
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of the structure–function relationship of a novel salt-resistant antimicrobial peptide, RR12†
Ping-Sheng Wua,
Shu-Jung Laibd,
Kit-Man Fungb and
Tien-Sheng Tseng *c
*c
aDivision of Infectious Diseases, Department of Pediatrics, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei, Taiwan
bInstitute of Biological Chemistry, Academia Sinica, Taipei 115, Taiwan
cInstitute of Molecular Biology, National Chung Hsing University, Taichung, Taiwan
dGraduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan
First published on 22nd June 2020
Abstract
Antimicrobial peptides (AMPs) are potential candidates in designing new anti-infective agents. However, many AMPs show poor bactericidal activities in physical salt and serum solutions. Here, we disclosed the structure–function relationships of a novel salt-resistant antimicrobial peptide, RR12, which could further explain its mode of action and show its applicability in developing new antibacterial agents.
Introductions
The rapid emergence of antibiotic-resistant bacteria is now a severe health problem all around the world.1,2 Therefore, developing new bactericidal agents with less probability to induce evolved resistance is urgently needed. In nature, organisms genetically obtain abilities from progenitors to defend themselves against pathogens. Their immune resistant abilities are connected to the development of specific immune responses. Antimicrobial peptides (AMPs) are the natural element of the innate immune system observed in most living organisms.3–5 AMPs target and disrupt the plasma membrane of bacteria leading to death, and have a minimal inhibitory concentration against bacteria in the μM range.6–8 Thus, AMPs are feasible candidates in developing potent therapeutics against the infections of antibiotic-resistant bacteria.Nowadays, thousands of AMPs are characterized and deposited in the Antimicrobial Peptide Database (AMSDb, http://aps.unmc.edu/AP/main.php). The database can be used to predict the structure, function, and antibacterial activities on the basis of peptide sequence.9–11 AMPs mostly present as amphipathic peptides (12–30 amino acids) containing positive charges from +2 to +9 with some hydrophobic residues.7,12,13 Structurally, AMPs are observed in four types: α-helical, β-sheet, loop, and extended peptides.13,14 In particular, the α-helical conformation is the most effective structural arrangement observed at the bacterial surface in innate defense.15 Functionally, AMPs possess two important features: (1) the capability of assuming an amphipathic helical conformation—most AMPs are linearly unstructured and fold into an amphipathic helix when targeting to bacterial membrane—and (2) a net cationicity—cationic AMPs bind to the negative-charged bacterial surface.13,16–20 With these properties, AMPs can disrupt the transmembrane potential, interfere the balance of ion gradients, and cause the leakage of cell contents, finally leading to microbial death.
To be molded into potent bactericidal agents, AMPs should have less hemolytic activities and effective bactericidal abilities. However, high amphipathicity and hydrophobicity of AMPs are reported to be correlated with high hemolytic activity.21 Additionally, natural AMPs show low bioavailability and are easily degraded by proteases.22,23 Therefore, shorter peptides were modified to enhance the antimicrobial activity but decrease hemolytic activity and cytotoxicity.24–27 While many natural AMPs still exhibit poor bactericidal abilities in physical salt concentrations.28,29 This significantly hindered the pharmaceutical development of AMPs as new therapeutic agents. Thus, investigation and development of AMPs with strong salt tolerance and antibacterial activities are essential in combating antibiotics-resistant bacteria.
Recently, Harini Mohanram et al. reported a peptide, RR12 (RRLIRLILRLLR), exerting bactericidal activity and is not affected by high concentration of sodium chloride (150–300 mM) or 10% human serum containing media.30 Although RR12 was observed to interfere membrane integrity of E. coli cells in the presence of salt, detailed characterizations of its structure–function relationships have not been thoroughly investigated. The development of biocidal AMPs is usually hindered by poorly understanding of information on their physicochemical characteristics, which are highly associated with their mode of actions.31,32 Therefore, to disclose the bactericidal function of RR12, we investigated its structural properties, capability of interacting with membranes, and the solution structure on binding with membrane-mimetic micelles.
Here, we employed biochemical and biophysical techniques to study the characteristics of RR12 in interacting with sodium dodecyl sulfate (SDS) micelles. We performed Far-UV CD, dye-leakage fluorescence assay, 2D and spin-labeling NMR to investigate the secondary structure content, membrane-permeabilizing activity, solution structure, and orientation in SDS micelles, respectively. The observed structural and functional insights could explain the mechanism of action of RR12 against microbes and provide valuable information for development of new anti-infective agents with salt-resistant abilities.
Materials and methods
Materials
RR12 peptide (RRLIRLILRLLR-amide) was synthesized by Yao-Hong Biotechnology Inc (http://www.yh-bio.com.tw/en/index.asp). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) were from Avanti Polar Lipids (Alabaster, AL, USA). Calcein, MnCl2 and 5-, and 12-doxyl stearic acids were from Sigma-Aldrich. SDS-d25, methanol-d4 and D2O were from Cambridge Isotope Laboratories. Sodium dodecyl sulfate was from Merk (Darmstadt, Germany).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DPC = 1:100 (molar ratio), and RR12:SDS = 1:100 (molar ratio)). Far-UV CD experiments were conducted by using a 1 mm path length quartz cuvette at 25 °C with the observed wavelength range between 190 and 260 nm. All spectra were averaged over three scans and converted to mean residue ellipticity [θ]. The helical contents of all spectra were estimated by using BESTSEL (http://bestsel.elte.hu/).000 rpm for 10 min) three times by using isosmotic buffer (10 mM Tris and 100 mM NaCl, pH 7.4). The size of the generated LUVs was confirmed by dynamic light scattering on a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).:100, containing 10 mM sodium phosphate and 10% D2O at pH 5.0. All NMR experiments involved Bruker Avance 600 and 800 MHz spectrometry at 310 K. 2D-NOESY spectra were acquired at two distinct mixing times of 150 and 300 ms. TOCSY spectra were recorded with mixing time 60 ms at 2048 points in t2 and 320 points in t1. A natural-abundance (1H, 15N) HSQC spectrum34 was obtained at 2048 points in t2 and 400 points in t1. The spectra were processed by using TopSpin 3.1 (Brucker Spectrospin) and NMRPipe.35 Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco) was employed for spectra analysis.:100) were acquired at pH 5.0, 310 K and then subjected to manually assignments to obtain distance constraints. Based on the peak intensity, the NOE cross-peaks were clustered into strong, medium, and weak, corresponding to a distance range of 1.8–2.8, 1.8–3.4, and 1.8–5.0 Å, respectively. The backbone dihedral angle constraints were deduced from the 1H and 15N chemical shifts of natural-abundance HSQC spectra by using TALOS.36,37 The solution structures were calculated by using CNS 1.2 for restrained molecular dynamic simulations.38,39 The 20 lowest energy structures were examined by using PROCHECK-NMR40 and MOLMOL.41 PyMol (http://www.pymol.org) was employed for molecular visualization.:100) and equilibrated for 15 min before 1H–1H TOCSY spectra recording. For the PRE experiments of DSA, the DSA powders were dissolved in deuterated methanol (D4-MeOH). The DAS solution was then mixed with SDS (in 10 mM sodium phosphate, pH 5.0) at a molar ratio of 1:60. Subsequently, RR12 was added to the prepared DSA–SDS mixed micelle solution. The final concentrations of DSA, SDS and RR12 were 2.5, 150, and 1.5 mM, respectively. These NMR samples were equilibrated for 15 min before 1H–1H TOCSY experiments. The 1H–1H TOCSY spectra were acquired with the same parameters (310 K with 2048 data points in t2 and 320 points in t1) except for the probe tuning and field shimming. The cross-peak intensities of RR12 were measured and calculated based on the protocol.42
:DPC = 1:100 (molar ratio), and RR12:SDS = 1:100 (molar ratio)). Far-UV CD experiments were conducted by using a 1 mm path length quartz cuvette at 25 °C with the observed wavelength range between 190 and 260 nm. All spectra were averaged over three scans and converted to mean residue ellipticity [θ]. The helical contents of all spectra were estimated by using BESTSEL (http://bestsel.elte.hu/).000 rpm for 10 min) three times by using isosmotic buffer (10 mM Tris and 100 mM NaCl, pH 7.4). The size of the generated LUVs was confirmed by dynamic light scattering on a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).:100, containing 10 mM sodium phosphate and 10% D2O at pH 5.0. All NMR experiments involved Bruker Avance 600 and 800 MHz spectrometry at 310 K. 2D-NOESY spectra were acquired at two distinct mixing times of 150 and 300 ms. TOCSY spectra were recorded with mixing time 60 ms at 2048 points in t2 and 320 points in t1. A natural-abundance (1H, 15N) HSQC spectrum34 was obtained at 2048 points in t2 and 400 points in t1. The spectra were processed by using TopSpin 3.1 (Brucker Spectrospin) and NMRPipe.35 Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco) was employed for spectra analysis.:100) were acquired at pH 5.0, 310 K and then subjected to manually assignments to obtain distance constraints. Based on the peak intensity, the NOE cross-peaks were clustered into strong, medium, and weak, corresponding to a distance range of 1.8–2.8, 1.8–3.4, and 1.8–5.0 Å, respectively. The backbone dihedral angle constraints were deduced from the 1H and 15N chemical shifts of natural-abundance HSQC spectra by using TALOS.36,37 The solution structures were calculated by using CNS 1.2 for restrained molecular dynamic simulations.38,39 The 20 lowest energy structures were examined by using PROCHECK-NMR40 and MOLMOL.41 PyMol (http://www.pymol.org) was employed for molecular visualization.:100) and equilibrated for 15 min before 1H–1H TOCSY spectra recording. For the PRE experiments of DSA, the DSA powders were dissolved in deuterated methanol (D4-MeOH). The DAS solution was then mixed with SDS (in 10 mM sodium phosphate, pH 5.0) at a molar ratio of 1:60. Subsequently, RR12 was added to the prepared DSA–SDS mixed micelle solution. The final concentrations of DSA, SDS and RR12 were 2.5, 150, and 1.5 mM, respectively. These NMR samples were equilibrated for 15 min before 1H–1H TOCSY experiments. The 1H–1H TOCSY spectra were acquired with the same parameters (310 K with 2048 data points in t2 and 320 points in t1) except for the probe tuning and field shimming. The cross-peak intensities of RR12 were measured and calculated based on the protocol.42Results
The secondary structure of RR12 in membrane mimic environment
We employed Far-UV CD spectroscopy to investigate the secondary structure of RR12. The CD spectra of RR12 showed a random coil conformation in buffer (10 mM sodium phosphate, pH 5.0). While, the spectra for RR12 in 50% TFE displayed strong signals at 208 and 222 nm (α-helical content = 51.6%) (Fig. 1). When interacting with DPC micelles (molar ratio of peptide:DPC = 1:100), RR12 showed a bit increased helicity (α-helical content = 57.3%), compared to that in 40% TFE. When interacting with SDS micelles (molar ratio of peptide:SDS = 1:100), RR12 exhibited comparable α-helical content (62%) to that in DPC micelles.
 | ||
| Fig. 1 Far-UV CD spectra for RR12 in TFE, DPC and SDS micelles. | ||
Solution structure of RR12 in complex with SDS micelle
We conducted NMR experiments to determine the solution structure of RR12. RR12 showed well dispersed and high resolved spectra (2D-NOESY and TOCSY) in SDS micelles at pH 5.0 and 310 K (Fig. S1†). Therefore, the solution structure of RR12 was determined in SDS micelles. The sequential assignments were completed by using 2D-TOCSY and NOESY spectra. The 200 NOE-derived distance constraints contained 96 intra-residue, 49 sequential, and 55 medium-range distance restraints. Besides, 18 backbone dihedral angle restrains derived from 1H, 15N- HSQC spectrum (Fig. 2A) were employed for structure calculation of the SDS micelle-bound RR12 (Table 1). The superimposed ensemble of the 20 lowest-energy structures is shown in Fig. 2B. The root mean square deviations (RMSD) of top10 lowest-energy coordinates are 0.7 ± 0.17 Å for all heavy atoms and 0.3 ± 0.13 Å for backbone heavy atoms (Table 1). Moreover, a Ramachandran plot of PROCHECK analysis demonstrated that 70% and 30% of RR12 residues are located at the most favored and additionally allowed regions, respectively. The structure of RR12 is composed of a two-turn α-helix (L3-L11) with the loop conformations at N- and C-termini (Fig. 2C). Notably, the structure along with the electrostatic surface (Fig. 2D) of RR12 revealed that positive-charged side chains of arginine residues (R1, R2, R5, R9, and R12) are on one side of the helix and the hydrophobic side chains are on the other side, revealing the apparent amphipathicity of RR12.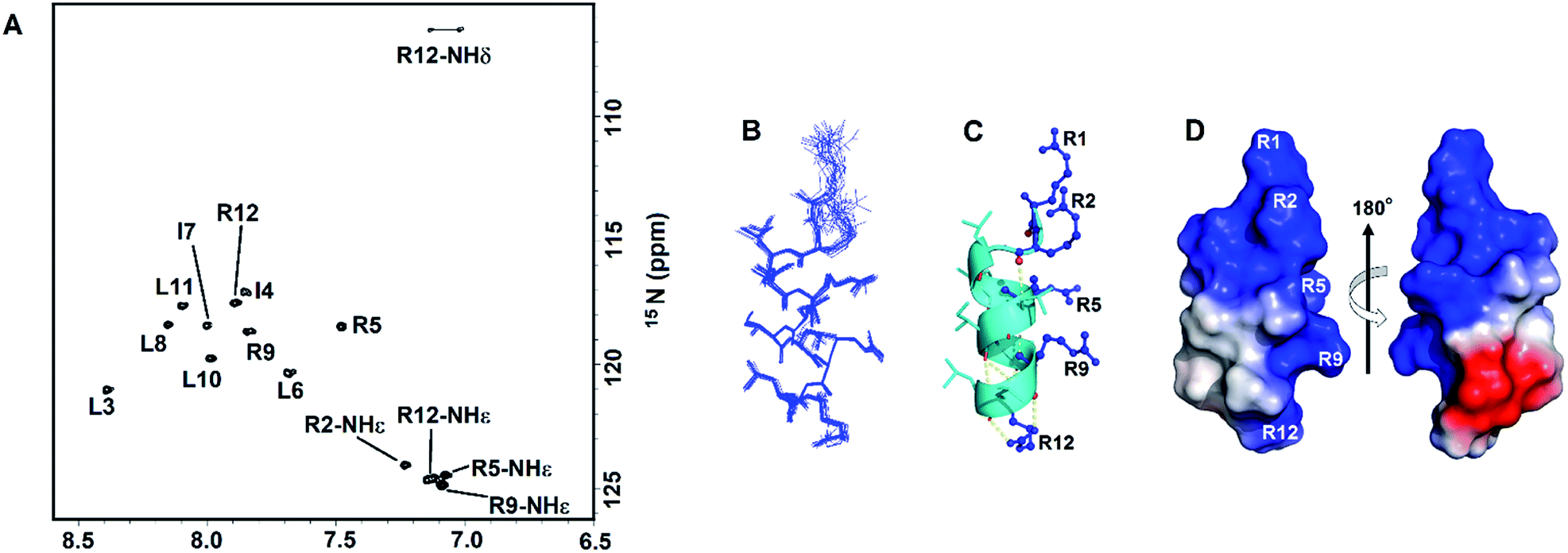 | ||
| Fig. 2 NMR spectra and solution structure of RR12 in SDS micelles (PDB ID: 6J9P). (A) The 1H–15N HSQC spectrum of RR12 in SDS micelles. (B) Superposition of the 20 lowest energy structures of RR12. The backbones and heavy chains are shown in blue lines. (C) Ribbon representation of the averaged SDS-bound solution structure. Arginine residues in blue are shown as balls and sticks. (D) The electrostatic surface of SDS-bound RR12.The positive charge is in blue, negative charge in red, and neutral positions in white. | ||
| a The default parameters and force constants of protein allhdg.param and anneal.inp in CNS1.2 are employed for the structure calculations. There are no NOE and dihedral angle violations observed during the structure calculations. | |
|---|---|
| NOE restraints | |
| Intra-residue NOEs | 96 |
| Sequential NOEs [(i − j) = 1] | 49 |
| Medium-range NOEs (|i − j| ≤ 4) | 55 |
| Total NOEs | 200 |
| Dihedral angle restraints | 18 |
|
|
| Ramachandran plot summary (%) | |
| Most favored | 70 |
| Additionally allowed | 30 |
| Generally allowed | 0 |
| Disallowed | 0 |
|
|
| Average RMSD from the mean structure | |
| Back atoms | 0.3 ± 0.13 |
| All heavy atoms | 0.7 ± 0.17 |
Membrane-permeabilizing ability of RR12 against synthetic LUVs
The peptide-induced leakage of calcein entrapped in LUVs of different surface-charge densities was employed to evaluate the membrane-permeabilizing ability of RR12. The highly negative-charged POPG and neutral POPC LUVs were used to mimic the bacterial and eukaryotic cell membranes, respectively. The minimal concentration of RR12 causing 100% leakage of calcein from the LUVs is termed as LC100. RR12 induced The LC100 of RR12 against POPG LUVs was 0.008 μM (Fig. 3). However, the LC100 of RR12 against POPC LUVs was 0.2 μM. These indicated that RR12 has stronger disrupting ability against negative-charged bacterial membranes but much weaker activity against neutral charged eukaryotic cell membranes.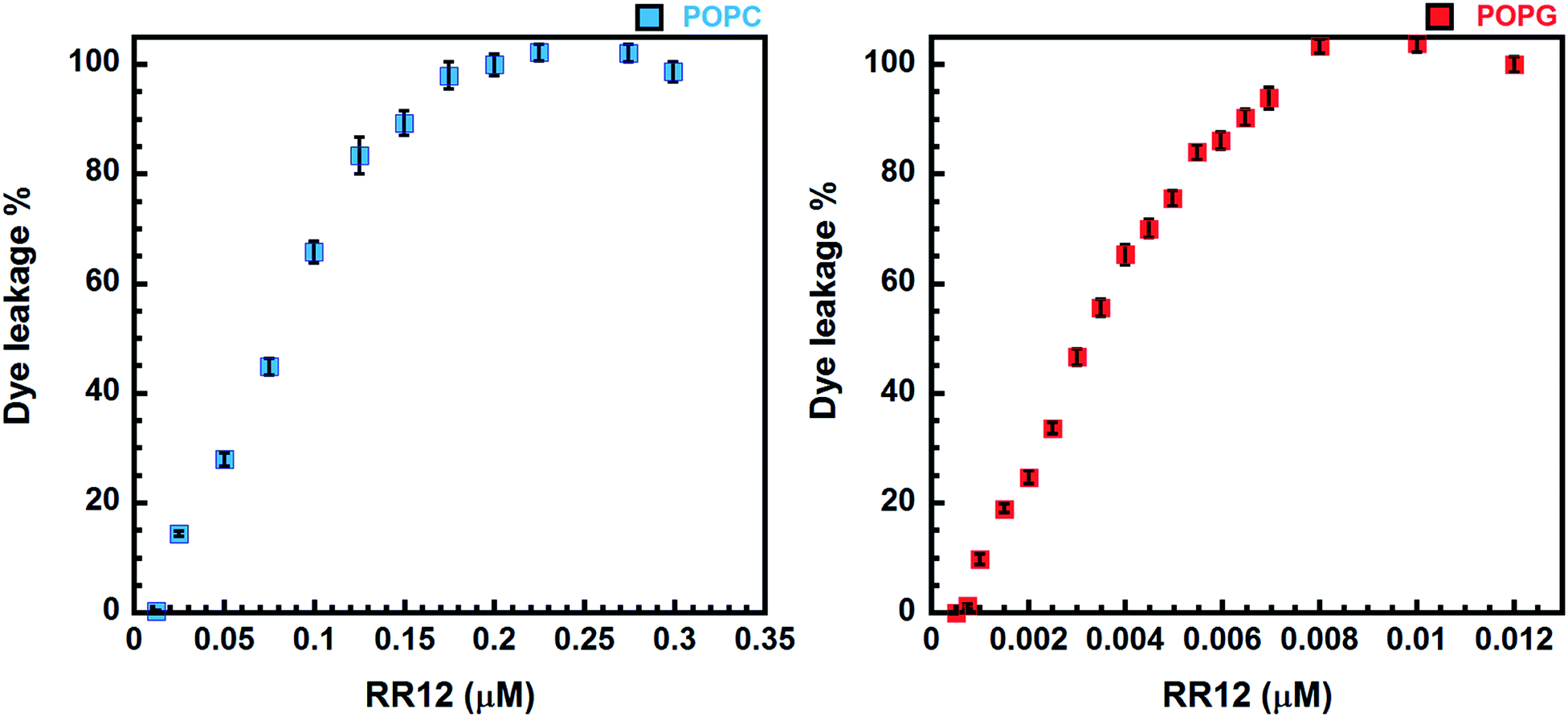 | ||
| Fig. 3 Calcein leakage caused by RR12 against the POPC and POPG LUVs. | ||
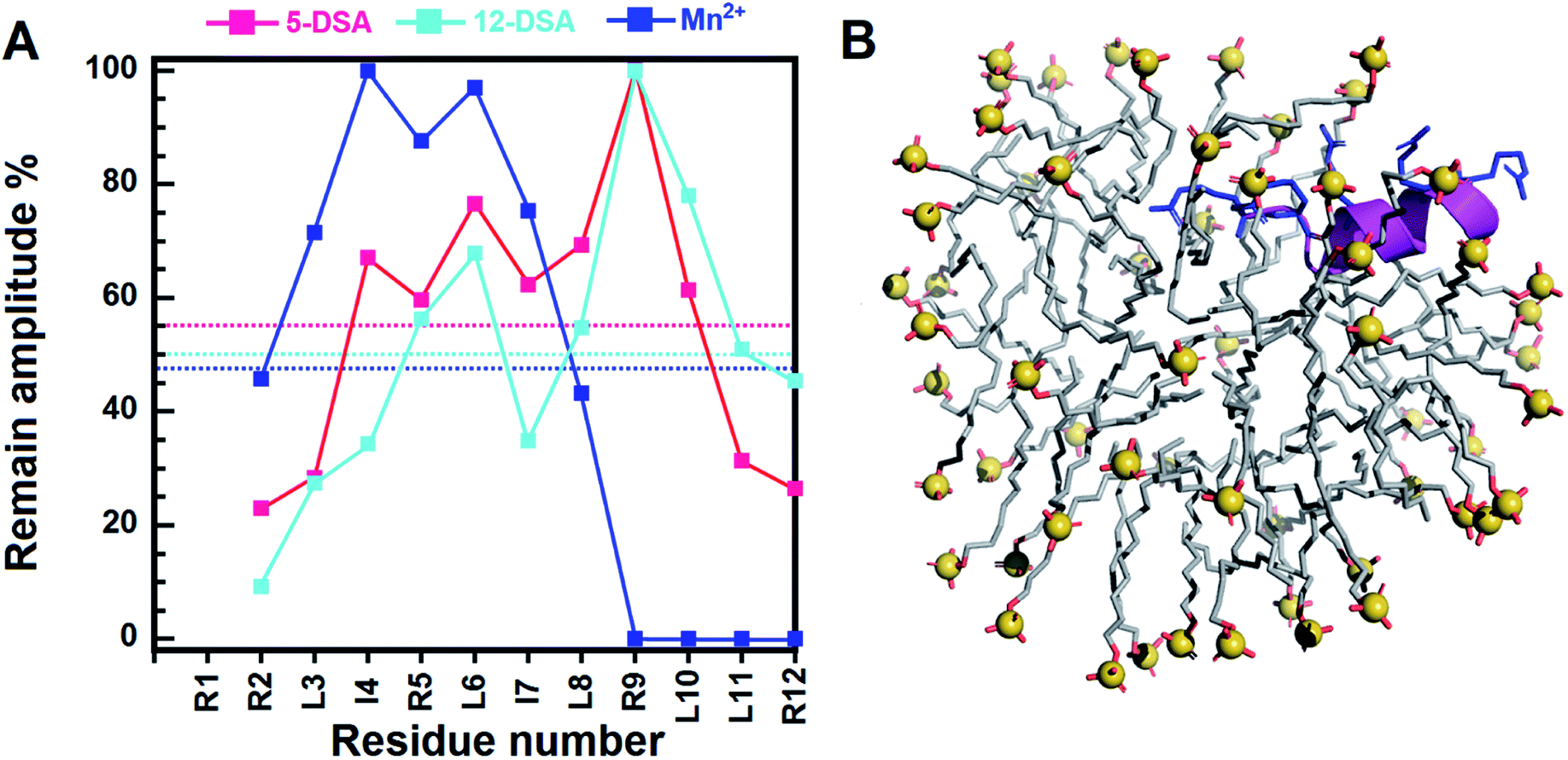 | ||
| Fig. 4 The position of RR12 in SDS micelles. (A) The remaining amplitudes of NH–CαH cross peaks derived from the TOCSY spectra of RR12 due to 5- and 12-DSAs. The concentration of each DSA corresponds to a spin label per micelle. The magenta, cyan, and blue dotted lines indicate the mean remaining amplitudes of RR12 in the presence of 5-, 12-, and (0.5 mM) Mn2+ ions, individually. (B) The model structure of RR12 in complex with SDS micelle deduced from PRE results. The RR12 peptide was shown as magenta ribbon with arginine residues and R1, R2, R5, R9 and R12 presented in blue sticks. The SDS lipids were displayed as light-grey sticks with the sulphur atoms shown as spheres (yellow) and oxygen atoms were colored in red. | ||
Discussion
The broad-spectrum antibiotics have been the major components for antibacterial therapy. While the rapid emergence of antibiotic-resistant bacteria forced to reconsider new strategies in combating pathogenic microbes. The AMPs are promising candidates in developing new anti-infective agents.43 Nevertheless, many AMPs exert limited bactericidal activities in physical salt and serum solutions.44 Thus, AMPs with salt-resistant capabilities are urgently required for developments of future antibiotics. A short AMP, RR12, was demonstrated exhibited antibacterial activity in the presence of NaCl (150 and 300 mM) and human serum, but its characteristics in terms of structure and function are not clearly investigated. Here, we used biochemical and biophysical methods to investigate the structure–function relationship of RR12 in SDS micelles to understand the possible mode of action.We characterized the structural properties of RR12 associated with its antimicrobial function through CD spectroscopy. RR12 is a random coil in phosphate buffer solution but shows apparent α-helical structure in 50% TFE. Fluorinated alcohol can change the electrostatic interactions of the peptides and affect the carbonyl and amide groups, enhancing the hydrogen bond formation to stabilize helical formation.45–47 RR12 also adapted the α-helical conformations in both zwitter-ionic DPC and negative-charged SDS micelle systems (Fig. 1), indicating the bioactive conformational transition (from unstructured to α-helical conformation) of RR12 is required for binding to phospholipid membranes. This observation is consistent with previous report that AMPs are disordered in aqueous conditions but become structured upon interacting with phospholipid membranes.19,48,49 To understand the structural basis for the mechanism of action of RR12 when interacting with bacterial membranes, we determined its solution structure by solution NMR. The synthetic peptide, RR12, was soluble and stable at acidic environment (pH 5.0) with high concentration (>2 mM). However it easily precipitated when dissolved or titrated to more basic environments (pH 7.0). The RR12 peptide solution was observed clear without precipitates at 1–300 μM but gradually get aggregated when the concentration was over 500 μM at pH 6.5–7.0. Thus, we acquired 1D and 2D NMR spectra of RR12 in zwitterionic DPC and negatively charged SDS micelles at pH 5.0 (Fig. 2A and S2–S4†). SDS micelles consists of a negatively charged outer surface and a hydrophobic inner core, thus mimicking the bacterial cell membranes.50,51 The spectra (2D-TOCSY and NOESY) of RR12 in SDS micelles are well-dispersed and highly resolved than those of in DPC micelles. To directly and successfully investigate the interactions between RR12 and micelles, we simplified the solvent condition to a phosphate buffer at pH 5.0 (10 mM) without salt effect. The obtained solution structure represents an initial scaffold and conformation of RR12 which is able to exert its salt-resistant ability. Thus, we finally determined the solution structure of RR12 in complex with SDS micelles. Structurally, residues L3–L11 of RR12 display an amphipathic α-helical conformation with hydrophobic and positive charged side chains positioned at opposite sides (Fig. 2).
Generally, short and small AMPs exert the bactericidal activities through binding and disrupting the integrity of microbial membrane.7,52,53 Especially, the cationic residues of amphipathic helical AMPs are critical in interacting with the negatively charged and lipidic components of microbial membranes.19,54 Therefore, we compared the structure of RR12 with those of known short AMPs (Fig. 5) to understand the possible mode of action of RR12. The AMPs, KR12 (from human cathelicidin LL-37 (PDB ID:2NA3)), P3967 (from medicinal leech Hirudo medicinalis (PDB ID:6RRL)), CM15 (a hybrid peptide of Cecropin1-8 + Melittin3-9 (PDB ID:2JMY)), and are 12, 12, and 15 in lengths. The structures of these AMPs are mostly amphipathic helices (Fig. S5†), however the distributions of their cationic residues are varied. RR12, KR12, P3967, and CM15 show similar 2–3 turns α-helical conformations with the positively charged resides (arginine and lysine) extrude outwards. In KR12, K1, R6, and R12 are on the same side while the side chains of R2 and K8 slightly orient in distinct directions. Similar characters were observed in the structure of P3967 (R2, R5, and R8 are nearly on the same side; K11 extrudes oppositely) and CM15 (K1 and K10 orient in the same direction; K3 and K7 are on the same side; K6 positions in between). In contrast, the side chains of all the positive charged residues of RR12 (R1, R2, R5, R9, and R12) orient in the same direction, exhibiting high amphipathicity. It has been reported that the designed, arginine-based peptides showed improved salt resistance bactericidal activity55 and the cation–π interaction of arginine is highly associated with.56 Accordingly, arginine residues aligned on the cationic side of RR12 is of great potential to render its salt resistant ability-due to the possible ionic and hydrogen bonds interactions towards the bacterial outer membranes.
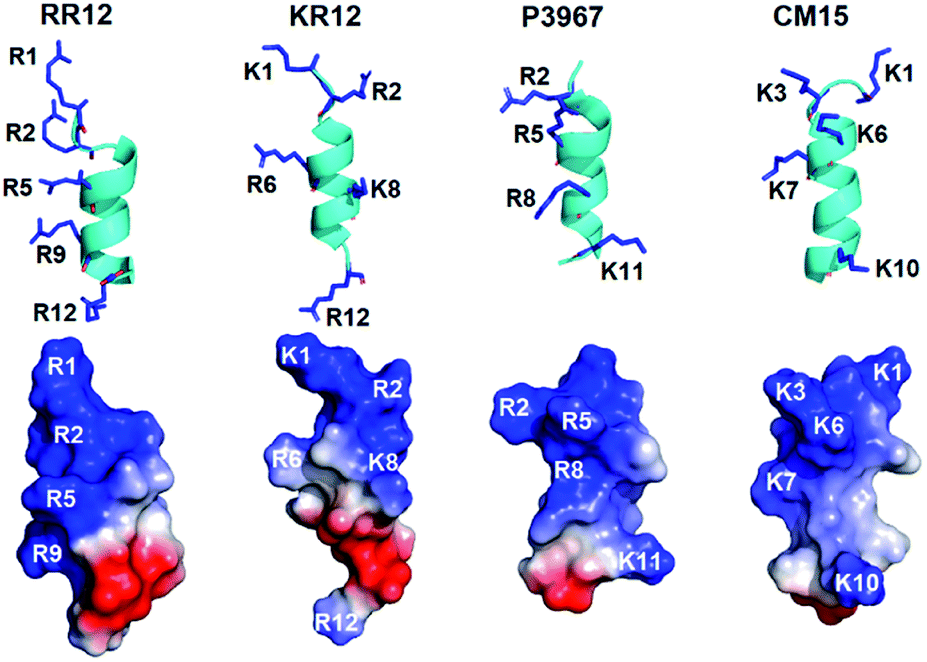 | ||
| Fig. 5 Structural comparisons of RR12 with the known AMPs. | ||
Functionally, RR12 exerts stronger ability in disrupting negatively charged phospholipid vesicles (POPG) as shown in our dye leakage assay (Fig. 3). RR12 can cause 100% calcein leakage at peptide concentration of 0.008 and 0.2 μM against POPG and POPC LUVs, respectively, revealing the selectivity of RR12 against bacterial membranes instead of mammalian cells. Consequently, the α-helix (L3–L11) induced by the presence of SDS micelles is essential in membraned permeability as well as the bactericidal ability. Consistently, the strong membrane disrupting activity of RR12 can also be seen from its broad bactericidal abilities against Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus (ATCC25923), and Staphylococcus aureus (ATCC25923)) and Gram-positive bacteria (Salmonella enterica (ATCC14028), Pseudomonas aeruginosa (ATCC27853), and Klebsiella pneumoniae (ATCC13883)).30
Most AMPs are prone to bind to the interfacial region of bacterial membranes with its polar face exposed to the solvent and the hydrophobic face embedded in the membranes.15,57–59 Our NMR paramagnetic experiments were performed with 5-DSA, 12-DSA, and Mn2+ ions (Fig. 4 and S6–S8†). Mn2+ can cause broadening of resonances of residues which are near the surface of micelles or exposed to the solvents. For the 5-DSA, it broadens the signals of resonances of residues situated near the micellar boundary, whereas the spin label of 12-DSA will result in the reduction of peak intensity of resonances near to the center of the micelle.60–62 In SDS micelles, the C-terminal residues of RR12 were considerably affected by Mn2+ ions, with cross peaks of R9, L10, L11, and R12 completely disappearing in the 2D-TOCSY spectrum (Fig. S8†), indicating that the C-terminus lies at the surface or lies outside the micelles. Apart from the R9–R12, the remaining residues (R2–L8) were obviously affected by both 5-and 12-DSAs. This could be explained by the fact that micelles are mobile and continuously change, altering their positions and shapes in the solutions. The flexibility of micelles and the hydrophobic tails of 5- and 12-DSAs make it difficult to clearly distinguish the effects of the two probes.63 This resulted in similar broadening effects for most of the resonances of residues buried inside the SDS micelles. Accordingly, the segment (R2–L8) of RR12 is more buried, probably with the positively charged residues (R2, R5, and R9) facing the inner surface of micelle and the hydrophobic residues oriented into the micelle's interior. It has been reported that the α-helicity of AMPs is important for the dye-leakage abilities and antimicrobial activities.64 Thus, the induced α-helical segment (R2–I7) of RR12 upon interacting with SDS micelles is essential for membrane permeabilization and bacteriostatic activity.
Conclusions
In this study, we characterized the bioactive structure and function of the salt-resistant AMP, RR12 by biophysical and biochemical examinations. RR12 shows conformational transition (disordered coil to well-defined α-helix structure) when interacting with SDS micelles. We determined the detail solution structure of RR12 in complex with SDS micelles. RR12 orients itself into SDS micelles, with R2–I7 buried and C-terminus (R9–R12) exposed to the solvent. RR12 shows well selectivity against negative-charged bacterial membranes, particularly, its α-helix segment (L3–L11) could significantly contribute in membrane permeabilization and bactericidal activity. Taken together, our study disclosed the structural and functional properties/relationships of RR12 which could further explain its possible mode of action and shows the applicability in developing new antibacterial agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the use of the Chirascan-Plus CD Spectrometer in the Biophysics Core Facility, Department of Academic Affairs and Instrument Service at Academia Sinica. We also acknowledge the acquisition of NMR spectra at the High-field Biomacromolecular NMR Core Facility, Academia Sinica. The study was supported by the Ministry of Science and Technology, Taiwan, ROC (MOST 108-2320-B-005-007).Notes and references
- C. L. Ventola, Pharm. Ther., 2015, 40, 277–283 Search PubMed.
- C. L. Ventola, Pharm. Ther., 2015, 40, 344–352 Search PubMed.
- M. Pasupuleti, A. Schmidtchen and M. Malmsten, Crit. Rev. Biotechnol., 2012, 32, 143–171 CrossRef CAS PubMed.
- S. Wang, P. A. Thacker, M. Watford and S. Qiao, Curr. Protein Pept. Sci., 2015, 16, 582–591 CrossRef CAS PubMed.
- L. Chopra, G. Singh, V. Choudhary and D. K. Sahoo, Appl. Environ. Microbiol., 2014, 80, 2981–2990 CrossRef PubMed.
- M. N. Melo, R. Ferre and M. A. Castanho, Nat. Rev. Microbiol., 2009, 7, 245–250 CrossRef CAS PubMed.
- M. Pushpanathan, P. Gunasekaran and J. Rajendhran, Int. J. Pept. Res. Therapeut., 2013, 2013, 675391 Search PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2009, 37, D933–D937 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- Z. Wang and G. Wang, Nucleic Acids Res., 2004, 32, D590–D592 CrossRef CAS PubMed.
- P. Staubitz, A. Peschel, W. F. Nieuwenhuizen, M. Otto, F. Gotz, G. Jung and R. W. Jack, J. Pept. Sci., 2001, 7, 552–564 CrossRef CAS PubMed.
- H. Jenssen, P. Hamill and R. E. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- A. A. Bahar and D. Ren, Pharmaceuticals, 2013, 6, 1543–1575 CrossRef PubMed.
- A. Tossi, L. Sandri and A. Giangaspero, Biopolymers, 2000, 55, 4–30 CrossRef CAS PubMed.
- R. Ferre, M. N. Melo, A. D. Correia, L. Feliu, E. Bardaji, M. Planas and M. Castanho, Biophys. J., 2009, 96, 1815–1827 CrossRef CAS PubMed.
- H. G. Boman, J. Intern. Med., 2003, 254, 197–215 CrossRef CAS PubMed.
- R. E. Hancock, Lancet Infect. Dis., 2001, 1, 156–164 CrossRef CAS PubMed.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS PubMed.
- Y. Shai, Biopolymers, 2002, 66, 236–248 CrossRef CAS PubMed.
- H. T. Chou, T. Y. Kuo, J. C. Chiang, M. J. Pei, W. T. Yang, H. C. Yu, S. B. Lin and W. J. Chen, Int. J. Antimicrob. Agents, 2008, 32, 130–138 CrossRef CAS PubMed.
- B. J. Moncla, K. Pryke, L. C. Rohan and P. W. Graebing, Adv. Biosci. Biotechnol., 2011, 2, 404–408 CrossRef CAS PubMed.
- I. M. Torcato, Y. H. Huang, H. G. Franquelim, D. Gaspar, D. J. Craik, M. A. Castanho and S. Troeira Henriques, Biochim. Biophys. Acta, 2013, 1828, 944–955 CrossRef CAS PubMed.
- M. L. Mangoni and Y. Shai, Biochim. Biophys. Acta, 2009, 1788, 1610–1619 CrossRef CAS PubMed.
- M. L. Mangoni and Y. Shai, Cell. Mol. Life Sci., 2011, 68, 2267–2280 CrossRef CAS PubMed.
- I. Zelezetsky and A. Tossi, Biochim. Biophys. Acta, 2006, 1758, 1436–1449 CrossRef CAS PubMed.
- E. Badosa, R. Ferre, M. Planas, L. Feliu, E. Besalu, J. Cabrefiga, E. Bardaji and E. Montesinos, Peptides, 2007, 28, 2276–2285 CrossRef CAS PubMed.
- J. Turner, Y. Cho, N. N. Dinh, A. J. Waring and R. I. Lehrer, Antimicrob. Agents Chemother., 1998, 42, 2206–2214 CrossRef CAS PubMed.
- H. L. Chu, H. Y. Yu, B. S. Yip, Y. H. Chih, C. W. Liang, H. T. Cheng and J. W. Cheng, Antimicrob. Agents Chemother., 2013, 57, 4050–4052 CrossRef CAS PubMed.
- H. Mohanram and S. Bhattacharjya, Biopolymers, 2016, 106, 345–356 CrossRef CAS PubMed.
- G. Laverty, S. P. Gorman and B. F. Gilmore, Int. J. Mol. Sci., 2011, 12, 6566–6596 CrossRef CAS PubMed.
- N. Y. Yount and M. R. Yeaman, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 337–360 CrossRef CAS PubMed.
- S. Y. Wei, J. M. Wu, Y. Y. Kuo, H. L. Chen, B. S. Yip, S. R. Tzeng and J. W. Cheng, J. Bacteriol., 2006, 188, 328–334 CrossRef CAS PubMed.
- P. K. Lewis Kay and E. Tim Saarinen, J. Am. Chem. Soc., 1992, 114, 10663–10665 CrossRef.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- G. Wang, Biochim. Biophys. Acta, 2007, 1768, 3271–3281 CrossRef CAS PubMed.
- G. Cornilescu, F. Delaglio and A. Bax, J. Biomol. NMR, 1999, 13, 289–302 CrossRef CAS PubMed.
- A. T. Brunger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef CAS PubMed.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr. Sect. D Biol. Crystallogr., 1998, 54, 905–921 CrossRef CAS PubMed.
- R. A. Laskowski, J. A. Rullmannn, M. W. MacArthur, R. Kaptein and J. M. Thornton, J. Biomol. NMR, 1996, 8, 477–486 CrossRef CAS PubMed.
- R. Koradi, M. Billeter and K. Wuthrich, J. Mol. Graph., 1996, 14(51–55), 29–32 Search PubMed.
- A. Bhunia, H. Mohanram and S. Bhattacharjya, Biochim. Biophys. Acta, 2012, 1818, 1250–1260 CrossRef CAS PubMed.
- Y. J. Gordon, E. G. Romanowski and A. M. McDermott, Curr. Eye Res., 2005, 30, 505–515 CrossRef CAS PubMed.
- H. Kim, J. H. Jang, S. C. Kim and J. H. Cho, J. Antimicrob. Chemother., 2014, 69, 121–132 CrossRef CAS PubMed.
- F. D. Sonnichsen, J. E. Van Eyk, R. S. Hodges and B. D. Sykes, Biochemistry, 1992, 31, 8790–8798 CrossRef CAS PubMed.
- A. P. Subasinghage, J. M. Conlon and C. M. Hewage, Biochim. Biophys. Acta, 2010, 1804, 1020–1028 CrossRef CAS PubMed.
- I. Alana, J. P. Malthouse, F. P. O'Harte and C. M. Hewage, Proteins, 2007, 68, 92–99 CrossRef CAS PubMed.
- E. Schrank, G. E. Wagner and K. Zangger, Molecules, 2013, 18, 7407–7435 CrossRef CAS PubMed.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- E. F. Haney, H. N. Hunter, K. Matsuzaki and H. J. Vogel, Biochim. Biophys. Acta, 2009, 1788, 1639–1655 CrossRef CAS PubMed.
- M. Lindberg and A. Graslund, FEBS Lett., 2001, 497, 39–44 CrossRef CAS PubMed.
- J. Lee and D. G. Lee, J. Microbiol. Biotechnol., 2015, 25, 759–764 CrossRef CAS PubMed.
- S. Bourbigot, E. Dodd, C. Horwood, N. Cumby, L. Fardy, W. H. Welch, Z. Ramjan, S. Sharma, A. J. Waring, M. R. Yeaman and V. Booth, Biopolymers, 2009, 91, 1–13 CrossRef CAS PubMed.
- Z. Raja, S. Andre, C. Piesse, D. Sereno, P. Nicolas, T. Foulon, B. Oury and A. Ladram, PLoS One, 2013, 8, e70782 CrossRef CAS PubMed.
- I. Y. Park, J. H. Cho, K. S. Kim, Y. B. Kim, M. S. Kim and S. C. Kim, J. Biol. Chem., 2004, 279, 13896–13901 CrossRef CAS PubMed.
- R. Saravanan, X. Li, K. Lim, H. Mohanram, L. Peng, B. Mishra, A. Basu, J. M. Lee, S. Bhattacharjya and S. S. Leong, Biotechnol. Bioeng., 2014, 111, 37–49 CrossRef CAS PubMed.
- K. Putsep, C. I. Branden, H. G. Boman and S. Normark, Nature, 1999, 398, 671–672 CrossRef CAS PubMed.
- P. Bulet, R. Stocklin and L. Menin, Immunol. Rev., 2004, 198, 169–184 CrossRef CAS PubMed.
- A. Giangaspero, L. Sandri and A. Tossi, Eur. J. Biochem., 2001, 268, 5589–5600 CrossRef CAS PubMed.
- E. Barany-Wallje, A. Andersson, A. Graslund and L. Maler, FEBS Lett., 2004, 567, 265–269 CrossRef CAS PubMed.
- J. Jarvet, J. Zdunek, P. Damberg and A. Graslund, Biochemistry, 1997, 36, 8153–8163 CrossRef CAS PubMed.
- M. Lindberg, H. Biverstahl, A. Graslund and L. Maler, Eur. J. Biochem., 2003, 270, 3055–3063 CrossRef CAS PubMed.
- A. P. Subasinghage, D. O'Flynn, J. M. Conlon and C. M. Hewage, Biochim. Biophys. Acta, 2011, 1808, 1975–1984 CrossRef CAS PubMed.
- H. T. Chou, H. W. Wen, T. Y. Kuo, C. C. Lin and W. J. Chen, Peptides, 2010, 31, 1811–1820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04299d |
| This journal is © The Royal Society of Chemistry 2020 |