
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrathiafulvalene-based covalent organic frameworks for ultrahigh iodine capture†
Jianhong
Chang‡
a,
Hui
Li‡
a,
Jie
Zhao
b,
Xinyu
Guan
a,
Cuimei
Li
c,
Guangtao
Yu
 cd,
Valentin
Valtchev
ef,
Yushan
Yan
g,
Shilun
Qiu
a and
Qianrong
Fang
*a
cd,
Valentin
Valtchev
ef,
Yushan
Yan
g,
Shilun
Qiu
a and
Qianrong
Fang
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: qrfang@jlu.edu.cn
bSINOPEC Research Institute of Petroleum Processing, P. R. China
cLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, P. R. China
dCollege of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350007, P. R. China
eQingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, 189 Songling Road, Laoshan District, Qingdao, Shandong 266101, P. R. China
fNormandie Univ., ENSICAEN, UNICAEN, CNRS, Laboratoire Catalyse et Spectrochimie, 6 Marechal Juin, 14050 Caen, France
gDepartment of Chemical and Biomolecular Engineering, Center for Catalytic Science and Technology, University of Delaware, Newark, DE 19716, USA
First published on 13th May 2021
Abstract
To safeguard the development of nuclear energy, practical techniques for capture and storage of radioiodine are of critical importance but remain a significant challenge. Here we report the synergistic effect of physical and chemical adsorption of iodine in tetrathiafulvalene-based covalent organic frameworks (COFs), which can markedly improve both iodine adsorption capacity and adsorption kinetics due to their strong interaction. These functionalized architectures are designed to have high specific surface areas (up to 2359 m2 g−1) for efficient physisorption of iodine, and abundant tetrathiafulvalene functional groups for strong chemisorption of iodine. We demonstrate that these frameworks achieve excellent iodine adsorption capacity (up to 8.19 g g−1), which is much higher than those of other materials reported so far, including silver-doped adsorbents, inorganic porous materials, metal–organic frameworks, porous organic frameworks, and other COFs. Furthermore, a combined theoretical and experimental study, including DFT calculations, electron paramagnetic resonance spectroscopy, X-ray photoelectron spectroscopy, and Raman spectroscopy, reveals the strong chemical interaction between iodine and the frameworks of the materials. Our study thus opens an avenue to construct functional COFs for a critical environment-related application.
Introduction
Nuclear power is one of the most important energy sources due to its high power density and low carbon emission.1,2 However, an urgent safety issue related to nuclear power is the administration of waste produced from nuclear fission.3,4 Radioiodines (129I and 131I) are typical pollutants that accompany nuclear fission, and attract a particular attention. 129I has a half-life of ∼17 million years and is a concern for repository and environmental conditions, and 131I has a half-life of 8 days and is of grave concern for human metabolic processes.5 For example, radioactive iodine that leaked out from the Chernobyl nuclear accident has entered the human metabolic system and resulted in human diseases like thyroid cancer. To safeguard the future development of atomic energy, efficient strategies for adsorption and removal of radioiodine are necessary but remain elusive.The traditional technology to remove radioactive iodine is based on a chemical transformation, that is, using silver-doped adsorbents to produce harmless AgI; however, these materials show a low efficiency. For instance, their theoretical and practical adsorption capacity is 1.18 g g−1 and 0.10–0.31 g g−1 (per gram Ag), respectively, which is far from meeting practical needs.6 In recent years, porous materials including inorganic porous materials,7,8 metal–organic frameworks (MOFs),9,10 and amorphous porous organic frameworks (POFs)11,12 have been shown to be a suitable choice for the removal of radioiodine. In particular, covalent organic frameworks (COFs),13–21 as a unique class of crystalline organic polymers with ordered porous channels, have been studied extensively for the uptake of iodine. These COF materials with specific pore environments and tunable chemistry can be easily functionalized to obtain active sites for more efficient iodine capture by electrostatic forces, Lewis acid–base interactions and coordinative sites. Nevertheless, most studies are still limited to exploitation of physical interaction, i.e., iodine uptake capacity in porous materials is driven by pore volume and uptake kinetics is dominated by pore connectivity and size. For example, although a state-of-the-art COF, TPB-DMTP COF, has achieved remarkable iodine adsorption capacity (6.26 g g−1), its adsorption kinetics is quite slow (0.13 g g−1 h−1).22 In principle, the combination of high physisorption and strong chemical interaction in a material will be beneficial to improving both iodine adsorption capacity and adsorption kinetics. However, this avenue is still not properly explored.
Herein, we report the synergistic effect of physical and chemical iodine adsorption in tetrathiafulvalene (TTF)-based COFs, JUC-560 (JUC = Jilin University China) and JUC-561, leading to ultrahigh iodine adsorption capacity and ultrafast adsorption kinetics. These architectures are designed to have large specific surface areas (1815 m2 g−1 for JUC-560 and 2359 m2 g−1 for JUC-561) for high iodine uptakes through the physical process, and plentiful TTF functional groups for powerful iodine chemisorption. As a result, JUC-561 exhibits an unprecedented iodine uptake of 8.19 g g−1 and adsorption kinetics of 0.70 g g−1 h−1 at 75 °C under ambient pressure. These values are much higher than those of other materials reported so far, including silver-doped adsorbents, inorganic porous materials, MOFs, POFs, and other COFs. The strong interaction between the COF framework and iodine has been probed by DFT calculations on model molecules, TTF·I2, TTF+·I2, TTF+·(I−)2, TTF+·(I3−), and TTF+·(I5−), and confirmed by experimental studies including X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) spectroscopy, and Raman spectroscopy.
Results and discussion
To achieve high iodine adsorption, we introduced a TTF-based derivative, tetrathiafulvalene-tetrabenzaldehyde (TTF-TBA, Fig. 1b), as a planar 4-connected building unit. TTF or TTF-based derivatives are known to form radical cations with electron acceptors (e.g., I2).23 Therefore, by the incorporation of TTF-based units, the resulting COF materials will promote iodine adsorption based on this strong interaction. We further chose two typical monomers, 2′,5′-dimethyl-[1,1′:4′,1′′-terphenyl]-4,4′′-diamine (DTDA, Fig. 1a) as a linear building unit and 2,4,6-tris(4-aminophenyl)amine (TAPA, Fig. 1c) as a 3-connected building unit. Thus, we developed two novel structures, two-dimensional (2D) JUC-560 with sql topology24 and three-dimensional (3D) JUC-561 with ffc topology (Fig. 1d–i),25 constructed from the condensation of TTF-TBA and linear DTDA or non-coplanar 3-connected TAPA (Fig. S1, ESI†), respectively.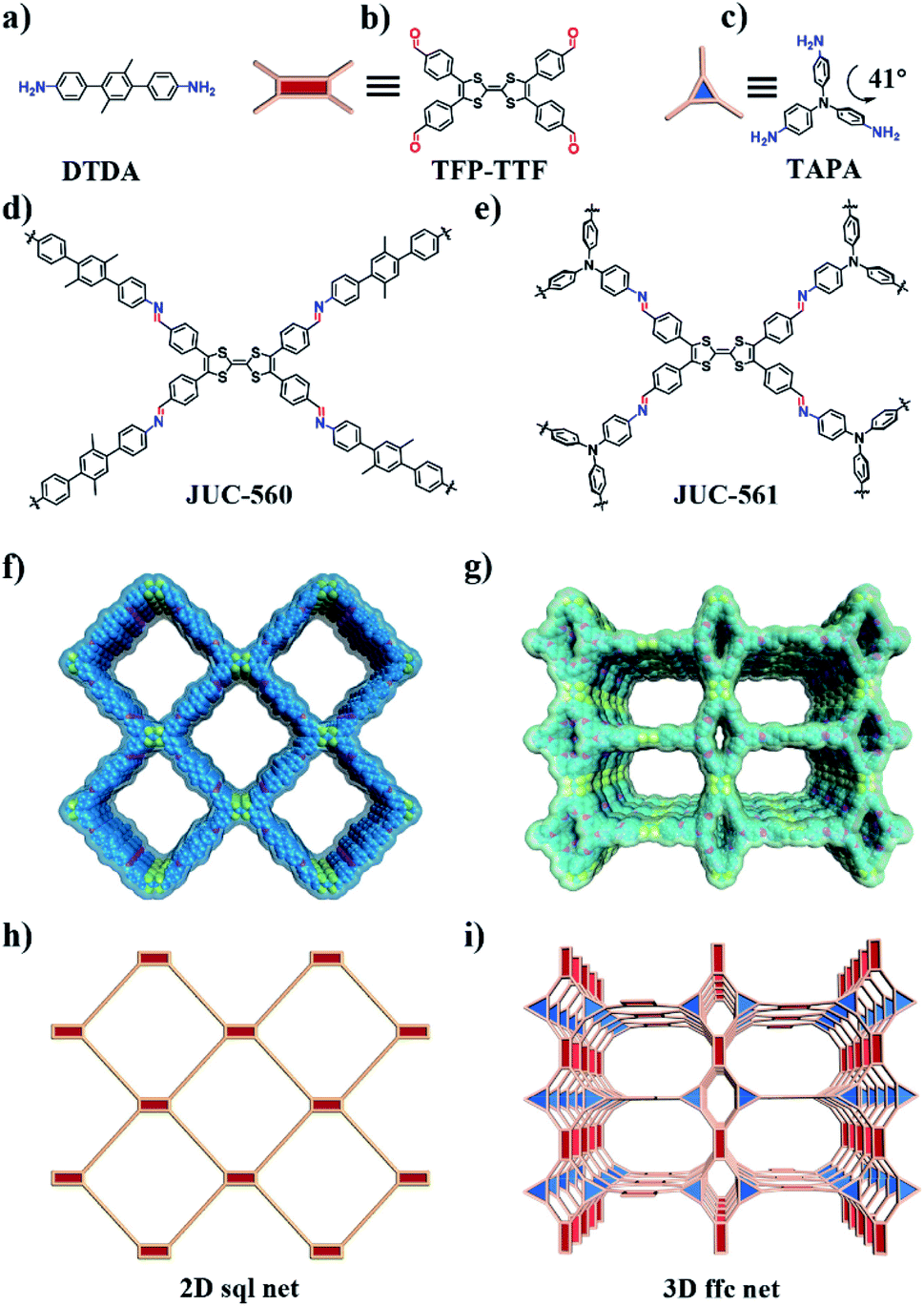 | ||
| Fig. 1 Schematic representation of the strategy for preparing TTF-based COFs. (a–c) Molecular structures of DTDA as a linear building unit, TFP-TTF as a 4-connected building unit, and TAPA as a 3-connected non-coplanar building unit. (d and e) Two novel TFF-based COFs, denoted as JUC-560 and JUC-561, are constructed by the condensation reaction of TTF and DTDA or TAPA. (f and g) Extended structures of mesoporous 2D JUC-560 (f) and 3D JUC-561 (g). (h and i) 2D sql and 3D ffc net for JUC-560 (h) and JUC-561 (i) respectively. | ||
The targeted COFs were synthesized by suspending TFP-TTF and DTDA or TAPT in a mixture of mesitylene/1,4-dioxane or o-dichlorobenzene/n-butanol in the presence of acetic acid, followed by heating at 120 °C for 3 days (Fig. S2–S9, ESI†). JUC-560 shows powder X-ray diffraction (PXRD) peaks at 2.69°, 3.48°, 5.38°, 5.60°, and 8.07° for 2θ, which are assigned to the (110), (200), (220), (310), and (330) facets, respectively. This structure adopts a 2D sql net with an eclipsed AA-stacking mode that yields a PXRD pattern similar to the experimentally observed one (Fig. 2a and S10–S12, ESI†). On the other hand, JUC-561 exhibits PXRD peaks at 1.99°, 3.21°, 4.01°, 5.20°, 6.06°, 6.89°, 7.77°, 8.44°, and 22.00° for 2θ, which correspond to the (020), (001), (040), (041), (060), (061), (042), (110), and (313) facets, respectively. JUC-561 assumes a 3D ffc net that exhibits a PXRD profile that is consistent with the experimental one (Fig. 2b and S13–S16, ESI†). PXRD patterns based on Pawley refinements26 also confirm the assignments of PXRD peaks, as evidenced by their negligible differences (residuals Rp = 1.31%, Rwp = 2.53% for JUC-560, and residuals Rp = 3.07%, Rwp = 4.51% for JUC-561; Tables S5–S9, ESI†). Based on these results, both TTF-based COFs have mesoporous frameworks with a diameter of about 2.62 nm for JUC-560 and about 2.55 nm for JUC-561.
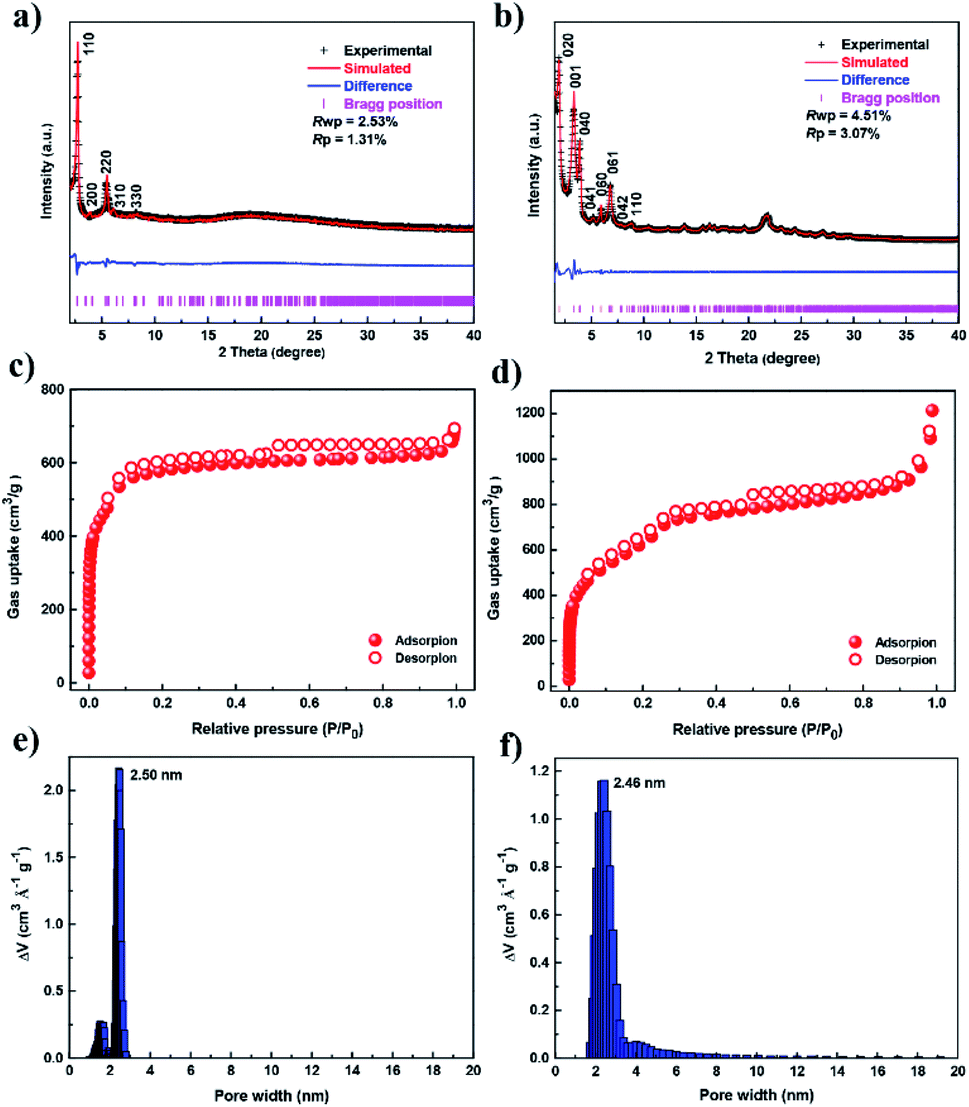 | ||
| Fig. 2 PXRD profiles and porosity. (a and b) PXRD patterns of JUC-560 (a) and JUC-561 (b). (c and d) N2 adsorption–desorption isotherms of JUC-560 (c) and JUC-561 (d) at 77 K. (e and f) Pore-size distribution of JUC-560 (e) and JUC-561 (f) calculated by fitting on the NLDFT model to the adsorption data. | ||
Both COFs were observed to be chemically stable in different organic solvents, including acetone, ethanol, cyclohexanone, hexane, tetrahydrofuran, N,N-dimethylformamide and dimethylsulfoxide. After being treated in these solvents for 24 h, JUC-560 and JUC-561 maintained their crystallinity (Fig. S17 and S18, ESI†). Furthermore, both COFs retained their structures in boiling water (100 °C) and even in a strong acid (3 M HCl, 25 °C) and strong base (3 M NaOH, 25 °C) for 24 h (Fig. S19 and S20, ESI†). According to the thermogravimetric analysis (TGA), these COFs did not decompose until 400 °C under nitrogen (Fig. S21 and S22, ESI†). Therefore, these TTF-based COFs are chemically and thermally stable, crystalline, porous materials.
The porosities of both COFs were determined by N2 adsorption and desorption isotherms at 77 K (Fig. 2c and d). For each COF, a sharp uptake below P/P0 = 0.05 and a step, indicating the presence of mesopores, was observed. The inclination of isotherms between P/P0 = 0.9–1.0 and slight desorption hysteresis is related to the presence of textural mesopores from the agglomeration of COF crystals.27–31 The Brunauer–Emmett–Teller (BET) specific surface areas were calculated to be 1815 m2 g−1 for JUC-560 and 2359 m2 g−1 for JUC-561, respectively (Fig. S23–S26, ESI†). The pore size distribution obtained by the nonlocal density functional theory (NLDFT) showed monomodal mesopores, 2.50 nm for JUC-560 and 2.46 nm for JUC-561 (Fig. 2e and f), which are in good agreement with those of the proposed model (2.62 nm for JUC-560 and 2.55 nm for JUC-561). Furthermore, the pore volume was determined to be 1.11 cm3 g−1 for JUC-560 and 1.92 cm3 g−1 for JUC-561, matching well with the simulated ones (1.16 cm3 g−1 for JUC-560 and 2.04 cm3 g−1 for JUC-561).
We conducted the iodine adsorption experiment by exposing COFs to iodine vapor at 75 °C under ambient pressure with the humidity concentration of 38%. JUC-560 exhibited a quick iodine uptake, showing a nearly linear increment in 7 h and then reaching adsorption saturation within 14 h (Fig. 3a). Similarly, JUC-560 showed a rapid adsorption in 8 h and achieved saturation in 18 h (Fig. 3b). JUC-560 showed iodine uptakes as high as 5.20 g g−1. Remarkably, JUC-561 showed much higher iodine uptakes of 8.19 g g−1 due to its 3D framework with higher specific surface areas and interconnected channels. On the basis of the pore volume together with the iodine density (4.93 g cm−3), their theoretical capacities (4.93 g cm−3 × pore volume) upon the full occupation of pores by iodine are 5.47 g g−1 for JUC-560 and 9.47 g g−1 for JUC-561 (Table S1, ESI†). The channels of both COFs are almost entirely occupied by iodine (95% for JUC-560 and 86% for JUC-561 based on their theoretical maxima), which is consistent with the high weight lost (Fig. S27 and S28, ESI†) for iodine-loading samples.
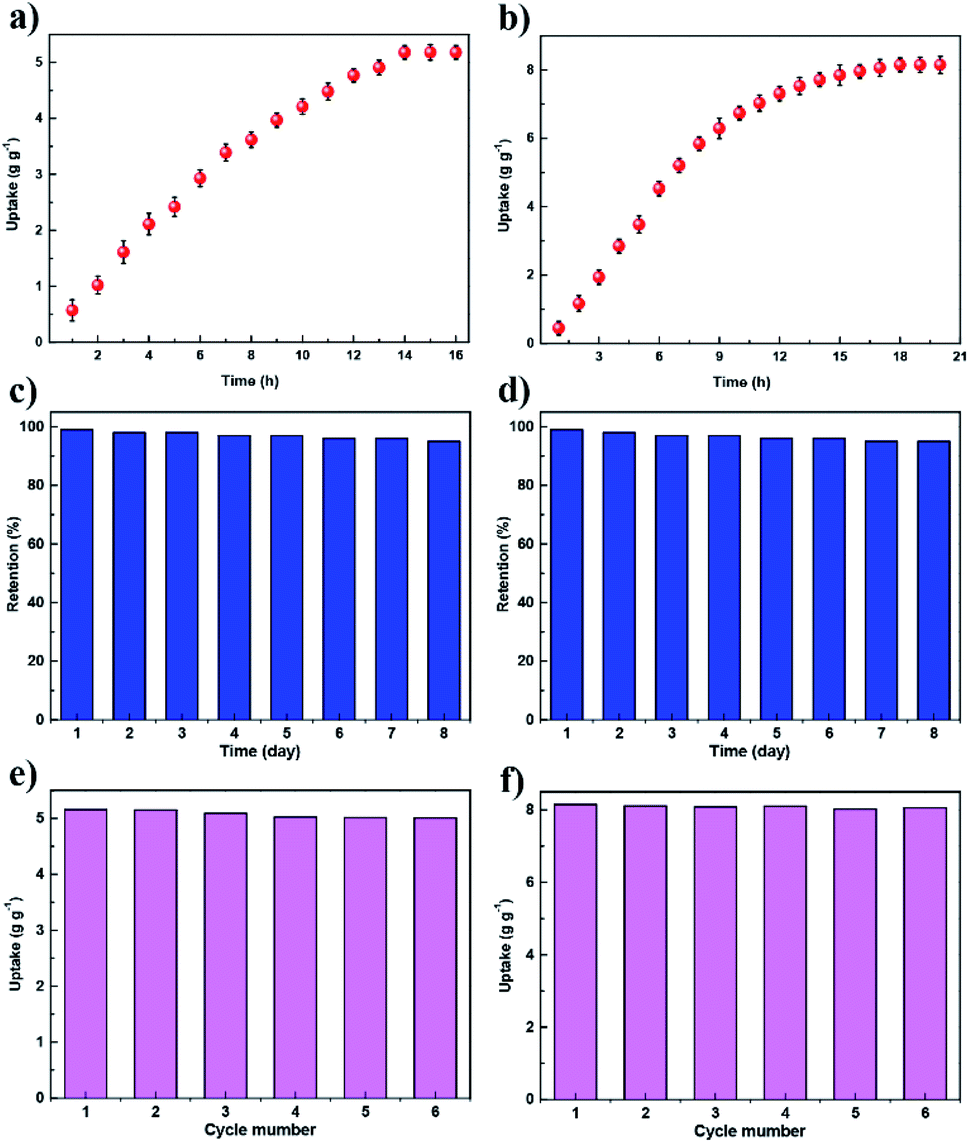 | ||
| Fig. 3 Study of iodine capture. (a and b) Uptake of iodine of JUC-560 (a) and JUC-561 (b) as a function of exposure time at 75 °C and ambient pressure. (c and d) Iodine retention of the iodine-captured JUC-560 (c) and JUC-561 (d) upon exposure to air at 25 °C and ambient pressure. (e and f) Recyclability of JUC-560 (e) and JUC-561 (f) in iodine adsorption. | ||
We summarized the iodine uptakes of typical adsorbents in Fig. 4 and Table S2, ESI†. As can be seen, the adsorption capacity of JUC-561 (8.19 g g−1) is far superior to those reported previously, such as 29-fold higher than that of the typical silver-doped zeolite mordenite (Ag-MOR, 0.28 g g−1),32 two orders of magnitude higher than those of nonporous materials, and much better than those of representative porous materials, including zeolitic imidazolate framework-8 (ZIF-8, 1.20 g g−1),33 porous aromatic framework (PAF-24, 2.76 g g−1),34 azo-bridged porphyrin–phthalocyanine network (AzoPPN, 2.90 g g−1),35 covalent organic polymers (COP10, 3.80 g g−1),36 and a state-of-the-art COF (TPB-DMTP COF, 6.26 g g−1).22 Notably, although the specific surface area of JUC-561 (2358 m2 g−1) is lower than those of some of the reported porous materials, such as COF-320 (2400 m2 g−1), NiP-conjugated microporous polymer (NiP-CMP, 2630 m2 g−1), and porous carbon (Uassis-PC800, 3053 m2 g−1), its iodine adsorption is higher (COF-320, 4.00 g g−1;37 NiP-CMP, 2.02 g g−1;38 and Uassis-PC800, 2.25 g g−1)39 due to the synergistic effect of physical and chemical adsorption of iodine in JUC-561.
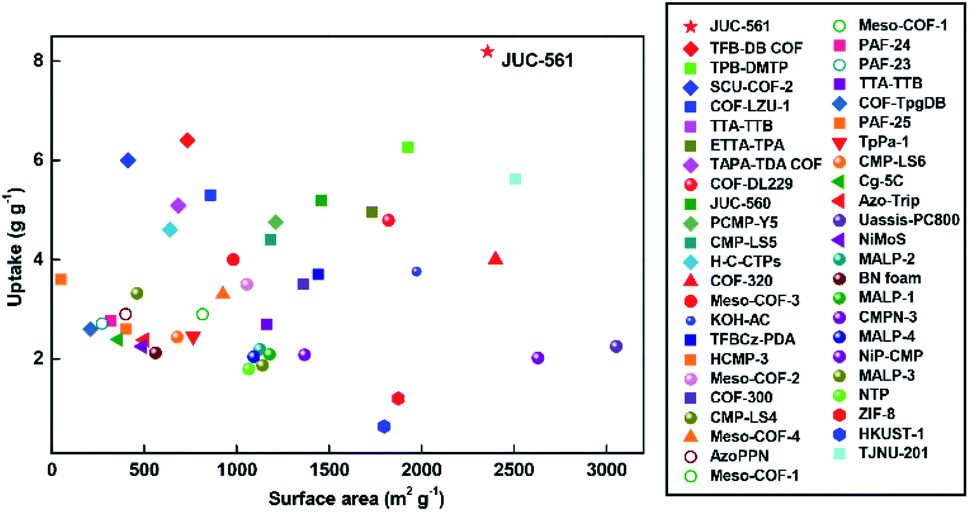 | ||
| Fig. 4 Comparison of iodine adsorption capacities against specific surface areas in different adsorbents. | ||
The adsorption kinetics of iodine in various materials are also summarized in Table S3, ESI.† In fact, the available relevant reports are limited due to low adsorption kinetics in most of the previously reported materials. Remarkably, the adsorption kinetics of TTF-based COFs (0.49 g g−1 h−1 for JUC-560 and 0.70 g g−1 h−1 for JUC-561) are superior to those of most of the adsorbents reported, such as the nitrogen-rich triptycene-based porous polymer (NTP, 0.11 g g−1 h−1),40 TTA-TTB COF (0.15 g g−1 h−1),22 NiP-CMP (0.17 g g−1 h−1),38 AzoPPN (0.18 g g−1 h−1),35 and even more than 34 times higher than that (0.02 g g−1 h−1) of Ag-loaded zeolites applied in real environments.32 These results confirm that the strong physical and chemical interaction between TTF units and iodine is favourable to enhance iodine adsorption ability.
The iodine-adsorbed COFs can preserve iodine upon exposure to air under ambient conditions (25 °C and 1 bar), and do not display iodine escape from the frameworks (Fig. 3c and d). Adsorbed iodine could be removed by immersing these samples in ethanol solution at room temperature. Iodine release was monitored through optical images and UV/Vis spectroscopy, leading to a linear increase based on the absorbance over time (Fig. S29–S36, ESI†). After ethanol rinse, these COFs were recyclable and retained similar iodine uptakes (Fig. 3e and f). FT-IR spectroscopy verified the chemical integrity of these COFs after cycled application (Fig. S37 and S38, ESI†). These regenerated COFs maintained their crystallinity and porosity, as evidenced by their unchanged PXRD patterns and retained specific surface areas (Fig. S39–S42, ESI†). These excellent cycling performances suggest that these materials are robust against oxidative iodine over long-term exposure.
Electron paramagnetic resonance (EPR) studies of the solid samples at 110 K confirmed the generation of TTF radical cations after iodine adsorption (Fig. 5a and b). Clearly, the pristine samples showed a very weak EPR signal (black curve) while there is an approximately two orders of magnitude increase in paramagnetic intensity for the iodine-doped samples (red curve). After I2 doping, sharp peaks were observed at g = 2.0182 for JUC-560 and 2.0189 for JUC-561 respectively, clearly demonstrating the presence of TTF radical cations from oxidation by iodine.41 Furthermore, we tested the electrochemical behaviour of JUC-560 and JUC-561 by cyclic voltammetry (CV). Two clear redox processes at 0.72 and 1.15 V vs. Ag/AgCl for JUC-560 and 0.69 and 1.08 V vs. Ag/AgCl for JUC-561 were observed in the reversible CV profiles (Fig. S47 and S48, ESI†), revealing that TTF units retain their redox activities in the frameworks and can be oxidized by iodine which has a reduction potential at 0.78 V.42
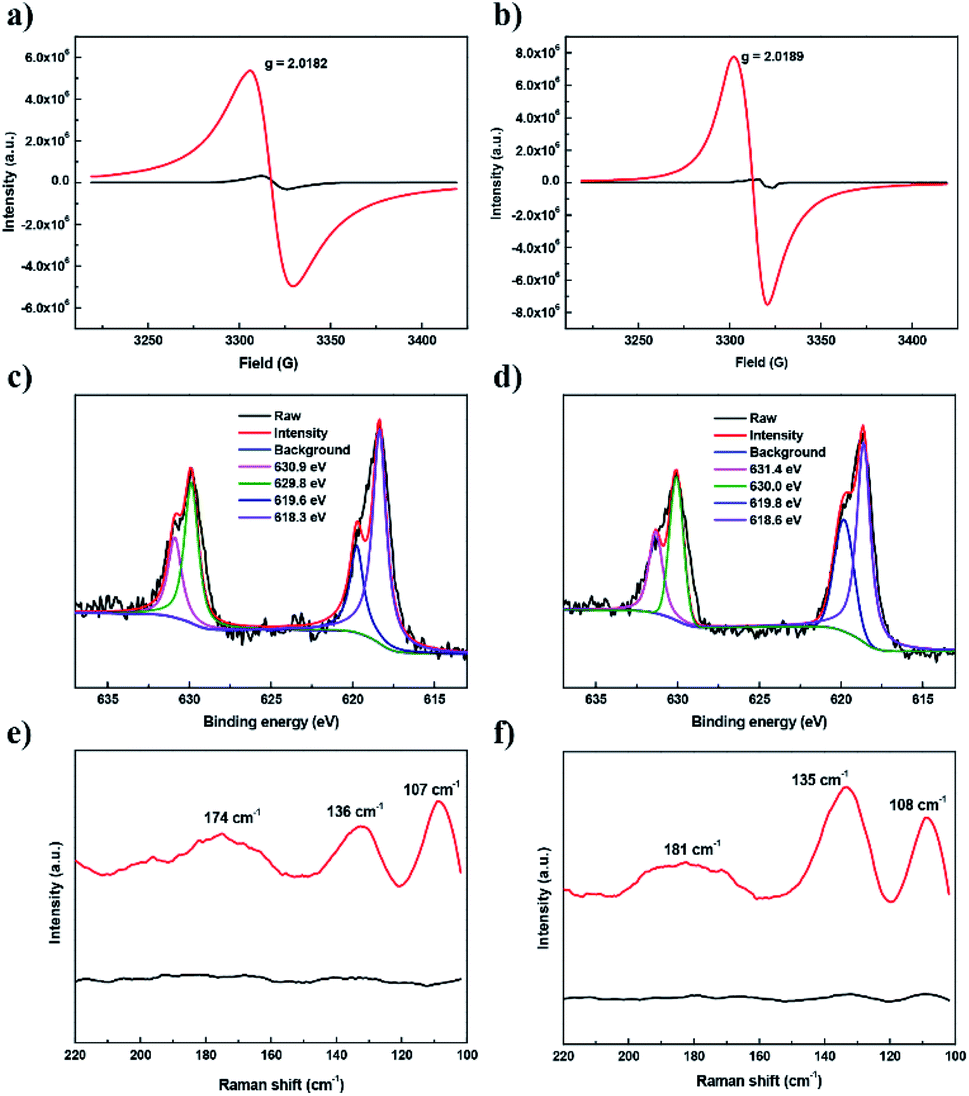 | ||
| Fig. 5 Spectroscopy study. (a and b) EPR spectra of JUC-560 (a) and JUC-561 (b) before (black curve) and after (red curve) iodine uptake. (c and d) XPS of iodine for JUC-560 (c) and JUC-561 (d) after iodine uptake. (e and f) Raman spectra of JUC-560 (e) and JUC-561 (f) before (black curve) and after (red curve) iodine uptake. | ||
To investigate the existing state of iodine captured in TTF-based COFs, we conducted X-ray photoelectron spectroscopy (XPS). In Fig. 5c and d, two conspicuous peaks located at 629.8 eV and 618.3 eV for JUC-560 as well as 630.0 eV and 618.6 eV for JUC-561, belonging to I 3d3/2 and I 3d5/2 orbitals of iodine molecules respectively, were observed, which indicates that the captured iodine partially exists as a molecule. Furthermore, another two peaks at 630.9 eV and 619.6 eV for JUC-560 as well as 631.4 eV and 619.8 eV for JUC-561 were found, which are attributed to the formation of polyiodine anions (such as I3− and I5−).43 In addition, the species of captured iodine were detected by Raman spectroscopy. As shown in Fig. 5e and f, the Raman spectra of TTF-based COFs showed no distinct peaks before iodine adsorption (black curve). However, after iodine capture, the low-frequency spectral region was dominated by three peaks at 107, 136 and 174 cm−1 for JUC-560 and 108, 135 and 181 cm−1 for JUC-561 (red curve). The peaks can be assigned to the symmetric and asymmetric stretching vibrations of I3− as well as the stretching vibrations of I5−, respectively.44 Thus the results show that the adsorbed iodine species in TTF-based COFs exist as both iodine molecules and polyiodine anions, and the process is the combination of physisorption and chemisorption.
To gain insight into how the COF frameworks bind iodine, we performed DFT calculations with the M06-2X-D3 functional based on the Gaussian 09 package (Revision D.01).45 Full geometry optimizations of model compounds, TTF·I2, TTF˙+·I2, TTF˙+·(I−), TTF˙+·(I3−), and TTF˙+·(I5−), offered related systems with the I2 molecule, and I−, I3− or I5− anions situated at the centre of the TTF or TTF˙+ unit (Fig. 6 and Table S4, ESI†). No symmetry constraints were imposed during the optimizations. The standard def2-SVP basis set was used for geometry optimizations, and the vibration analysis and def2-TZVP basis set were applied for calculating single point energy. The binding energy of the I2 molecule to the TTF or TTF˙+ unit calculated at the M06-2X-D3 level was low (−8.3 kcal mol−1 and −7.1 kcal mol−1 respectively). However, the association of the I− anion with the TTF˙+ unit showed a large binding energy as high as −74.6 kcal mol−1, which is the result of the electrostatic interaction between the anions and the positively charged viologen. Similarly, the I3− or I5− anion also displayed a relatively high binding energy of −70.6 kcal mol−1 or −65.3 kcal mol−1 to the TTF˙+ unit, respectively. Thus, these results suggest that TTF units in these COFs are subjected to a charge-transfer from TTF to iodine, forming oxidized TTF+ radical-cations and (poly-)iodides, which at last are tightly bound to the cationic framework through electrostatic interactions.
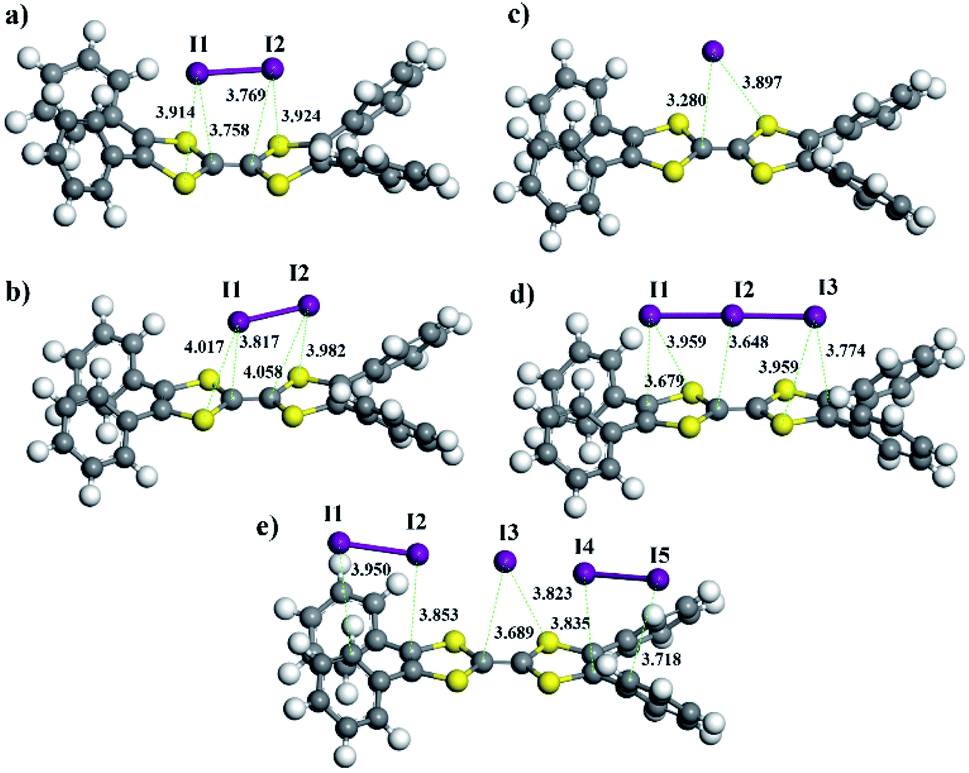 | ||
| Fig. 6 Optimized geometries of TTF·I2 (a), TTF˙+·I2 (b), TTF˙+·I− (c), TTF˙+·I3− (d), and TTF˙+·I5− (e). | ||
Conclusions
By developing TTF-based COF materials, we have successfully obtained high iodine adsorption capacity and quick iodine adsorption kinetics. These results point out that the integration of virtues of physical and chemical adsorption is of paramount importance in the designed synthesis of porous materials for the very challenging capture of radioiodine. The pre-designed COF frameworks exhibiting synergy between physisorption and chemisorption showed a dual impact on the iodine adsorption: substantially improved total iodine capacity, in particular the 3D COF (JUC-561); and accelerated transport rate due to their strong interaction between functionalized units and iodine. Consequently, the TTF-based COFs achieved the highest iodine uptake (∼8.19 g g−1) and adsorption kinetics (∼0.70 g g−1 h−1), which are far superior to those of other materials reported so far, such as silver-doped adsorbents, inorganic porous materials, MOFs, POFs, and other COFs. The set of experimental results thus demonstrate the potential of functionalized COF materials as platforms for the removal of I2 ejected into the environment through environmental and accidental events.Author contributions
J. C. and H. L. performed the synthesis and characterization of COF samples and measurements of nitrogen and iodine adsorption. X. G., C. L. and G. Y. performed the DFT calculations. Q. F. led the overall design and direction of the project. Q. F., J. Z., V. V., Y. Y., and S. Q. prepared the manuscript with help from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the SINOPEC Research Institute of Petroleum Processing, the National Natural Science Foundation of China (22025504, 21621001, and 21390394), the “111” project (BP0719036 and B17020), the China Postdoctoral Science Foundation (2020TQ0118 and 2020M681034), and the Program for JLU Science and Technology Innovative Research Team. V. V., Q. F. and S. Q. acknowledge the Collaboration in the Framework of China-French Joint Laboratory “Zeolites”.Notes and references
- E. Kintisch, Science, 2005, 310, 1406 CrossRef CAS PubMed
.
- J. D. Vienna, Int. J. Appl. Glass Sci., 2010, 1, 309 CrossRef CAS
- R. C. Ewing and F. N. von Hippel, Science, 2009, 325, 151 CrossRef CAS PubMed
- N. R. Soelberg, T. G. Garn, M. R. Greenhalgh, J. D. Law, R. Jubin, D. M. Strachan and P. K. Thallapally, Sci. Technol. Nucl. Install., 2013, 2013, 702496 Search PubMed
- M. I. Ojovan and W. E. Lee, Phys. Chem. Glasses, 2005, 46, 7 CAS
- B. J. Riley, J. D. Vienna, D. M. Strachan, J. S. McCloy and J. L. Jerden Jr, J. Nucl. Mater., 2016, 470, 307 CrossRef CAS
- T. Hertzsch, F. Budde, E. Weber and J. Hulliger, Angew. Chem., Int. Ed., 2002, 41, 2281 CrossRef CAS
- L. Mohanambe and S. Vasudevan, Inorg. Chem., 2004, 43, 6421 CrossRef CAS PubMed
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed
- T. Hasell, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 14920 CrossRef CAS PubMed
- C. Pei, T. Ben, S. Xu and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179 RSC
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228 CrossRef CAS PubMed
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC
- K. Y. Geng, T. He, R. Y. Liu, S. Dalapati, K. T. Tan, Z. P. Li, S. S. Tao, Y. F. Gong, Q. H. Jiang and D. L. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS PubMed
- Y. Yusran, X. Y. Guan, H. Li, Q. R. Fang and S. L. Qiu, Natl. Sci. Rev., 2020, 7, 170–190 CrossRef CAS
- X. Y. Guan, F. Q. Chen, Q. R. Fang and S. L. Qiu, Chem. Soc. Rev., 2020, 49, 1357 RSC
- X. Y. Guan, H. Li, Y. C. Ma, M. Xue, Q. R. Fang, Y. S. Yan, V. Valtchev and S. L. Qiu, Nat. Chem., 2019, 11, 587 CrossRef CAS PubMed
- Y. Z. Liu, Y. J. Wang, H. Li, X. Y. Guan, L. K. Zhu, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, Chem. Sci., 2019, 10, 10815 RSC
- L. W. He, L. Chen, X. L. Dong, S. T. Zhang, M. X. Zhang, X. Dai, X. J. Liu, P. Lin, K. F. Li, C. L. Chen, T. T. Pan, F. Y. Ma, J. C. Chen, M. J. Yuan, Y. G. Zhang, L. Chen, R. H. Zhou, Y. Han, Z. F. Chai and S. A. Wang, Chem, 2021, 7, 699 CAS
- P. Wang, Q. Xu, Z. Li, W. Jiang, Q. Jiang and D. Jiang, Adv. Mater., 2018, 30, 1801991 CrossRef PubMed
- H. Li, J. Chang, S. Li, X. Guan, D. Li, C. Li, L. Tang, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 13324 CrossRef CAS PubMed
- http://rcsr.net/nets .
- Y. Lan, X. Han, M. Tong, H. Huang, Q. Yang, D. Liu, X. Zhao and C. Zhong, Nat. Commun., 2018, 9, 5274 CrossRef PubMed
-
Materials Studio ver. 7.0, Accelrys Inc., San Diego, CA, 2013 Search PubMed
- X. Y. Guan, Y. C. Ma, H. Li, Y. Yusran, M. Xue, Q. R. Fang, Y. S. Yan, V. Valtchev and S. L. Qiu, J. Am. Chem. Soc., 2018, 140, 4494 CrossRef CAS PubMed
- S. C. Yan, X. Y. Guan, H. Li, D. H. Li, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, J. Am. Chem. Soc., 2019, 141, 2920 CrossRef CAS PubMed
- Y. J. Wang, Y. Z. Liu, H. Li, X. Y. Guan, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, J. Am. Chem. Soc., 2020, 142, 3736 CrossRef CAS PubMed
- H. Li, J. H. Ding, X. Y. Guan, F. Q. Chen, C. Y. Li, L. K. Zhu, M. Xue, D. Q. Yuan, V. Valtchev, S. L. Qiu and Q. R. Fang, J. Am. Chem. Soc., 2020, 142, 13334 CrossRef CAS PubMed
- Y. C. Ma, Y. J. Wang, H. Li, X. Y. Guan, B. J. Li, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, Angew. Chem., Int. Ed., 2020, 59, 19633 CrossRef CAS PubMed
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897 CrossRef CAS PubMed
- D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398 CrossRef CAS PubMed
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733 CrossRef CAS PubMed
- H. Li, X. Ding and B. H. Han, Chem.–Eur. J., 2016, 22, 11863 CrossRef CAS PubMed
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Plataslglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361 RSC
- S. An, X. Zhu, Y. He, L. Yang, H. Wang, S. Jin, J. Hu and H. Liu, Ind. Eng. Chem. Res., 2019, 58, 10495 CrossRef CAS
- A. Sigen, Y. Zhang, Z. Li, H. Xia, M. Xue, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 8495 RSC
- K. Xiao, H. Liu, Y. Li, G. Yang, Y. Wang and H. Yao, Chem. Eng. J., 2020, 382, 122997 CrossRef CAS
- H. Ma, J. J. Chen, L. Tan, J. H. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039 CrossRef CAS
- R. Berridge, P. J. Skabara, C. Pozo-Gonzalo, A. Kanibolotsky, J. Lohr, J. J. W. McDouall, E. J. L. McInnes, J. Wolowska, C. Winder, N. S. Sariciftci, R. W. Harrington and W. Clegg, J. Phys. Chem. B, 2006, 110, 3140 CrossRef CAS PubMed
- P. H. Qi and J. B. Hiskey, Hydrometallurgy, 1993, 32, 161 CrossRef CAS
- S. Hsu, A. Signorelli, G. Pez and R. Baughman, Highly Conducting Iodine Derivatives of Polyacetylene: Raman, XPS and X-ray Diffraction Studies, J. Chem. Phys., 1978, 69, 106 CrossRef CAS
- D. K. L. Harijan, V. Chandra, T. Yoon and K. S. Kim, J. Hazard. Mater., 2018, 344, 576 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and characterization, optimization of molecular structures, SEM and TEM images, FT-IR spectra, solid-state 13C NMR spectra, TGA analysis, BET plots, stability test, PXRD patterns and structures, and unit cell parameters. See DOI: 10.1039/d1sc01742j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |