
Seawater electrocatalysis: activity and selectivity
Sakila
Khatun
ab,
Harish
Hirani
ac and
Poulomi
Roy
 *ab
*ab
aCSIR – Central Mechanical Engineering Research Institute (CMERI), Mahatma Gandhi Avenue, Durgapur 713209, West Bengal, India. E-mail: poulomiroy@yahoo.com; p.roy@cmeri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, Uttar Pradesh, India
cDepartment of Mechanical Engineering, Indian Institute of Technology Delhi, New Delhi 110016, India
First published on 14th November 2020
Abstract
Seawater is considered to be a major hydrogen reservoir. However, the presence of multielements in seawater and their interference in electrochemistry, especially the chlorine chemistry, makes the electrocatalytic water splitting of seawater very challenging and still not completely understandable. To make seawater electrolysis sustainable, the activity of electrocatalysts may not be the only parameter, but the selectivity of the efficient oxygen evolution reaction suppressing the corrosive chlorine chemistry is highly desirable. Thereby, the current review not only focuses on fundamentals to understand the mechanisms involved in the anode and cathode, but also discusses different electrocatalysts, factors affecting their performance, and finally the rational design of electrolyzers finding the possibilities towards commercialization.
 Sakila Khatun | Sakila Khatun received her BSc (2011–2014) and MSc (2014–2016) degrees in Physics from the University of Kalyani, West Bengal, India. She qualified SET (State Level Eligibility Test), NET (National Level Eligibility Test) with JRF (Junior Research Fellowship) from UGC, Delhi. Currently, she is pursuing her PhD degree under the supervision of Dr Poulomi Roy at CSIR-CMERI, and her research mainly focuses on mixed metal oxide based nanostructures in electrocatalytic water splitting for hydrogen production. |
 Harish Hirani | Prof. Harish Hirani is presently the Director of the CSIR-Central Mechanical Engineering Research Institute, Durgapur since 2016. Previously, he served as a Professor at IIT Delhi. For his research achievements, Prof. Hirani received the prestigious “BOYSCAST fellowship” at MIT, USA. Having extensive knowledge in Materials and Mechanical Engineering, his R&D focuses more on the Green Technology realizing substantial socio-economic impact. He has a long list of 150+ research publications and 20+ IPR to his credit. His book on “Fundamentals of Engineering Tribology with Applications” has proven to be a success. |
 Poulomi Roy | Dr Poulomi Roy is a Senior Scientist at CSIR – Central Mechanical Engineering Research Institute. Dr Roy is a Fulbright Fellow under Fulbright-Nehru Academic and Professional Excellence Award 2016–2017, and visited the University of Wisconsin – Madison, USA. Dr Roy obtained her PhD in Inorganic Nanomaterials from the Indian Institute of Technology Kharagpur, India in 2007. She spent 2008–2011 at the Friedrich–Alexander-Universität Erlangen-Nürnberg, Germany, as a postdoctoral research fellow. Her research interests comprise nanoscale materials and their applications in energy conversion and storage devices. She has published more than 50 research articles in various high impact international journals to her credit. |
1. Introduction
In the era of exponentially raising global energy demand, the limited availability of fossil fuels and environmental pollution associated with it brings us a major challenge. To address this, hydrogen technologies may play a major role in satisfying the electricity demand with zero carbon emission. The recent international modeling study indicates the fact that electricity production must involve decarbonization by 2020, and must continue in the coming years to establish the balance in the carbon footprint.1 Hydrogen gas, owing to its high gravimetric energy density (142 MJ kg−1 at 25 °C), is considered to be a potential fuel for the future.2,3 The concept of hydrogen economy was proposed long ago in 1972 by the electrolysis of water.4,5 However, the current status for hydrogen production indicates that fossil fuels, like natural gas and coal or biomass gasification, are still the primary source for hydrogen production and only 4% of hydrogen production occurs from water electrolysis.5 The low production percentage from water electrolysis is due to the low system efficiency of 70% involving slow electrochemical kinetics.2 Such a low system efficiency makes the water electrolysis process expensive and challenging for commercialization.6 The process can be made practical not only by choosing the cost-effective electrocatalysts, but also by using impure water or seawater, knowing the scarcity of usable fresh water with growing population. It is a well-known fact that 97% of the earth surface water is seawater, which can be recommended for use in electrolyzers. However, the presence of salts up to 3.5% in seawater composed of different elements, as shown in Fig. 1, and their involvement in various competitive electrochemical reactions create much complexity, limiting the efficiency of electrolysis.7 Therefore, it is very important to understand all the possible electrochemical reactions in seawater, and to carry out selective electrochemical reactions facilitating hydrogen production. It is needless to say that the process is quite difficult and challenging. Even though seawater electrolysis was pioneered by Trasatti in 1984,8 very few studies have been reported so far. Knowing these facts and the importance of the field, the present review focuses not only on the possible electrochemistry involved in the process, but also provides thermodynamic insights identifying the possibilities on the selective electrocatalysis and gaps towards the industrial commercialization of the process.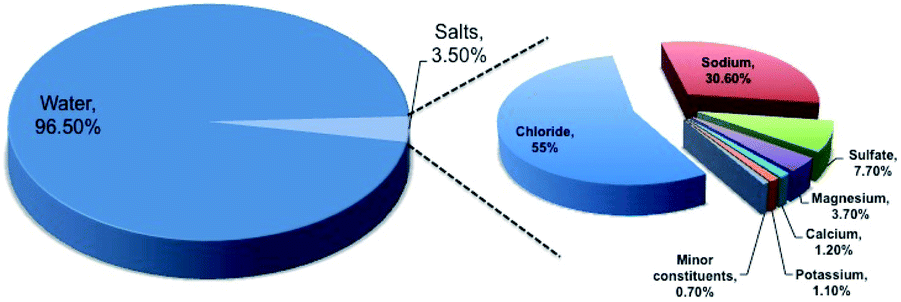 | ||
| Fig. 1 Elementary distribution of seawater. The percentages have been calculated in wt/wt unit. | ||
2. Fundamental aspects
Reaction mechanism
The splitting of fresh water into hydrogen and oxygen is a thermodynamically uphill process with a positive Gibbs free energy (ΔG) value of +237.2 kJ mol−1 for one mole of water. This indicates that a thermodynamic electrical potential of 1.23 V is required for reversible electrocatalysis. However, in reality, an extra cell voltage of over 1.23 V, known as the overpotential, is necessary to carry out the overall water splitting involving cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER) to make the overall reaction reasonably progressive.9 Amongst OER and HER, OER is known to be a kinetically sluggish reaction involving multi-electron transfer, which also depends on the nature of the electrolyte solution. The possible cathodic and anodic reactions for fresh water in alkaline and acidic medium are described in eqn (1)–(4).In alkaline media
| Anode: 4OH− = 2H2O + O2 + 4e−E0a = +0.4 V | (1) |
| Cathode: 2H2O + 2e− = 2OH− + H2E0c = −0.83 V | (2) |
In acidic media
| Anode: 2H2O = 4H+ + O2 +4e−E0a = +1.23 V | (3) |
| Cathode: 2H+ + 2e− = H2E0c = 0 V | (4) |
In alkaline media, the kinetics for OER are more favourable due to the availability of hydroxyl ions in the solution, while the HER mechanism struggles.
However, the electrochemical scenario for seawater electrolysis will be different with the interference of many side reactions. In his recent review article, Strasser listed the possible redox reactions with the species present in the seawater.10 The presence of chloride in the form of NaCl is the major concern, which mostly interfere, causing the chlorine evolution reaction (CER) and compete with the OER at the anode. Indeed, the reactions at the anode will depend upon the pH of the electrolyte, as per the Pourbaix diagram of chlorine, represented in Fig. 2.
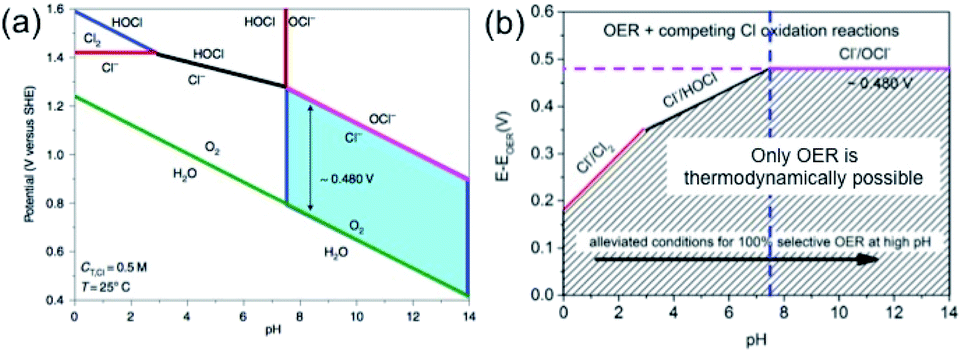 | ||
| Fig. 2 (a) Pourbaix diagram for artificial seawater with 0.5 M NaCl aqueous solution representing the oxygen and chlorine systems. The green line indicates the thermodynamic equilibrium between H2O/O2, and the red line in the alkaline side shows the equilibrium between Cl−/OCl− and in acidic side Cl−/Cl2. The blue filled area indicates a fixed potential difference of 480 mV at pH > 7. The black and blue lines in the acidic side show the equilibrium of Cl−/HOCl and Cl2/HOCl, respectively. (b) Representation, derived from the Pourbaix diagram, showing the overpotential ranges for OER and other chlorine oxidation reactions at different pH values. Reproduced with permission from ref. 11. | ||
In acid media
| 2Cl− → Cl2 + 2e−E0 = +1.36 V | (5) |
In alkaline media
| Cl− + 2OH− → ClO− + H2O + 2e−E0 = +0.89 V | (6) |
Amongst other species present in seawater, the redox potential of the bromide–bromine (1.331 V) conversion is very close to the chloride–chlorine redox reaction, giving a high possibility of interference. However, the probability can be ignored on the practical level, due to its presence in trace amounts in seawater. Considering the other species to be less problematic based on their redox potential values, one must compare the redox reactions of the OER and CER depending on the pH of the medium. Eqn (5) and (6) indicate that while in acid media, chlorine evolution occurs, in the alkaline media, chloride is converted into the hypochlorite species. It is important to note that in both cases, the redox reactions involve two electron transfers and the formation of a single intermediate, which will be discussed in further detail later. The involvement of a four-electron transfer in OER, as stated in eqn (1) and (3), thereby indicates faster kinetics of CER than OER. However, the Pourbaix diagram (Fig. 2(a)) reveals OER as a thermodynamically much more favorable mechanism than CER. It can be noted that while at alkaline pH, an electrode potential difference between the OER and CER is found to be 0.480 V. Conversely, in acidic pH, the difference decreases. This suggests that an overpotential value of less than 0.480 V needs to be maintained in order to generate O2 by OER, avoiding hypochlorite formation by CER in alkaline media. However, the seawater oxidation becomes challenging in acidic medium, especially in the range of pH < 3, where the overpotential window becomes 180–350 mV for making OER favorable, as shown in Fig. 2(b).11
In order to understand the selectivity of the electrocatalyst for OER and CER, critical thermodynamic and kinetic assessments are necessary. Considering RuO2 for OER and Pt for HER as conventional electrocatalysts, Over and his group12 have carried out extensive studies in determining the free energies, and identifying the transition states (TS) and reaction intermediates (RI) during OER or CER and HER in alkaline or acidic medium on the single crystal models RuO2(110) and Pt(111). The single crystalline RuO2(110) is composed of uncoordinated Ru sites (Rucus) readily capped with the on-top oxygen (Oot) and bridging oxygen Obr, partially saturated by hydrogen as OHbr, depending on the overpotential as represented in Fig. 3(a).13,14 The Pourbaix diagram, showing OER and CER over a pH range in Fig. 3(a), indicates that the O-caped RuO2 surface is stable over a wide potential and pH range. To be specific, the diagram shows that above 1.36 V, both OER and CER may take place. Above 1.49 V at pH < 4, chlorine adsorption on Oot can be preferred, making CER mechanism favourable.15 The RuO2 surface with (1Obr1OHbr + 2Oot) combination is recognized as the thermodynamically most stable phase for CER reaction. The CER reaction can be explained by three mechanisms: Volmer–Tafel, Volmer–Heyrovsky, and Krishtalik mechanism, among which the Volmer–Heyrovsky mechanism is mostly favored. The mechanism involves the adsorption and discharge of the chloride ion (Volmer step) to form Ru–OotCl as the reaction intermediate, followed by the recombination of adsorbed chlorine with free chloride (Heyrovsky step), and thereby release of Cl2 as described in eqn (7) and (8).
| Ru–Oot + 2Cl− → Ru–OotCl + e− + Cl− (Volmer step) | (7) |
| Ru–OotCl + Cl− → Ru–Oot + Cl2 + e− (Heyrovsky step) | (8) |
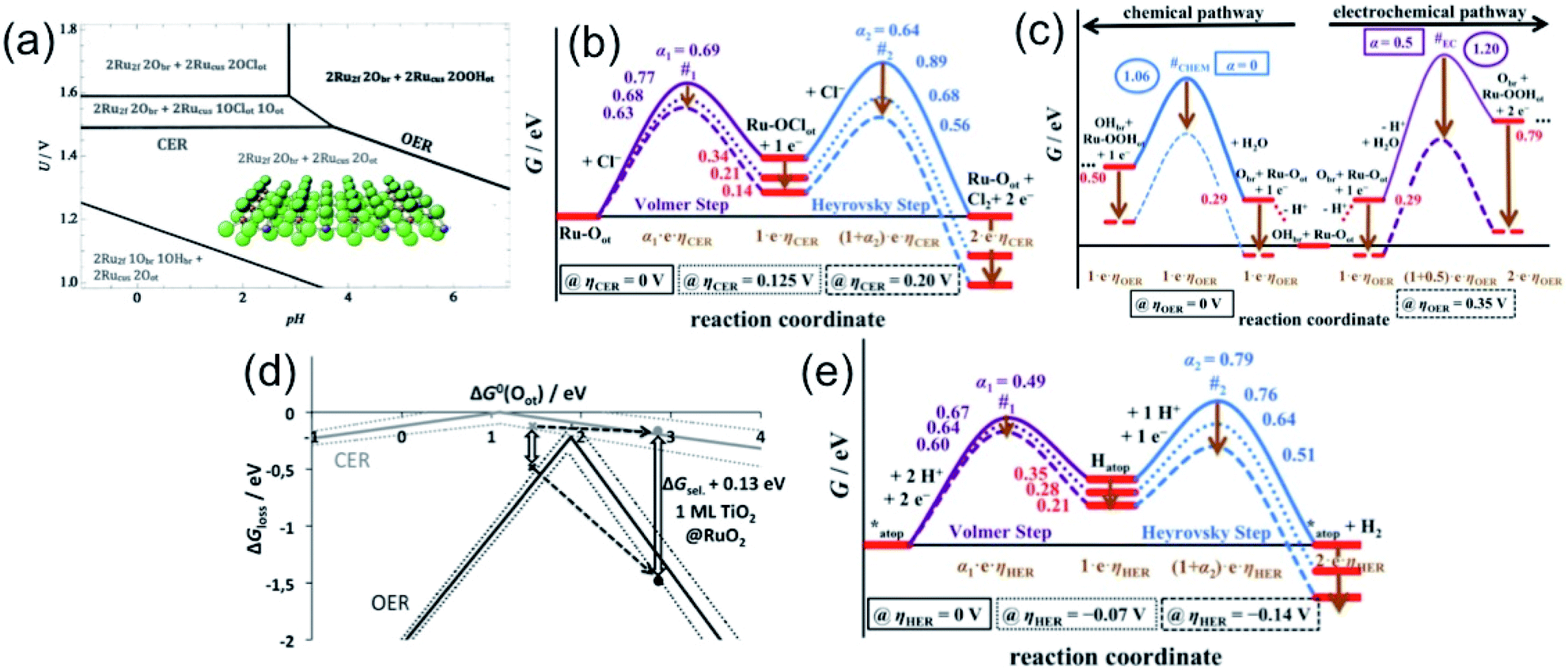 | ||
| Fig. 3 (a–d) Thermodynamics of RuO2(110) as the model catalyst: (a) Pourbaix diagram in equilibrium with H+, Cl− and H2O at T = 298 K and a (Cl−) = 1, indicating the different surface combinations of RuO2(110). Reproduced with permission from ref. 15. (b and c) Free energy diagram for CER (b) via Volmer–Heyrovsky mechanism at different overpotentials; and for OER (c) depicting two competing reactions, chemical and electrochemical, and their pathways at different overpotentials. Reproduced with permission from ref. 12. (d) Volcano diagram for the CER (gray) and for the OER (black). The gray cross denotes the catalytic activity (ΔGloss) for the CER, and the black cross is for the OER. Manipulation of the ΔGsel values, with TiO2(110)@RuO2(110) as an example, demonstrating the selectivity amongst the OER and CER. Reproduced with permission from ref. 15. (e) Free energy diagram for HER on Pt(111) as a model electrocatalyst describing the Volmer–Heyrovsky mechanism pathways at different overpotential values. Reproduced with permission from ref. 12. | ||
The mechanism can be described in more detail by varying the overpotential value ηCER > 0 V in light of the Tafel plot obtained in acidic pH, as represented in the free energy diagram (Fig. 3(b)), based on the study carried out by Over and his group. The free energy diagram indicates the formation of the single reaction intermediate, Ru–OotCl, and the high TS free energy value in the Heyrovsky step, thereby making the step as the rate determining step. The TS free energy can be decreased upon increasing the overpotential, especially in the Heyrovsky step (#2), lowering more than that in the Volmer step (#1) at ηCER > 0.20 V.12
Unlike CER, OER involves a four-electron transfer, making the process kinetically slower than CER. The OER is considered on the completely O-capped RuO2(110) surface by capping the uncoordinated RuCUS atoms coordinatively with Oot as oxygen on the top under acidic pH. The mechanism can be described in four steps as formulated in eqn (9)–(12).16,17
| 2Oot + H2O → 1Oot1OOHot + H+ + e− | (9) |
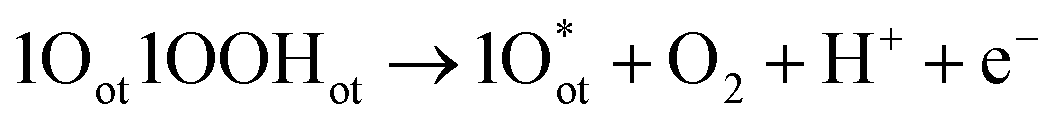 | (10) |
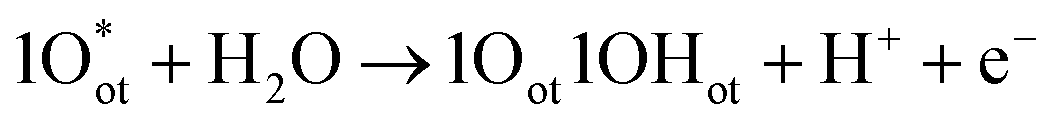 | (11) |
| 1Oot1OHot → 2Oot + H+ + e− | (12) |
As described by Over et al.,12 upon splitting of water, the adsorption of OH may happen on the active Oot site or the H+ may adsorb onto Obr to form OHbr, or may release H+ into the electrolyte. The free energy diagram for OER in Fig. 3(c) depicts the two competing reaction pathways, one chemical and another electrochemical, showing the formation of the corresponding RI's. The diagram illustrates the change in pathways for both at different overpotential values, at the reversible electrode potential, i.e., ηOER = 0 V and at ηOER > 0.3 V. It can be seen that at low overpotentials (ηOER < 0.3 V), the OER mechanism initiates through the active (OHbr + Ru–Oot) surface combination, and proceeds through the chemical pathway shown in blue line (Fig. 3(c)). However, at ηOER > 0.3 V, the active site combination switches to the (Obr + Ru–Oot) surface combination, and the reaction proceeds through the electrochemical pathways shown in the violet line in Fig. 3(c). It is important to note that at ηOER > 0.3 V, the TS for the electrochemical pathway (#EC) becomes lower than that of the chemical pathway (#CHEM), making the electrochemical path favourable.
The activity for OER and CER can be expressed with ΔGloss, representing the unfavourable Gibbs free energy change at the equilibrium potential. Based on the computational studies, it has been found that for the thermodynamic barrier in both cases, CER and OER on the model RuO2(110) surface are not zero, which suggests that there is enough chance of manipulation on the O-capped RuO2 surface for selective catalytic reactions.15 The hypothesis can be established with the help of a volcano diagram as a function of the free oxygen adsorption energy, represented in Fig. 3(d). In the volcano plot, while the higher values of ΔG0(Oot) indicate limited Cl− desorption, smaller values lead to limited Cl− adsorption. On the other hand, in the case of OER, higher ΔG0(Oot) values indicate OOHot decomposition, and the smaller values lead to the restricted formation of OHot. It is important to note that both cases proceed with maximum activity at the middle of the higher and smaller values. It is obvious to see that the volcano plot for CER is quite flat compared to OER, and the difference in the slopes reflects the different numbers of reaction intermediates, 1 for CER and 3 for OER. The difference in ΔGloss values at a certain ΔG0(Oot) defines the selectivity, and is quantified by ΔGsel. The ΔGsel value can be manipulated by weakening the Ru–O bond, i.e., lowering the OER catalytic activity, and in another way, allowing for substantial selectivity, as exampled by the TiO2(110) monolayer on RuO2(110).
The cathodic reaction was studied on the single crystalline Pt(111) surface as a model electrocatalyst. The process can be described by the Volmer–Tafel or Volmer–Heyrovsky mechanism, among which the latter one has been preferred. The mechanism can be described in two steps, by adsorbing a proton on the Pt surface, known as the Volmer step, followed by the recombination of the adsorbed hydrogen with the H+ ion, recognized as the Heyrovsky step.
| Pt–H Pt + 2H+ + 2e− → Pt–H Pt–H + H+ + e− (Volmer step) | (13) |
| Pt–H Pt–H + H+ + e− → Pt–H·Pt + H2 (Heyrovsky step) | (14) |
Like RuO2 in OER or CER, the Pt(111) surface is covered with hydrogen atoms with H-atom occupancy in the fcc hollow sites, while the Pt atom sites remain unoccupied. As described in eqn (13) and (14), the Pt atom sites mostly take part in the mechanism. The free energy diagram for HER (Fig. 3(e)) via Volmer–Heyrovsky mechanism reveals that the pathways for both steps change, depending on the applied overpotential values. It has been found that the Heyrovsky step becomes the rate-determining step at ηHER > −0.07 V, and the Volmer step is kinetically limiting for ηHER < −0.07 V.12
3. Electrolytes and effect of pH
In seawater electrolysis, the pH of seawater has been considered as a critical parameter as the OER and HER are accompanied with local pH changes at the electrodes. There has been a continuous increase in pH at the cathode due to hydrogen evolution, and a decrease in pH at the anode during oxygen evolution observed.18 Such pH fluctuation is found to be in the order of pH 5–9 during the electrocatalysis, causing severe damages to the electrocatalysts even at a low current density of <10 mA cm−2.6 In the anodic reaction, the continuous decrease in pH turns the electrolyte acidic at the electrode surface, causing severe corrosion at the electrode. It is known from the Pourbaix diagram (Fig. 2(b)) that OER becomes unfavourable and chlorate formation becomes most favourable as the local pH decreases, considering the current density unchanged.19 However, the hypochlorite formation seems to be pH-independent in the pH range of 6.5–10.5, and rather depends on the convective diffusion of ions.20 In order to carry out the selective OER mechanism avoiding CER at the anode, the pH of the medium must be controlled and should be >7.5, as indicated in the Pourbaix diagram (blue dotted line in Fig. 2(b)).10 Such control on the pH at the electrode surface can only be possible using certain buffer solutions. Thereby, the addition of buffer solution to consume protons, formed during OER, is highly desirable. While the electrolyte composed of 1 M KOH and 0.5 M NaCl is generally considered as the synthetic seawater to study the electrocatalytic performances, the addition of borate buffer (pH 9.2), carbonate buffer (pH 8.6) or phosphate buffer (pH 7) have been attempted by various researchers.11,21 Even though trace amounts of borate and carbonate are present in the seawater and may act as a buffer, their presence in a negligible amount does not alter the situation.The fact was examined by observing a noticeable difference in the OER overpotential values at a certain current density at different pH values obtained with and without borate buffer solution using NiFe LDH nanoplates supported on carbon as the electrocatalyst, reported by Strasser and his group11 (Fig. 4(a)). A shift of the ∼87 mV overpotential vs. RHE per pH was identified at a current density of 1 mA cm−2. A similar trend in the anodic peak shifted over a range of pH was observed on hydrous Ni oxyhydroxide thin films, as shown in the CV plots presented in Fig. 4(c).22 The presence of NaCl in the electrolyte definitely has an effect in lowering the solution resistance, which can be reflected by the enhancement in the current density in the LSV plot with borate buffer electrolyte at pH 9.2 in Fig. 4(a). However, the membrane-containing electrolysers exhibit some drop in performance in NaCl-containing electrolyte due to the interference of Cl− in the OH− transportation through the membrane, as observed by Dresp et al.23 The chlorine evolution can be ruled out by fixing the overpotential value to below 480 mV. Nonetheless, the local pH changes significantly. This phenomena was confirmed by measuring the quadrupole mass spectrometer ion currents for O2 and Cl2 during chronopotentiometric experiments at overpotential < 480 mV, showing no spikes for Cl2 evolution using an electrolyte composed of borate buffer and NaCl of pH 9.2 (Fig. 4(b)). Indirectly, it also confirms the fact that borate buffer can efficiently consume protons generated at the electrode surface, and thereby make OER favourable. In another report, Esswein et al. showed Co-borate compound as an OER catalyst exhibiting 550 mV overpotential at low current density of 1 mA cm−2 in borate buffer electrolyte with pH 9.2.24 Here, the metal-based catalyst retained catalytic activity at near neutral pH buffered seawater electrolyte by reducing the formation of passive oxide layers of metals. The use of phosphate buffer at near neutral pH has also been studied. In general, the overpotential for OER has been found to be in the higher end compared to the highly acidic or alkaline media.24–26 Nocera and his group27,28 further explored the cobalt-based phosphate/borate compound as Co–Pi, Co–MePi or Co–Bi catalysts prepared by electrodeposition method, and found them to be highly efficient for OER in seawater irrespective of the presence of chloride. The catalysts in phosphate electrolyte established proton-accepting characteristics, with phosphate as a proton carrier preserving a stable local pH. This unique phenomena was believed to be responsible for generating high current density with the oxygen faradaic efficiency of 100%. Alternatively, the OER of the simulated seawater has also been examined without the addition of any buffer solution with the CoFe-LDH supported on a Ti mesh as a catalyst at a near neutral pH value of 8. Under this condition, it exhibited 530 mV overpotential at a current density of 10 mA cm−2. It is believed that the presence of multiple ions in real seawater may affect the performance by mediating the proton transfer reactions, and thereby make the process kinetically faster as can be observed with the smaller Tafel slope value of 140 mV dec−1 with the seawater compared to NaCl, Na2SO4 or MgCl2-based electrolytes, keeping the pH value of 8.0 for all cases, shown in Fig. 4(d).25
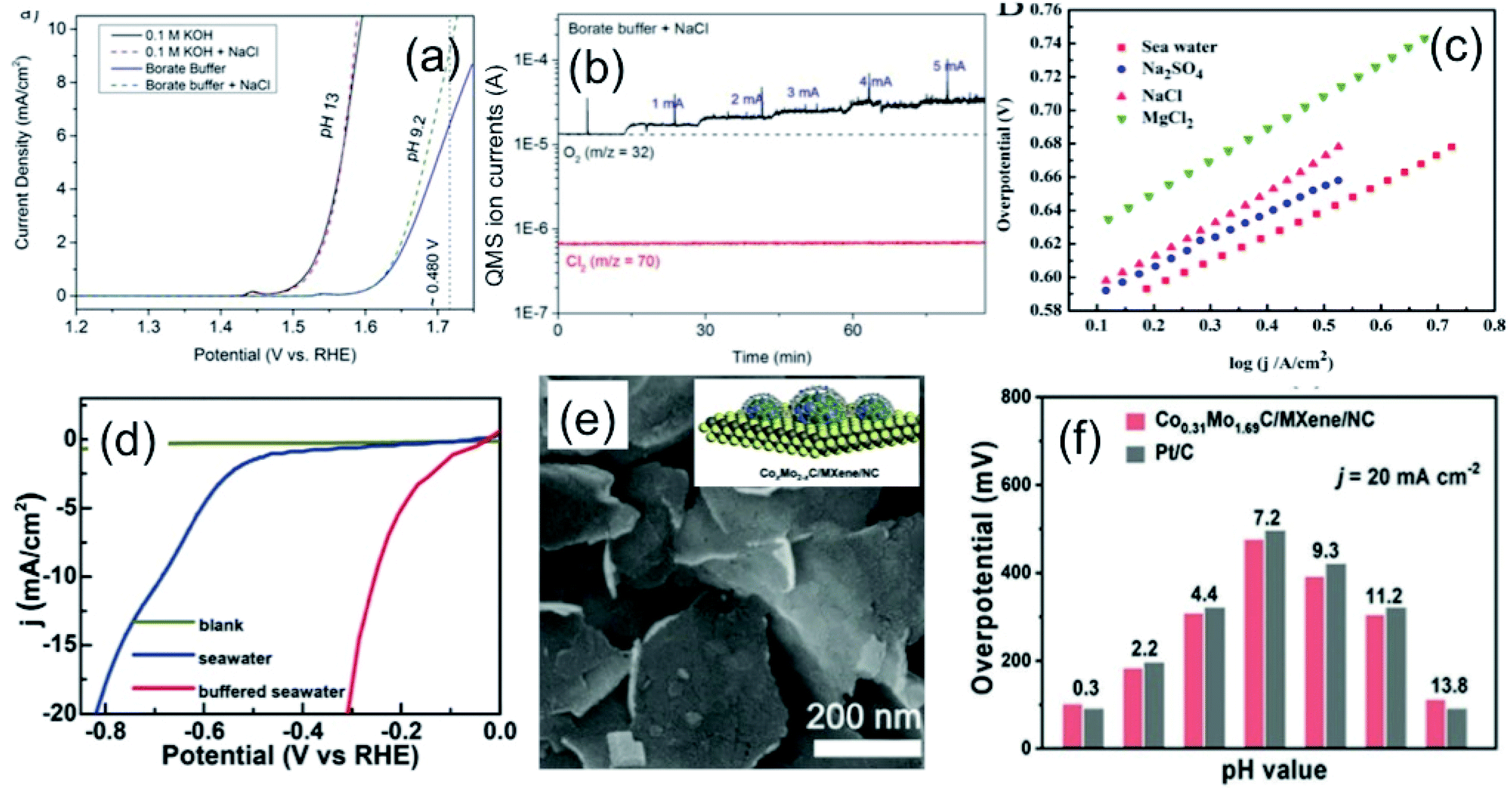 | ||
| Fig. 4 (a) LSV plots of NiFe LDH nanoplates supported on carbon, showing the OER activities in four different electrolytes. (b) The quadrupole mass spectrometer ion currents for O2 and Cl2 detected during chronopotentiometric experiments with borate buffer + NaCl electrolyte (pH 9.2). Reproduced with permission from ref. 11. (c) Tafel plots (scan rate: 1 mV s−1) of Co–Fe LDH/GCEs in seawater and other aqueous solutions, like MgCl2, NaCl or Na2SO4. The concentration and pH of the electrolytes are the same. Reproduced with permission from ref. 25. (d) Current density vs. applied voltage without catalyst (blank), and with CNT-based catalyst (U-CNT-900) in buffered seawater (pH 7) and natural seawater. Reproduced with permission from ref. 29. (e) SEM image of Co0.31Mo1.69C/MXene/NC with 2D-sheet like nanostructure (inset: schematic representation), and (f) its HER performance comparison with 20% Pt/C under pH 0.3–13.8. Reproduced with permission from ref. 30. | ||
Few reports are available on the catalytic performance of the non-precious Pt-free catalyst for HER at different pH values in seawater. Zou and his group29 carried out an extensive study with cobalt-embedded N-doped carbon nanotubes as an alternative catalyst to Pt for HER in seawater at different pH values. They performed the HER performance in acidic (pH 0), neutral using phosphate buffer (pH 7), and alkaline (pH 14) media, which indicated faster HER Tafel kinetics at neutral pH than acidic or alkaline media. While comparing in natural seawater and buffered seawater, the buffered seawater showed undoubtedly better performance than the natural seawater, along with great stability as shown in Fig. 4(e). The MXene or metal carbide and nitride-based electrode materials showed noticeable HER performance at a wide pH range in seawater, sometimes outperforming the conventional Pt electrode.30–32 Generally, the fast kinetics was observed under highly acidic or basic conditions. Under such corrosive conditions, the Mxene-based materials withstand its catalytic activity exhibiting outstanding stability unlike Pt.
4. Design of anode materials
An extensive investigation has been carried out on developing novel and highly selective anodic electrode towards OER suppressing CER in chloride-containing electrolyte solutions, including seawater. Transition metal oxides and hydroxides are well known for their promising electrocatalytic properties for OER in alkaline water due to their effective active sites created by defects, and oxygen vacancies tuning the electronic structure of the material.33–35 The selectivity of the anodic OER electrode depends on the active adsorption sites, electronic conductivity, and binding energy of the reaction intermediates species (M–OOH, M–OH) formed during the reaction.36–38 As discussed in the previous sections, anodic reactions for seawater splitting deal with the keen competition between OER and CER (as under normal condition reaction kinetics) makes the CER dominate over OER. The selectivity of the OER electrodes can be enhanced to manipulate the electronic structure by doping with heteroatoms,38 metals (like Mo, Co, Fe, Ni or Mn39,40), or by increasing the number of active sites.38 The use of titanium as an inner substrate can be commonly noticed in an anode for seawater electrolysis due to its corrosion resistance nature against chloride, or any other impurities as also identified by chloro-alkali industries.6 This section will focus on discussing the selectivity and design of highly efficient OER electrocatalysts over CER in seawater electrolysis.Bennett, in 1980, reported on the selective OER reaction in seawater using a MnOx-based electrocatalyst.41 The study identified MnO2 as a selective electrocatalyst for OER over CER. In commercial chlorine production, the TiO2/RuO2 substrate has been commonly used as an anode due to its high stability in chlorine water. The electrochemical deposition of the porous MnO2 coating on the TiO2/RuO2 substrate increases the oxygen evolution efficiency to 99+% in seawater, and 95% in saturated NaCl brine. The mechanism involved in this work was highly influenced by the mass transfer limitation of chlorine at the electrode surface, where the MnO2 coating acted as a diaphragm and allowed OER as the dominant anodic reaction without any significant chlorine evolution. During this period, a few other reports also investigated MnOx-based electrodes to understand the mechanism behind the selective OER anodic reaction in the chlorine-containing electrolyte.42,43 Trasatti8 in 1984 compared the catalytic reactivity of various oxide materials for both anodic OER and CER, and proposed the fact that the mechanism for OER (by adsorption of hydroxides on a metal oxide) involving an increase in the valence state of metal can also be applicable for CER. He suggested that although the electronic structure of the metal oxide determines the kinetics and thereby activity of the catalyst, the selectivity of chlorine may not depend on the electrode material, as confirmed from a linear slope in the plot of the potential for OER vs. the same for CER, as shown in Fig. 5(a). Later on, Hashimoto and his co-workers developed an electrode composed of MnOx supported on titanium (Ti) substrate with intermediate IrO2 coating by thermal decomposition technique, and extended the work by incorporating a series of metal elements, like transition metals (Ni, Co, Fe, Mo, Sn), noble metals (Pt, Ir, Ru) and rare-earth metals (Ce, La), to evaluate the effect on OER in seawater.40 The study in 0.5 M NaCl and 0.1 M NaOH aqueous electrolyte solution (pH = 8) showed that although the addition of non-noble metals up to 10 mol% enhanced the oxygen evolution efficiency, noble metals (Pt, Ru, Ir) had a detrimental effect on the OER activity in chloride-containing solution. The results are compiled and represented in Fig. 5(b), reflecting the influence on the OER activity at different doping concentrations of metal additives. Among all, Mo as an additive in the MnOx/IrO2/Ti electrodes exhibited the maximum oxygen evolution efficiency of up to 90% with low (10 mol%) doping concentration, above which the efficiency decreased considerably. The reason of such an enhancement of OER activity was due to the formation of a single α-Mn2O3 at the low dopant concentration. At the high Mo dopant concentration, the formation of MnMoO4 and double-oxide composite made OER unfavorable in chloride-containing solution. Hashimoto further explored the OER activity in seawater by incorporation of various mixed metal oxide coatings via electrochemical anodization, such as Mn–Zn,44 Mn–W,45 Mn–Mo–Fe,46–48 Mn–Mo,48–50 Mn–Mo–W,51–54 Mn–Fe–V,48 and Mn–Mo–Sn55,56 on IrOx/Ti electrodes. In all of these cases, the IrOx acted as the protective layer to prevent the oxidation of the Ti substrate during the anodic deposition of mixed metal oxides. Based on theses studies, it was established that while the MnOx coating on the IrOx/Ti electrodes led to oxygen evaluation selectively by preventing CER mechanism in seawater, the incorporation of the triple oxide composition with a single-phase γ-MnO2 structure steered the enhancement of the oxygen evolution efficiency further. Among all, the incorporation of Mo(VI) or W(VI) in MnOx with their high valence states was found to be the most beneficial, as up to 100% oxygen evolution efficiency was achieved.55 Moreover, with this approach, the stability of the electrodes was addressed by preventing the oxidation of the inner Ti substrate responsible for the detachment of the electrocatalyst from the surface. Recently, Vos et al.57 further extended this concept by electrodepositing MnOx onto glassy carbon supported hydrous iridium oxide (IrOx/GC) for selective OER over CER in acidic chlorine containing solution. Their observations also revealed that although MnOx is believed to be catalytically inactive, it acted as a diffusion barrier. It prevented Cl− from reacting on the IrOx catalyst underneath, but allowed for the water, protons, and O2 transportation between IrOx and the electrolyte, which is necessary for OER activity.
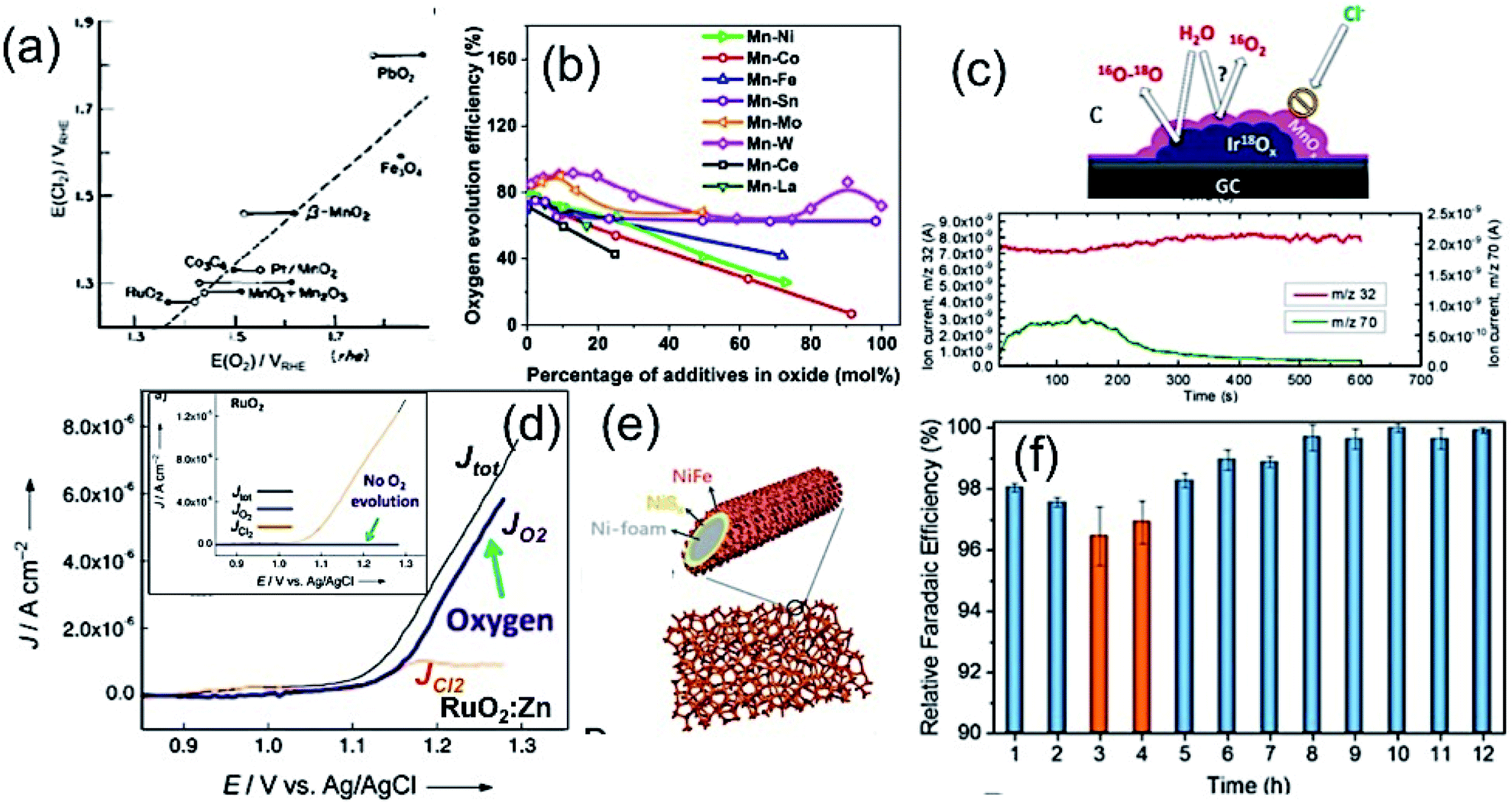 | ||
Fig. 5 (a) Plot of the potential for Cl2 against the potential of O2 evolution at the same current density for metal oxides in (○) alkaline and (●) acidic solution. Reproduced with permission from ref. 8. (b) Compilation of oxygen evolution efficiencies of (Mn–M)Ox/IrO2/Ti, M = Ni, Co, Fe, Sn, Mo, W, Ce and La electrode as a function of different M contents measured in 0.5 M NaCl at pH 8. Adapted from ref. 40. (c) Online electrochemical mass spectrometry (OLEMS) measurements for O2 (m/z 32) and Cl2 (m/z 72) with MnOx coated IrOx showing CER restriction, but allowing OER as a potential selective electrocatalyst (inset: schematic representation). Reproduced with permission from ref. 57. (d) LSV plots of Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RuO2 and RuO2 (inset) in 0.1 M HClO4/0.15 M NaCl solution, showing highly selective OER upon Zn incorporation. Reproduced with permission from ref. 59. (e) Schematic representation of the dual-layer NiFe/NiSx–Ni foam anode and (f) its relative faradaic efficiency for O2 production. Reproduced with permission from ref. 3. :RuO2 and RuO2 (inset) in 0.1 M HClO4/0.15 M NaCl solution, showing highly selective OER upon Zn incorporation. Reproduced with permission from ref. 59. (e) Schematic representation of the dual-layer NiFe/NiSx–Ni foam anode and (f) its relative faradaic efficiency for O2 production. Reproduced with permission from ref. 3. | ||
The conventional RuO2 or IrO2-based electrodes are well known electrocatalysts for effective OER, as well as CER as confirmed theoretically.12,58 The DFT study with the single crystal RuO2(110) surface indicated the formation of a peroxo group with Ocus atoms on the neighboring Ru atoms as surface intermediates, which might act as the active site for CER. Thereby, the selectivity of the oxygen evolution suppressing the chlorine evolution was found to be a challenging task with RuO2 or IrO2. This suggested the necessity in manipulating the electronic structure of the catalyst by doping. Krtil and his group59 developed a Zn-doped ruthenium oxide deposited on a Ti mesh by freeze-drying method to study the selective OER over CER in the chloride-containing acidic solution. Interestingly, the LSV showed that while pristine RuO2 led to chlorine evolution without any O2 evolution at low pH media as a favourable electrolyte for CER, the RuO2:Zn indicated the O2 evolution suppressing chlorine evolution considerably under the same condition (Fig. 5(c)). The mechanism of such selective OER was anticipated with the prevention of the peroxo-bridged intermediate formation due to the rearrangement of the RuO2 lattice, creating defects upon insertion of Zn.
Strasser and his co-workers11 designed a noble metal-free NiFe-LDH electrocatalyst by solvothermal method, which exhibited high selectivity towards OER with close to 100% efficiency at <480 mV in an artificial seawater electrolyte at pH 13. However, the activity and selectivity of the electrocatalyst became limited at near neutral pH in chlorine-containing buffer electrolyte, indicating the suppression of CER as a difficult task at this condition. Following the same track, Cheng et al.25 developed an inexpensive layered structure of CoFe-LDH nanomaterials, and loaded them onto a Ti substrate as an electrode to investigate the selective OER electrocatalyst for seawater splitting without using any buffer solution. The enhanced catalytic activity and sufficient faradaic O2 efficiency of CoFe-LDH nanoparticles was found at 530 mV overpotential in an electrolyte of pH 8 due to the synergistic effect of multiple metal cations. Kuang and co-workers3 designed a corrosion-resistant anode for seawater splitting with an amorphous coating of NiFe-hydroxide by electrodeposition onto the NiSx-layered Ni substrate prepared by surface sulfuration. While the top amorphous layer of NiFe-hydroxide acted as an effective electrocatalyst for OER, the lower NiSx layer not only enhances the conductivity of the electrode, but also generates cation-selective polyatomic anion-rich electrode-resisting corrosion caused by the chlorine present in seawater. It is also operable at high current density, as mostly required in industries. Recently, the highly porous S-doped NiFe-oxyhydroxide was developed via an ultrafast room temperature and scalable process, which was found to be highly effective for OER in seawater, reaching a very high current density at a reasonable overpotential.38
5. Design of cathode materials
It is well-known that the noble metal Pt is considered a conventional electrocatalyst for HER due to its lower kinetic energy barrier for the dissociation of a water molecule, which is 0.89 eV with the Pt(111) surface.60,61 However, the scenario for seawater is very challenging due to the presence of impurities, which may hinder the activity and cause catalyst poisoning.62,63 Therefore, in practical application, seawater splitting needs to find an alternative way to establish a corrosion-resistive, but efficient cathode material.In this regard, the Pt-based alloys with transition metals can be considered as the substitute for pristine Pt, showing promising electrocatalytic activity with increased stability towards HER mechanism. The competitive dissolution reaction rate in alloys is believed to assist the increased anti-corrosion properties of the electrode. It is very important to understand the fundamental concepts of the Brewer–Engel bond theory for developing such alloy-based active HER catalyst, as it describes the possible d-orbital overlapping of metals in intermetallic, and thereby predicting a stable system for kinetically favorable HER mechanism.64 Guided by the theory, different Pt-based alloys have been developed for alkaline water electrolysis. The approach was found to be more suitable, while being applied in seawater splitting, as reported by Li et al.65 They developed the Pt–Ru–M (M = Fe, Mo, Co, Ni, Cr) alloy by incorporation of Ru and 3d transition metals (M) as guest metals into FCC platinum (Pt) as the host metal without any apparent change in the lattice structure of the host metal deposited on the Ti mesh by electrodeposition method. The lattice distortions due to the insertion of guest metals offer effective active sites for enhanced H+ adsorption. Among different transition metals, Mo in the Pt–Mo alloy showed high Mo–H bond strength, leading to high catalytic performance (Fig. 6(a)). Importantly, such alloy formation exhibited much desired stability over 172 h in seawater electrolysis due to the outstanding d band interaction of the PtMo alloys, according to Brewer theory, along with the increased surface area of the alloys. Later on, a similar approach was reported by Zheng et al.,66 exhibiting the long stability of the Pt–M alloy over 170 h in HER for seawater splitting. Such works were well supported by the thermodynamic study on the corrosion behavior of Ni and Mo in a chlorine-containing environment, as reviewed by Galetz and his co-workers.67 It can be noted that compared to Ni, Mo undergoes a reaction with chlorine in a poor oxygen environment in a much slower rate, which was reflected in the outstanding stability of the Pt–Mo alloy material in chlorine-containing seawater. Even though the cost of cathode material can be reduced to some extent by making alloys with Pt achieving good stability, the cost of electrode still remain higher limiting their use in scaled up hydrogen production from seawater.
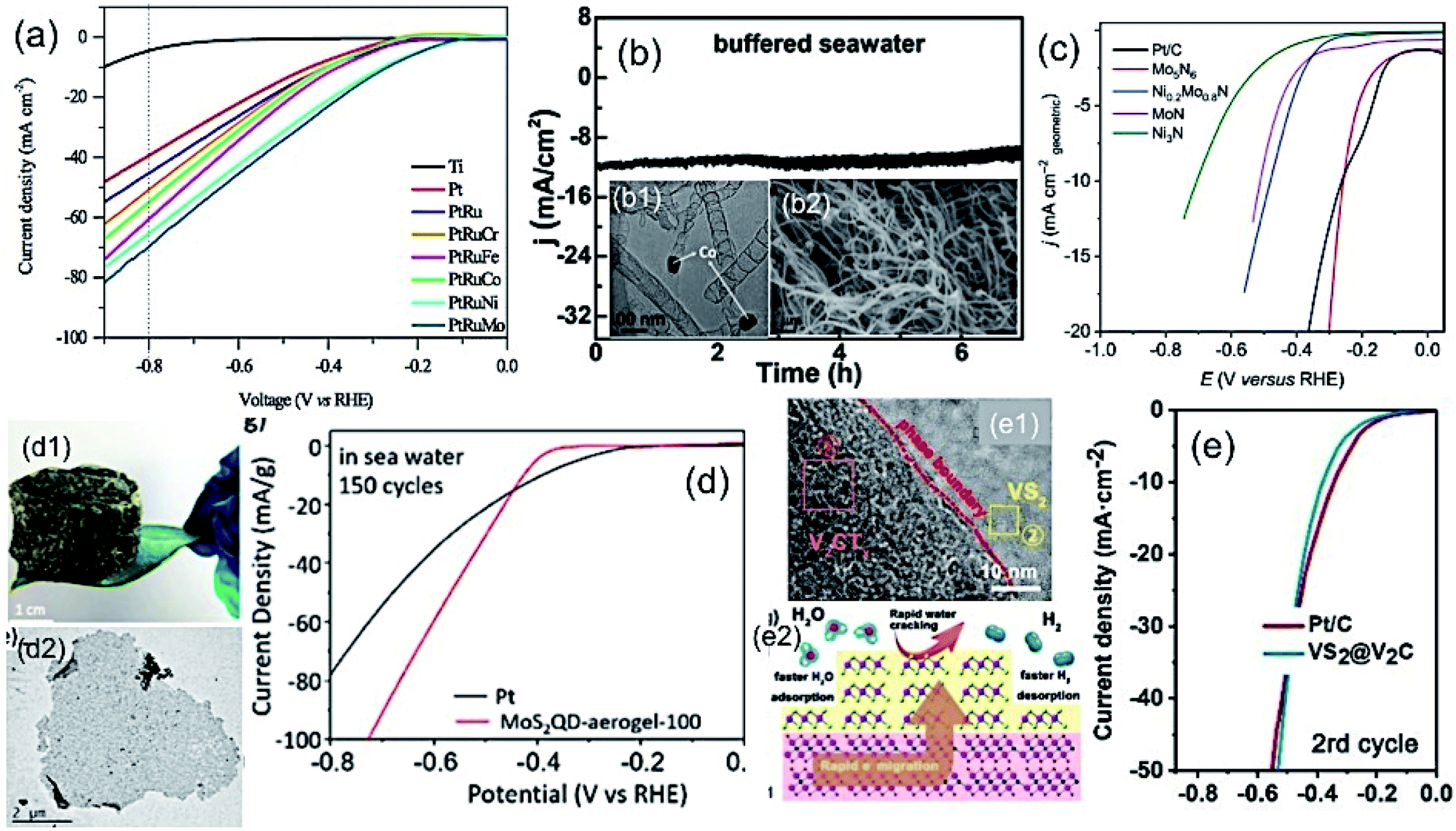 | ||
| Fig. 6 (a) LSV plots of different Pt-based alloy cathodes for HER. Reproduced with permission from ref. 65. (b) Current vs. time plot for Co-incorporated CNTs as an efficient cathode at η = 270 mV (b1 and b2 are corresponding TEM and FESEM images). Reproduced with permission from ref. 29. (c) LSV plots of different metal nitride cathodes for HER in Ar-saturated seawater. Reproduced with permission from ref. 73. (d) LSV plot of lightweight MoS2 aerogel as cathode for HER in seawater recorded after 150 cycles showing superior performance over the conventional Pt electrode (d1 and d2 are the corresponding optical and TEM images). Reproduced with permission from ref. 71. (e) LSV plot of VS2@V2C as the HER electrocatalyst, showing similar performance as the conventional Pt/C in seawater (e1: TEM image showing the interface, e2: schematic representation showing the H2 production process). Reproduced with permission from ref. 31. | ||
To find an alternative solution to the use of expensive Pt-based cathodes for seawater splitting, a number of materials have been developed by different research groups with great catalytic activity and non-corrosive nature in chlorine-containing electrolyte. One of the useful ways to protect the electrocatalyst against corrosion in seawater during water splitting is to cover the efficient electrocatalyst with a protective carbon layer. Gao et al.29 developed a Co-embedded N-doped carbon nanotube as an efficient cathode for seawater splitting, as examined at different pH conditions (Fig. 6(b)). Even though the catalytic activity may not be as good as that for the pure Pt as electrode, the material exhibited good stability at three different pH conditions, including the buffered seawater and untreated seawater. Ma et al.68 fabricated CoMoP nanocrystals coated by few layers of N-doped carbon shell by the pyrolysis process. The electrocatalyst showed promising HER activity over a wide pH range, and exhibited a faradaic efficiency as high as 92.5% in real seawater. The findings suggest that the carbon coating on CoMoP promoted the HER activity not only by providing protection to CoMoP against the impurities in seawater and improving the adsorption free energy of H, but also by increasing the adsorption of water. In another approach, Lu and his co-workers39 observed the deposition of Na+, Ca2+ or Mg2+ salts on the surface of their developed Mn-doped Ni/NiO on Ni foam, blocking the active sites and thereby affecting the performance. They suggested a mild acid treatment as an effective solution to regain the activity of the electrocatalyst.
Molybdenum disulfide with exposed S active sites has been considered to be a very effective catalyst for the HER mechanism due to its 2D layered structure allowing easy access of electrolytes to the active sites.69 Identifying MoS2 as an efficient electrocatalyst for HER, attempts have been made on seawater splitting as well. Miao and co-workers70 developed a hierarchical Ni–Mo–S nanosheet on carbon cloth by hydrothermal method. As per their investigation, the incorporation of Ni atoms into MoS2 created defects at the catalytic active sites, thereby regulating the electronic structure of the layered MoS2. The material with an optimum Ni:Mo ratio of 1:1 exhibited promising catalytic activity for HER in neutral buffer electrolyte, as well as in real seawater, owing to its mesoporous nature and better exposed active sites. Very recently, Chen and his co-workers71 developed a 3D porous MoS2 quantum dot aerogel as an efficient cathode for seawater splitting. The aerogel exhibited high surface area. It was found to be an excellent electrocatalyst that was as good as conventional Pt over a wide pH range, as well as in seawater with outstanding stability over 10000 cycles. Like metal phosphides and sulfides, metal nitrides have great potential as an electrocatalyst for HER due to their inherent electronic structure and electrical conductivity. However, the stability issue remains a challenging factor due to the low valence state of the metal.72 The problem can be resolved by N-enrichment in the metal nitrides, increasing the valence state of the metal atoms, and thereby improving the corrosion resistance property. Jin et al.73 developed Mo5N6 nanosheets, a N-enriched metal nitride via Ni-induced salt template method. Their study reveals that apart from the high surface area, the N incorporation into MoN altered the d band center closest to Pt metal leading to higher HER catalytic activity as good as Pt along with good stability (100 h). Very recently, Yu et al.74 developed a nickel phosphide (NixP)-based electrode sandwiched by a NiCoN nanostructure for efficient HER in natural seawater. The electrocatalyst performed close to the benchmarking performance of Pt at higher current density with good stability. The corrosion resistance behavior of NixP, as well as NiCoN, was believed to be responsible for the stability of the electrocatalyst, maintaining the performance in natural seawater. To achieve the benchmarking catalytic activity of 20 wt% Pt for HER, Xiu and his co-workers75 developed a hollow Mxene-tailored low-Pt as a catalyst, especially for seawater operable in full pH range. The hollow morphology composed of low-Pt exhibited better catalytic activity for HER in seawater due to the enhanced surface area and conductivity. Wu and his co-workers30 developed 2D CoxMo2−xC/MXene/NC electrocatalysts with low overpotential, faster HER kinetics in the full pH range, as well as in natural seawater. It was believed that the precise interfacial engineering was responsible for such high catalytic activity, exceptional durability and faradaic efficiency in natural seawater. Furthermore, in a recent study, the combination of metal sulphide and metal carbide as VS2@V2C exhibited superior catalytic activity than the conventional Pt over all pH ranges and in natural seawater.31 The reason for such high activity was attributed to the tight wrapping of V2C by VS2, allowing the rapid charge transfer between the two materials (Fig. 6(e)) and lower free energy for hydrogen adsorption exhibiting excellent electrocatalytic activity.
6. Bifunctional electrocatalysts and complete cell designing
The design of a bifunctional electrocatalyst for both anodic oxygen evolution and cathodic hydrogen evolution with high activity and long stability is as challenging as necessary.76 For alkaline water splitting, different types of bifunctional catalysts including metal chalcogenides or nitrides with necessary alterations in their electronic properties and morphologies have been reported. Only a few reports are available for direct seawater splitting, considering the selectivity and stability of the catalysts. Zhao and co-workers77 developed a series of cobalt selenide electrodes with a charge state manipulation by one-step calcinations controlling the Co and Se mass ratio. In this approach, the high Co charge state favors OER and the low Co charge state promotes the HER activity, giving rise to the overall seawater splitting achieving 10.3 mA cm−2 current density at 1.8 V. Zhao et al.32 fabricated a NiNS electrode with an interface among Ni3N and Ni3S2, and highly exposed electrochemical active sites for the dissociation of water molecules. Such a design of materials with ubiquitous interfaces was found to be beneficial for the dissociative adsorption of water molecules, leading to the overall seawater splitting achieving current density of 48.3 mA cm−2 at 1.8 V. Hsu et al.78 designed a PV-driven seawater splitting device using transition metal hexacyanometallate with basic cobalt carbonate (BCC) as a conductive core grown on a pretreated carbon cloth (MHCM-z-BCC) as the anode and NiMoS as the cathode. The electrode combination was found to be very effective for neutral seawater splitting with no trace of Cl2 evolution even after 100 h. The authors claimed that the superior selectivity of the MHCM-z-BCC towards OER over CER played a key role in the effective seawater splitting into H2 and O2.7. Electrolyzer designing and challenges
Principle
The basic understanding on seawater splitting reveals the fact that while CER dominates at low pH values, one needs to deal with the competition between OER and hypochlorite formation at the anode at high pH values. The natural seawater, with a slightly alkaline pH of 8.2, leads to the consideration of the selective OER over the hypochlorite formation as the major challenge. The suppression of hypochlorite can only be possible by keeping the anodic reaction <1.72 V vs. RHE or the overpotential < 480 mV at alkaline pH, as discussed in earlier sections.79 Furthermore, the cathodic reaction of natural seawater splitting accompanies the deposition of cationic species, like Ca2+ or Mg2+, in the form of hydroxides on the cathode, covering the active sites for HER. Such phenomena may not be observed on the laboratory scale, as either purified seawater or NaCl containing artificial seawater have been used.6,10 To protect the cathode from impurities, different membrane technologies, like proton exchange membrane alkaline water electrolyser (PEMAWE), and anion exchange membrane alkaline water electrolyser (AEMAWE), have been developed and allow for selective ion diffusion through the membrane. Moreover, the rational design and alloy-based HER electrode are believed to be the most important ways, as discussed in the previous section, for selective HER restricting the probable corrosion at the electrode surface.6 The use of a membrane in the electrolyser also has some detrimental effects, as they restrict ion diffusion, causing reduction in ionic conduction in water affecting the performance.79,80On practical ground considering the large-scale hydrogen production from seawater, different technologies have been conceived. The energetic assessment of the electrolyzer can be done on the basis of the specific energy required to produce 1 kg hydrogen or per volume of hydrogen. In a PEM electrolyzer for seawater, the electrolysis one needs to carry out pre-purification by reverse osmosis (RO) technique prior to the electrolysis. This extra step requires extra energy, which may increase the specific energy values for the plant. It has been found that the energy required for RO treatment of 9 kg of seawater is about 0.03 kW h, which is considered to be a negligible addition to the specific energy required (47–66 kW h kg−1) for the stack electrolyzer for fresh water.10 However, the single-step RO technique prior to the electrolysis may not be enough to reach the required purification level of water for use in a PEM electrolyzer, compelling some extra purification steps to maintain a sustainable process. This indicates the considerable enhancement of specific energy consumption for seawater electrolyzer. Furthermore, an extensive feasibility study on hydrogen production by direct seawater electrolysis from offshore marine farm reveals the requirement of very high specific energy, making the process less practical in terms of energy and economy.81
Therefore, the design of the electrolyser reactor is one of the most important aspects, which needs to be properly considered. In general, the symmetric reactor chamber has been mostly used for seawater, similar to alkaline water electrolyser (AWE), where 20–30% KOH solution is used in both compartments.7 The choice of membrane remains an issue in the case of the seawater electrolyser. The use of PEM can be ignored due to its acidic electrolyte, making the seawater splitting more challenging. Although the problem in PEMAWE can be overcome in AWE and AEMAWE using alkaline electrolyte, the migration of anions including Cl− and OH− through AEM keeps the competition between them active limiting the performance. The use of AWE may be preferred over AEAWE due to the use of Zirfon as the diaphragm material in AWE, which is much more robust and more defensive against blockages, giving rise to a sustainable reactor for seawater splitting.6
Progress
Recently, Strasser and his group customized the conventional electrolyser reactor, and proposed the asymmetric electrolyser in keeping two different electrolyte feedings in the anode and cathode compartments using AEM in between.79 The asymmetric combination with seawater in the cathode and KOH electrolyte in the anode at the limited cell potential of 1.7 V exhibited performance as good as KOH electrolyte containing AEMAWE for fresh water, and interestingly superior to the symmetric seawater electrolyser (Fig. 7(a)). The observation suggested that a very low level of Cl− migration into the KOH analyte with such asymmetric feeding combination (Fig. 7(a2)) led to the high performance, as well as good stability.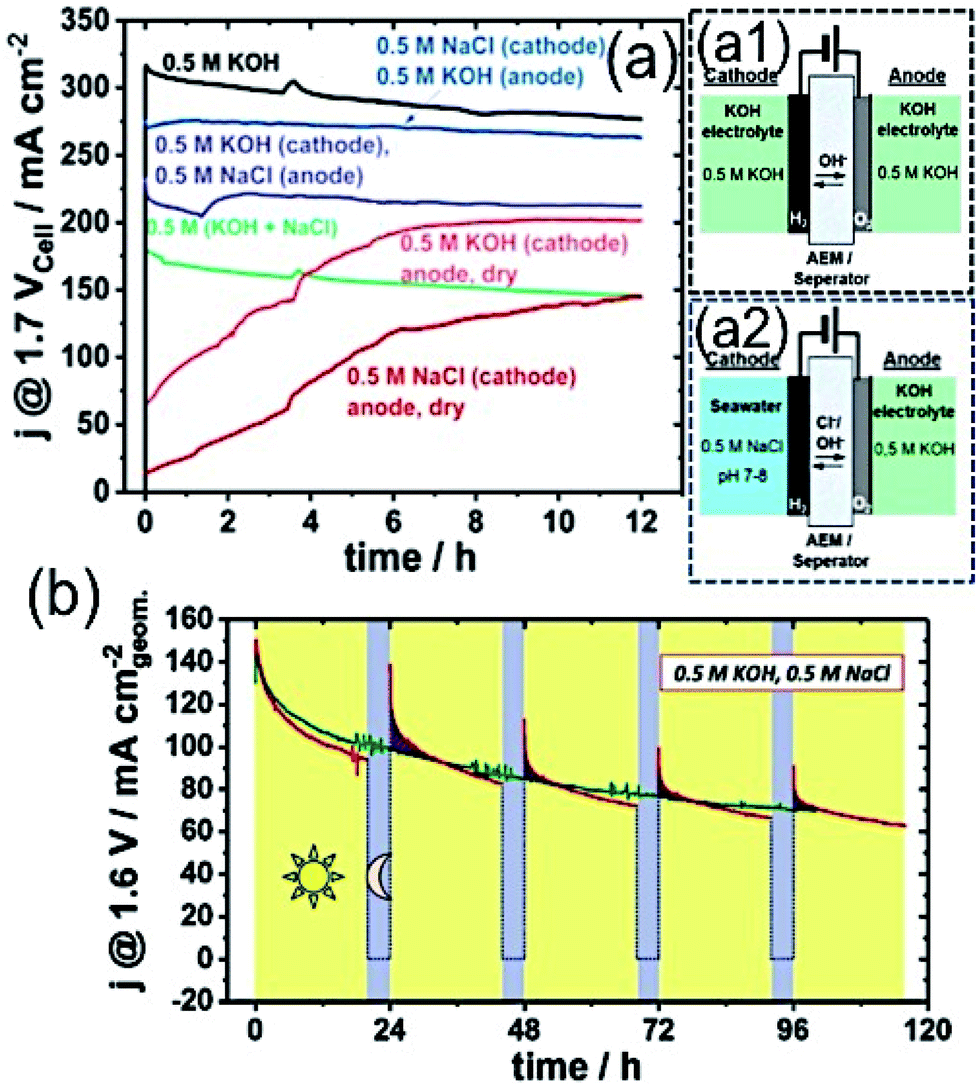 | ||
| Fig. 7 (a) Stability measurement over 12 h of electrolyzer with various electrolyte feeder combinations at 1.7 Vcell–cell potential with an active area of 5 cm2 using commercial Pt/C (48.5 wt%) of 0.5 mg cm−2 loading as the cathode and crystalline Ni0.66Fe0.34-LDH of 2.0 mg cm−2 loading as the anode catalyst. Schematic representations of AEMAWE with (a1) symmetric (0.5 M KOH/0.5 M KOH) and (a2) asymmetric (0.5 M NaCl/0.5 M KOH) electrolyte feeder combinations. Reproduced with permission from ref. 79. (b) Sustainable electrolyzer performance over 100 h with 20 h (V = 1.6 V) + 4 h (V = OCP) alterations. Electrolyzer composed of Pt/C (46.7 wt%) with 0.5 mg cm−2 as the cathode and NiFe-LDH with 2.5 mg cm−2 loading as the anode, and Tokuyama A201 as the membrane in 0.5 M KOH and 0.5 M NaCl electrolyte. Reproduced with permission from ref. 23. | ||
Knowing the difficulties of seawater splitting, apparently the choice of electrocatalyst as an anode and cathode is equally important as the design of electrolyser. The conventional AEMAWE for seawater splitting usually is composed of IrOx-based anode due to its stability, and Pt/C as the cathode. Strasser and his co-workers later identified NiFe-LDH as an effective and stable electrocatalyst for OER not only in the symmetric electrolyzer reactor, but also in the asymmetric combinations.23,79,82 The detailed investigation on the selective OER/ClOx mechanism with NiFe-LDH revealed that the material achieved a very high faradaic efficiency of 94% (FEO2) at a very high cell potential of 2.4 V with high current up to 4 A. In addition, a reduced FEO2 value to 84% was achieved upon increase in the cell voltage to 4 V. The absence of any ClO− traces indicated the high selectivity of the material towards OER.79 The performance degradation over time and recovery of activity was also investigated. It was believed that the accumulation of gas bubbles at the electrode surface might cover many active sites, causing the degradation of the performance over a longer period of time. The performance of the electrolyzer at a cell voltage of 1.6 V for 20 h was followed by a break of 4 h at the open circuit potential, and was found to exhibit sustainable performance with a good recovery rate over 100 h run, as shown in Fig. 7(b).23 Although these recent studies indicate the feasibility of the seawater electrolyzer, the practical implication is yet to be proven.
8. Conclusion and future scope
Seawater is considered to be a major reservoir for hydrogen. The presence of impurities and their interference, especially Cl−, makes the seawater electrocatalysis tougher than the freshwater by competing with OH− for anodic reactions. The design of an electrocatalyst with selective OER is therefore highly desirable, diminishing the possibility of CER. A number of electrocatalysts have been designed, showing not only OER selectivity but also the stability over a longer duration of electrolysis. The Pourbaix diagram of the oxygen and chlorine system for seawater reveals the selectivity criterion of η < 480 mV for efficient OER, as the anodic chlorine reaction requires at least 1.73 V. Even though seawater with a slightly alkaline pH of 8.2 is considered to be OER favourable, the local pH change at the vicinity of the electrode surface indicated the requirement of the use of buffer solution in order to achieve the steady performance. In the case of the cathode, however, the conventional Pt has been proved to be a good HER catalyst, but not a sustainable solution due to its vulnerable reactions with Cl− in the electrolyte. Therefore, Pt-free seawater cathode HER catalysts will become important in the future. The Pt-based alloys seem to be a possible solution of corrosion resistance, but efficient.The challenges of seawater electrolysis keep it away from the commercial implementation like freshwater. Moving towards hydrogen-based fuel technology, the difficulties with seawater must be addressed to utilize the major hydrogen source in order to satisfy the future energy demand. The filtration or removal of undesired ionic species from seawater may not be a fruitful way, as it requires expensive membranes with their questionable sustainability, and extra energy that makes the hydrogen production from seawater costly. Therefore, the development of novel electrocatalysts with selective active sites for OER will not only be enough to address the challenge, but the design of electrolyzers also needs to be explored. It is evident that seawater electrolysis will be much more different than the conventional AWE. The reactor and the use of membranes (PEM or AEM) need to be rationally designed, making the membrane the least affected. However, performance-wise, it should be sustainable and similar to freshwater electrolysis in AWE. To achieve this, recent findings suggest the asymmetric reactor design as a promising technique over the symmetric reactor with two different electrolyte feeders, alkaline water at the anode and seawater at the cathode chambers, restricting Cl− ion diffusion towards the anode. Such a design not only protects the electrocatalyst at the anode, but also exhibits high and sustainable performance. However, it is important to note here that the approach may not be considered as the ultimate solution, as it offers 50% use of seawater combined with 50% of alkaline fresh water.
Even though much effort has been focused on the seawater electrolysis, the scaled up production of hydrogen by seawater electrolysis still remains a way far from the commercialization. The extensive market survey for a sustainable industrial hydrogen production plant from seawater indicates a target efficiency of 70–80% for the electrolyzer. Nevertheless, it is apparent that extensive studies in this field will be in high demand, as 97% of the earth is covered by seawater as a potential hydrogen source. The present review aims to provide not only the fundamental theories related to the seawater splitting, but also discussed the design of electrocatalysts for the selective anodic reactions followed by recent studies on rational electrolyzer designing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. R. gratefully acknowledges financial support from the Department of Science and Technology (DST/TMD/HFC/2k18/60), New Delhi, India and the Council of Scientific and Industrial Research (CSIR) for this work. S. K. thanks CSIR and University Grant Commission (UGC, India) for providing fellowship to carry out her PhD program.References
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS.
- J. O. Bockris, Science, 1972, 176, 1323 CrossRef CAS.
- A. Ursua, P. Sanchis and L. M. Gandia, Proc. IEEE, 2012, 100, 410–426 CAS.
- W. Tong, M. Forster, F. Dionigi, S. r. Dresp, R. S. Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- S. Fukuzumi, Y. M. Lee and W. Nam, ChemSusChem, 2017, 10, 4264–4276 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- J. Li and G. Zheng, Adv. Sci., 2017, 4, 1600380 CrossRef.
- S. r. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS.
- K. S. Exner, I. Sohrabnejad-Eskan and H. Over, ACS Catal., 2018, 8, 1864–1879 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., 2016, 55, 7501–7504 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Electrochim. Acta, 2014, 120, 460–466 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., 2014, 53, 11032–11035 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, ChemElectroChem, 2015, 2, 707–713 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- A. T. Kuhn and C. Y. Chan, J. Appl. Electrochem., 1983, 13, 189–207 CrossRef CAS.
- R. K. B. Karlsson and A. Cornell, Chem. Rev., 2016, 116, 2982–3028 CrossRef CAS.
- L. Hammar and G. Wranglen, Electrochim. Acta, 1964, 9, 1–16 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. G. Li, J. Z. Wu, J. Feng, M. Lanza and H. J. Dai, Science, 2013, 342, 836–840 CrossRef CAS.
- M. E. G. Lyons, A. Cakara, P. O'Brien, I. Godwin and R. L. Doyle, Int. J. Electrochem. Sci., 2012, 7, 11768–11795 CAS.
- S. r. Dresp, F. Dionigi, S. Loos, J. F. d. Araujo, C. Spöri, M. Gliech, H. Dau and P. Strasser, Adv. Energy Mater., 2018, 1800338 CrossRef.
- A. J. Esswein, Y. Surendranath, S. Y. Reece and D. G. Nocera, Energy Environ. Sci., 2011, 4, 499–504 RSC.
- F. Cheng, X. Feng, X. Chen, W. Lin, J. Rong and W. Yang, Electrochim. Acta, 2017, 251, 336–343 CrossRef CAS.
- J. B. Gerken, J. G. McAlpin, J. Y. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS.
- Y. Surendranath, M. Dinca and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS.
- S. Gao, G. D. Li, Y. Liu, H. Chen, L. L. Feng, Y. Wang, M. Yang, D. Wang, S. Wang and X. Zou, Nanoscale, 2015, 7, 2306–2316 RSC.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 1901333 CrossRef.
- Z. Wang, W. Xu, K. Yu, Y. Feng and Z. Zhu, Nanoscale, 2020, 12, 6176–6187 RSC.
- Y. Zhao, B. Jin, A. Vasileff, Y. Jiao and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 8117 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., 2014, 53, 102–121 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS.
- H. Liang, F. Meng, M. Caban-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- X. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Mater. Trans., 1997, 38, 899–905 CrossRef CAS.
- J. E. Bennett, Int. J. Hydrogen Energy, 1980, 5, 401–408 CrossRef CAS.
- M. Morita, C. Iwakura and H. Tamura, Electrochim. Acta, 1977, 22, 325–328 CrossRef CAS.
- M. Morita, C. Iwakura and H. Tamura, Electrochim. Acta, 1979, 24, 639–643 CrossRef CAS.
- K. Izumiya, E. Akiyama, H. Habazaki, A. Kawashima, K. Asami, K. Hashimoto and N. Kumagai, J. Appl. Electrochem., 1997, 27, 1362–1368 CrossRef CAS.
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Electrochim. Acta, 1998, 43, 3303–3312 CrossRef CAS.
- N. A. A. Ghany, N. Kumagai, S. Meguro, K. Asami and K. Hashimoto, Electrochim. Acta, 2002, 48, 21–28 CrossRef.
- N. A. A. Ghany, S. Meguro, N. Kumagai, K. Asami and K. Hashimoto, Mater. Trans., 2003, 44, 2114–2123 CrossRef.
- N. Jiang and H.-m. Meng, Surf. Coat. Technol., 2012, 206, 4362–4367 CrossRef CAS.
- K. Fujimura, T. Matsui, H. Habazaki, A. Kawashima, N. Kumagai and K. Hashimoto, Electrochim. Acta, 2000, 45, 2297–2303 CrossRef CAS.
- K. Fujimura, T. Matsuia, K. Izumiya, N. Kumagai, E. Akiyama, H. Habazaki, A. Kawashima, K. Asami and K. Hashimoto, Mater. Sci. Eng., A, 1999, 267, 254–259 CrossRef.
- A. A. El-Moneim, N. Kumagai and K. Hashimoto, Mater. Trans., 2009, 50, 1969–1977 CrossRef CAS.
- H. Habazaki, T. Matsui, A. Kawashima, K. Asami, N. Kumagai and K. Hashimoto, Scr. Mater., 2001, 44, 1659–1662 CrossRef CAS.
- T. Matsui, H. Habazaki, A. Kawashima, K. Asami, N. Kumagai and K. Hashimoto, J. Appl. Electrochem., 2002, 32, 993–1000 CrossRef CAS.
- A. A. El-Moneim, N. Kumagai, K. Asami and K. Hashimoto, Mater. Trans., 2005, 46, 309–316 CrossRef CAS.
- A. A. El-Moneim, J. Bhattarai, Z. Kato, K. Izumiya, N. Kumagai and K. Hashimoto, ECS Trans., 2010, 25, 127–137 CrossRef CAS.
- Z. Kato, M. Sato, Y. Sasaki, K. Izumiya, N. Kumagai and K. Hashimoto, Electrochim. Acta, 2014, 116, 152–157 CrossRef CAS.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. M. Koper, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS.
- H. A. Hansen, I. C. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Angew. Chem., 2010, 49, 4813–4815 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, e1802880 CrossRef.
- D. W. Kirk and A. E. Ledas, Int. J. Hydrogen Energy, 1982, 7, 925–932 CrossRef CAS.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef.
- M. M. Jakšić, Electrochim. Acta, 1984, 29, 1539–1550 CrossRef.
- H. Li, Q. Tang, B. He and P. Yang, J. Mater. Chem. A, 2016, 4, 6513–6520 RSC.
- J. Zheng, Y. Zhao, H. Xi and C. Li, RSC Adv., 2018, 8, 9423 RSC.
- M. C. Galetz, B. Rammer and M. Schuetze, Mater. Corros., 2015, 66, 1206 CrossRef CAS.
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS.
- J. Miao, F. X. Xiao, H. B. Yang, S. Y. Khoo, J. Chen, Z. Fan, Y. Y. Hsu, H. M. Chen, H. Zhang and B. Liu, Sci. Adv., 2015, 1, e1500259 CrossRef.
- I. P. Chen, C. H. Hsiao, J. Y. Huang, Y. H. Peng and C. Y. Chang, ACS Appl. Mater. Interfaces, 2019, 11, 14159–14165 CrossRef CAS.
- S. Wang, H. Ge, S. Sun, J. Zhang, F. Liu, X. Wen, X. Yu, L. Wang, Y. Zhang, H. Xu, J. C. Neuefeind, Z. Qin, C. Chen, C. Jin, Y. Li, D. He and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4815–4822 CrossRef CAS.
- H. Jin, X. Liu, A. Vasileff, Y. Jiao, Y. Zhao, Y. Zheng and S.-Z. Qiao, ACS Nano, 2018, 12, 12761–12769 CrossRef CAS.
- L. Yu, L. Wu, S. Song, B. McElhenny, F. Zhang, S. Chen and Z. Ren, ACS Energy Lett., 2020, 5, 2681–2689 CrossRef CAS.
- L. Xiu, W. Pei, S. Zhou, Z. Wang, P. Yang, J. Zhao and J. Qiu, Adv. Funct. Mater., 2020, 1910028 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev., 2017, 1, 0003 CAS.
- Y. Zhao, B. Jin, Y. Zheng, H. Jin, Y. Jiao and S.-Z. Qiao, Adv. Energy Mater., 2018, 1801926 CrossRef.
- S.-H. Hsu, J. Miao, L. Zhang, J. Gao, H. Wang, H. Tao, S.-F. Hung, A. Vasileff, S. Z. Qiao and B. Liu, Adv. Mater., 2018, 30, 1707261 CrossRef.
- S. Dresp, T. N. Thanh, M. Klingenhof, S. Bruckner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- S. Ravichandran, R. Balaji, B. S. Kannan, S. Elamathi, D. Sangeetha, J. Lakshmi, S. Vasudevan and G. Sozhan, ECS Trans., 2011, 33, 157–166 CrossRef.
- R. d'Amore-Domenech and T. J. Leo, ACS Sustainable Chem. Eng., 2019, 7, 8006–8022 CrossRef.
- S. Dresp, F. Luo, R. Schmack, S. Kühl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020–2024 RSC.
| This journal is © The Royal Society of Chemistry 2021 |