
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral drug discovery: preparing for the next pandemic
Catherine S.
Adamson
 a,
Kelly
Chibale
b,
Rebecca J. M.
Goss
c,
Marcel
Jaspars
d,
David J.
Newman
e and
Rosemary A.
Dorrington
*f
a,
Kelly
Chibale
b,
Rebecca J. M.
Goss
c,
Marcel
Jaspars
d,
David J.
Newman
e and
Rosemary A.
Dorrington
*f
aSchool of Biology, Biomedical Sciences Research Complex, University of St Andrews, St Andrews KY16 9ST, Scotland, UK
bDrug Discovery and Development Centre (H3D) and South African Medical Research Council Drug Discovery and Development Research Unit, Department of Chemistry and Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch 7701, South Africa
cSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife, UK
dMarine Biodiscovery Centre, Department of Chemistry, University of Aberdeen, Old Aberdeen, AB24 3DT, Scotland, UK
eRetired Chief, Natural Products Branch, NIH/NCI, Frederick National Laboratory, MD, USA
fDept. of Biochemistry and Microbiology, Rhodes University, Makhanda (Grahamstown), South Africa. E-mail: r.dorrington@ru.ac.za
First published on 1st February 2021
Abstract
Clinically approved antiviral drugs are currently available for only 10 of the more than 220 viruses known to infect humans. The SARS-CoV-2 outbreak has exposed the critical need for compounds that can be rapidly mobilised for the treatment of re-emerging or emerging viral diseases, while vaccine development is underway. We review the current status of antiviral therapies focusing on RNA viruses, highlighting strategies for antiviral drug discovery and discuss the challenges, solutions and options to accelerate drug discovery efforts.
 The Author Team during their inaugural Zoom meeting during Lockdown: Top (left to right): David Newman, Rebecca Goss and Gordon Cragg; Middle (left to right): Rosemary Dorrington, Catherine Adamson and Kelly Chibale; Bottom: Marcel Jaspars | Catherine Adamson is a Lecturer in Molecular Medicine, University of St Andrews, Scotland, UK. Catherine’s laboratory focuses on virology with special interest in antiviral drugs and strategies. Her internationally recognized work encompasses multiple important aspects of the drug discovery process including cell-based assay design; execution of high-throughput screens to discover antiviral compounds against novel viral targets; hit antiviral compound characterization; determination of lead compound mechanism-ofaction, antiviral activity, evolution of drug resistance, and understanding performance in clinical trials; and using antiviral compounds as chemical tools to provide new fundamental knowledge in virology to drive further drug discovery. Her work has included antiviral research targeting HIV-1, RSV, CMV and SARS-CoV-2. Kelly Chibale is a Professor of Organic Chemistry at the University of Cape Town (UCT) where he holds the Neville Isdell Chair in African-centric Drug Discovery & Development. He is also a Full Member of the UCT Institute of Infectious Disease & Molecular Medicine, a Tier 1 South Africa Research Chair in Drug Discovery, founding Director of the South African Medical Research Council (SAMRC) Drug Discovery & Development Research Unit at UCT and the Founder and Director of the UCT Drug Discovery and Development Centre (H3D). His main research focus is global health drug discovery. Rebecca Goss is Professor of Organic/Biomolecular Chemistry at the University of St Andrews. Her team’s research is on medicinally relevant bioactive compounds, particularly natural products, and in hyphenating Synthetic Biology – Synthetic Chemistry to enable access analogues of these compounds. The team also focus on discovering and developing enzymes as tools for synthesis, and working toward sustainable antibiotic production for Africa. She is founder of St Andrews Multidisciplinary Anti-infective Research and Therapeutics Centre (SMART Centre) and Scottish Health and Innovation Network for Entrepreneurship (SHINE). Marcel Jaspars is Professor of Organic Chemistry at the University of Aberdeen, Scotland, UK. His main research focus is on the discovery, characterisation, utilisation and biosynthesis of marine natural products. This forms the core of the marine biodiscovery pipeline, and Marcel has frequent contact with people operating at all stages of this pipeline, from the collection and identification of the organisms to their testing in whole animal models. Marcel has been active at national and international levels to develop the science, its applications/industrial uptake and associated policy involved in marine biodiscovery and biotechnology. He is a co-founder of GyreOx Therapeutics. David Newman retired as Chief, Natural Products Branch, NCI in January 2015, after 24 years at NCI (10 as Chief, plus an earlier 25 years in pharma mainly in the US) initially being responsible for collection of microbes and marine invertebrates (and plants from January 2005) under the NCI’s long standing program for bioactive agents from Nature. With Gordon Cragg, the NCI’s Open Repository program was established from 1993 for any researcher to use. He was trained (UK) as an analytical, organic, and then microbial chemist, David still publishes extensively, frequently with Gordon in spite of “both being nominally retired”. Rosemary Dorrington is Professor of Microbiology and holds and DST/NRF SARChI Chair of Marine Natural Products Research at the Rhodes University, South Africa and leads the SA-UK MRC Antimicrobial Drug Discovery Partnership Hub. Her main research focus is on bioactive secondary metabolites from marine invertebrates and their associated microbiota. In addition, Rosemary’s group also works on small insect RNA viruses and developing their potential applications for the discovery of broad-spectrum antiviral compounds. She is the current chair of the Tetravirus study Group of the International Committee for the Taxonomy of Viruses. |
Introduction
Viruses, and in particular, RNA viruses, dominate the World Health Organisation (WHO)'s current list of ten global health threats. The list includes AIDS caused by the Human Immunodeficiency virus (HIV), a global influenza pandemic, Dengue virus (DENV), and viral diseases caused by the Ebola virus (EBOV) and other high threat pathogens: various haemorrhagic fevers, Zika virus (ZIKV), Nipah virus, Middle Eastern Respiratory Syndrome coronavirus (MERS-CoV), Severe Acute Respiratory Syndrome coronavirus (SARS-CoV-1) and Disease X (“an unknown pathogen that could cause a serious epidemic”).1 Today Disease X is unquestionably COVID-19, caused by SARS-CoV-2, a coronavirus closely related to SARS-CoV-1 that almost certainly emerged as a zoonosis from horseshoe bats in China.2,3 While planning for an influenza pandemic is an ongoing global priority, the 2014 EBOV disease (EBVD) outbreak in West Africa and the emergence in 2015 of ZIKV-caused diseases in Brazil that spread rapidly to 84 countries around the world are a stark warning of the potential threat and the lack of preparedness for dealing with RNA virus outbreaks.4–6 The SARS-CoV-2/COVID-19 pandemic has further exposed the lack of antiviral compounds that can be rapidly mobilized and deployed for the treatment of re-emerging or emerging viral diseases.7 Here we review the current status of antiviral therapies, highlighting strategies for combatting EBOV-causing disease (EBOD) and COVID-19, targets for antiviral drug discovery and discuss the challenges, solutions and options to accelerate antiviral drug discovery efforts.Antiviral strategies
The development of antiviral therapies requires a fundamental understanding of the chemical biology of the virus and in particular its interaction with the host cell. As obligate parasites, all viruses are dependent on the cellular processes of their host cells and as such, share the basic features of their infectious lifecycle. For the purposes of this review the focus will be on EBOV and SARS-CoV-2 representing negative (−) and positive (+) sense single-stranded RNA viruses, respectively (Fig. 1).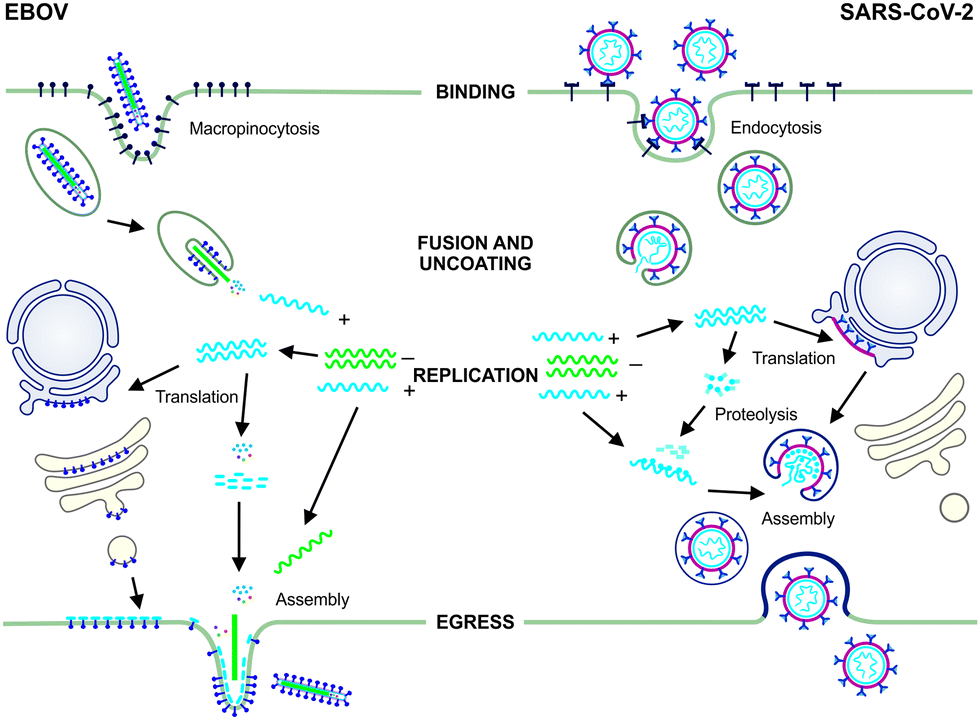 | ||
| Fig. 1 The infectious lifecycles of EBOV and SARS-CoV-2. EBOV binding to host cells occurs via interaction between the viral glycoproteins (GPs) and C-type lectins and cellular phosphatidylserine (PtdSer) receptors that interact with PtdSer in the viral envelope. The virus enters host cells by macropinocytosis and is trafficked through early and late endosomes, where acidification triggers fusion and uncoating followed by synthesis of the (+) genome and its release into thecytoplasm.8,9 EBOV replicates in cytoplasmic inclusion bodies.10 Using the host cell machinery, viral mRNAs encoding the GPs are translated into the endoplasmic reticulum (ER) and trafficked to the cell surface. Nucleocapsid proteins and non-structural proteins required for (+) RNA synthesis during infection, are translated in the cytoplasm and assembly of particles occurs at the cell surface.11 SARS-CoV-1 and likely SARS-CoV-2 binding and entry involves interaction between the virus surface spike (S) protein and the host angiotensin-converting enzyme 2 (ACE2) receptor. Proteolytic cleavage by host serine proteases mediates entry via endocytosis12 and the virus is trafficked within endosomes until fusion with mature lysosomes. This results in SARS-CoV-2 uncoating, fusion and release of the (+) RNA genome into the cytoplasm13 followed by translation of the viral replicase. Replication likely occurs in association with membrane-bound vesicles derived from the ER. mRNAs are translated into viral polyproteins that are cleaved into structural and non-structural proteins by viral proteases (3CLpro, also known as the main protease, Mpro, and PLpro).14 Assembly occurs in the intermediate compartment between the ER and Golgi with packaged virions transported to the cell surface and released via exocytosis to infect new cells.12 | ||
The strategies for combatting viral diseases are prevention (vaccines) and treatment (antiviral drugs and antibodies). More than 220 viruses are known to infect humans, 80% of which are naturally persistent in animals.15 In September 2019 there was a total of 87 clinically approved vaccines against 14 human viruses (mostly for influenza).16,17 The time “normally” required for the successful development of a vaccine against any disease be it bacterial, parasitic or viral is usually between 3 and 10 years and in many cases, longer. Not all vaccines that elicit protection against infection prove to be successful. The licenced Dengue fever vaccine, Dengvaxia®, led to antibody-dependent enhancement (ADE) of infection by different DENV serotypes in vaccinated patients18 and ZIKV vaccines have also been unsuccessful due to ADE. However, a significant milestone was reached in December 2019, when the US FDA approved the first EBOV vaccine that was extensively deployed to suppress a major EBVD outbreak in the Democratic Republic of the Congo (DRC).19 Still, while the first EBVD vaccine trials in primates had already begun in 2000, it took the 2014 West African outbreak to galvanize a global effort to develop an effective vaccine.
The declaration of COVID-19 as a Public Health Emergency of International Concern on January 30, 2020 has been followed by an unprecedented mobilisation of global resources to develop a protective vaccine for SARS-CoV-2, building on experience gained from the SARS-CoV-1 and MERS-CoV epidemics.20,21 However, developing a safe, effective vaccine for a rapidly-emerging, highly virulent novel pathogen like SARS-CoV-2, to stem a global pandemic is extremely challenging.22 Furthermore, producing the > 5.6 billion doses of a single dose vaccine to establish COVID-19 herd immunity world-wide will require an unprecedented, coordinated effort.
In the absence of an effective vaccine, the availability of safe and effective treatments is critically important in the early stages of an emerging, highly pathogenic, viral outbreak. The isolation and identification of neutralizing antibodies from blood samples donated by recovered patients provides opportunities for refining vaccine design and the development of antibody therapies, including neutralizing monoclonal antibodies and nanobodies (therapeutic proteins based on single-domain antibody fragments), for the treatment of COVID-19.23,24 There are numerous, monoclonal antibody therapies, including LyCoV555 (Eli Lilly) and REGN-COV2 (Regeneron) in clinical trials. But while early data show promise, the success of antibody therapies for SARS-CoV-2 will depend upon overcoming resistance through mutations in the S protein and the potential problem of ADE.24
Antiviral drug development can be considered a success story, but currently for only 10 human viral pathogens with almost half of these compounds being antiretrovirals.16,17 Typically the paucity of antiviral drugs stems from the fact that a number of key criteria are required to be met before substantial funding is invested into drug development programmes. First, there must be sufficient medical need, chiefly influenced by high disease incidence and severity. Investment can be influenced by the availability and effectiveness of a vaccine (although effective vaccine availability does not always preclude antiviral development). There must be sufficient technical knowhow and knowledge about the target virus to facilitate antiviral drug development. Importantly, economic incentives driven by high development costs and market value render diseases with a long duration, more financially attractive targets. It is thus no coincidence that the majority of clinically approved antiviral drugs target viruses that cause persistent and/or latent infections e.g. herpes viruses, HIV, Hepatitis C Virus (HCV) and Hepatitis B Virus. Influenza viruses, which cause acute respiratory infections, are the key exception. However, the threat posed by influenza pandemics is ever present and anti-influenza drugs are stockpiled by individual governments as part of their pandemic preparedness plans.4 Thus, with the exception of HIV/AIDS, Influenza and now COVID-19, most of the viral diseases on the WHO list of global health threats can be considered to be “neglected viral diseases”.25
Antiviral drugs and targets
The SARS-CoV-2 pandemic has brought the need for antiviral drug development into sharp focus, not only to respond to the current global health crisis but also to prepare for future outbreaks caused by other viruses for which antiviral drugs are unavailable. Antiviral drug development can be steered by building upon existing successful strategies. This section will review key antiviral targets that have led to clinically approved antivirals, using examples against herpes, HIV, HCV and influenza viruses and how these successful strategies can be applied to SARS-CoV-2 and EBOV drug discovery. The key drug classes discussed primarily target the entry, replication, proteolytic processing and particle egress steps of the infectious virus lifecycle (Fig. 1).Most approved antiviral drugs target a viral enzyme that plays an essential role in virus replication.16 Viral (DNA or RNA) polymerases are highly successful drug targets. Polymerase inhibitors can be divided into two broad classes (i) nucleos(t)ide analogues and (ii) non-nucleos(t)ide allosteric inhibitors. Allosteric inhibitors bind the polymerase, but not at the catalytic active site, causing conformational changes that impair polymerase function.16 Examples include the HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs), the most recent to obtain FDA approval being doravirine 1 (Fig. 2). Nucleos(t)ide analogue inhibitors are chemically synthesized purine and pyrimidine derivatives with chemical alterations in base or sugar components that abrogate the growing nucleotide chain during replication.16 AZT (azidothymidine) 2 (Fig. 2), the first anti-HIV nucleoside reverse transcriptase inhibitor (NRTI), is a thymidine analogue that contains an azide group in place of the usual nucleoside OH group, to which subsequent nucleosides cannot bind.16 Nucleos(t)ide analogue inhibitors are administered as prodrugs, which upon cell up-take are metabolized by host and/or viral kinases to their active triphosphate form. Acyclovir 3; an acyclic guanosine analogue (Fig. 2) used to treat Herpes simplex virus (HSV) infection, has high selectivity due to specific phosphorylation by the HSV-encoded thymidine kinase confining activity to virus-infected cells.16subsequent nucleosides cannot bind.16 Nucleos(t)ide analogue inhibitors are administered as prodrugs, which upon cell up-take are metabolized by host and/or viral kinases to their active triphosphate form. Acyclovir 3; an acyclic guanosine analogue (Fig. 2) used to treat Herpes simplex virus (HSV) infection, has high selectivity due to specific phosphorylation by the HSV-encoded thymidine kinase confining activity to virus-infected cells.16
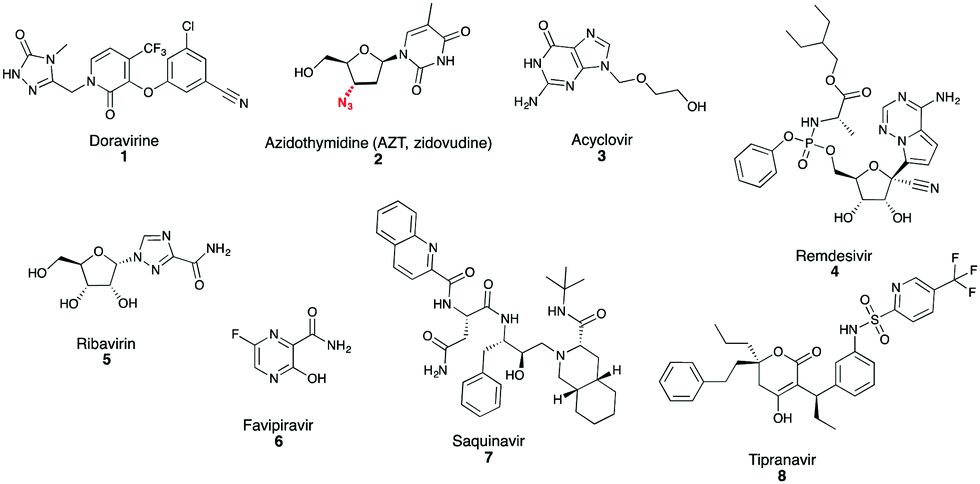 | ||
| Fig. 2 Representative examples of polymerase and protease inhibitors highlighted in this review. | ||
Development of an effective antiviral drug is typically followed by expansion of the successful strategy with numerous chemical variations providing improvements in parameters including affinity, pharmacology, toxicity and drug resistance profiles. Expansion of the successful structure(s) to other viruses beyond the initial clinical target is also possible, especially for polymerase inhibitors because all RNA viruses encode an RNA-dependent RNA polymerase (RdRp). Drugs targeting RdRps may have the capacity for activity against different viruses for which antiviral drugs are not currently available. Indeed, remdesivir 4 (Fig. 2), a monophosphate prodrug of a 1′-cyano adenosine nucleoside analogue, has in vitro and in vivo activity against multiple RNA virus families including filoviruses (EBOV) and coronaviruses (SARS-CoV-1, SARS-CoV-2, MERS-CoV)26–28 and has recently been granted FDA emergency use authorization for COVID-19 treatment.29 Another example of a broadly acting antiviral is the guanosine nucleoside analogue inhibitor ribavirin.30 Ribavirin 5 (Fig. 2) is often combined with other antiviral agents and is clinically approved e.g. against HCV, but is also used, unlicensed, against some viral hemorrhagic fevers.30 Also in this category is Favipiravir 6 (Fig. 2), a prodrug that is ultimately converted to its ribofuranosyl 5′-triphosphate metabolite (favipiravir-RTP), incorporated as a pseudopurine by the viral RdRp.25 Favipiravir is clinically approved in Japan for treating influenza, is also broadly active in vitro against other RNA viruses including SARS-CoV-1 and MERS-CoV21 and exhibits potential protective effects against EBOV in patients with low to moderately high viremia.5,25 Favipiravir is incorporated at high rates resulting in lethal mutagenesis of SARS-CoV-2 viral RNA during replication in vitro31 and the compound is currently in clinical trials for treating COVID-19.
Protease inhibitors are another major class of approved antiviral drugs developed primarily to treat HIV and then HCV infections. All approved HIV protease inhibitors are designed with a peptidomimetic hydroxyethylene scaffold e.g. Saquinavir 7, with the exception of tipranavir 8, which has a coumarin scaffold (Fig. 2).16 Coronaviruses encode two proteases, chymotrypsin-like protease (3CLpro, also known as the main protease Mpro) and papain-like protease (PLpro), which are very attractive antiviral targets for candidate antiviral molecules, particularly following the emergence of SARS-CoV-2.32,33 Repurposing of HIV-1 protease inhibitors (e.g. Lopinavir/Rotinavir) for the treatment of Covid-19, has been proposed on the basis of in vitro and animal model studies showing inhibitory activity against SARS-CoV-1, SARS-CoV-2 and MERS-CoV by targeting 3CLpro, however clinical trials do not support the use of Lopinavir/Ritonavir for the treatment of Covid-19 patients as no clinical benefit has been observed.34 Following the SARS-CoV-1 and MERS-CoV outbreaks, numerous 3CLpro and PLpro inhibitors have been identified. Whilst these inhibitors have not been developed clinically, they represent an excellent resource to kick-start the development of SARS-CoV-2 protease inhibitors, particularly because SARS-CoV-1 and SARS-CoV-2 3CLpro and PLpro share 96% and 83% sequence identity respectively.32,33 Further, It would be prudent to test candidate protease inhibitors against SARS-like coronaviruses identified in bats that have zoonotic potential, to enable the development of pan-coronavirus inhibitors to future proof against further coronavirus outbreaks. Polymerase inhibitors likely have greater capacity than protease inhibitors for broadly acting activity because not all viruses encode a protease; EBOV lacks a protease and thus protease inhibitors cannot be part of any future arsenal of anti-EBOV drugs.34
Other viral enzymes have been successfully targeted, but the resultant drug classes tend to be specific to a particular virus or related viruses. Important examples are the influenza neuraminidase (NA) inhibitors, oseltamivir, zanamivir and peramivir, which are the standard-of-care therapeutic treatment for influenza infection.4 NA inhibitors competitively target the NA active site, blocking cleavage of sialic acid residues, preventing influenza virus particle egress. In 2018, the first new anti-influenza drug class in two decades was approved for clinical use. The first-in-class drug, baloxavir marboxil, targets the polymerase acidic endonuclease activity of the influenza polymerase complex, inhibiting the mRNA cap-snatching mechanism.4 The highly conserved coronavirus helicase and the exonuclease nsp14, which play an essential role in viral RNA synthesis, are also attractive potential drug targets for SARS-CoV-2.36,37
Entry/fusion inhibitors are an important example of antiviral drugs that target non-enzymatic viral processes; for example three very different anti-HIV agents have been approved (i) Enfuvirtide, a biomimetic peptide that inhibits fusion,16 (ii) Maraviroc, an antagonist of the host cell co-receptor CCR5 required for HIV entry16 and (iii) Ibalizumab, a non-immunosuppressive humanized monoclonal antibody that acts as a post attachment inhibitor that binds the host CD4 primary receptor.38 Cocktails of therapeutic antibodies such as REGN-EB3 are being trialed against EBOV35 and the search is on for therapeutic antibodies against SARS-CoV-2.39 SARS-CoV-2 entry requires the S protein, which recognizes the host cell ACE2 receptor (Fig. 1) and is primed by host cell proteases (e.g. cathepsin L proteinase, furin, TMPRSS2), offering multiple potential antiviral strategies,33,40 indeed a clinically proven TMPRSS2 (protease) inhibitor, A1AT, has recently been shown to block SARS-CoV-2 entry.40
In addition to antiviral drugs that directly target host-virus interactions, host cell pathways, such as immunomodulatory drugs, are being used as indirect antiviral targets. Extensive discussion of this class of agent is beyond the scope of this review, but a classic example is interferon (IFN), which enhances the immune response and elicits an antiviral state. IFN is clinically approved for use in HCV and HBV infections and is being trialed in COVID-19 patients and a number of other immunomodulatory agents are being explored as anti-COVID-19 strategies.33,36 Importantly, the readily available and affordable immunosuppressant and anti-inflammatory drug, dexamethasone, reported by the Oxford University RECOVERY trial to reduce death in hospitalized COVID-19 patients with severe respiratory complications,41 is routinely being employed in patients in intensive care units.
A major challenge facing antiviral drug development is resistance. Indeed, drug resistance is a commonly reported issue affecting approved antiviral drugs that directly act against a viral target or virus-host interaction. Drug resistance is particularly problematic with respect to RNA viruses due to the error-prone nature of RdRps, the rapid rate of viral replication and frequent recombination events. Existing drug classes are subject to ongoing programs to improve the genetic barrier to resistance and the availability of drug classes with different mechanisms of action facilitates salvage or combination therapy. For example, HIV combination therapy consists of a minimum of two drugs from different mechanistic classes and typically contains three of more different drugs. This combination therapy does not provide a cure thus patients are required to take antivirals for life, increasing problems associated with drug resistance. Although not typically as challenging, drug resistance issues have also been documented for acute infections. Overall, problems with drug resistance significantly increase costs and time associated with antiviral drug development, exacerbating a lack of willingness to invest in neglected viral diseases.
Alternative antiviral strategies that target host cell factors and/or pathways required for virus replication are widely predicted to make acquisition of resistance less likely, but the trade-off is likely to be a higher chance of undesirable side effects.42,43 However this may not be as problematic for acute viral diseases, such as EBVD and Covid-9 since toxicity is often linked to the duration of treatment. Approaches targeting common cellular pathways exploited by multiple viruses offer the potential for development of broadly-acting antivirals that have activity across a family of viruses or ideally multiple virus families or viruses with a similar “life-style” e.g. respiratory viruses. The development of host-targeted antivirals has lagged behind that of directly-acting antivirals with very few, such as IFN, approved for clinical use. Hydroxquinolines are an example of a class of molecules under development, which like IFN, modulate immune responses to change infection outcome in favour of the host by activating the IFN pathway component IRF3.44 Several other host targets are being investigated for their utility to develop broadly acting antivirals, including cyclophilins, glucosidase, heat shock proteins, inosine-5′-monphospahte dehydrogenase, eukaryotic initiation factor 2α and various kinases.42 Ideally, targets should be host factors that are critical for the replication of several types of viruses but dispersible for the host. Significant further research is needed to identify the most appropriate cellular factors, pathways and processes to target. As discussed above, broadly-acting antivirals can also be developed against highly conserved viral targets, such as RdRps, which may have a high natural genetic barrier to drug resistance; e.g. coronavirus resistance to remdesivir has been reported, but is associated with an in vitro fitness cost and in vivo attenuation.45 Thus polymerase inhibitors are likely to be the best option for developing broad spectrum directly-acting antivirals.
Directing research effort and investment into the development of broadly-acting antivirals,46 rather than the contemporary one-virus-one-drug approach, is urgently needed to tackle the fact that antivirals are not currently available for hundreds of viruses that cause human disease and the conundrum of predicting which deadly virus will be the next to emerge into the human population. Development of broadly acting antivirals is very challenging requiring substantial, sustained research investment. Such an investment would be a shrewd response to the SARS-CoV-2 pandemic, which has starkly highlighted the importance of having antiviral drugs that can be rapidly deployed to treat a newly emerged viral disease.
Approaches to antiviral drug discovery
One of the main technological limitations of antiviral testing on emerging RNA viruses is the need for biosafety containment facilities (e.g. SARS-CoV-2 and EBOV require Category 3 and 4 containment levels, respectively). These facilities are expensive to build, commission and maintain and understandably require a high level of oversight. As a result, their global distribution is limited, reducing opportunities for testing compounds against live viruses. A high level of training and skill is required to ensure safe working inside these facilities and to prevent accidental infection of researchers and escape of viruses. It is essential that solutions are sought to these issues to provide a larger pipeline of potential antiviral candidates. One option is to use pseudoviruses that are safe to use in Category 2 facilities. Pseudoviruses are chimeric particles produced with envelope proteins from the target pathogenic virus displayed on the surface of a benign carrier virus (e.g. Vesicular Stomatitis Virus that can be useful for antiviral discovery with respect to inhibitors that act at entry/fusion steps of virus replication). Pseudoviruses may utilize the envelope proteins of the carrier virus for binding and entry, but retain the native functions required to initiate infection and viral replication of the target virus, which is detected via the activity of a reporter gene e.g. luciferase. Viral mini replicons expressing reporter genes, that do not produce infectious particles, are also alternatives for isolating replication inhibitors. The sensitivity of these ex vivo systems is particularly useful for screening natural product extracts to identify potential antiviral candidates to progress to phenotypic screening with live virus, an approach used by the NIH National Cancer Institute in the early 1990s with HIV.Phenotypic screening in host cells, however, reveals a great deal more about the targets and mechanism of action of assay compounds compared to high throughput assay technology. Throughput is being accelerated by improvements in automated image analysis amongst other approaches. An alternative approach is to test the effect of compounds in vitro on target viral proteins to determine if they can inhibit enzymes such as RdRps and proteases or block key protein–protein interactions. Recombinant viral proteins can safely be produced in larger amounts and used in a variety of assays using biophysical methods such as surface plasmon resonance.
Screening natural products for antiviral activity is particularly attractive given their biological relevance as well as intrinsic chemical and structural diversity. One way to introduce more natural products and/or derivatives into antiviral screening is to use a virtual screening approach against a number of different viral proteins. The approach is summarised in Fig. 3, starting with calculating minimised structures of natural product databases, followed by docking these against crystal structures of viral proteins. Using docking scores and calculated physicochemical parameters allows the prioritisation of compounds for antiviral assays and crystal soaking experiments to verify activity and to assess whether the docking calculations were accurate. The first step is to have a comprehensive database of all described natural products. Although many open-access and subscription databases have been created, most focus on specific types of compounds, or have not been maintained over a number of years. A recent attempt, ‘COlleCtion of Open NatUral producTs’ (COCONUT), contains structures and sparse annotations for over 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 non-redundant natural products.47
000 non-redundant natural products.47
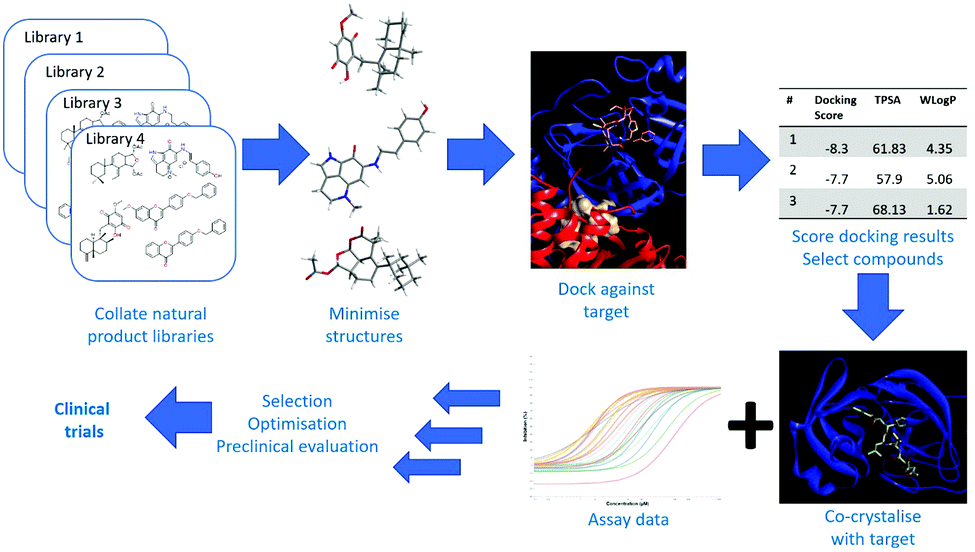 | ||
| Fig. 3 A possible route for virtual screening of natural product libraries followed by the prioritisation of hits based on assay, co-crystallisation and physicochemical data before structural optimisation, preclinical and clinical evaluation can occur. | ||
The ZINC database,48 containing commercially available compounds that have been minimised for docking simulations, has a subset of natural products. Docking of minimised natural product structures against X-ray structures of target proteins can be achieved using open source software such as AutoDock,49 which gives docking scores that can be used to select the best candidates, although it is good practice to visually inspect top candidates to check if they actually maintain essential interaction with target residues. There are issues around the conformational flexibility of the compounds and proteins, but these can be addressed using multiple conformers of the compounds or a fully flexible docking approach in which both the compound and protein are treated dynamically. Docking scores can be complemented by calculated physicochemical properties and predictions of whether compounds are likely to be absorbed in the gastrointestinal tract or whether they cross the blood–brain barrier.50 Care must also be taken to ensure that binding to the intended active site is stronger than potential alternative off-target binding sites. A recent publication reviews existing natural products active against several coronavirus targets and also presents an example of the virtual screening approach above to select natural products that may inhibit SARS-CoV-2 Mpro and have drug-like properties.51 A much larger scale effort is ‘deep-docking’ of the entire 1.3 billion ZINC library against SARS-CoV-2 Mpro, which gave a list of 1000 chemically diverse, commercially available compounds that have better docking scores than existing protease inhibitors.52 Large scale open projects such as the Covid Moonshot53 are attempting to use a combination of approaches including crystal structures of Mpro with drug-like ‘fragments’ to enable crowd-sourced drug design, rapid synthesis, followed by crystal soaking and structure determination of the protein–ligand complex and enzyme assays.
Challenges, solutions and options
Given that drug discovery and development is an inherently lengthy and costly undertaking, this becomes even more apparent when considering the high attrition rate of compounds during preclinical research. Furthermore, only three out of ten drugs that make it to the clinic recover their capital investment.54 Therefore, an efficient drug discovery pipeline is essential for ensuring profitability to sustain the business and maintain continued investment in R&D by the pharmaceutical industry. Furthermore, agitation for cost containment of drug supplies advocated by governments, commercial players and civil society activism can weigh heavily on profit margins. This partly explains why relatively speaking, infectious disease drug discovery is not prominent in the innovative pharmaceutical R&D industry. Within the context of antivirals, drug discovery efforts have targeted viruses that are economically important in developed countries. However, COVID-19 has reminded us that an emerging virus can wreck world economies and significantly disrupt life in unprecedented ways.As mentioned above, a dearth of clinically approved antivirals, focussed toward a very limited series of viruses are available. It is imperative that this precarious situation is addressed. Funding within the value chain of antiviral drug discovery and development, from blue skies research and preclinical drug discovery to clinical trials, is fragmented. The area of antiviral drug development needs to be viewed as a continuum so funding one component will not suffice and is therefore futile. There is limited funding of academic research projects targeting neglected viruses that can replenish the pipeline. Furthermore, the short-term nature of the funding means that promising molecules do not progress far along the value chain Thus funders should coordinate their activities to ensure that there is appropriate focus on each component of the value chain. For any small molecule drug discovery project regardless of approach (validated target- or phenotypic whole cell screening-based), the chemistry starting point is critical. With the same established screening approach not delivering much, there is an urgent need to find new sources of starting points. In this regard underexplored natural product sources provide an opportunity to find unique biologically-relevant starting points.
Fortunately, it is not all bad news as far as COVID-19 is concerned. The pandemic is catalysing and promoting collaboration and innovation in R&D between funding bodies from both the public and private sectors. An example of this is the COVID-19 Therapeutics Accelerator, first announced by the Bill and Melinda Gates Foundation (BMGF), which attracted additional funding from a range of governments, philanthropies and individuals who would not ordinarily fund infectious disease research. The second is the announcement of a partnership between the BMGF and 15 Pharma companies who have committed their expertise and assets to the fight against COVID-19. Needless to say, these focused efforts on COVID-19 could be a blueprint for preparedness against future outbreaks of yet-to-be identified or re-emerging infectious agents and could provide avenues to address unmet medical needs where an economic/business case for Pharma is absent. Also encouraging is the agreement between Gilead and Cipla to provide generic remdesivir at an affordable cost to developing countries e.g. South Africa.
An innovative pharmaceutical industry should view antivirals targeting neglected viruses as an opportunity to address an unmet medical need of public health significance. In addition, they present the opportunity to boost the pipeline of new medicines, given that the chances of discovering a novel druggable target are greater in this field, relative to the more widely explored therapeutic areas. A novel druggable target identified from a neglected virus could have a human equivalent in a lucrative therapeutic area where there are attractive commercial returns.55 Furthermore, collaborations between industry and academia in a precompetitive space would facilitate mutually beneficial sharing of knowledge in innovative strategies for improving drug discovery efficiency.56 The announcement of a partnership between the BMGF and 15 Pharma companies who have committed their expertise and assets to the fight against COVID-19 are excellent examples of this.
COVID-19 has been a real wake-up call particularly in Africa and has exposed the fundamental limitations of many sectors. The devastation caused by the 2014 EBVD outbreak is still fresh in people's minds. With sporadic outbreaks still being reported in countries like the DRC, there is genuine fear that COVID-19 will exacerbate the situation. The impact of COVID-19 on other infectious agents, e.g. those responsible for Ebola, malaria, tuberculosis and HIV/AIDS (and vice versa), is bringing renewed focus on the importance of not neglecting endemic infectious diseases given the risk of shifting resources away from other longstanding endemic diseases to fighting COVID-19. Historically, pharmaceutical R&D has mostly occurred in high-income countries, where technology, infrastructure and a critical mass of skilled scientists and clinicians are available. There is an urgent need for Africans to build drug discovery and development capacity in close proximity to African patient populations. For any developed vaccine or drug to benefit the African patient population in terms of treatment outcomes, there is therefore an urgent need to increase the number of clinical trials happening in Africa and the number of Africans enrolled into clinical trials. Amongst other interventions, this would require African governments to insist on local clinical trial data before a product can be licensed in their country. To make this a reality, there needs to be increased support for building capacity in clinical research.
From a resource mobilization perspective in Africa, Ebola has provided the necessary blueprint in view of the inequalities and high poverty levels on the continent. The 2014 outbreak exposed the poor health systems in West Africa, whose failure contributed to more deaths. COVID-19 has offered opportunities for funders, including Foundations and High Net Worth Individuals, to be innovative and collaborative even as they partner with governments to provide continent-specific solutions. For example, during the 2014 Ebola crisis a fund was set up through the African Union to fight the pandemic. Individuals as well as philanthropies could donate money through digital platforms and banking innovations.
Fexinidazole, the first oral treatment for African sleeping sickness, provides an excellent example for the antiviral field. During a search for compounds with anti-parasitic activity in 2005, the not-for-profit Drugs for Neglected Diseases initiative (DNDi,) in collaboration with the Swiss Tropical and Public Health Institute, rediscovered fexinidazole, a drug previously abandoned by its pharmaceutical industry developer, Hoechst (now Sanofi) in the 1980s. A formal partnership between the DNDi and Sanofi led to the development and subsequent approval of fexinidazole by the European Medicines Agency for the treatment of African sleeping sickness.by The drug is currently in use in the Democratic Republic of Congo.
Conclusions
SARS-CoV-2/COVID-19 pandemic has underscored the critical need for antiviral compounds that can be rapidly deployed when a previously neglected or new virus is abruptly catapulted to global emergency status. The one-virus-one-drug approach to antiviral discovery and development has been successful, but only if the viral disease satisfies the medical and economic criteria required to drive the substantial research effort, financial investment and political will, essential for delivering an effective clinically approved antiviral drug. The global campaign to find SARS-CoV-2 antivirals will undoubtedly result in the development innovative approaches to screening, accelerated clinical testing and rapid manufacturing scale-up to bring these compounds to the market as quickly as possible.Are we prepared for the next viral pandemic? The answer is still “no”. The catastrophic impact of the SARS-CoV-2 pandemic has the potential to divert attention away from other viruses that cause human disease and have the potential to cause the next pandemic. To mitigate this risk, there needs to be continued surveillance and modelling to support the prediction of which deadly virus will be the next to emerge into the human population. Of critical importance is directing research effort and investment into the development of broadly-acting antivirals that can be mobilised as the first line of defence upon emergence of new viral pathogens.
Developing antivirals to treat emerging viral diseases will require consideration of new consortium models, including public-private partnerships, exemplified by the product development partnership, DNDi, as well as open and precompetitive consortia models. The open consortium model would involve joint collaboration between the pharmaceutical industry, academic and public-funded organizations whose defining features would include consortium members collectively sharing the funding burden, resources and expertise. Alternatively, the precompetitive consortium model would involve industry partners, who ordinarily would be commercial competitors, and other non-industry partners in multidisciplinary collaborative research. Several precompetitive consortia have been formed in specialized healthcare-related sectors, including the recently established Bill and Melinda Gates Foundation-supported Tuberculosis Drug Accelerator, a ground-breaking partnership between several Pharma companies, a biotech, research institutions and a prescription drug plan that seeks to develop a new TB drug regimen through collaboration in early-stage drug discovery research. Other examples include the Structural Genomics Consortium and Innovative Medicines Initiative. The aforementioned approaches would have greater impact on antiviral drug discovery efforts and efficiency than the currently fragmented venture.
Conflicts of interest
MJ is a founder of, consultant to and shareholder in GyreOx Therapeutics, a company that uses molecular design and chemoenzymatic synthesis to develop compounds for complex human diseases.Acknowledgements
The authors also gratefully acknowledge financial support from the South African Medical Research Council (MRC) with funds received from the South African National Department of Health and the UK Government's Newton Fund (R. A. D., RL. M. G., K. C.), the UK Engineering and Physical Sciences Research Council EQATA (R. J. M. G.), the UK Global Challenge Research Fund (R. J. M. G., R. A. D.), the University of Cape Town (K. C.) and the South African Research Chairs Initiative of the Department of Science and Innovation, administered through the South African National Research Foundation (NRF) to K. C. (UID: 64767) and R. A. D. (UID: 87583). C. S. A. acknowledges financial support for SARS-CoV-2/Covid-19 research from UKMRC (CVG-1725-2020) and UKRI-DHSC (MR/Vo28464/1). The authors acknowledge Bronwyn Tweedie of the Rhodes University Print Services Unit who provided the graphics for Fig. 1 and thank Gordon Cragg for his insightful comments and encouragement during the preparation of this manuscript.Notes and references
- M. J. Friedrich, JAMA, J. Am. Med. Assoc., 2019, 321, 1041 Search PubMed.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu and P. Niu, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS.
- M. F. Boni, P. Lemey, X. Jiang, T. T. Y. Lam, B. Perry, T. Castoe, A. Rambaut and D. Robertson, Nat. Med., 2020, 5, 1408–1417 CAS.
- A. C. Hurt, Tropicalmed., 2019, 4, 67 CrossRef.
- M. Rojas, D. M. Monsalve, Y. Pacheco, Y. Acosta-Ampudia, C. Ramírez-Santana, A. A. Ansari, M. E. Gershwin and J. M. Anaya, J. Autoimmun., 2020, 106, 102375 CrossRef CAS.
- V. Koppolu, T. Shantha and T. Raju, J. Neurovirol., 2018, 24, 255–272 CrossRef CAS.
- P. I. Andersen, A. Ianevski, H. Lysvand, A. Vitkauskiene, V. Oksenych, M. Bjørås, K. Tellingc, I. Lutsard, U. Dumpise, Y. Iriec, T. Tensonc, A. Kantelef and D. E. Kainova, Int. J. Infect. Dis., 2020, 93, 268–276 CrossRef CAS.
- S. Moller-Tank and W. Maury, PLoS Pathog., 2020, 11, e1004731 CrossRef.
- M. F. Saeed, A. A. Kolokoltsov, T. Albrecht and R. A. Davey, PLoS Pathog., 2010, 6, e1001110 CrossRef.
- A. Nanbo, S. Watanabe, P. Halfmann and Y. Kawaoka, Sci. Rep., 2013, 3, 1206 CrossRef.
- Y. Noda, T. Ebihara, H. Muramoto, K. Fujii, A. Takada, H. Sagara, J. H. Kim, H. Kida, H. Feldmann and Y. Kawaoka, PLoS Pathog., 2006, 2, e99 CrossRef.
- E. E. A. Osman, P. L. Toogood and N. Neamati, ACS Infect. Dis., 2020, 6, 1548–1552 CrossRef CAS.
- D. Ballout, R. A. Sviridov, M. I. Bukrinsky and A. T. Remaley, FASEB J., 2020, 34, 7234–7264 CrossRef.
- C. Liu, Q. Zhou, Y. Li, L. V. Garner, S. P. Watkins, L. J. Carter, J. Smoot, A. C. Gregg, A. D. Daniels, S. Jervey and D. Albaiu, ACS Cent. Sci., 2020, 6, 315–331 CrossRef CAS.
- M. K. Parvez and S. Parveen, Intervirology, 2017, 60, 1–7 CrossRef.
- E. De Clercq and G. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 CrossRef.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- A. Wilder-Smith, E. E. Ooi, O. Horstick and B. Wills, The Lancet, 2019, 393, 350–363 CrossRef.
- https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-ebola-virus-disease-marking-critical-milestone-public-health .
- N. Lurie, M. Saville, R. Hatchett and J. Halton, N. Engl. J. Med., 2020, 382, 1969–1972 CrossRef CAS.
- E. De Wit, N. Van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS.
- M. S. Diamond and T. C. Pierson, Cell Host Microbe, 2020, 27, 699–703 CrossRef CAS.
- J. Huo, A. Le Bas, R. R. Ruza, H. M. Duyvesteyn, H. Mikolajek, T. Malinauskas, T. K. Tan, P. Rijal, M. Dumoux, P. N. Ward and J. Ren, Nat. Struct. Mol. Biol., 2020, 1–9 Search PubMed.
- A. Renn, Y. Fu, X. Hu, M. D. Hall and A. Simeonov, Trends Pharmacol. Sci., 2020, 41, 815–829 CrossRef CAS.
- L. Delang, R. Abdelnabi and J. Neyts, Antiviral Res., 2018, 153, 85–94 CrossRef CAS.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648–1661 CrossRef CAS.
- M. K. Lo, R. Jordan, A. Arvey, J. Sudhamsu, P. Shrivastava-Ranjan, A. L. Hotard, M. Flint, L. K. McMullan, D. Siegel, M. O. Clarke, R. L. Mackman, H. C. Hui, M. Perron, A. S. Ray, T. Cihlar, S. T. Nichol and C. F. Spiropoulou, Sci. Rep., 2017, 7, 43395 CrossRef.
- J. J. Malin, I. Suárez, V. Priesner, G. Fätkenheuer and J. Rybniker, Clini. Microbiol. Rev., 2020, 34, e00162–20 Search PubMed.
- C. Liang, L. Tian, Y. Liu, N. Hui, G. Qiao, H. Li, Z. Shi, Y. Tang, D. Zhang, X. Xie and X. Zhao, Eur. J. Med. Chem., 2020, 201, 112527 CrossRef CAS.
- J. D. Graci and C. E. Cameron, Rev. Med. Virol., 2006, 16, 37–48 CrossRef CAS.
- A. Shannon, B. Selisko, J. Huchting, F. Touret, G. Piorkowski, V. Fattorini, F. Ferron, E. Decroly, C. Meier, B. Coutard and O. Peersen, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS.
- C. Gil, T. Ginex, I. Maestro, V. Nozal, L. Barrado-Gil, M. A. Cuesta-Geijo, J. Urquiza, D. Ramirez, C. Alonso, N. E. Campillo and A. Martinez, J. Med. Chem., 2020, 63, 12359–12386 CrossRef CAS.
- P. W. Horby, M. Mafham, J. L. Bell, L. Linsell, N. Staplin, J. Emberson, A. Palfreeman, J. Raw, E. Elmahi, B. Prudon and C. Green, Lancet, 2020, 396, 1345–1352 CrossRef CAS.
- T. Hoenen, A. Groseth and H. Feldmann, Nat. Rev. Microbiol., 2019, 17, 593–606 CrossRef CAS.
- H. A. Elshabrawy, Vaccines, 2020, 8, 335 CrossRef CAS.
- N. S. Ogando, J. C. Zevenhoven-Dobbe, Y. van der Meer, P. J. Bredenbeek, C. C. Posthuma and E. J. Snijder, J. Virol., 2020, 94, e01246–20 CrossRef CAS.
- M. V. Beccari, B. T. Mogle, E. F. Sidman, K. A. Mastro, E. Asiago-Reddy and W. D. Kufel, Antimicrob. Agents Chemother., 2019, 63, e00110–19 CrossRef CAS.
- S. Xiu, A. Dick, H. Ju, S. Mirzaie, F. Abdi, S. Cocklin, P. Zhan and X. Liu, J. Med. Chem., 2020, 63, 12256–12274 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Kruger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Muller, C. Drosten and S. Pohlmann, Cell, 2020, 181(271-280), e278 Search PubMed.
- RECOVERY Collaborative Group, New Eng. J. Med., 2020.
- X. Ji and Z. Li, Med. Res. Rev., 2020, 40, 1519–1557 CrossRef CAS.
- N. Kumar, S. Sharma, R. Kumar, B. N. Tripathi, S. Barua, H. Ly and B. T. Rouse, Clin. Microbiol. Rev., 2020, 33, e00168–19 CrossRef CAS.
- S. Pattabhi, C. R. Wilkins, R. Dong, M. L. Knoll, J. Posakony, S. Kaiser, C. E. Mire, M. L. Wang, R. C. Ireton, T. W. Geisbert and K. N. Bedard, J. Virol., 2016, 90, 2372–2387 CrossRef CAS.
- M. L. Agostini, E. L. Andres, A. C. Sims, R. L. Graham, T. P. Sheahan, X. Lu, E. C. Smith, J. B. Case, J. Y. Feng, R. Jordan, A. S. Ray, T. Cihlar, D. Siegel, R. L. Mackman, M. O. Clarke, R. S. Baric and M. R. Denison, mBio, 2018, 9, e00221–18 CrossRef CAS.
- A. L. Totura and S. Bavari, Exp. Opin. Drug Discovery, 2019, 14, 397–412 CrossRef.
- M. Sorokina and C. Steinbeck, J. Cheminfor., 2020, 12, 1–51 CrossRef.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 5–461 Search PubMed.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11, 1117 CrossRef CAS.
- A. M. Sayed, A. R. Khattab, A. M. AboulMagd, H. M. Hassan, M. E. Rateb, H. Zaid and U. R. Abdelmohsen, RSC Adv., 2020, 10, 19790–19802 RSC.
- A. T. Ton, F. Gentile, M. Hsing, F. Ban and A. Cherkasov, Mol. Inf., 2020, 39, 2000028 CrossRef CAS.
- https://postera.ai/covid .
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711–715 CrossRef CAS.
- S. Nwaka and A. Hudson, Nat. Rev. Drug Discovery, 2006, 5, 941–955 CrossRef CAS.
- https://info.perkinelmer.com/covid19-analytics .
| This journal is © The Royal Society of Chemistry 2021 |