
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom-temperature synthesis of water-dispersible sulfur-doped reduced graphene oxide without stabilizers†
Jianqiang Guo *abc,
Weimiao Wangd,
Yue Liab,
Jiafeng Liangab,
Qiaosi Zhuab,
Jiongli Li*abc and
Xudong Wangabc
*abc,
Weimiao Wangd,
Yue Liab,
Jiafeng Liangab,
Qiaosi Zhuab,
Jiongli Li*abc and
Xudong Wangabc
aAECC Beijing Institute of Aeronautical Materials, Beijing 100095, China. E-mail: guojianqiang2010@163.com
bBeijing Institute of Graphene Technology, Beijing 100094, China
cBeijing Engineering Research Centre of Graphene Application, Beijing 100095, China
dSchool of Materials and National Graphene Institute, The University of Manchester, Manchester M13 9PL, UK
First published on 14th July 2020
Abstract
Sulfur-Doped graphene has attracted significant attention because of its potential uses in sensors, catalysts, and energy storage applications. In conventional approaches, the sulfur-doped graphene is fabricated with graphene oxide and sulfur-containing compounds through thermal annealing or hydrothermal process, which generally involves special equipment and heat treatment, and requires additional stabilizers to make it solution-processable. In this work, we report a facile one-step approach to synthesize water-dispersible sulfur-doped reduced graphene oxide (S-rGO). Graphene oxide (GO) could be readily reduced and converted to S-rGO simultaneously by directly mixing GO dispersion with hydrosulfide hydrate (NaSH·xH2O) at room temperature. The sulfur doping is confirmed by high resolution S 2p XPS spectrum and element mapping. The colloidal dispersion state of S-rGO is confirmed by the investigation of Tyndall effect, the zeta potential and particle size distribution measurement. Compared with previously reported strategies, NaSH can initiate the reduction and sulfur doping at room temperature, demand no heat treatment, require no equipment and form stable aqueous S-rGO dispersion without using any stabilizer. These advantages will facilitate large-scale production of water-dispersible (sulfur doped) graphene and further boost their applications in sensors, catalysts and energy storage devices.
Introduction
Due to its remarkable physical properties, graphene has attracted intensive research interests since its discovery.1,2 To fully exploit the practical applications of graphene, a prerequisite is scalable production of processable graphene-based materials. There are four main routes toward mass production of graphene: chemical vapour deposition (CVD),3 epitaxial growth,4 liquid phase exfoliation,5 and chemical reduction of graphene oxide.6 Both CVD and epitaxial growth approaches are conducive for high-quality graphene, however, they have disadvantages such as high production cost, high energy consumption and difficulty in transferring graphene.7 Liquid phase exfoliation technique provides a facile and environmentally friendly route to produce high-quality graphene, but requires long sonication time and can only obtain a low-concentration graphene dispersion.5 Chemical reduction of graphene oxide has been commonly used for large-scale production of graphene, albeit limited to structural defects in the reduced graphene oxide (rGO).Graphene is a single-layer nanosheet composed of sp2 hybridized carbon atoms,8 while as a derivative of graphene, GO additionally contains sp3 hybrid regions and oxygen-containing functional groups such as hydroxyl, epoxy, carboxyl, carbonyl, etc.9 Owing to severe structural damage in the sp2 region, GO behaves completely different with graphene in terms of electrical, mechanical and surface morphological properties. By removing oxygen-containing functional groups and restoring the conjugate structure, GO could be (partially) reduced to graphene.10 It has proven to be one of the most feasible approaches for fabricating graphene-based composites, sensors, catalysts, and energy storage devices due to its low cost and versatility in chemical functionalization.11
The carbon atoms of graphene can be substituted or covalently bonded by heteroatoms, such as boron, nitrogen, sulfur, phosphor, etc. These foreign atoms could tune the properties of graphene by donating or withdrawing electrons. Sulfur-doped graphene-based materials have attracted considerable interest because of a wider band gap derived from the electron-withdrawing character of sulfur atoms.12 For example, sulfur-doped reduced graphene oxide (S-rGO) has been applied to sensors for cancer biomarker detection.13 S-rGO can exhibit superior catalytic activity as metal-free electrocatalysts in oxygen reduction reaction for fuel cells,14 and nitrogen reduction reaction for industrial NH3 synthesis,15 respectively. They can also be used to fabricate electrode materials for lithium batteries and pseudocapacitors.16
In the past decade, the preparation of sulfur-doped graphene have been intensively investigated.15,17,18 Generally, sulfur-doped graphene (sulfur-doped reduced graphene oxide) is fabricated from graphite/graphene oxide with sulfur-containing compounds by heat treatment.19 For example, Pumera's group synthesized S-rGO with electrocatalysis via thermal exfoliation of graphite oxide in SO2, H2S or CS2 atmospheres at 600 or 1000 °C.20 Xia et al. prepared the S-rGO for electrocatalytic N2-to-NH3 fixation by directly annealing GO sheet and benzyl disulfide at 1050 °C in argon.15 However, these thermal annealing approaches often requires special instruments and rigorous conditions, and the scale-up production is limited.
Sulfur-containing compounds such as Na2S, thioacetamide,21 and phosphorus pentasulfide have been investigated to reduce GO and synthesize S-rGO at lower temperature by wet chemical methods.22,23 Tian et al. proposed a one-pot approach to fabricate S-rGO with GO and Na2S under hydrothermal conditions.17 Wang et al. synthesized S-rGO via the hydrothermal process with GO and thioacetamide at 180 °C.21 Klingele et al. prepared S-rGO by refluxing GO with phosphorus pentasulfide.23 Basically, these synthetic methods also require high reaction temperature and long reaction time to convert GO to S-rGO.
Additionally, the rGO sheets usually show irreversible restacking tendency in solution due to the reduction in surface charge density,24 which severely limits the practical applications of S-rGO. In this context, sonication and extra stabilizers, such as polymers and surfactants, are usually involved to prevent the precipitation of S-rGO sheets.25 However, the sonication process is likely to cause further damage to S-rGO sheets,26 and the presence of such stabilizers is also undesirable in most applications.27 In short, the fabrication of S-rGO often involves special instruments and energy-consuming processes, and requires additional stabilizers to make it solution-processable.
In this study, we report a facile one-step approach to fabricate stable aqueous S-rGO dispersion without the assistance of heat, sonication and stabilizers. GO could be readily converted to S-rGO with NaSH at room temperature. The C/O ratio of GO increases from 2.2 to 6.7, and approximately 2.5 wt% sulfur is doped into S-rGO framework. The sulfur doping is confirmed by high resolution S 2p XPS spectrum and element mapping. S-rGO sheets could be well-dispersed in water at least 1 week without any obviously sediment. It is worth noting that NaSH has significant advantages over the previously reported sulfur-containing compounds such as Na2S, thiourea dioxide, etc. It could initiate the reaction at room temperature, require little to no equipment, and form stable aqueous S-rGO dispersion.
Materials and methods
Natural graphite flakes with an average size of ∼2 μm were supplied from Qingdao Xinghua Graphite Products Co., Ltd. NaSH·xH2O (68–72%) was purchased from Aladdin-reagent Inc. and used as received. Concentrated sulfuric acid (H2SO4, 95–98%), hydrochloric acid (HCl, 36–38%), potassium permanganate (KMnO4), sodium nitrate (NaNO3), and 30% hydrogen peroxide (H2O2) were provided from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China) and used directly without further purification.GO was prepared from natural graphite flakes using modified Hummers method.28 Stable S-rGO dispersion can be obtained from the GO dispersion in only one step at room temperature. Typically, 20 mg NaSH·xH2O was added into 20 ml GO dispersion at a concentration of 0.5 mg ml−1 under stirring. Without further process, the GO dispersion turned to be black in 1 hour. To establish the optimal conditions for fabricating S-rGO dispersion, both the loadings of NaSH·xH2O and the reaction time were investigated. 10 mg, 20 mg, 40 mg, 100 mg, 200 mg, 400 mg, and 800 mg NaSH·xH2O was added into the GO dispersions, respectively. The dispersion state of all the samples were recorded for 1 week.
The chemical structures of the GO and S-rGO samples were characterized by Fourier transformed infrared (FTIR, Nicolet 6700) spectra and X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALab 250Xi). The morphology and element analysis of as-prepared GO and S-rGO samples were performed under a field-emission scanning electron microscope (SEM, HITACHI S-4800) equipped with an energy dispersive spectrometer (EDS). Transmission electron microscope (TEM) images were obtained on a JEM-2100F microscope. Atomic force micrograph (AFM) images were collected using a JPK Nanowizard microscope in tapping mode. The zeta potential and particle size distribution of GO/S-rGO dispersions were measured by a Malvern Zetasizer. Thermogravimetric analysis (TGA) data was collected on a STA 409 PC analyzer (NETZSCH), at a heating rate of 10 °C min−1. Samples were heated up to 800 °C under nitrogen. The standard uncertainty of weight loss temperature is ±1 °C. Raman spectra was taken on a SENTERRA Micro Raman Spectrometer (Bruker Instruments) with 514 nm laser excitation. X-ray powder diffraction (XRD) was performed at room temperature on a Rigaku Model D/max-2B diffractometer using Cu/Kα radiation (λ = 0.154 nm). All the testing data were collected from 3 to 80° at a scanning rate of 5° min−1.
Results and discussion
Different loadings of NaSH·xH2O were added and gradually dissolved into the GO dispersions at room temperature, respectively. As shown in Fig. 1, all the GO dispersions eventually turned to be black, demonstrating the reduction reaction occurred. It takes around one day to finish the reaction with small amount of NaSH·xH2O (NaSH·xH2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GO = 0.5:1 by weight). The reaction significantly accelerates with the amount of NaSH·xH2O increases. When the mass ratio of NaSH·xH2O to GO reaches 20:1, the reduction could complete within 10 minutes. However, either insufficient or excessive addition of NaSH·xH2O decreases the stability of S-rGO dispersions.
:GO = 0.5:1 by weight). The reaction significantly accelerates with the amount of NaSH·xH2O increases. When the mass ratio of NaSH·xH2O to GO reaches 20:1, the reduction could complete within 10 minutes. However, either insufficient or excessive addition of NaSH·xH2O decreases the stability of S-rGO dispersions.
 | ||
| Fig. 1 Digital photographs of aqueous GO dispersions after adding NaSH·xH2O at 10 min, 1 h, 1 d and 7 day, respectively. The mass ratio of NaSH·xH2O to GO is 0:1, 0.5:1, 1:1, 2:1, 5:1, 10:1, 20:1 and 40:1, respectively. | ||
Generally, a mass ratio of NaSH·xH2O:GO from 2:1 to 5:1 enables rapid reduction of GO and stable dispersion of S-rGO. No sediment was observed even after 10 minutes centrifugation at 4000 rpm. For clarifying more detailed chemical structure of as-prepared S-rGO, GO reduced with a mass ratio of 2:1 (NaSH·xH2O:GO) was characterized and discussed.
Chemical-structural characterization
FTIR and Raman spectra were collected to characterize the structural changes of GO before and after the reaction with NaSH·xH2O. Fig. 2a shows the FTIR spectra of GO and S-rGO, a serials of typical characteristic peaks of GO were observed: broad O–H stretching vibrations at 2900–3500 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations from carbonyl and carboxyl vibrations at 1715 cm−1, CC skeletal stretching vibrations at 1620 cm−1, C–O stretching peaks of epoxy and alkoxy at 1215 cm−1 and 990 cm−1.29 For the S-rGO sample, it can be observed that the peaks at around 990, 1215, 1715, and 3300 cm−1 almost disappear, suggesting the oxygen-containing groups of GO were significantly reduced. However, no pronounced signal about sulfur was detected, either S–H or C–S bond.
O stretching vibrations from carbonyl and carboxyl vibrations at 1715 cm−1, CC skeletal stretching vibrations at 1620 cm−1, C–O stretching peaks of epoxy and alkoxy at 1215 cm−1 and 990 cm−1.29 For the S-rGO sample, it can be observed that the peaks at around 990, 1215, 1715, and 3300 cm−1 almost disappear, suggesting the oxygen-containing groups of GO were significantly reduced. However, no pronounced signal about sulfur was detected, either S–H or C–S bond.
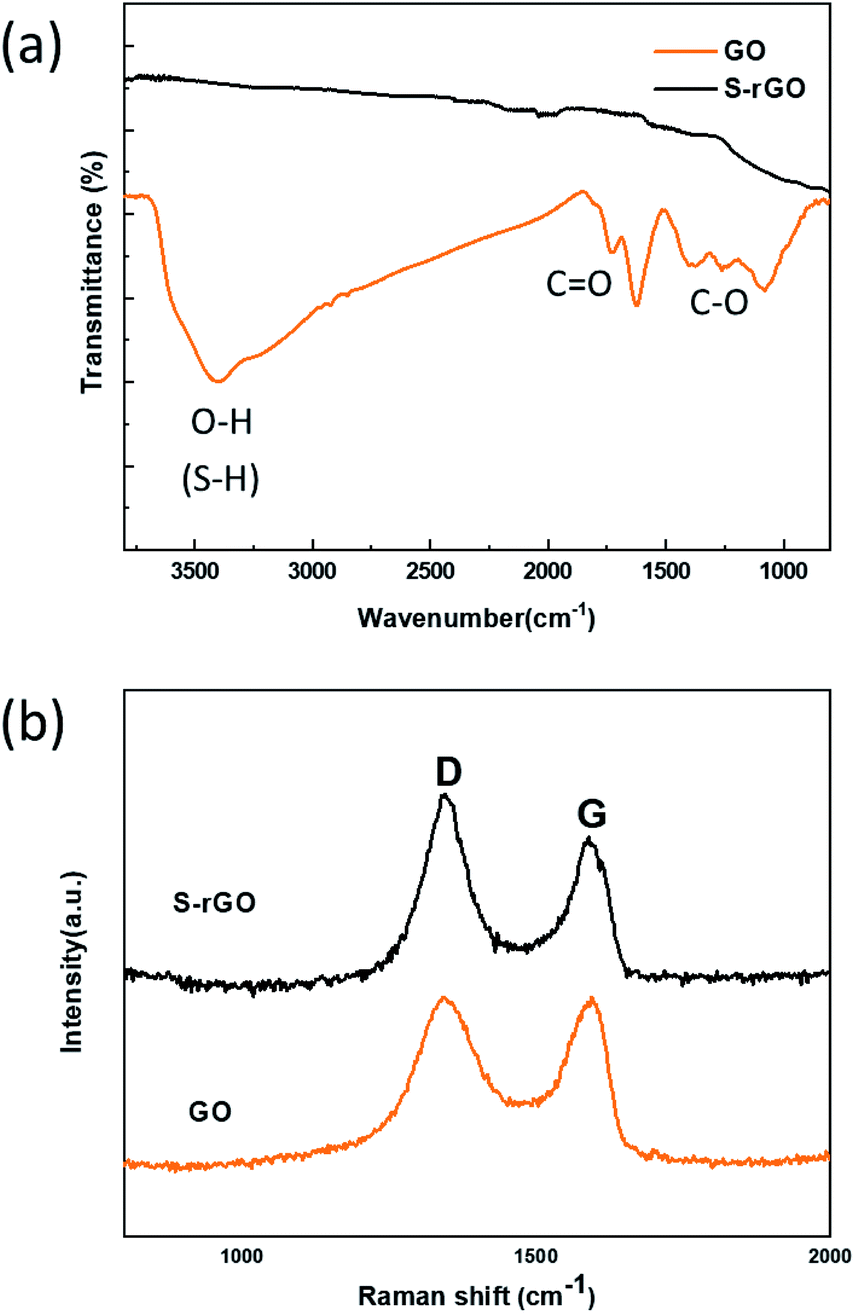 | ||
| Fig. 2 (a) FT-IR spectra of GO and S-rGO sheets; (b) Raman spectra of GO and S-rGO sheets. | ||
GO displays two characteristic peaks on its Raman spectrum (Fig. 2b): D band at 1342.1 cm−1 and G band at 1594.7 cm−1.30 D band indicates the structural defects and disorders, whereas G band represents the graphitic component in the structure.31 The intensity ratio of D to G band (ID/IG) is a measure of disordered carbon.32 From Fig. 2b, it is observed that the D and G band shift to 1340.3 cm−1 and 1589.1 cm−1 respectively, and the intensity ratio of ID/IG increases from 0.99 to 1.13 after reduction. This change suggests an increase in number of sp2-hybridized domains but a decrease in the average size after reduction.33 It is deduced that a large number of small graphitic domains are created after reduction, but the formation of large conjugated regions is prevented.
XPS characterization provides direct evidence of the reduction and sulfur doping of GO. In the XPS scan of GO (Fig. 3a), two peaks appear at 286.3 eV (C 1s) and 533.1 eV (O 1s), while the S-rGO sample shows an enhanced C 1s peak and a suppressed O 1s peak, with a C/O ratio increasing from 2.2 to 6.7. The C 1s spectrum of GO (Fig. 3c) can be fitted into five main components with binding energies at 284.8, 285.6, 286.7, 287.3, and 288.8 eV, which are assigned to the aromatic C, C–O, C–O–C, CO and O–CO species, respectively. In the C 1s spectrum of S-rGO (Fig. 3d), the intensities of peaks of C–O, C–O–C, and CO functional groups decreased dramatically, revealing that most oxygen-containing functional groups were removed.
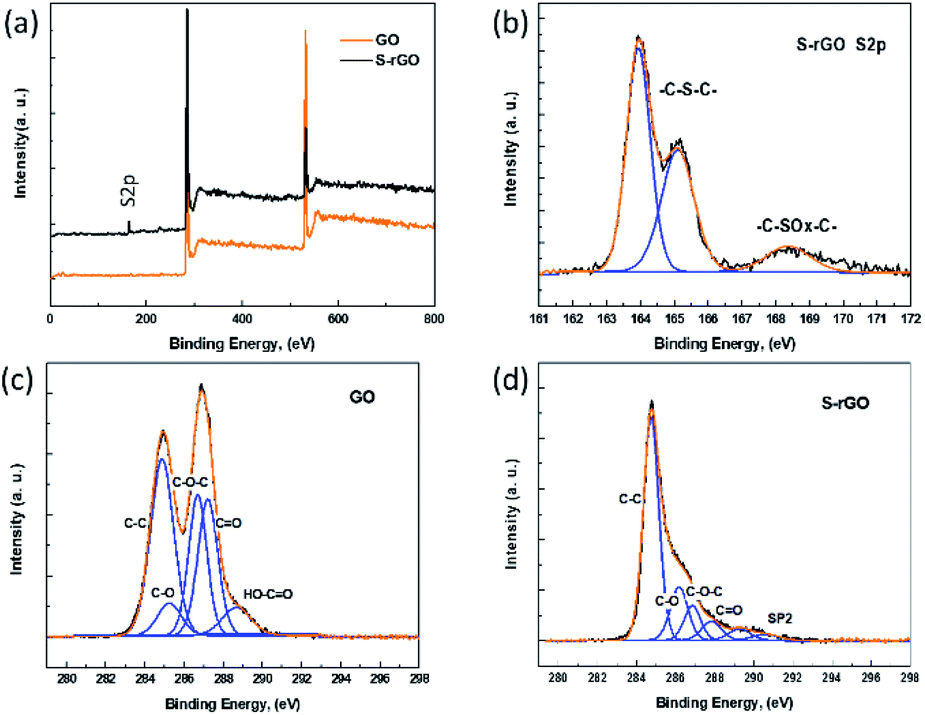 | ||
| Fig. 3 (a) XPS spectra of GO and S-rGO; (b) S 2p XPS spectrum of S-rGO; (c) C 1s XPS spectrum of GO sheets; (d) C 1s XPS spectrum of S-rGO sheets. | ||
Meanwhile, an additional S 2p signal is observed in the S-rGO sample (Fig. 3a), demonstrating the existence of sulfur element on the S-rGO surface. As summarized in Table S1,† around 2.50 wt% sulfur is doped on S-rGO sheets. The introduction of sulfur moieties on the S-rGO surface was further validated via EDS elemental analysis (Fig. 4b). The sulfur-elemental mapping image shows that the distribution of sulfur atoms is homogenous, rather than forming aggregations or clusters on the surface of S-rGO.
 | ||
| Fig. 4 (a) SEM image of S-rGO sheets; (b) the sulfur elemental mapping of S-rGO (purple, S atoms); (c) TEM image of S-rGO sheets; (d) AFM image of S-rGO sheets. | ||
The high resolution S 2p XPS spectrum of S-rGO shows an obvious doublet peak and a broad weak peak (Fig. 3b). The former peak consists two distinct components at 164.0 and 165.1 eV, which in agreement with the reported –C–S–C– covalent bond of thiophene-sulfur.21 The latter peak at 168.4 eV could be ascribed to some oxidized sulfur. However, the thiol (SH) signal at around 162.0 eV cannot be detected in the S 2p XPS spectrum. Moreover, no S–H stretching vibration signal could be observed in the FTIR spectrum of S-rGO sample (Fig. 2a). Therefore, it can be concluded that the sulfur atoms are doped into the rGO skeleton, rather than being attached onto the rGO surface via covalent bonds.
It is worth noting that the doped sulfur mainly presents as thiophene-sulfur (–C–S–C–), and a few of them exists as oxidized sulfur groups (–C–SOx–C–, X = 2, 3) (Fig. 3b), which is similar to the S 2p XPS spectrum of S-rGO obtained by either thermal annealing or hydrothermal method.15,17
For comparison, details of S-rGO prepared using different methods are summarized in Table S2.† The S content of S-rGO in this work is 2.50 wt%, which is comparable to that of S-doped rGO synthesized by conventional strategies such as CVD method, thermal annealing and hydrothermal treatment. The C/O ratio is 6.71, which is close to the value of S-rGO prepared by either hydrothermal treatment or thermal annealing at around 650 °C,34 but lower than that of S-rGO prepared using a 1000 °C annealing temperature.34 Generally, both the CVD method and thermal annealing process will inevitably involve special equipment, high energy input and expensive precursors.35 The hydrothermal method requires high reaction temperature and long reaction time. In comparison with these high energy-consuming methods, the NaSH-induced reaction is very simple and mild, which enable the reduction and sulfur doping of GO at room temperature, without additional experimental equipment and harsh reaction conditions.
Mechanism analysis
The chemical structure of S-rGO has been well studied that the thiophene-like structure can only be formed at the defects and edges of rGO sheets (Scheme 1a), even in the case of high-temperature thermal annealing.34,36 Since the hydroxyl and epoxide groups of GO are mainly located in the basal plane, while the carbonyl and carboxyl groups are located at the defect sites and edges, it is reasonable to expect that the sulfur doping is related to carbonyl and/or carboxyl groups. However, it is still challenging to propose an explicit reaction path for the conversion from GO to S-rGO because it involves the chemical reduction and sulfur doping mechanisms of GO, and neither of them has been well elaborated so far.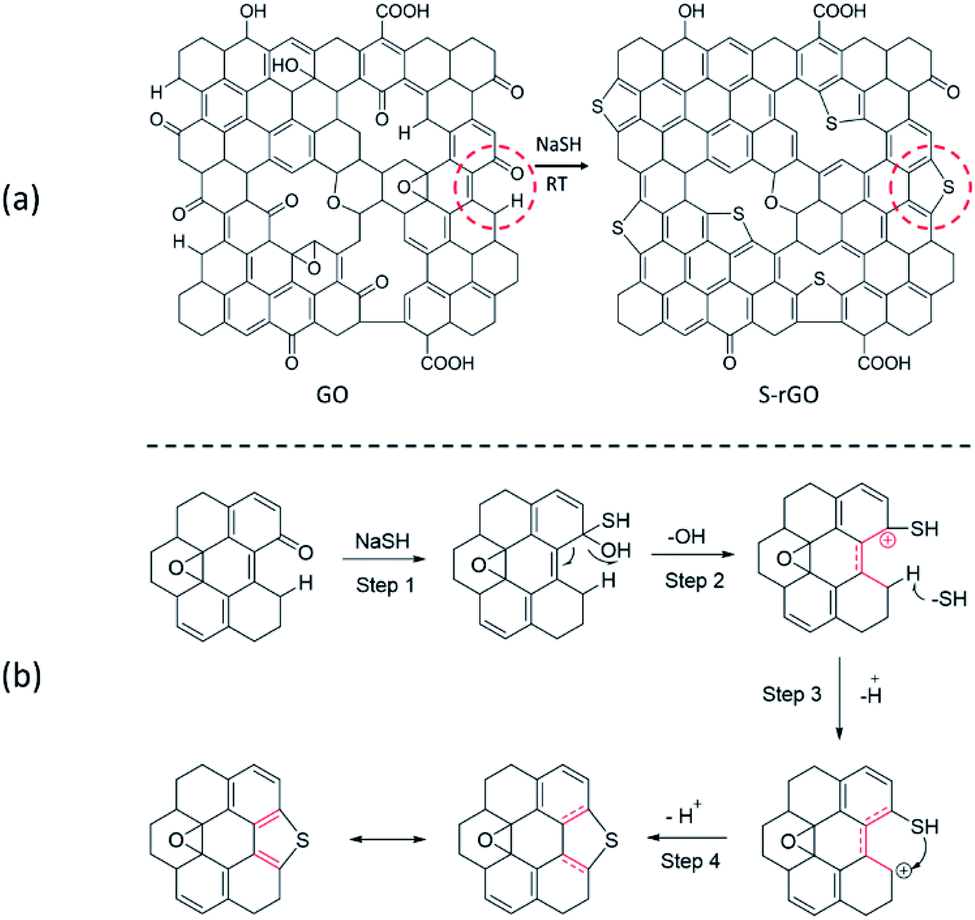 | ||
| Scheme 1 (a) Schematic structure of GO and S-rGO; (b) a possible mechanism for the formation of thiophene-like structure. | ||
For chemical reduction process, the hydroxyl and epoxy groups on GO are significantly reduced and sp2 regions are partially reconstructed. It is well accepted that the epoxy groups of GO could be readily opened by ring-opening reaction.37 Considering the strong nucleophilicity of sulfur compounds, it is speculated that the groups can open the epoxy and attached to GO surface via nucleophilic reaction, and the epoxy is therefore converted to C–OH and C–SH. Given that the binding energy of C–S is lower than that of C–O, the thiol groups are easier to remove compare with hydroxyl groups on GO basal plane. Unfortunately, to our knowledge, the dehydroxylation of GO is often attributed to a thermal-induced process.38,39
In this work, the NaSH induced a room-temperature reaction to remove the effects of heat treatment. We thus propose the reaction route for the sulfur doping of GO based on the carbonyl groups located at the defects and edges of GO sheets (Scheme 1b): (1) –SH attacks the carbonyl group to generate a hydroxyl and a thiol group; (2) the hydroxyl group leaves to form a stabilized carbocation (p–π conjugation), which will enhance the acidity of C–H; (3) the C–H bond breaks to give a new carbocation; (4) C–SH attacks the carbocation to form a thiophene-like structure.
Morphology and colloidal properties
In addition to the room-temperature reduction and sulfur doping of GO, it is also found that the as-prepared S-rGO could be well-dispersed in water without using any stabilizer. A dispersant-free aqueous S-rGO dispersion will has remarkable advantages in preparing graphene-based composite materials, sensors, catalysts and energy storage devices (Fig. S3†). To investigate the dispersion state of S-rGO dispersions, we carefully characterized the surface morphology and colloidal properties of S-rGO. The accurate morphology of S-rGO is recorded by SEM, TEM and AFM images. Both the SEM and TEM images (Fig. 4a and c) show that the S-rGO samples are thin and highly transparent with wrinkles on the sheets. AFM results (Fig. 4d) show that the S-rGO sheets are flat, with a thickness of ∼1 nm. These results demonstrate that as-prepared S-rGO sheets haven't aggregated after reduction.Since the as-prepared S-rGO sheets remain separated after reduction, there is a possibility that they were well-dispersed in water before. It is well accepted that GO sheets could be highly negatively charged in water and form well-dispersed aqueous colloids as a result of electrostatic repulsion.24 The negative surface charge of GO sheets is attributed to ionization of the hydroxyl and carboxyl groups.40 Considering that these groups are not completely removed at given conditions as presented in FT-IR and XPS spectrum, S-rGO sheets could still be negatively charged, and the electrostatic repulsion would enable the formation of stable S-rGO dispersions.
The colloidal nature of S-rGO dispersion was initially confirmed by the investigation of Tyndall effect (Fig. 5a), in which a laser optical path was observed due to the light scattering. The colloidal stability of S-rGO dispersions is further supported by zeta potential analysis, and the surface charge of S-rGO are highly pH-dependent.24,27 Zeta potential results (Fig. 5b) show that S-rGO sheets are highly negatively charged under alkaline conditions, with zeta potential values below −30 mV. The zeta potential can reach −42 mV when the pH approaches to 9.5. In this regard, to maintain high surface charge density, earlier studies usually introduce extra ammonia or sodium hydroxide to create an alkaline environment.27 It is noted that when the amount of NaSH·xH2O is as twice as that of GO, the pH of rGO dispersion is 8.67, and the corresponding zeta potential is −32 mV. If the mass ratio of NaSH·xH2O to GO increased to 5:1, the pH and zeta potentials would be 10.02 and −35 mV, respectively. Consequently, the S-rGO dispersion could keep stable without the need of ammonia/sodium hydroxide.
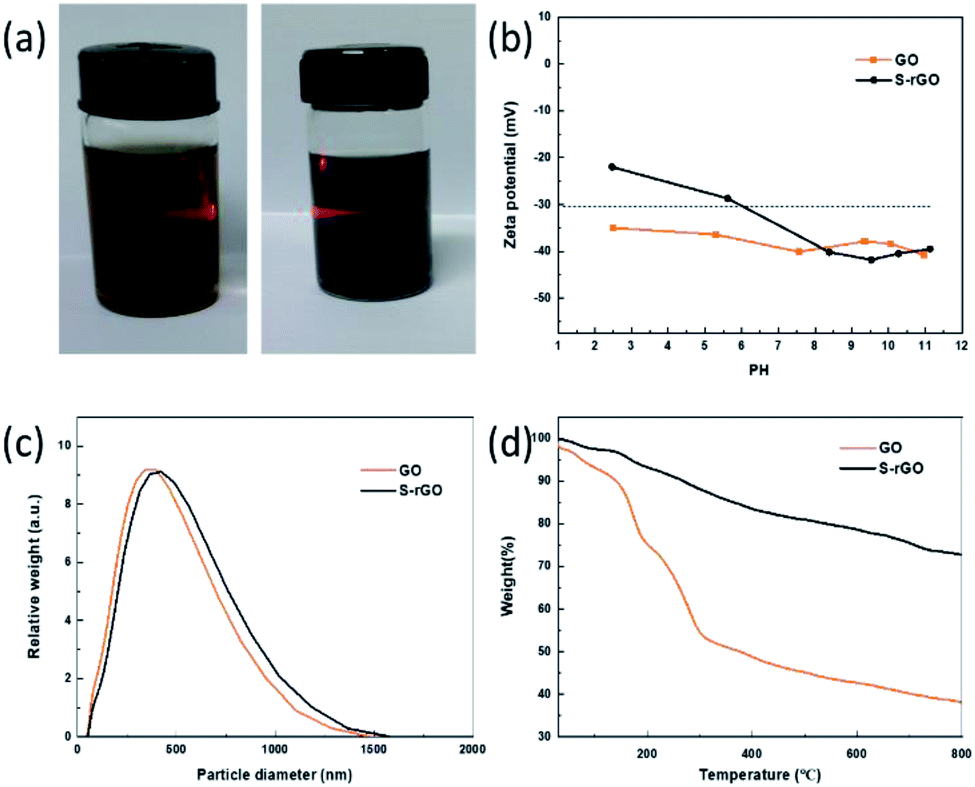 | ||
| Fig. 5 (a) The Tyndall effect of GO and S-rGO dispersions (GO: yellow, S-rGO: black); (b) zeta potential of GO and S-rGO dispersions as a function of PH, at a concentration of 0.02 mg ml−1; (c) particle size distribution of GO and S-rGO dispersions; (d) TGA curves of GO and S-rGO. | ||
The dispersion state of S-rGO sheets in water was also monitored by measuring their average particle size (Fig. 5c). Both GO and S-rGO sheets range between 100 and 1500 nm (maxima at 500 nm). There is no significant increase in the particle size during the reduction of GO.
These results show that the as-prepared S-rGO colloidal aqueous solution could keep stable without using any extra reagent. However, it is worth pointing out that, in addition to pH, the stability of S-rGO colloid is also strongly dependent on the electrolyte concentration. Reducing agents/sulfur compounds can generate ionic species and act as electrolytes in aqueous solution. Therefore, the overuse of NaSH will also lead to destabilization of S-rGO colloids (Fig. 1).
The thermal stability of GO and S-rGO was characterized by TGA. Fig. 5d illustrates the TGA traces of GO and S-rGO. There are two main steps for the mass loss of GO: the first mass loss step around 100 °C is attributed to the removal of residual moisture, while the second step between 200 and 300 °C come from the decomposition of hydroxyl and epoxy groups. The mass loss of GO is ∼45.7% at 300 °C. In contrast, obvious mass loss before 300 °C no longer presents in the TGA curve of S-rGO sample. The S-rGO exhibits a significantly high thermal stability with a mass loss of merely ∼11.8% at 300 °C, which is attributed to the effective reduction of GO.
Conclusions
To the best of our knowledge, for the first time, we report a one-step room-temperature approach for preparing sulfur doped reduced graphene oxide. Compared with the conventional thermal annealing and hydrothermal methods, the NaSH-induced reduction and sulfur doping approach requires little to none equipment, involves no energy-consuming processes (either heat treatment or sonication), and can form stable aqueous S-rGO dispersion without using any stabilizer. This simple and mild reaction thus shows great potential for efficient and large-scale production of water-dispersible S-rGO. In addition, as-prepared dispersant-free S-rGO dispersion also has advantages in preparing graphene-based composite materials, sensors, catalysts and energy storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was supported by Beijing Municipal Science and Technology Commission (Z171100002017016) and National Natural Science Foundation of China (51802296).Notes and references
- Z. F. Yang, J. R. Tian, Z. F. Yin, C. J. Cui, W. Z. Qian and F. Wei, Carbon, 2019, 141, 467–480 CrossRef CAS
.
- X. J. Lee, B. Y. Z. Hiew, K. C. Lai, L. Y. Lee, S. Gan, S. Thangalazhy-Gopakumar and S. Rigby, J. Taiwan Inst. Chem. Eng., 2019, 98, 163–180 CrossRef CAS
- M. Chen, R. Haddon, R. Yan and E. Bekyarova, Mater. Horiz., 2017, 4(6), 1054–1063 RSC
- G. Yazdi, T. Iakimov and R. Yakimova, Crystals, 2016, 6, 53 CrossRef
- Y. Xu, H. Cao, Y. Xue, B. Li and W. Cai, Nanomaterials, 2018, 8, 942 CrossRef PubMed
- K. K. H. De Silva, H. H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS
- Y. Chen, X.-L. Gong and J.-G. Gai, Adv. Sci, 2016, 3, 1500343 CrossRef PubMed
- L. Dong, J. Yang, M. Chhowalla and K. P. Loh, Chem. Soc. Rev., 2017, 46, 7306–7316 RSC
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS
- A. B. Alayande, H. D. Park, J. S. Vrouwenvelder and I. S. Kim, Small, 2019, 15, 12 CrossRef PubMed
- K. Thakur and B. Kandasubramanian, J. Chem. Eng. Data, 2019, 64, 833–867 CrossRef CAS
- W. Wang, Z. Wang, J. Liu, Z. Luo, S. L. Suib, P. He, G. Ding, Z. Zhang and L. Sun, Sci. Rep., 2017, 7, 46610 CrossRef PubMed
- F. Shahzad, S. A. Zaidi and C. M. Koo, Sens. Actuators, B, 2017, 241, 716–724 CrossRef CAS
- M. A. Hoque, F. M. Hassan, D. Higgins, J.-Y. Choi, M. Pritzker, S. Knights, S. Ye and Z. Chen, Adv. Mater., 2015, 27, 1229–1234 CrossRef CAS PubMed
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC
- W. Ai, Z. Luo, J. Jiang, J. Zhu, Z. Du, Z. Fan, L.-H. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed
- Z. S. Tian, J. T. Li, G. Y. Zhu, J. F. Lu, Y. Y. Wang, Z. L. Shi and C. X. Xu, Phys. Chem. Chem. Phys., 2016, 18, 1125–1130 RSC
- X. Ren, H. Ma, T. Zhang, Y. Zhang, T. Yan, B. Du and Q. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 37637–37644 CrossRef CAS PubMed
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC
- H. L. Poh, P. Šimek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS PubMed
- Y. Wang, M. Hu, D. Ai, H. Zhang, Z.-H. Huang, R. Lv and F. Kang, Nanomaterials, 2019, 9, 752 CrossRef CAS PubMed
- W. Chen, L. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CrossRef CAS
- M. Klingele, C. Pham, K. R. Vuyyuru, B. Britton, S. Holdcroft, A. Fischer and S. Thiele, Electrochem. Commun., 2017, 77, 71–75 CrossRef CAS
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed
- Z. Lin, Y. Liu, U. Halim, M. Ding, Y. Liu, Y. Wang, C. Jia, P. Chen, X. Duan and C. Wang, Nature, 2018, 562(7726), 254–258 CrossRef CAS PubMed
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS
- K. Hatakeyama, K. Awaya, M. Koinuma, Y. Shimizu, Y. Hakuta and Y. Matsumoto, Soft Matter, 2017, 13, 8353–8356 RSC
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef
- G. Santamaría-Juárez, E. Gómez-Barojas, E. Quiroga-González, E. Sánchez-Mora, M. Quintana-Ruiz and J. D. Santamaría-Juárez, Mater. Res. Express, 2020, 6, 125631 CrossRef
- S. N. Alam, N. Sharma and L. Kumar, Graphene, 2017, 6, 1–18 CrossRef CAS
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS
- E. Dervishi, Z. Q. Ji, H. Htoon, M. Sykora and S. K. Doorn, Nanoscale, 2019, 11, 16571–16581 RSC
- L. A. Perez, N. Bajales and G. I. Lacconi, Appl. Surf. Sci., 2019, 495, 11 CrossRef
- F. Shahzad, P. Kumar, S. Yu, S. Lee, Y.-H. Kim, S. M. Hong and C. M. Koo, J. Mater. Chem. C, 2015, 3, 9802–9810 RSC
- L. Feng, Z. Qin, Y. Huang, K. Peng, F. Wang, Y. Yan and Y. Chen, Sci. Total Environ., 2020, 698, 134239 CrossRef CAS PubMed
- N. I. Ikhsan and A. Pandikumar, Doped-Graphene Modified Electrochemical Sensors, Elsevier Science Bv, Amsterdam, 2019 Search PubMed
- M. Fallah-Mehrjardi, A. R. Kiasat and K. Niknam, J. Iran. Chem. Soc., 2018, 15, 2033–2081 CrossRef CAS
- C. K. Chua and M. Pumera, Chem. Commun., 2016, 52, 72–75 RSC
- K. K. H. De Silva, H.-H. Huang, R. Joshi and M. Yoshimura, Carbon, 2020, 166, 74–90 CrossRef CAS
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: The PH of GO and S-rGO dispersions, the 13C NMR spectra of S-rGO, and the XRD pattern of GO and S-rGO sheets. See DOI: 10.1039/d0ra04838k |
| This journal is © The Royal Society of Chemistry 2020 |