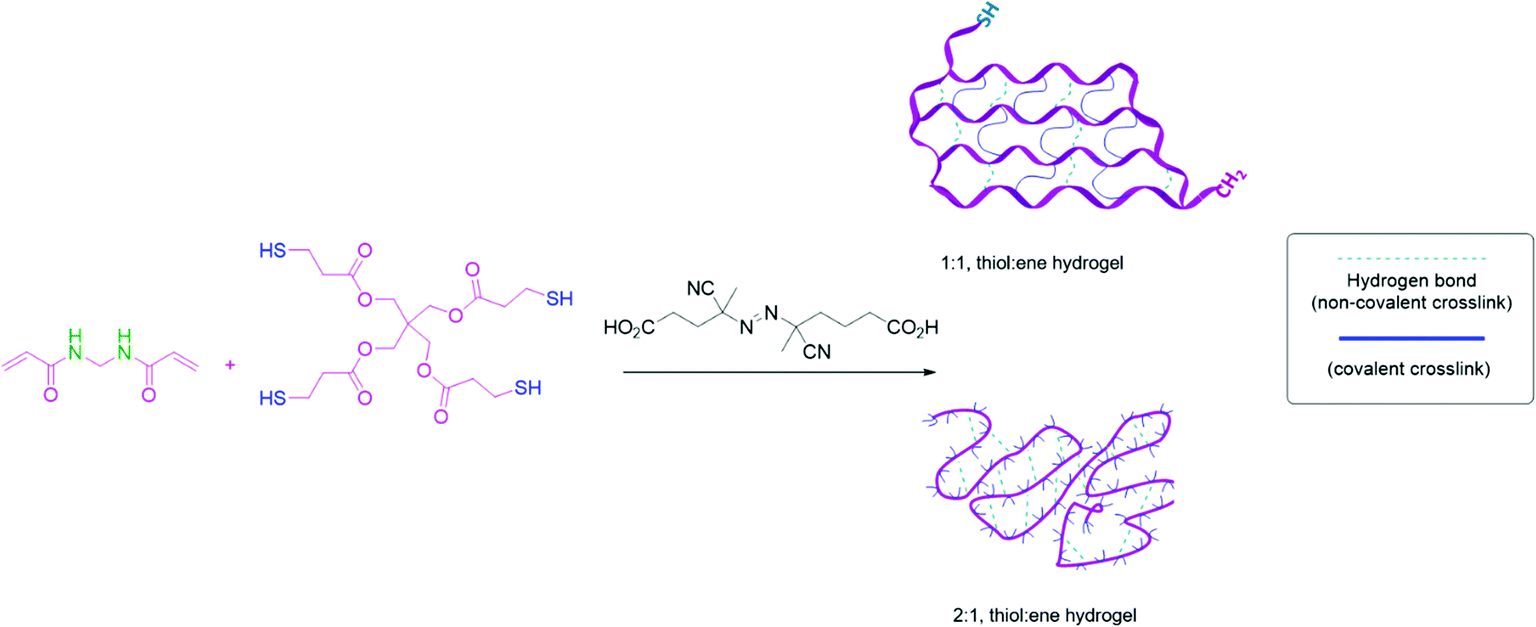 | ||
| Fig. 1 The copolymerization of N,N-methylene bis(acrylamide) with pentaerythritol tetrakis(3-mercaptopropionate) to form controlled and tunable hydrogels. | ||
The gels were synthesized via one-step polymerization and cross-linking procedure. Three different gels were synthesized that differed in their ratio of functional groups (thiol:ene). The procedure was a one-pot process that copolymerized a diene monomer N,N-methylene bis(acrylamide) (MBA) with a tetra thiol monomer pentaerythritol tetrakis(3-mercaptopropionate) (PTK). These reactions formed hydrogels without further crosslinking steps.
Evidence of gel network formation
The first evidence of gel network formation was provided by solubility tests for the gels after polymerization for the samples. The monomers were soluble in mixtures of methanol and dichloromethane (1:1, v/v), while the corresponding gels were only dispersible in these solvents. The gels were insoluble in all solvents tested.
Differential scanning calorimetry (DSC) analysis was performed on both gel compositions to determine if the solvent water molecules exhibited a freezing point depression. The DSC thermograms for the gels showed the crystallization exotherm occurred at (−15 °C) for the gels, which suggests water molecules are trapped in the gel network interior.15 Additionally, there is only one sharp peak in the heating cycle of the thermograms, which indicates one type of bound water in each gel network, as shown in the S2 in the ESI.†15 It should be noted that these curves exhibit super-cooling of water at the measured scan rate.
Additionally, infrared analysis of the dry and swollen samples for the 2:1 (thiol:ene) gel composition supports the existence of hydrogen bonding between water and the network (in S3 in the ESI†). The O–H stretch becomes significantly more pronounced upon swelling in water. This finding is consistent with the observation of one type of water state (bound) in the DSC thermogram.
Structural characterization of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1, thiol:ene gel networks
:1, thiol:ene gel networks
(1:1, thiol:ene)
In the 1H NMR spectrum of the sample of the 1:1 composition of thiol:ene groups, the vinyl end group is present in the spectrum at ∼5–6 ppm. The NMR solvent for the sample overlaps with the methylene protons of the MBA monomer; therefore, those protons cannot be distinguished (see ESI†). The NH protons from MBA are also missing in the spectrum, likely due to the base-catalyzed exchange of the amide protons with the deuterated solvent.17 In order to establish whether the initial gel network was composed of blocks of MBA monomer, two-dimensional NMR analysis with the gHMBC experiment shows the strongest evidence for no branching in the MBA portions of the network. The correlations between the protons labeled “A” and “B” at 2.42 and 2.16 ppm in Fig. 2B are consistent with the following thiol–ene adduct shown below. Additionally, the methylene proton at the “A” position correlates with the carbon at position “C” and the carbonyl carbon of the MBA monomer. The assignment of chemical shift for carbons at the “B” and “C” positions was made based on the proximity to the moderately deshielding sulfur atom. The spectrum also shows cross peaks for the methylene protons from MBA at 4.79 ppm that correlate with the amidic carbonyl carbon at 204.7 ppm.
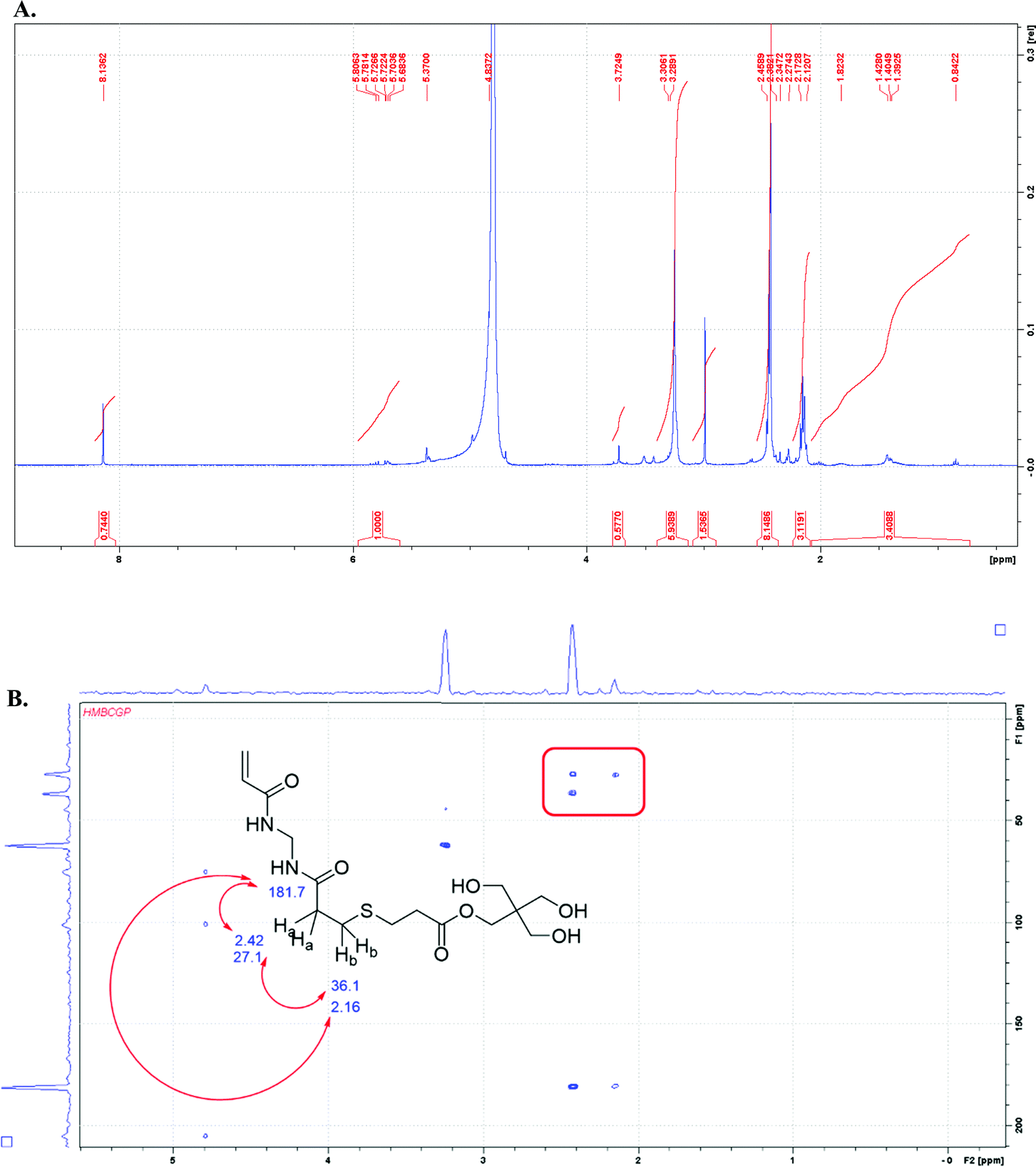 | ||
| Fig. 2 The NMR analysis for 1:1 (thiol:ene) gel after basic degradation with sodium hydroxide in deuterium oxide solvent (D2O, 400 MHz): (A) the 1H NMR spectrum of the hydrolysis product; (B) the gHMBC spectrum for the hydrolysis product with key correlations highlighted. | ||
Additionally, the diffusion coefficients from DOSY NMR analysis shed additional insight into the structure of the gel network.18 It was hypothesized that if MBA homo-polymerized prior to reacting with the thiol groups of PTK, there would be large blocks of unreactive (to hydroxide base) regions of the gel. Because only the ester groups of the PTK portions of the gel are base-labile, those portions of the network would fragment into smaller components. When subjected to a basic degradation, three different scenarios are possible for the products studied. In the first scenario, one could observe polymer fragments for blocks of MBA. Subsequently, one would observe two types of diffusion coefficients for small fragments from PTK and the large polymeric blocks for MBA.19 In the second scenario, if the original network is comprised of precisely arranged alternating PTK and MBA units, then only small fragments will result. Depending on the resolution of the spectrometer, one would see only one diffusion coefficient for both small fragments. In the third scenario, there might be a combination of the first two scenarios, where the product of a basic degradation in water would produce a mixture of MBA blocks, as well as tracts of alternating copolymer. This scenario would likely produce different sized fragments after a basic degradation from the tracts of MBA and alternating copolymer having their own unique reactivity.
To distinguish the three possible scenarios, a sample of the 1:1 gel was dissolved completely in a solution of sodium hydroxide in deuterium oxide (pH ∼ 10). For the 1:1 gel composition, the NMR spectrum from the DOSY experiment showed one new diffusion constant for the signal at δ 4.24. This is consistent with the presence of fragments of similar size, which would likely rule out the third scenario. For the 2:1 gel composition, the DOSY experiment showed one new diffusion coefficient at δ 3.45 that was larger than the signal for the 1:1 composition (see S1B in the ESI†). This suggests that the network structure was different between the two gel samples. For this gel composition, it is unclear how many components comprise the signal at −9.7 (log (m2 s−1)) because the signal is very broad.
To distinguish the first and second scenarios, the molecular weight of the resulting fragments was approximated with a calibration curve of polystyrene standard compounds. The solution in basic deuterium oxide was extracted with deuterated chloroform, so the diffusion NMR spectrum could be compared with polystyrene standards diffusion NMR spectra in the same solvent. From the Stokes–Einstein relationship between the diffusion coefficients and the hydrodynamic radius of a polymer, the value of the diffusion coefficient is consistent with a small-molecular fragment, as shown in Fig. 3. The fragmented product has a diffusion coefficient similar to the solvent (deuterium oxide); therefore, the two compounds have a similar sized hydrodynamic radius.
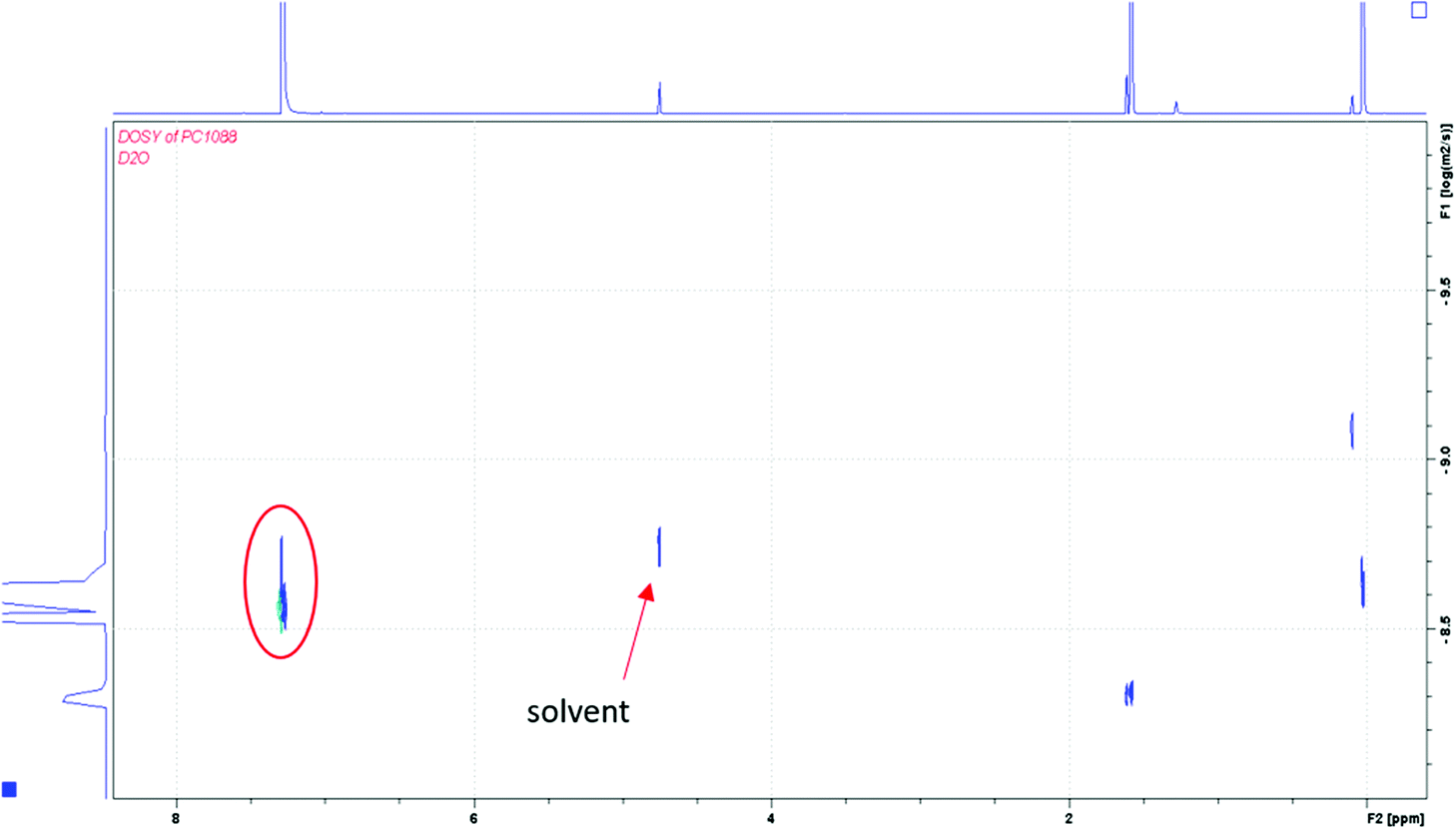 | ||
| Fig. 3 The DOSY NMR (D2O, 400 MHz) spectrum of the 1:1 (thiol:ene) gel after a basic degradation in deuterium oxide. The signal at ∼7.5 ppm suggests the presence of only small fragments in the system. | ||
After extraction of the sample with CDCl3, another DOSY NMR spectrum was obtained. A standard curve analysis in CDCl3 for the DOSY NMR spectrum confirmed the diffusion coefficient has Mw < 1000 g mole−1 (see ESI†). When considering the analysis from gHMBC NMR data, there must be incorporation of PTK into the fragments analyzed and there are no large blocks of polymerized MBA. Therefore, the diffusion data is consistent with a small molecular weight fragments that originated from a larger controlled network. Thus, homo-polymerization of MBA is suppressed in the network. However, this experiment does not rule out other types of homo-polymerization or cross-linking between the thiol groups of PTK units to form disulfide bonds. Disulfide bonds are also base-labile and would produce small fragments as well. However, there are reports of photo-initiated thiol–ene polymerization chemistry with anhydrides that do not show evidence of homo-polymerization of PTK.20 These reports are consistent with an alternating monomeric network in our analogous system.
Rheological measurements
In order to understand the rheo-mechanical behavior of the gels, the three compositions were characterized with frequency sweep measurements. The storage and loss moduli, G′ and G′′ in Fig. 4A, show the elastic and viscous response of the hydrogels, respectively. The elastic modulus of both the 1:1 and 2:1 (thiol:ene) samples shows a plateau in a wide range of angular frequency and confirms a highly solid-like behavior of the hydrogels. The storage modulus for the 1:1 (thiol:ene) composition is higher than other samples because by carefully controlling the stoichiometry of the monomers and positioning of crosslinks, the gel structure approaches that of an ideal network polymer. The ideal network should be relatively defect-free with minimum physical chain entanglements. Additionally, an ideal network allows for efficient stress dissipation and high strength.6 The 4:1 (thiol:ene) sample does not result in a stable hydrogel and the storage modulus increases with angular frequency.
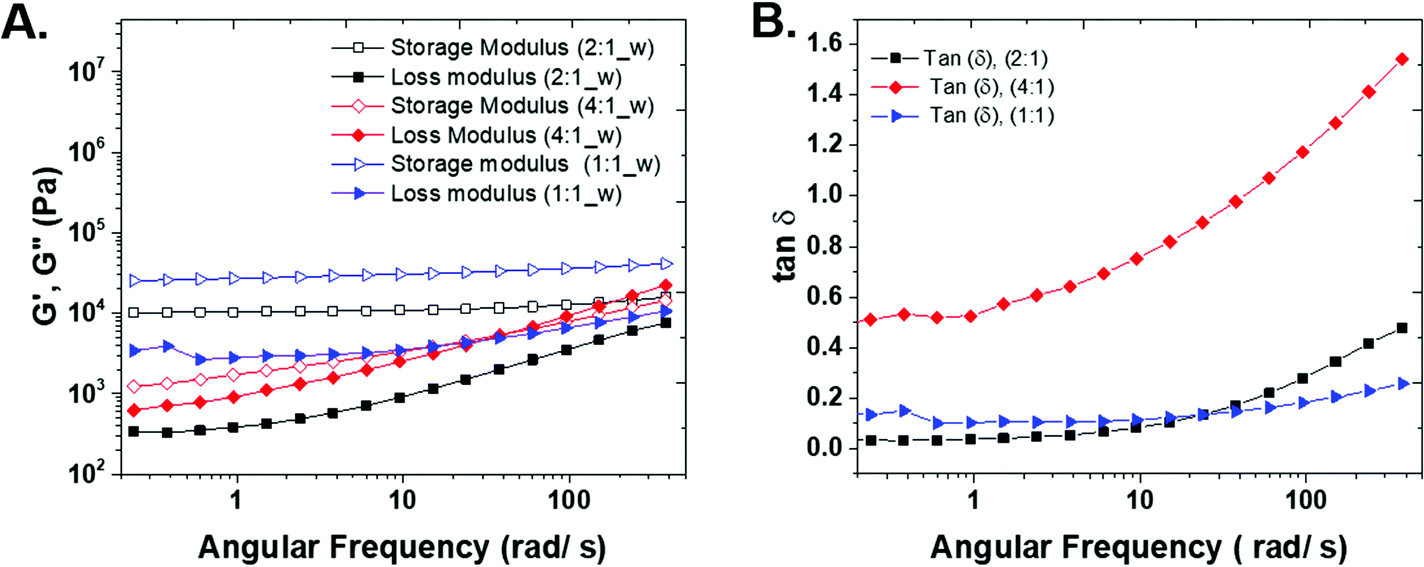 | ||
| Fig. 4 (A) Comparative storage and loss moduli for representative samples from each gel composition of MBA and PTK with respect to thiol:ene functionality from frequency sweep measurements; (B) plot of tan (δ) as a function of angular frequency. | ||
The loss modulus shows the viscous contribution of each sample. For 1:1 sample, there is a shallow minimum in the G′′ plot. The minimum in loss modulus shows the presence of two relaxation behaviors in the system and the transition from α-relaxation (long time, related to low frequency) to β-relaxation (short time, high frequency).21 We believe the minimum in G′′ is present in 2:1 and 4:1 samples and would occur in lower frequencies than the measured range in this study. As the gel becomes stronger, the characteristic minimum frequency for relaxation crossover shift to higher frequencies, because smaller segments connect the network together.22,23
There is a crossover between G′ and G′′ in 4:1 sample that shows the transition from solid-like (at low angular frequencies) to liquid-like behavior (at high angular frequencies).
The tangent of the phase angle (Fig. 4B) is calculated as follows:
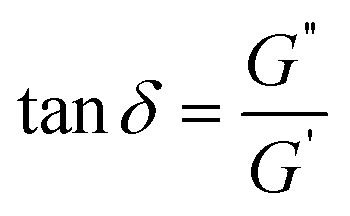 | (1) |
δ for 4:1 sample is higher than that of 2:1 and 1:1 samples, indicating the sample is close to the gel point, and a three-dimensional network has not been formed. For 1:1 and 1:2 samples, the tanδ values approach zero in all frequencies that confirm the solid-like behavior of this samples (see S5 in ESI†).
Fig. 5 shows the frequency sweep results of 2:1 and 4:1 samples swollen in chloroform. Hydrogel samples show a plateau in G′ versus frequency curve that indicates high stability of 3-D crosslinked networks. In order to discuss the difference between the order of magnitude of G′ in water (Fig. 5) and chloroform swollen hydrogels, we should keep in mind that there are two types of crosslinking involved in the mechanical properties of hydrogels, chemical crosslinks and physical crosslinks. The latter is a result of hydrogen bonding between chains.
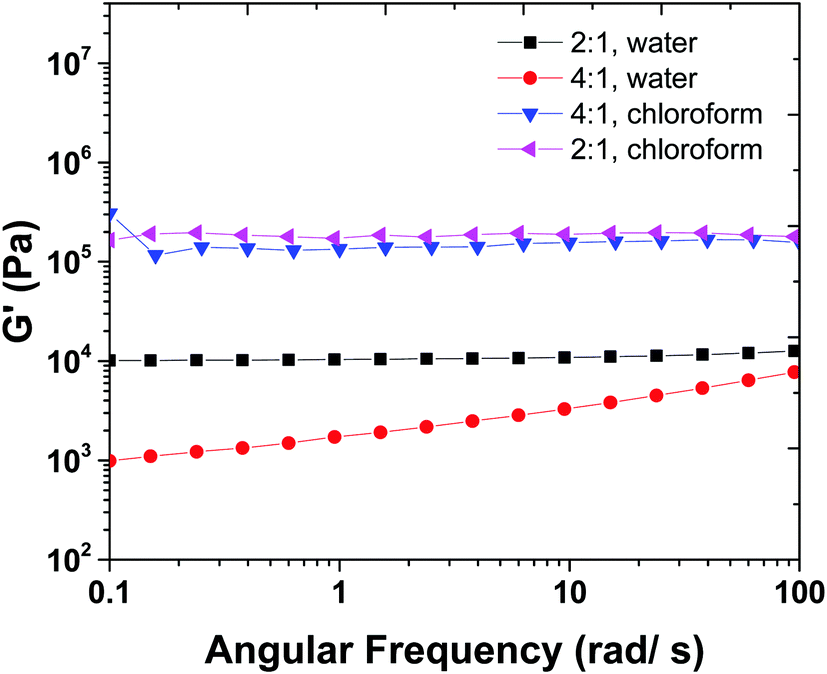 | ||
| Fig. 5 Frequency sweep measurements for gels in water and chloroform allow for physical and chemical crosslink density to be calculated. | ||
The 2:1 and 4:1 samples (thiol:ene) were swollen in water and chloroform solvents. The mechanical properties of 4:1 (thiol:ene) sample in water are not high enough and it is close to the chemical gel point with low mechanical properties. When swollen in water, physical networks dissociate, and hydrogel has lower crosslink density and mechanical properties. In contrast, when the hydrogels are swollen in chloroform, in addition to chemical crosslinks, physical crosslinks can exist between the amidic hydrogens (as confirmed from FTIR, see ESI,† S3) in the MBA fragments.
The cross-link density for the 1:1, thiol:ene network was calculated from the theory of rubber elasticity to quantify the number of crosslinks, as follows:
| G = nRT | (2) |
:1 and 4:1, thiol:ene) gel compositions because of the presence of topological defects in those materials, such as dangling chain ends and loops from an excess of PTK in the network. These network defects are reflected in the equilibrium swelling measurement data in the ESI† S6. A similar discrepancy between the rheological and swelling ratio data has been reported for hemicellulose gels.24 In the present work, the excess of PTK in the gel composition leads to an increase in the swelling capacity in water. This would suggest the gel network is more flexible and is consistent with the smaller elastic modulus from rheological measurements when compared with the ideal network 1:1 (thiol:ene).
Stress–strain curves-discussion
The 1:1 (thiol:ene) hydrogel composition sustains more stress compared to the 2:1 composition, as shown in Fig. 6. Thus, the mechanical properties of the 1:1 (thiol:ene) sample are higher than that of 2:1 (thiol:ene) sample. This result agrees with the rheological data, as the storage modulus of sample with the 1:1 (thiol:ene) composition was higher than that of the sample with 2:1 (thiol:ene) composition. We believe that the network in the 2:1 composition is less ideal than that of the 1:1 composition. As discussed before, the ideal network structure allows for efficient stress dissipation and high strength.6 The 2:1 sample has free branches, which makes it more flexible than the 1:1 sample. Therefore, the 2:1 sample has a higher strain at break. The 1:1 ratio has an ideal network, which provides a higher stress at break than the 2:1 sample. The area under compressive stress–strain curve is attributed to the compressive toughness of the hydrogels, as shown in Table 1, along with some hydrogels from the literature. Sample 4:1 does not provide a robust gel and therefore the mechanical strength results are not reported for this composition. These results show that the 2:1 gel's compressive toughness is higher than that of the 1:1 sample, which indicates that having a more flexible (non-ideal) network can provide higher toughness. In this case, the role of topological defects might also be advantageous for mechanical properties when dangling chain ends become entangled.
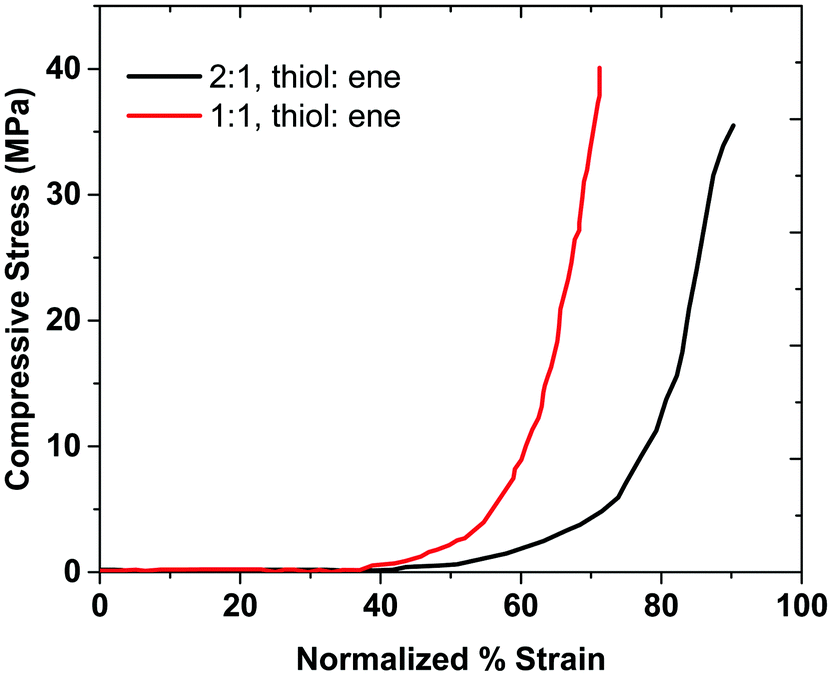 | ||
| Fig. 6 The compressive stress–strain curves for the hydrogels. | ||
| Hydrogel | Compressive toughness (MPa) |
|---|---|
| 2:1 (thiol:ene) |
509.5 ± 32.9 |
| 1:1 (thiol:ene) |
413.0 ± 51.5 |
| Macromolecular microsphere composite (MMC) hydrogel25 | 34.4 |
| Chitin whiskers modified gelatin hydrogels26 | 0.3 |
| Cellulose modified polyNIPAM hydrogels27 | 0.6 |
| Polyurethane modified polyacrylamide hydrogels28 | 0.1 |
The synthesized hydrogels have toughness values greater than the literature for conventional hydrogels, as shown in Table 1. The results show that the 2:1 gel has higher compressive toughness than that of the 1:1 sample, which indicates that having a more flexible (non-ideal) network can provide higher toughness. These measurements support the value of incorporating monomers with hydrogen-bond donating groups into hydrogel networks as a way to access materials with a range of toughness values.
Conclusion
The work described detailed efforts toward the synthesis and characterization of controlled gel networks with tunable mechanical properties. By incorporating cross-linkable groups into the monomers, thiol–ene chemistry provides access to a range of materials with high compressive strength and tunable rheo-mechanical properties. The relationship between the spatial connectivity afforded by thiol–ene polymerization chemistry and the network structure needs to be further investigated. Our results suggest that the ideal network has a higher mechanical strength, whereas the non-ideal network with topological defects provides higher toughness. Future work in this area will expand the approach to produce structure–mechanical property studies on a range of hydrogels with varying monomer compositions. Additionally, by incorporating acrylamide monomers with thiol–ene chemistry, the combination of physical and chemical crosslinking might also allow for self-healing behavior in these hydrogels.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge Nathan Schramm (Stockton University) and project funding provided by the Provost's Faculty Opportunity Fund at Stockton University. PGC would also like to acknowledge and thank the Research and Engineering Apprenticeship Program of the Army Educational Outreach Program for sponsoring an internship for PB, as well as Dr. Qiong Wu (University of Illinois at Urbana-Champaign) for helpful discussions in the NMR analysis.References
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. I. Chung, Design and Fabrication of a High-Strength Hydrogel with Ideally Homogeneous Network Structure from Tetrahedron-like Macromonomers, Macromolecules, 2008, 41(14), 5379–5384, DOI:10.1021/ma800476x.
- M.-G. Kim, Y. Shon, W. Miao, J. Lee and Y.-K. Oh, Biodegradable Graphene Oxide and Polyaptamer DNA Hybrid Hydrogels for Implantable Drug Delivery, Carbon, 2016, 105, 14–22, DOI:10.1016/j.carbon.2016.04.014.
- H. H. Lee-Wang, I. Blakey, T. V. Chirila, H. Peng, F. Rasoul, A. K. Whittaker and B. L. Dargaville, Novel Supramolecular Hydrogels as Artificial Vitreous Substitutes, Macromol. Symp., 2010, 296, 229–232, DOI:10.1002/masy.201051032.
- L. A. Sharpe, A. M. Daily, S. D. Horava and N. A. Peppas, Therapeutic Applications of Hydrogels in Oral Drug Delivery, Expert Opin. Drug Delivery, 2014, 11(6), 901–915, DOI:10.1517/17425247.2014.902047.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Highly Stretchable and Tough Hydrogels, Nature, 2012, 489(7414), 133–136, DOI:10.1038/nature11409.
- M. Shibayama, Structure-Mechanical Property Relationship of Tough Hydrogels, Soft Matter, 2012, 8(31), 8030, 10.1039/c2sm25325a.
- Y. Sun, G. Gao, G. Du, Y. Cheng and J. Fu, Super Tough, Ultrastretchable, and Thermoresponsive Hydrogels with Functionalized Triblock Copolymer Micelles as Macro-Cross-Linkers, ACS Macro Lett., 2014, 3(5), 496–500, DOI:10.1021/mz500221j.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Tough and Self-Healing Hydrogels Formed via Hydrophobic Interactions, Macromolecules, 2011, 44(12), 4997–5005, DOI:10.1021/ma200579v.
- Q. Chen, D. Wei, H. Chen, L. Zhu, C. Jiao, G. Liu, L. Huang, J. Yang, L. Wang and J. Zheng, Simultaneous Enhancement of Stiffness and Toughness in Hybrid Double-Network Hydrogels via the First, Physically Linked Network, Macromolecules, 2015, 48(21), 8003–8010, DOI:10.1021/acs.macromol.5b01938.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, Materials Chemistry B Fundamentals of Double Network Hydrogels, J. Mater. Chem. B, 2015, 3(3), 3645–3886, 10.1039/C5TB00123D.
- J. Xu, E. Feng and J. Song, Bioorthogonally Cross-Linked Hydrogel Network with Precisely Controlled Disintegration Time over a Broad Range, J. Am. Chem. Soc., 2014, 136(11), 4105–4108, DOI:10.1021/ja4130862.
- Z. Qiao, Z. Wang, C. Zhang, S. Yuan, Y. Zhu and J. Wang, PVAm–PIP/PS Composite Membrane with High Performance for CO2/N2 Separation, AIChE J., 2012, 59(4), 215–228, DOI:10.1002/aic.
- B. J. Sparks, E. F. T. Hoff, L. P. Hayes and D. L. Patton, Mussel-Inspired Thiol-Ene Polymer Networks: influencing Network Properties and Adhesion with Catechol Functionality, Chem. Mater., 2012, 24(18), 3633–3642, DOI:10.1021/cm302301e.
- K. A. Fredriksen, T. E. Kristensen and T. Hansen, Combined Bead Polymerization and Cinchona Organocatalyst Immobilization by Thiol-Ene Addition, Beilstein J. Org. Chem., 2012, 8, 1126–1133, DOI:10.3762/bjoc.8.125.
- P. Yang, Thermal Analysis to Determine Various Forms of Water Present in Hydrogels, 2014, pp. 1–4.
- F. Halverson, J. E. Lancaster and M. N. O’Connor, Sequence Distribution of Carboxyl Groups in Hydrolyzed Polyacrylamide, Macromolecules, 1985, 18(6), 1139–1144, DOI:10.1021/ma00148a016.
- Y. Z. Zhang, Y. Paterson and H. Roder, Rapid Amide Proton Exchange Rates in Peptides and Proteins Measured by Solvent Quenching and Two-Dimensional NMR, Protein Sci., 1995, 4(4), 804–814 CrossRef CAS PubMed.
- W. Li, H. Chung, C. Daeffler, J. A. Johnson and R. H. Grubbs, Application of 1 H DOSY for Facile Measurement of Polymer Molecular Weights, Macromolecules, 2012, 45(24), 9595–9603, DOI:10.1021/ma301666x.
- S. A. Bencherif, J. A. Sheehan, J. O. Hollinger, L. M. Walker, K. Matyjaszewski and N. R. Washburn, Influence of Cross-Linker Chemistry on Release Kinetics of PEG-Co-PGA Hydrogels, J. Biomed. Mater. Res., Part A, 2009, 90(1), 142–153, DOI:10.1002/jbm.a.32069.
- K. L. Poetz, H. S. Mohammed, B. L. Snyder, G. Liddil, D. S. K. Samways and D. A. Shipp, Photopolymerized Cross-Linked Thiol-Ene Polyanhydrides: erosion, Release, and Toxicity Studies, Biomacromolecules, 2014, 15(7), 2573–2582, DOI:10.1021/bm500420q.
- S. Qavi and R. Foudazi, Rheological Characteristics of Mesophases of Block Copolymer Solutions, Rheol. Acta, 2019, 58(8), 483–498, DOI:10.1007/s00397-019-01162-y.
- Z. Emami, M. Ehsani, M. Zandi and R. Foudazi, Controlling Alginate Oxidation Conditions for Making Alginate-Gelatin Hydrogels, Carbohydr. Polym., 2018, 198, 509–517, DOI:10.1016/j.carbpol.2018.06.080.
- Z. Emami, M. Ehsani, M. Zandi and R. Foudazi, Corrigendum to “Controlling Alginate Oxidation Conditions for Making Alginate-Gelatin Hydrogels” [Carbohydr. Polym. 198 (2018) 509–517], Carbohydr. Polym., 2019, 208, 200, DOI:10.1016/j.carbpol.2018.12.038.
- L. Maleki, U. Edlund and A. C. Albertsson, Synthesis of Full Interpenetrating Hemicellulose Hydrogel Networks, Carbohydr. Polym., 2017, 170, 254–263, DOI:10.1016/j.carbpol.2017.04.091.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, A Novel Hydrogel with High Mechanical Strength: a Macromolecular Microsphere Composite Hydrogel, Adv. Mater., 2007, 19(12), 1622–1626, DOI:10.1002/adma.200602533.
- S. Ge, Q. Liu, M. Li, J. Liu, H. Lu, F. Li, S. Zhang, Q. Sun and L. Xiong, Enhanced Mechanical Properties and Gelling Ability of Gelatin Hydrogels Reinforced with Chitin Whiskers, Food Hydrocolloids, 2018, 75, 1–12, DOI:10.1016/j.foodhyd.2017.09.023.
- J. Wei, Y. Chen, H. Liu, C. Du, H. Yu and Z. Zhou, Thermo-Responsive and Compression Properties of TEMPO-Oxidized Cellulose Nanofiber-Modified PNIPAm Hydrogels, Carbohydr. Polym., 2016, 147, 201–207, DOI:10.1016/j.carbpol.2016.04.015.
- L. Wu, G. Mao, G. Nian, Y. Xiang, J. Qian and S. Qu, Mechanical Characterization and Modeling of Sponge-Reinforced Hydrogel Composites under Compression, Soft Matter, 2018, 14(21), 4355–4363, 10.1039/C8SM00678D.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb00482d |
| This journal is © The Royal Society of Chemistry 2021 |