
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen vacancy engineering in spinel-structured nanosheet wrapped hollow polyhedra for electrochemical nitrogen fixation under ambient conditions†
Feili
Lai
ac,
Jianrui
Feng
a,
Xiaobin
Ye
c,
Wei
Zong
a,
Guanjie
He
 *b,
Chao
Yang
d,
Wei
Wang
d,
Yue-E.
Miao
a,
Bicai
Pan
c,
Wensheng
Yan
c,
Tianxi
Liu
*a and
Ivan P.
Parkin
*b
*b,
Chao
Yang
d,
Wei
Wang
d,
Yue-E.
Miao
a,
Bicai
Pan
c,
Wensheng
Yan
c,
Tianxi
Liu
*a and
Ivan P.
Parkin
*b
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Innovation Center for Textile Science and Technology, Donghua University, Shanghai 201620, P. R. China. E-mail: txliu@dhu.edu.cn
bChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk; i.p.parkin@ucl.ac.uk
cHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
dDepartment of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, P. R. China
First published on 9th January 2020
Abstract
Electrochemical nitrogen-to-ammonia conversion by the nitrogen reduction reaction (NRR) under ambient conditions is regarded as a potential approach to tackle the energy-intensive Haber–Bosch process with excessive CO2 emission. However, the NRR is still restricted by low faradaic efficiency and NH3 yield, which is due to the chemical inertness of N-related groups for efficient adsorption/activation on the electrocatalysts. Here, a series of spinel-structured nanosheet wrapped hollow nitrogen-doped carbon polyhedra with abundant oxygen vacancies are constructed successfully. From theoretical aspects, these materials show increased charge density on their surface for enhanced capture and activation of N2 molecules. As a result, oxygen vacancy-rich NiCo2O4 on hollow N-carbon polyhedra (Vo-rich NiCo2O4@HNCP) shows outstanding electrocatalytic NRR performance with high production yield (NH3: 4.1 μg h−1 cm−2/17.8 μg h−1 mg−1; faradaic efficiency: 5.3%) and high stability under ambient conditions and is superior to the counterpart oxygen vacancy-poor electrocatalysts. Oxygen vacancy engineering introduces a new concept for rational design of advanced NRR catalysts for energy conversion systems.
Introduction
Ammonia (NH3) is one of the most important carbon-free energy intermediates with low liquefying pressure and high hydrogen density1,2 and is widely used in producing numerous chemicals, such as nitrogen fertilizers or pharmaceuticals, to satisfy the increasing demand from a booming world population.3–5 Up to now, over 1% of the world's energy supply is used to produce ammonia by the traditional Haber–Bosch process which is responsible for 1.6% of global CO2 emissions and requires harsh operating conditions (400–600 °C and 20–40 MPa) due to the chemical inertness of N2 with a high bond energy of 940.95 kJ mol−1.6–9 In contrast, the electrochemical N2 reduction reaction (NRR, N2 + 6H+ + 6e− → NH3) occurs at ambient temperature and pressure and is regarded as a more sustainable and energy-efficient process for ammonia generation.10–14 Although some electrocatalysts have recently been investigated for the NRR, including noble metal electrocatalysts (such as Ru, Au, and Rh),13–16 non-noble metal-based electrocatalysts (such as Fe-, Co-, and Mo-based materials),7,12,17,18 and conducting polymers (such as polyaniline and polypyrrole),19,20 their electrocatalytic properties are still limited by the low faradaic efficiency due to the confined electro-active sites in the corresponding bulk.10,11,21 Therefore, constructing multilevel nanostructures (such as one-dimensional nanofibers/nanotubes,22,23 two-dimensional ultrathin nanosheets,24,25 and three-dimensional hollow nanostructures26,27) has become an efficient solution to obtain outstanding NRR electrocatalysts with a high specific surface area, thus resulting to an increased number of active sites for efficient reactant adsorption.Apart from this, the impeded NRR process is deeply rooted in the chemical inertness of N2 molecules. Then, the question on how to capture and activate N2 gas much more efficiently using electrocatalysts arises. Atomic surface engineering (such as defect engineering28,29 and doping engineering30) is the best choice to regulate the electronic structure and alter the charge density distribution. Among various types of defects, the vacancy-type defect (such as oxygen vacancies) is regarded as a kind of point defect, which is caused by independent atoms escaping from the atomic surface.31–33 Up to now, several possible strategies have been discovered for generating oxygen vacancies in metal oxides, including chemical reduction,34 annealing in an oxygen poor atmosphere,35,36 and heterogeneous atom substitution.37 Furthermore, the oxygen vacancies created on the metal oxide surface could serve as trapping sites to capture and activate inert gas molecules of N2. However, to our knowledge, the proper control of the oxygen vacancy content in the electrocatalytic N2 reduction reaction is still in the rudimentary state.
To figure out the contribution of the oxygen vacancies in NRR applications, we conducted density functional theory (DFT) calculations by using NiCo2O4 with a perfect surface and oxygen vacancy-introduced surface as two models. As shown in Fig. 1, the introduction of oxygen vacancies in NiCo2O4 is beneficial for enhancing its stability towards NNH groups due to the presence of fewer coordination sites of Ni/Co cations around the vacancies. As further proved by the differential charge density of NiCo2O4 with oxygen vacancies, the bonded electrons can be delocalized to electron donors (Ni and Co elements) near the oxygen vacancies and be activated dramatically. As a result, the NiCo2O4 with oxygen vacancies exhibits a lower stabilization energy of 0.30 eV for NNH groups than NiCo2O4 with a perfect surface (0.61 eV), indicating the positive role of oxygen vacancies in the following nitrogen reduction reactions. In addition, the delocalized electrons on the surface of NiCo2O4 with oxygen-vacancies are easier to excite to the conduction band, which is favorable not only for enhanced conductivity35,38 but also for activation of N-related groups, by transferring its electrons into their antibonding orbitals. At the same time, the hollow carbon polyhedron is beneficial not only for electron transfer from buried carbon to the oxygen vacancy-rich NiCo2O4 surface for the fast NRR process, but also for increasing the number of active sites for the capture of NNH groups.
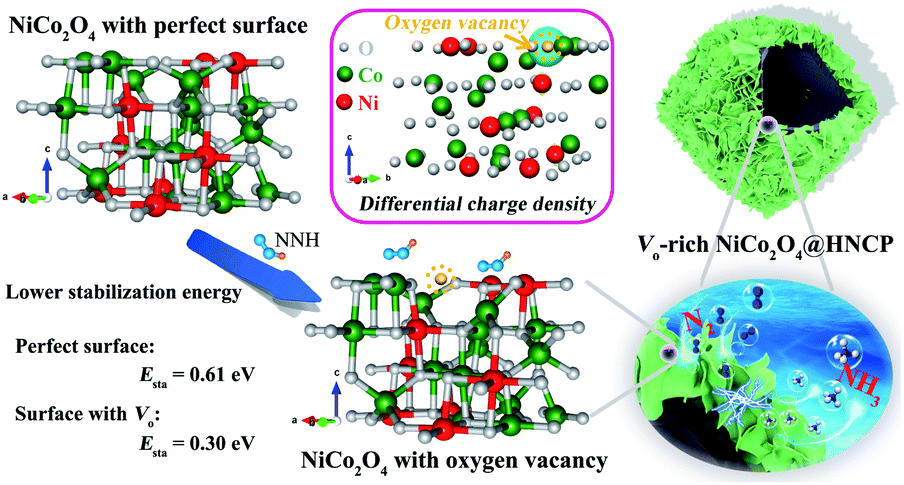 | ||
| Fig. 1 Schematic illustration of the stabilization of NNH groups on the NiCo2O4 with perfect and oxygen vacancy-introduced surfaces, as well as the partial charge density of Vo-rich NiCo2O4. | ||
In this study, we design a series of highly efficient oxygen vacancy-rich spinel-structured nanosheets on hollow N-carbon polyhedra to facilitate the electrocatalytic N2 reduction reaction. Taking the oxygen vacancy-rich NiCo2O4 on hollow N-carbon polyhedra (Vo-rich NiCo2O4@HNCP) as an example, it exhibited a high production yield (NH3: 4.1 μg h−1 cm−2/17.8 μg h−1 mg−1 and faradaic efficiency: 5.3%), good selectivity at −0.25 V versus RHE, and high stability during the NRR process and is much more outstanding than the corresponding oxygen vacancy-poor NiCo2O4 on hollow N-carbon polyhedra (Vo-poor NiCo2O4@HNCP). Furthermore, this oxygen vacancy engineering can also be applied in other spinel-structured nanosheets (such as ZnCo2O4 and Co3O4), which leads to a general solution for the design of advanced NRR electrocatalysts.
Results and discussion
According to the theoretical work, oxygen vacancy-rich NiCo2O4 on hollow N-carbon polyhedra (Vo-rich NiCo2O4@HNCP) was synthesized (Fig. S1†). As shown in Fig. 2a, the Vo-rich NiCo2O4@HNCP displays a regular geometrical shape with a maximum length of ∼ 850 nm. A blurry interface between HNCP and Vo-NiCo2O4 nanosheets can be observed in the corresponding HRTEM image (Fig. 2b), with mixed phases of amorphous carbon and crystalline NiCo2O4, which is beneficial for the charge accumulation and transfer. As a result, the two parts of HNCP and Vo-rich NiCo2O4 nanosheets can be well connected with a shared electron transfer region. Meanwhile, the fringe spacing of Vo-rich NiCo2O4 is revealed to be ∼0.23 nm in its HRTEM image (corresponding to the (311) plane), which demonstrates a negative role of oxygen vacancies in the crystalline NiCo2O4. As shown in the X-ray diffraction (XRD) pattern of Vo-rich NiCo2O4@HNCP (Fig. 2c), three typical peaks at 2θ = 36.9°, 43.0°, and 62.7° can be well indexed to the (311), (400) and (440) planes of spinel-structured NiCo2O4 (JCPDS card no. 73-1702), demonstrating its successful loading on the hollow nitrogen-doped carbon polyhedra. The nine-fold higher specific surface area (82.1 m2 g−1) of Vo-rich NiCo2O4@HNCP compared to the Vo-rich NiCo2O4 bulk (9.0 m2 g−1) (Fig. 2d) indicates its higher number of active sites, leading to shortened diffusion paths for reagents during the NRR process.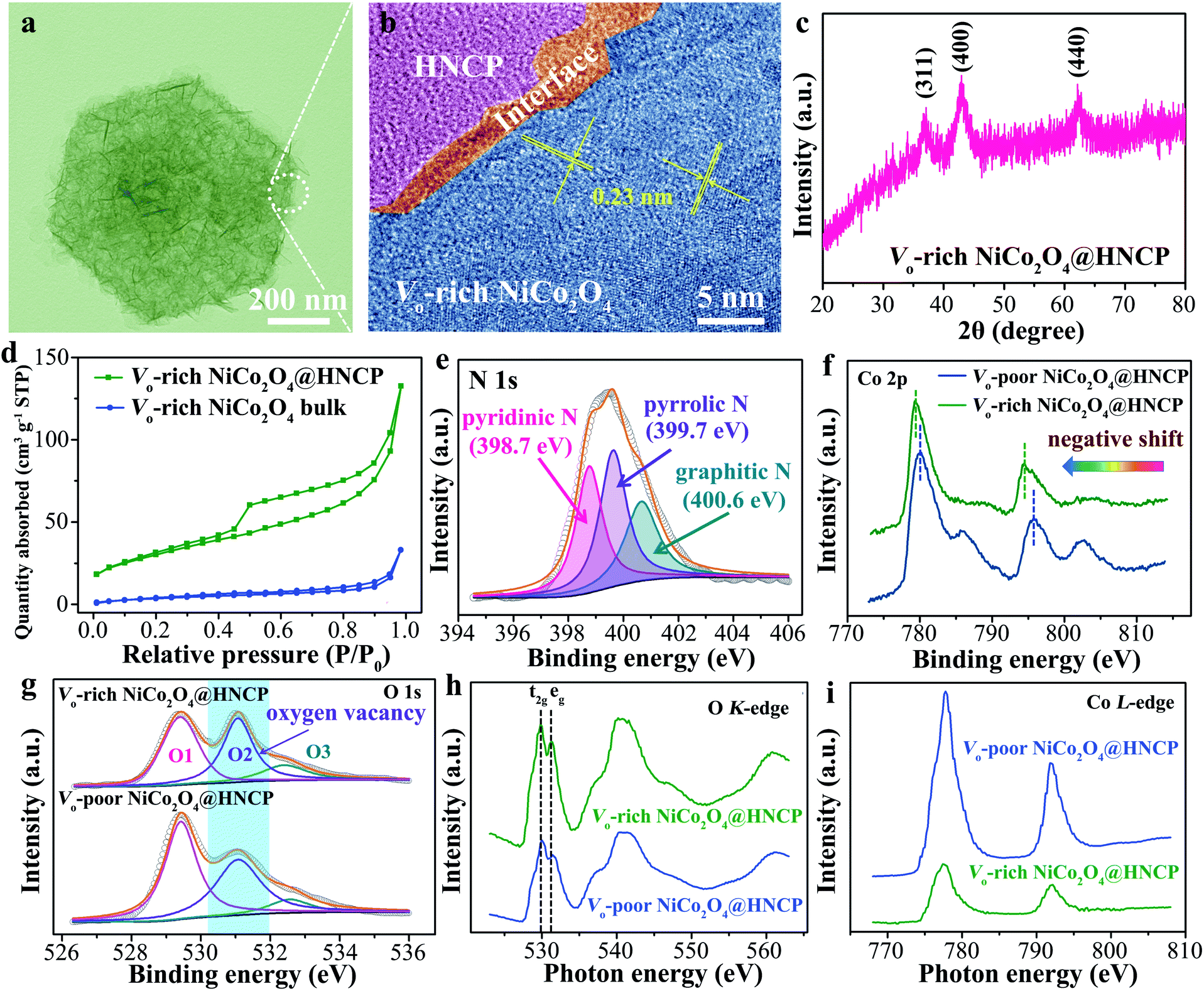 | ||
| Fig. 2 (a) TEM and (b) HRTEM images of Vo-rich NiCo2O4@HNCP. (c) XRD pattern of Vo-rich NiCo2O4@HNCP. (d) N2 adsorption isotherms of Vo-rich NiCo2O4@HNCP and the Vo-rich NiCo2O4 bulk. (e) N 1s spectrum of Vo-rich NiCo2O4@HNCP. (f) Co 2p and (g) O 1s spectra of Vo-rich NiCo2O4@HNCP and Vo-poor NiCo2O4@HNCP. (h) O K-edge and (i) Co L-edge XANES spectra of Vo-rich NiCo2O4@HNCP and Vo-poor NiCo2O4@HNCP. | ||
Further, for comparison, oxygen vacancy-poor NiCo2O4 on hollow N-carbon polyhedra (Vo-poor NiCo2O4@HNCP) was obtained by calcining the corresponding nickel–cobalt layered double hydroxide nanosheet wrapped hollow nitrogen-doped carbon polyhedra (Ni–Co LDH@HNCP) in an oxygen atmosphere, and a series of characterization experiments was conducted (Fig. S2–S8†). In the N 1s X-ray photoelectron spectroscopy (XPS) spectrum (Fig. 2e), the peaks at 400.6 eV, 399.7 eV, and 398.7 eV corresponded to the graphitic N, pyrrolic N, and pyridinic N of the HNCP template, which is beneficial for the efficient connection of both Vo-rich and Vo-poor NiCo2O4 nanosheets to form a charge-rich region. In addition, the nitrogen content was calculated to be 11.6 wt% (Fig. S9†). The binding energies of Co 2p3/2 (780 eV) and Co 2p1/2 (795 eV) for Vo-rich NiCo2O4@HNCP show negative shifts compared to those for Vo-poor NiCo2O4@HNCP (Fig. 2f). This indicates that some Co3+ ions in the NiCo2O4 nanosheets are reduced to Co2+ after introduction of oxygen vacancies,39–42 due to the charge transfer from the Vo to the Co(Ni)3+ with a decreased number of Co(Ni)–O bonds at the surfaces (Fig. S10†). In the O 1s XPS spectra (Fig. 2g), the peaks at 529.5, 531.1 and 532.5 eV are associated with Co(Ni)–O bonds (O1), oxygen vacancies or defects with lower oxygen coordination (O2), and surface adsorbed water molecules (O3), respectively.43–45 The proportion of the integral-area of the O2 peak in Vo-rich NiCo2O4@HNCP is much larger than that in Vo-poor NiCo2O4@HNCP, indicating the successful introduction of abundant oxygen vacancies in Vo-rich NiCo2O4@HNCP.35,39 In addition, the Vo (O2) proportion in Vo-rich NiCo2O4@HNPC increases from 33.0% to 68.2% as the annealing temperature increased from 200 to 300 °C (Fig. S11†), which means that the incorporated Vo amount can be well controlled by adjusting the annealing temperature. This finding could be further confirmed using the X-ray absorption near edge structure (XANES) of the O K-edge and Co L-edge. Fig. 2h shows the O K-edge XANES spectra of Vo-rich and Vo-poor NiCo2O4@HNCP with two main folds. In the first fold at lower energy, another two sub-folds are centered at about 529.7 and 531.4 eV, which are denoted as t2g and eg by the ligand field, respectively. A relatively high intense ratio of eg to t2g can be observed for Vo-rich NiCo2O4@HNPC, indicating the successful generation of abundant Vo, which can be further proved in the magnified version of t2g and eg at the O K-edge (Fig. S12–S13†). Meanwhile, attributed to the Co 2p to 3d transition and spin–orbit coupling, the Co L-edge spectra can be split into two parts, L3-edge and L2-edge (Fig. 2i).46,47 The lower peak intensity for both the Co L3-edge and L2-edge in Vo-rich NiCo2O4@HNCP indicates that more electrons would occupy the Co 3d orbitals, providing more crucial evidence for the presence of abundant Vo on the surface of Vo-rich NiCo2O4@HNCP. A similar phenomenon is observed in the Ni L-edge spectrum (Fig. S14†). The generation of oxygen vacancies is due to the inadequate oxidation of NiCo2O4 in an oxygen-deficient atmosphere as compared to an oxygen atmosphere, leading to abundant partially unoxidized metal domains in the NiCo2O4 structure.48,49
To reveal the role of oxygen vacancies in tuning the electronic structures of the NiCo2O4 nanosheets, DFT calculations were performed for different types of oxygen vacancies. Considering that there are two types of oxygen (e.g. surface and sub-surface oxygens) in the perfect crystallographic lattice of NiCo2O4 (Fig. 3a), we separately studied each situation regarding their density of states, formation possibility and adsorption ability towards the key NRR intermediates of NNH groups. The surface oxygen was removed first for building the surface oxygen vacancy (sur-Vo) model, as shown in Fig. 3b. We found that the d-band center of the sur-Vo model is −0.28 eV, much closer to the Fermi level compared with that of the pristine NiCo2O4 model (−0.34 eV). This result indicates that oxygen vacancies can induce more activity of the metal atoms, that is, the adsorption strength of molecules and radicals over the metal atoms will be enhanced. As shown in Fig. 3c, we tested another type of oxygen vacancy, the sub-surface oxygen vacancy (sub-Vo) model, which has the closest d-band center (−0.24 eV) to the Fermi level among the three models. In the subsequent calculation, we found that the possibility of the formation of these two types of oxygen vacancies is not the same (Fig. 3d). As a more positive formation energy denotes that a vacancy is more difficult to form, the results show that sub-Vo (2.47 eV) has a higher stability than sur-Vo (3.25 eV). Finally, we calculated the adsorption strength of the three models by using NNH as a probe and found that the sub-Vo model presents the most favorable energy (−0.03 eV). The adsorption energy of the sur-Vo (0.30 eV) is also more negative than that of the pristine model (0.61 eV), indicating its enhanced adsorption ability, which is consistent with the d-band center results. According to the trend of formation energy, we can deduce that the sub-Vo is preferentially formed when the vacancy density is relatively low. Therefore, the adsorption enhancement will be significantly dependent on the vacancy density in the region of low vacancy density.
 | ||
| Fig. 3 (a) Pristine NiCo2O4 structure and projected density of states (PDOS) of the surface transition metal. (b) The defect model with removal of a surface oxygen vacancy (sur-Vo) and its PDOS. (c) The defect model with removal of a sub-surface oxygen vacancy (sub-Vo) and its PDOS. (d) The formation energy of oxygen vacancies for the three catalyst models. (e) Adsorption energy of NNH groups in the three catalyst models. | ||
To illustrate the role of oxygen vacancies in the NRR, a two-compartment cell was assembled by employing Vo-rich NiCo2O4@HNCP as the cathode catalyst with continuous N2 bubbling in a 0.1 M Na2SO4 electrolyte. The highest average yields and corresponding faradaic efficiencies of Vo-rich NiCo2O4@HNCP are achieved when the negative potential increases to −0.25 V versus the reversible hydrogen electrode (RHE) and are calculated to be about 4.1 μg h−1 cm−2/17.8 μg h−1 mg−1 and 5.3%, respectively (Fig. 4a and S15†). This production yield at −0.25 V is also confirmed by the nuclear magnetic resonance (NMR) method as shown in Fig. S16–S18 and Table S1.† Beyond this negative potential, the NH3 yields and faradaic efficiencies decrease significantly because of the overwhelming competition from the hydrogen evolution reaction (HER). The NRR performance of the Vo-rich NiCo2O4@HNCP catalyst is much more outstanding than that of the Vo-poor NiCo2O4@HNCP catalyst (1.6 μg h−1 cm−2/6.9 μg h−1 mg−1 and 1.8% at −0.25 V), as well as other results under ambient conditions or at high temperatures and pressures (Table S2†). Note that no hydrazine is detected in the electrolyte (Fig. S19†). In addition, the charge amounts of the experimentally quantified NH3 and H2 gases and the calculated one (Fig. S20†) are roughly in agreement (S2 ≈ S3). These results indicate the high selectivity of the Vo-rich NiCo2O4@HNCP catalyst for NH3 generation except for H2 gas. For practical use, stability is another critical criterion to evaluate the NRR performance of a catalyst. As shown in Fig. 4b and S21–26,† the NH3 yield, faradaic efficiency, and current density of the Vo-rich NiCo2O4@HNCP catalyst are all stable without obvious fluctuation. As shown in Fig. S27,† the Vo-rich NiCo2O4@HNCP catalyst displays ultra-stable properties in both NH3 yield and faradaic efficiency even after working for 100 h and is comparable to the recently reported catalysts but with a much longer practical life.50 By using XRD and XPS analyses (Fig. S28†), the Vo-rich NiCo2O4@HNCP catalyst is found to exhibit an unchanged crystal structure and only slightly decreased oxygen vacancy content after working for 100 h, which demonstrates the relatively stable structure of Vo-rich NiCo2O4@HNCP as an NRR catalyst. Due to the similar results of NH3 yield and faradaic efficiency after varying the nitrogen flow rate at −0.25 V (Fig. 4c), the N2 diffusion process becomes a non-rate-determining step as it is an independent gas–solid interface. Moreover, by varying the reaction temperature of the NRR, both the NH3 yield and faradaic efficiency of the Vo-rich NiCo2O4@HNCP catalyst increase simultaneously (Fig. 4d). For instance, the NRR yield is about 2.5 times higher at 60 °C than at 0 °C, indicating that mass transfer plays a key role in enhancing the reaction rate of the Vo-rich NiCo2O4@HNCP catalyst. In accordance with the Arrhenius equation and Arrhenius plot (Fig. S29†), the apparent activation energy of Vo-rich NiCo2O4@HNCP for the NRR is calculated to be 11.4 kJ mol−1. The UV/Vis absorption spectra of various samples (Fig. 4e) and photographs of NH4+-containing solutions before and after staining with indophenol indicator (Fig. 4f) are exhibited. After comparing the EIS spectra (Fig. S30†) and the performance of the Vo-rich NiCo2O4 bulk (1.4 μg h−1 cm−2/6.1 μg h−1 mg−1 and 1.6% at −0.25 V) in Fig. S31,† it could be seen that the hollow nitrogen-doped carbon polyhedron template in Vo-rich NiCo2O4@HNCP is extremely vital for its enhanced NRR performance, due to the increased specific surface area (Fig. 2d) and well-formed interface between HNCP and Vo-rich NiCo2O4 nanosheets for charge accumulation/transfer. For the verification of the source of ammonia, a 15N isotope labeling experiment using NMR (600 MHz) was performed. As shown in Fig. S32,† the 1H NMR signals of 14NH4+ produced 14N triplets in the region of 6.8–7.1 ppm. Importantly, the 1H NMR signal of 15NH4+ only produced an 15N doublet without the appearance of 14N triplets in the same region, which demonstrates that the doped nitrogen atoms in the HNCP template are stable and do not escape from the structures. Therefore, the source of ammonia is the feed N2 gas rather than the electrocatalyst.
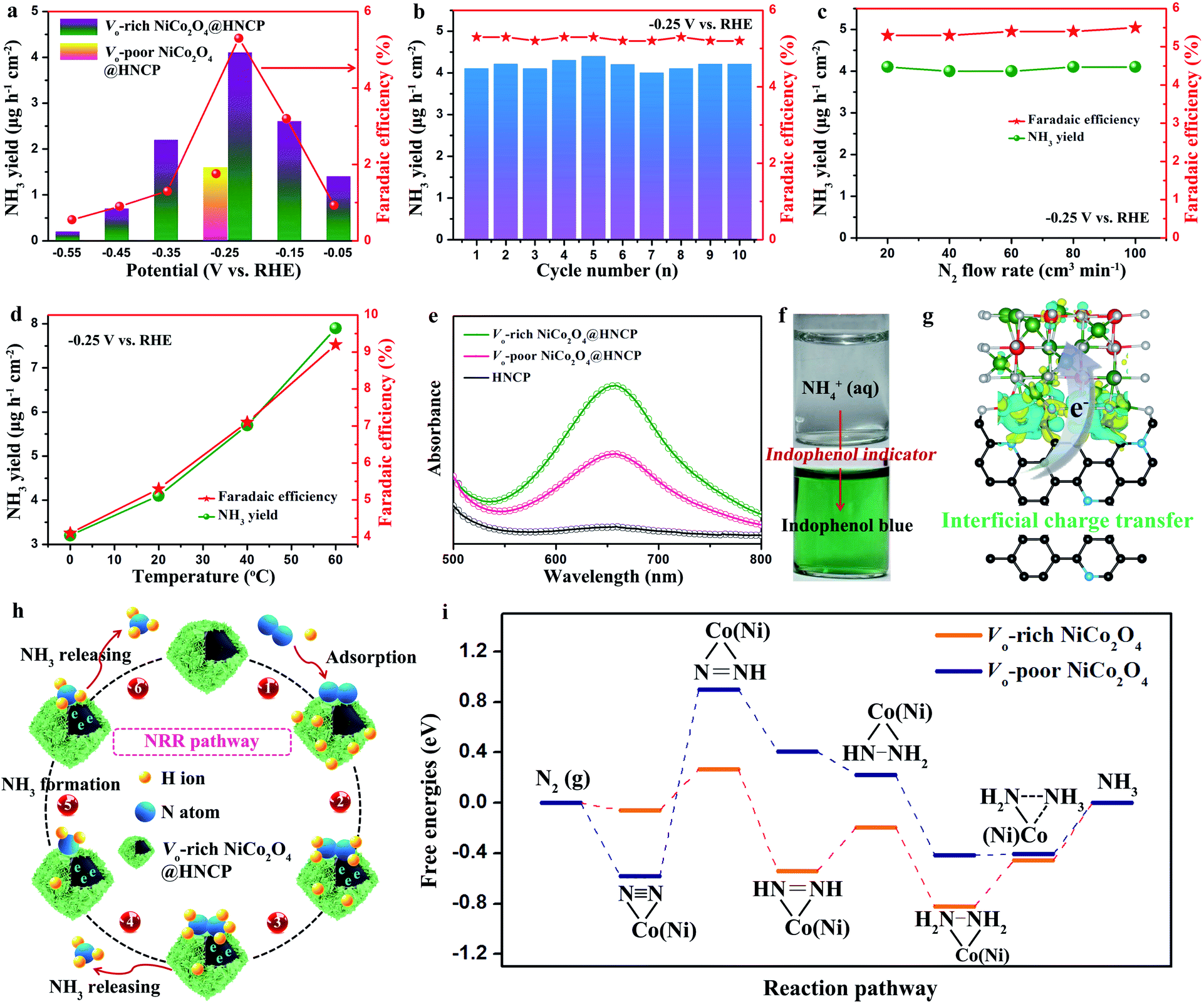 | ||
| Fig. 4 Electrocatalytic NRR of Vo-rich and Vo-poor NiCo2O4@HNCP in an aqueous solution of 0.1 M Na2SO4 under ambient conditions. (a) Yield of NH3 (bar graph) and faradaic efficiency (red) at each given potential. (b) Recycling test of Vo-rich NiCo2O4@HNCP at a potential of −0.25 V. Yield rate of NH3 (green) and faradaic efficiency at different (c) N2 flow rates and (d) temperatures at −0.25 V. (e) UV-Vis absorption spectra of various samples after chronoamperometry testing at −0.25 V versus RHE. (f) NH4+-containing solution stained with indophenol indicator. (g) Charge density distribution of Vo-rich NiCo2O4@HNCP around the interface and oxygen vacancy. (h) Schematic illustration of the proposed electrochemical NRR pathway by using the Vo-rich NiCo2O4@HNCP catalyst. (i) Free energy diagram of the electrochemical reduction of N2 to NH3 in an alternating pathway on the surface of the Vo-rich NiCo2O4@HNCP catalyst. | ||
For further detailed insights into the structure–activity relationship between the Vo-rich NiCo2O4@HNCP catalyst and its enhanced NRR properties, DFT calculation was carried out from the perspective of thermodynamic and kinetic acceleration. First, from the thermodynamic point of view, the electrons that previously occupied the O 2p orbital would partially delocalized to the neighboring Ni/Co cations after introduction of oxygen vacancies, which suggests that a surface with oxygen vacancies is much more activated. Meanwhile, a rational model for the Vo-rich NiCo2O4@HNCP catalyst was constructed by aligning optimized Vo-rich NiCo2O4 with highly conductive nitrogen-doped carbon (Fig. 4g and S33†). After analysis in detail, abundant charges accumulated on the blurry interface between Vo-rich NiCo2O4 and nitrogen-doped carbon, leading to the successful generation of highly active regions for the electrocatalytic process. As shown in Fig. S34 and S35,† the electrons are prone to transfer from nitrogen-doped carbon to Vo-rich NiCo2O4 due to their inconsistent charge distribution. In turn, the Ni/Co cations show increased electronic states of the d-orbital around the Fermi level, which is beneficial for activation of N2 and formation of an N-catalyst bond. From the kinetic point of view, the hollow nitrogen-doped carbon polyhedron template endows the Vo-rich NiCo2O4 nanosheets with a higher specific surface area with more exposed active sites for adsorption/activation of N-related species. Attributed to the structural merits of the Vo-rich NiCo2O4@HNCP catalyst, a possible NRR mechanism is depicted in Fig. 4h and i. Firstly, the N2 gas can be easily adsorbed on the surface of the Vo-rich NiCo2O4@HNCP catalyst to form chemisorbed Co(Ni)–N2 bonds, which can be denoted as 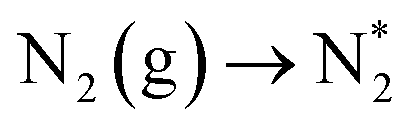 (here, the asterisk * denotes an adsorption site). After full structural relaxation, two energetically favorable configurations were found: end-on and side-on. On the Vo-poor NiCo2O4 surface, an N2 molecule was more likely to anchor in the end-on configuration with a Co–N bond length of 1.84 Å (Fig. S36a†), whereas, on the Vo-rich NiCo2O4 surface, two Co–N bonds (1.20 Å in length) in the side-on configuration are formed with an elongated N–N bond length from 1.12 Å to 1.16 Å (Fig. S36b†). When taking ΔEZPE and entropy into consideration, the ΔG values for N2 adsorption are −0.05 eV for the Vo-rich NiCo2O4 surface and −0.57 eV for the Vo-poor NiCo2O4 surface, respectively. Secondly, six consecutive protonation and reduction processes on the Vo-rich(poor) NiCo2O4 surfaces were further proposed with atomic configurations at various states of each elementary step (Fig. S37†). As discussed in Fig. 4i and Table S3,† the free energies of all these states were slightly downhill for the Vo-rich NiCo2O4 surface compared to the Vo-poor NiCo2O4 surface, which also proves that the successful introduction of oxygen vacancies in NiCo2O4 is beneficial for the nitrogen reduction reaction process theoretically.
(here, the asterisk * denotes an adsorption site). After full structural relaxation, two energetically favorable configurations were found: end-on and side-on. On the Vo-poor NiCo2O4 surface, an N2 molecule was more likely to anchor in the end-on configuration with a Co–N bond length of 1.84 Å (Fig. S36a†), whereas, on the Vo-rich NiCo2O4 surface, two Co–N bonds (1.20 Å in length) in the side-on configuration are formed with an elongated N–N bond length from 1.12 Å to 1.16 Å (Fig. S36b†). When taking ΔEZPE and entropy into consideration, the ΔG values for N2 adsorption are −0.05 eV for the Vo-rich NiCo2O4 surface and −0.57 eV for the Vo-poor NiCo2O4 surface, respectively. Secondly, six consecutive protonation and reduction processes on the Vo-rich(poor) NiCo2O4 surfaces were further proposed with atomic configurations at various states of each elementary step (Fig. S37†). As discussed in Fig. 4i and Table S3,† the free energies of all these states were slightly downhill for the Vo-rich NiCo2O4 surface compared to the Vo-poor NiCo2O4 surface, which also proves that the successful introduction of oxygen vacancies in NiCo2O4 is beneficial for the nitrogen reduction reaction process theoretically.
We further studied the NRR activity of other spinel-structured nanosheet wrapped HNCP, such as ZnCo2O4@HNCP and Co3O4@HNCP, in which oxygen vacancies were deliberately introduced. It should be noted that the XPS O 1s spectra (Fig. S38†) reveal that abundant oxygen vacancies were successfully introduced into the surface of both the Vo-rich ZnCo@HNCP and Vo-rich Co@HNCP catalysts. As shown in Fig. 5a, all the catalysts with abundant oxygen vacancies showed a higher NH3 yield rate and faradaic efficiency than their Vo-poor counterparts in 0.1 M Na2SO4. Taking Vo-rich ZnCo@HNCP and its Vo-poor counterpart as examples, the NH3 yield rate and faradaic efficiency for the Vo-rich ZnCo@HNCP catalyst were 3.7 μg h−1 cm−2/16.0 μg h−1 mg−1 and 3.6%, respectively, which are much more outstanding than those of the Vo-poor ZnCo@HNCP catalyst (1.6 μg h−1 cm−2/6.9 μg h−1 mg−1 and 1.7% at −0.25 V). Besides, the time-dependent curves of various catalysts at −0.25 V (Fig. 5b) exhibited excellent stability, indicating the stable vacancy structure in both the Vo-rich ZnCo@HNCP and Vo-rich Co@HNCP catalysts during the NRR process.
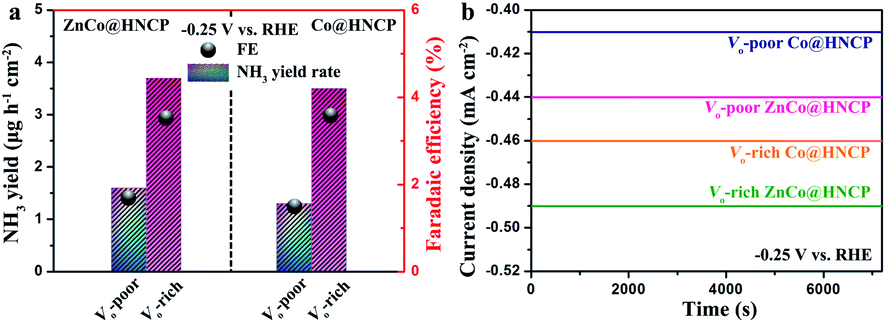 | ||
| Fig. 5 (a) Average NH3 yields and faradaic efficiency of other spinel-structured nanosheet wrapped HNCP (ZnCo@HNCP and Co@HNCP) with and without oxygen vacancies after chronoamperometry testing at −0.25 V versus RHE. (b) Time-dependent current density curves of various catalysts for the NRR at −0.25 V versus RHE. | ||
Conclusions and outlook
In summary, we uncovered the positive role of oxygen vacancies in spinel-structured nanosheets on hollow N-carbon polyhedra (e.g. Vo-rich NiCo2O4@HNCP, Vo-rich ZnCo2O4@HNCP, and Vo-rich Co3O4@HNCP) towards the electrocatalysis of the nitrogen reduction reaction. For example, the Vo-rich NiCo2O4@HNCP catalyst showed higher production yield (NH3: 4.1 μg h−1 cm−2/17.8 μg h−1 mg−1 and faradaic efficiency: 5.3%), good selectivity and high stability when compared with its Vo-poor counterpart (NH3: 1.6 μg h−1 cm−2/6.9 μg h−1 mg−1 and faradaic efficiency: 1.8%). As revealed by DFT calculations, the oxygen vacancies enhance the reactivity of the active sites, leading to reduced stabilization energy of NNH groups. Meanwhile, the HNCP template increased the number of active sites for facilitating the reaction on the surface of Vo-rich NiCo2O4 nanosheets and improved the conductivity for interfacial electron transfer between Vo-rich NiCo2O4 and HNCP. Systematic study from both theoretical and experimental aspects further confirmed the superior kinetics for NH3 production using Vo-rich NiCo2O4@HNCP. Therefore, this work provides a guideline for the rational design of novel and highly efficient catalysts towards N2 electrochemical reduction by increasing the surface area and introducing surface oxygen vacancies in spinel-structured nanosheets simultaneously.Experimental section
Details of the synthetic procedures, characterization and theoretical calculation methods can be found in the ESI†.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are very grateful for the financial support from the National Natural Science Foundation of China (51433001, 21674019, and 21604010), the Science and Technology Commission of Shanghai Municipality (16520722100), the Program of Shanghai Academic Research Leader (17XD1400100), the “Chenguang Program” supported by the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (16CG39) and the Engineering and Physical Sciences Research Council (EPSRC, EP/L015862/1). The computational center of the USTC is acknowledged for computational support.References
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- R. Lan, J. Irvine and S. W. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1492 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. de Groot and M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef CAS PubMed.
- M. Ali, F. L. Zhou, K. Chen, C. Kotzur, C. L. Xiao, L. Bourgeois, X. Y. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335 CrossRef CAS PubMed.
- Y. H. Lu, Y. Yang, T. F. Zhang, Z. Ge, H. C. Chang, P. S. Xiao, Y. Y. Xie, L. Hua, Q. Y. Li, H. Y. Li, B. Ma, N. J. Guan, Y. F. Ma and Y. S. Chen, ACS Nano, 2016, 10, 10507–10515 CrossRef CAS PubMed.
- C. X. Guo, J. R. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- G. F. Chen, X. R. Cao, S. Q. Wu, X. Y. Zeng, L. X. Ding, M. Zhu and H. H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS PubMed.
- Y. Nishibayashi, Inorg. Chem., 2015, 54, 9234–9247 CrossRef CAS PubMed.
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 170000133 Search PubMed.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 160655017 Search PubMed.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- H. M. Liu, S. H. Han, Y. Zhao, Y. Y. Zhu, X. L. Tian, J. H. Zeng, J. X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666–5669 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem. Int. Ed., 2016, 55, 14291–14295 ( Angew. Chem. , 2016 , 128 , 14503–14507 ) CrossRef CAS PubMed.
- F. Koleli and D. B. Kayan, J. Electroanal. Chem., 2010, 638, 119–122 CrossRef.
- F. Koleli and T. Ropke, Appl. Catal., B, 2006, 62, 306–310 CrossRef.
- R. Lan, J. Irvine and S. W. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- F. L. Lai, D. Y. Yong, X. L. Ning, B. C. Pan, Y. E. Miao and T. X. Liu, Small, 2017, 13, 16028667 Search PubMed.
- F. X. Ma, L. Yu, C. Y. Xu and X. W. Lou, Energy Environ. Sci., 2016, 9, 862–866 RSC.
- S. C. Chen, Z. X. Kang, X. Hu, X. D. Zhang, H. Wang, J. F. Xie, X. S. Zheng, W. S. Yan, B. C. Pan and Y. Xie, Adv. Mater., 2017, 29, 170168730 Search PubMed.
- J. Q. Xu, X. D. Li, W. Liu, Y. F. Sun, Z. Y. Ju, T. Yao, C. M. Wang, H. X. Ju, J. F. Zhu, S. Q. Wei and Y. Xie, Angew. Chem. Int. Ed., 2017, 56, 9121–9125 ( Angew. Chem. , 2017 , 129 , 9249–9253 ) CrossRef CAS PubMed.
- B. Y. Guan, S. L. Zhang and X. W. Lou, Angew. Chem. Int. Ed., 2018, 57, 6176–6180 ( Angew. Chem. , 2018 , 130 , 6284–6288 ) CrossRef CAS PubMed.
- L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. Lou, Angew. Chem. Int. Ed., 2018, 57, 172–176 ( Angew. Chem. , 2018 , 130 , 178–182 ) CrossRef CAS PubMed.
- Y. Tong, Y. Q. Guo, K. J. Mu, H. Shan, J. Dai, Y. Liu, Z. Sun, A. D. Zhao, X. C. Zeng, C. Z. Wu and Y. Xie, Adv. Mater., 2017, 29, 1703123 CrossRef PubMed.
- J. F. Xie and Y. Xie, ChemCatChem, 2015, 7, 2568–2580 CrossRef CAS.
- Y. Shi, Y. Zhou, D. R. Yang, W. X. Xu, C. Wang, F. B. Wang, J. J. Xu, X. H. Xia and H. Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Norskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
- C. van der Ham, M. Koper and D. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- H. K. Lee, C. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. M. Han, C. K. Tsung and X. Y. Ling, Sci. Adv., 2018, 4, eaar32083 Search PubMed.
- S. J. Peng, F. Gong, L. L. Li, D. S. Yu, D. X. Ji, T. R. Zhang, Z. Hu, Z. Q. Zhang, S. L. Chou, Y. H. Du and S. Ramakrishna, J. Am. Chem. Soc., 2018, 140, 13646–13653 Search PubMed.
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Angew. Chem. Int. Ed., 2015, 54, 7399–7404 ( Angew. Chem. , 2015 , 127 , 7507–7512 ) CrossRef CAS PubMed.
- H. Yuan, J. T. Li, W. Yang, Z. C. Zhuang, Y. Zhao, L. He, L. Xu, X. B. Liao, R. Q. Zhu and L. Q. Mai, ACS Appl. Mater. Interfaces, 2018, 10, 16410–16417 CrossRef CAS PubMed.
- Y. X. Zeng, Z. Z. Lai, Y. Han, H. Z. Zhang, S. L. Xie and X. H. Lu, Adv. Mater., 2018, 30, 1802396 CrossRef PubMed.
- Y. C. Wang, T. Zhou, K. Jiang, P. M. Da, Z. Peng, J. Tang, B. A. Kong, W. B. Cai, Z. Q. Yang and G. F. Zheng, Adv. Energy Mater., 2014, 4, 140069616 Search PubMed.
- W. J. Xu, F. L. Lyu, Y. C. Bai, A. Q. Gao, J. Feng, Z. X. Cai and Y. D. Yin, Nano Energy, 2018, 43, 110–116 CrossRef CAS.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Huang and L. M. Dai, Angew. Chem. Int. Ed., 2016, 55, 5277–5281 ( Angew. Chem. , 2016 , 128 , 5363–5367 ) CrossRef CAS PubMed.
- J. J. Wu, R. M. Yadav, M. J. Liu, P. P. Sharma, C. S. Tiwary, L. L. Ma, X. L. Zou, X. D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- Y. L. Ge, K. Kan, Y. Yang, L. Zhou, L. Q. Jing, P. K. Shen, L. Li and K. Y. Shi, J. Mater. Chem. A, 2014, 2, 4961–4969 RSC.
- V. M. Jimenez, A. Fernandez, J. P. Espinos and A. R. Gonzalezelipe, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 61–71 CrossRef CAS.
- M. Ma, K. Zhang, P. Li, M. S. Jun, M. J. Jeong and J. H. Park, Angew. Chem. Int. Ed., 2016, 55, 11819–11823 ( Angew. Chem. , 2016 , 128 , 11998–12002 ) CrossRef CAS PubMed.
- N. Kornienko, J. Resasco, N. Becknell, C. M. Jian, Y. S. Liu, K. Q. Nie, X. H. Sun, J. H. Guo, S. R. Leone and P. D. Yang, J. Am. Chem. Soc., 2015, 137, 7448–7455 CrossRef CAS PubMed.
- P. Z. Chen, T. P. Zhou, M. L. Chen, Y. Tong, N. Zhang, X. Peng, W. S. Chu, X. J. Wu, C. Z. Wu and Y. Xie, ACS Catal., 2017, 7, 7405–7411 CrossRef CAS.
- X. R. Li, J. L. Wei, Q. Li, S. S. Zheng, Y. X. Xu, P. Du, C. Y. Chen, J. Y. Zhao, H. G. Xue, Q. Xu and H. Pang, Adv. Funct. Mater., 2018, 28, 1800886 CrossRef.
- L. Hua, Z. Y. Hui, Y. Sun, X. Zhao, H. Xu, Y. J. Gong, R. Y. Chen, C. Y. Yu, J. Y. Zhou, G. Z. Sun and W. Huang, Nanoscale, 2018, 10, 21006–21012 RSC.
- H. Cheng, L. X. Ding, G. F. Chen, L. L. Zhang, J. Xue and H. H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, TEM, XRD, EDS, TGA, and XPS results, adsorptive properties of samples, XANES data, UV-Vis curves, electrochemical properties of samples, and theoretical calculations. See DOI: 10.1039/c9ta11408d |
| This journal is © The Royal Society of Chemistry 2020 |