
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel method to produce sustainable biocomposites based on thermoplastic corn-starch reinforced by polyvinyl alcohol fibers
Peng Yina,
Xin Donga,
Wen Zhoua,
Dongdong Zhaa,
Jie Xub,
Bin Guo *acd and
Panxin Licd
*acd and
Panxin Licd
aCollege of Science, Nanjing Forestry University, Nanjing 210037, China. E-mail: gbm@ustc.edu; Tel: +86-25-8542-7625
bState Key Laboratory for Hubei New Textile Materials and Advanced Processing Technology, College of Materials Science and Engineering, Wuhan Textile University, Wuhan 430200, China
cAgricultural and Forest Products Processing Academician Workstation of Henan Province, Luohe 462600, China
dPost-Doctoral Research Center of Henan Nanjiecun Group, Luohe 462600, China
First published on 22nd June 2020
Abstract
Adding reinforced fiber or cross-linking agent into thermoplastic starch (TPS) is an effective method to improve its performance. Herein, biodegradable polyvinyl alcohol fiber (PVAF) and sodium hexametaphosphate (SHMP) were not added into TPS directly; the PVAFs were preliminary treated (pre-soaking) by an SHMP solution, and then mixed with starch and glycerol to prepare 2 wt% PVAF/TPS composites through extrusion and injection molding. This process promoted crosslinking action between PVAFs and starch, and as a consequence enhanced the mechanic and dynamic mechanic behavior. The PVAFs with different immersion times were characterized by SEM, FTIR, and WAXD. The results confirmed that the SHMP coating was formed by a chemical bond connection on the surface of the PVAFs, particularly for the PVAFs soaked for 1.5 h, which produced a corresponding PVAF/TPS composite with a maximum tensile strength of 9.18 MPa and an impact strength of 21.29 kJ m−2. The corresponding tensile fractured cross-section images were shown by SEM. The DMA curves indicated that the pre-soaked PVAFs effectively improved the energy storage modulus and transition temperature of composites, and the activation energy of the starch macromolecules reached a maximum of 349.9 kJ mol−1 during the dynamic mechanic process. The contact angle attained a maximum of 66.25°. Compared with TPS, the pre-soaked PVAF-reinforced PVAF/TPS composites had better mechanical properties, good processability through traditional extrusion and injection molding, reduced water wettability, and potential applications for packaging and catering.
1. Introduction
Industrial corn starch is widely used in the textile, paper, medical, packaging, and other industries.1,2 The development of thermoplastic starch (TPS) has received considerable attention over the last two decades owing to its biodegradability, renewability, and low cost.3 However, the retrogradation (hydrolytic degradation) and relatively poor mechanical properties of TPS, particularly in wet or dry environments, still significantly restrict its further application.4,5A large number of studies have focused on improving the mechanical properties and water resistance of TPS, mainly through modification of the starch, addition of reinforcements, and formation of a crosslinking system.6,7 Modified starch is usually first produced through esterification,8,9 oxidation,10,11 or etherification,12 after which thermoplastic starch can then be obtained. However, the disadvantage of this method is the higher cost resulting from the complex modification process.
The reinforcements used in TPS can be divided into inorganic minerals and organic fibers. The inorganic minerals mainly include montmorillonite,13,14 bentonite,15,16 and kaolin.17,18 The organic fibers are mainly natural fibers,19,20 synthetic fibers,21–23 and micro/nanocellulose.24–27 Recently, we studied the effect of ultra-high molecular weight polyethylene (UHMWPE) fibers on the properties of TPS, and found that the mechanical properties and water resistance of UHMWPE/TPS composites were significantly improved. The resulting composite material was significantly improved with the inclusion of 2 wt% UHMWPE fibers (tensile strength = 8.78 MPa, contact angle = 80.2°).21 However, UHMWPE is not degradable. In contrast, polyvinyl alcohol fiber (PVAF) is a biodegradable synthetic fiber. Recently, we have prepared PVAF/TPS composites,23 and found that the addition of a small amount of PVAF can reduce the viscosity of the PVAF/TPS system and improve its processability. When the PVAF content is between 0.25 and 1.0 wt%, the tensile strength of the composite will increase almost linearly. At 2 wt%, the tensile strength can reach 5.2 MPa, and the elongation at breaking reaches a maximum of approximately 158% at a PVAF content of 1.5 wt%. Nevertheless, the mechanical properties are expected to be further improved in the future.
For the crosslinking system, the main crosslinking agents used are glutaraldehyde,28,29 epichlorohydrin,30,31 citric acid,9,32 boric acid,33,34 and sodium hexametaphosphate (SHMP).35,36 Because glutaraldehyde and epichlorohydrin have irritant and toxic properties,28,30 they are not considered in our experiments owing to the risk for health hazards in their preparation process. A small amount of citric acid can enhance the toughness of TPS, but its effect on the strength of the material is not obvious.37 The addition of boric acid can significantly improve the strength of TPS, but it also has a negative impact on its toughness and water resistance.33 SHMP is better choice than the other aforementioned materials, as it can be used as a safe and non-toxic crosslinking agent in the food field.38,39
In the preparation of thermoplastic starch and its composite film, the crosslinking agent is added mainly in two modes. In the first mode, it is added during the film preparation process. Citric acid and boric acid are added in this mode. Aritra et al.40 compared the effects of temperature and citric acid content on a PVA/starch/glycerol/citric acid composite membrane, and found that the highest tensile strength (approximately 4.12 MPa) was obtained when the citric acid content was 0.5 wt% and the temperature was 60 °C. Yin et al.34 first premixed PVA, starch, and glycerin, and then added boric acid during the gelation of starch to prepare a PVA/TPS composite film. They found that increasing the concentration of boric acid (from 0 to 1.5 wt%) improved the degree of crosslinking between the starch and PVA, thereby enhancing the bonding of the two molecules.
In the second mode for addition of the crosslinking agent, the addition is achieved through post-treatment of the prepared film. In this case, glutaraldehyde and SHMP are commonly used as crosslinking agents. Liu et al.28 adopted a method in which a PVA composite membrane was immersed in an aqueous solution of glutaraldehyde and then heated to achieve crosslinking modification. The results showed that the post-treatment crosslinking modification significantly reduced the hydrophilicity of the TPS/PVA composite film while increasing the tensile strength and modulus of elasticity. Liu35 also immersed a TPS/PVA composite membrane in aqueous solutions of sodium carbonate and SHMP for crosslinking treatment. It was found that the crosslinking modification markedly reduced the water sensitivity of the composite membrane, and the soaking time in the SHMP aqueous solution had a significant effect on the mechanical properties of the composite membrane.
To improve the mechanical properties and reduce the water wettability further, this study proposes a method for introducing the reinforcement (biodegradable PVAF) and cross-linking agent (SHMP) systems into the TPS simultaneously. Unlike the methods reported in previous studies, we pre-treated the PVAF with SHMP solution, and then mixed it with starch to prepare the PVAF/TPS composite through extrusion and injection molding. PVAF samples with different immersion times were characterized. At the same time, the tensile properties, impact properties, dynamic mechanical properties, and surface water wettability of different PVAF/TPS composites were systematically studied.
2. Materials and methods
2.1. Materials
Corn starch (moisture content = 13.6 wt%, amylose content = 27%) was obtained from the Shandong Hengren Industry and Trade Company (Tengzhou, Shandong, China). Glycerol (analytical grade) and sodium carbonate (analytical grade) were purchased from the Sinopharm Group Chemical Reagent Co., Ltd (Shanghai, China). Polyvinyl alcohol fiber (length 15–20 mm, fineness 1.1–4 D) was obtained from the Nanjing Pioneer Technology Industrial Co., Ltd. Sodium hexametaphosphate (analytical grade) was purchased from the Xilong Chemical Co., Ltd (Shantou, Guangdong, China).2.2. Preparation of modified PVAF
First, the PVAF was cut into lengths of 5 mm and divided into four groups. Then, the 9 g of PVAF comprising each group were immersed in a solution of 3 wt% Na2CO3 and 8 wt% SHMP with the pH value of 10.5, as shown in Fig. 1, and soaked for 0.5, 1.0, 1.5, or 2.0 h to obtain modified PVAF.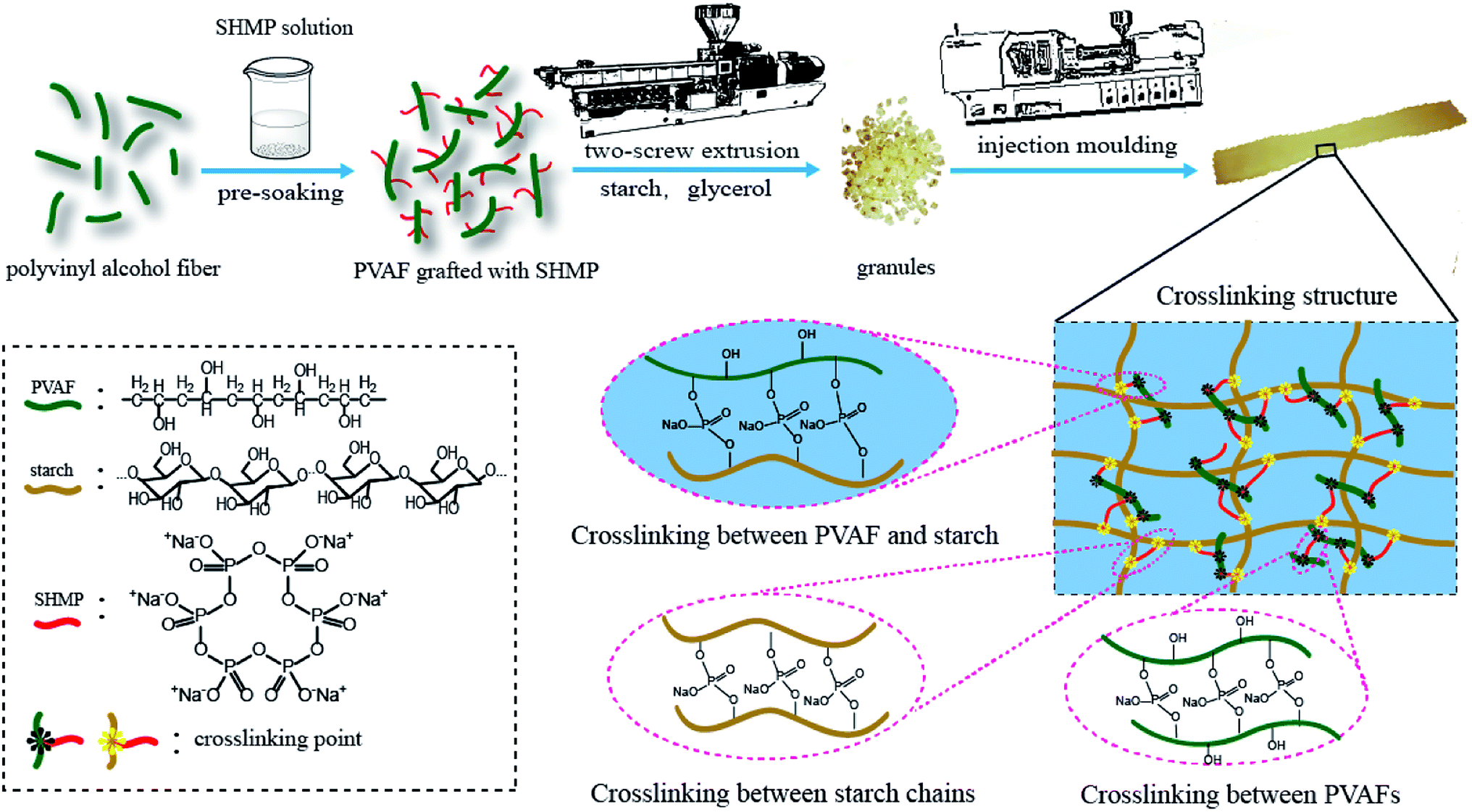 | ||
| Fig. 1 Schematic illustration of the fabrication of the PVAF/TPS composites reinforced with pre-soaked PVAFs. | ||
After soaking, the PVAF samples were dried in an oven and then weighed; the weight gain on the PVAF surface was 1.18, 1.49, 2.02, and 1.86 g, respectively, for soaking times of 0.5, 1.0, 1.5, and 2.0 h. To determine whether the soaking process for PVAF in the SHMP solution is a physical adsorption or chemical crosslinking process, some of the PVAF samples soaked for 1.5 h were cleaned ultrasonically in pure water with ultrasonic frequency of 40 kHz and ultrasonic time of 10 min for several times, and then characterized using infrared spectroscopy and wide-angle X-ray diffraction (WAXD) before and after cleaning.
2.3. Preparation of PVAF/TPS composites
The corn starch (450 g) and glycerol (150 g)were manually premixed at a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, based on our previous optimization work,7,21 in polyethylene bags and stored overnight. Then, mixture and 9 g of PVAF (2 wt% based on the mass of corn starch) soaked for varying amounts of time (referred to as 0.5, 1.0, 1.5, and 2.0 h ∼ PVAF/TPS) were further blended at 25000 rpm for 15 s using a QE-500 high-speed mixer (Zhejiang Yili Industry and Trade Co., Ltd, China).
:1, based on our previous optimization work,7,21 in polyethylene bags and stored overnight. Then, mixture and 9 g of PVAF (2 wt% based on the mass of corn starch) soaked for varying amounts of time (referred to as 0.5, 1.0, 1.5, and 2.0 h ∼ PVAF/TPS) were further blended at 25000 rpm for 15 s using a QE-500 high-speed mixer (Zhejiang Yili Industry and Trade Co., Ltd, China).
To confirm that the enhanced properties of the composite were a consequence of the reinforcement effect of the modified PVAF rather than a pure SHMP crosslinking effect with the starch macromolecules in the composites, two samples were prepared and compared: a reinforced PVAF/TPS composite composed of PVAF presoaked for 1.5 h, denoted 1.5 h ∼ PVAF/TPS, and a corresponding SHMP-loaded PVAF/TPS comprising the same PVAF (9 g) and SHMP (2.02 g) contents, denoted SHMP/PVAF/TPS.
After these steps, the mixtures were transferred into a twin-screw extruder (SHJ-20, Nanjing Giant Machinery Co., Ltd, China) with a screw speed of 180 rpm; the zone temperatures were 116, 118, 118, and 116 °C. The residual time of the material in the extruder's barrel was 83 s, and the length of the barrel was 80 mm, the throughput of the material at the set conditions was 0.6 kg h−1. After processing, the mixtures were cooled and cut into small particles. An injection molding machine (BV90, APOLLO, Shanghai, China) was used to inject a dumbbell shaped spline, and the temperatures in each zone were 135, 135, 125, 125, and 120 °C.
2.4. Characterization of PVAF
2.5. Characterization of PVAF/TPS composites
3. Results and discussion
3.1. Characterization of the fiber
 | ||
| Fig. 2 SEM images of (a) unmodified-PVAF; and PVAF soaked for (b) 0.5 h, (c) 1.0 h, (d) 1.5 h, and (e) 2.0 h. | ||
As shown in Fig. 2d, after a soaking time of 1.5 h, most of the PVAF surface was covered uniformly by SHMP. This layer of SHMP changes the surface structure of the fiber and serves to enhance the bonding force between the PVAF fiber and the starch matrix. When the soaking time was 2 h, too much SHMP maybe agglomerated and stacked on the PVAF surface, and this thicker SHMP layer cannot connect tightly with the PVAF surface, therefore, too much SHMP can easily fall off the fiber surface, leaving some residue on the surface of the fiber, as shown in Fig. 2e.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, PO stretching, and P–O–P stretching vibrations, respectively.36 In addition, five distinctive absorption bands at 1449, 1328, 1088, 836, and 610 cm−1 corresponding to C–H and O–H in-plane bending, C–O stretching, and C–H and O–H out-of-plane bending vibrations, respectively, were observed in the FTIR spectrum of the unmodified PVAF.42
O, PO stretching, and P–O–P stretching vibrations, respectively.36 In addition, five distinctive absorption bands at 1449, 1328, 1088, 836, and 610 cm−1 corresponding to C–H and O–H in-plane bending, C–O stretching, and C–H and O–H out-of-plane bending vibrations, respectively, were observed in the FTIR spectrum of the unmodified PVAF.42
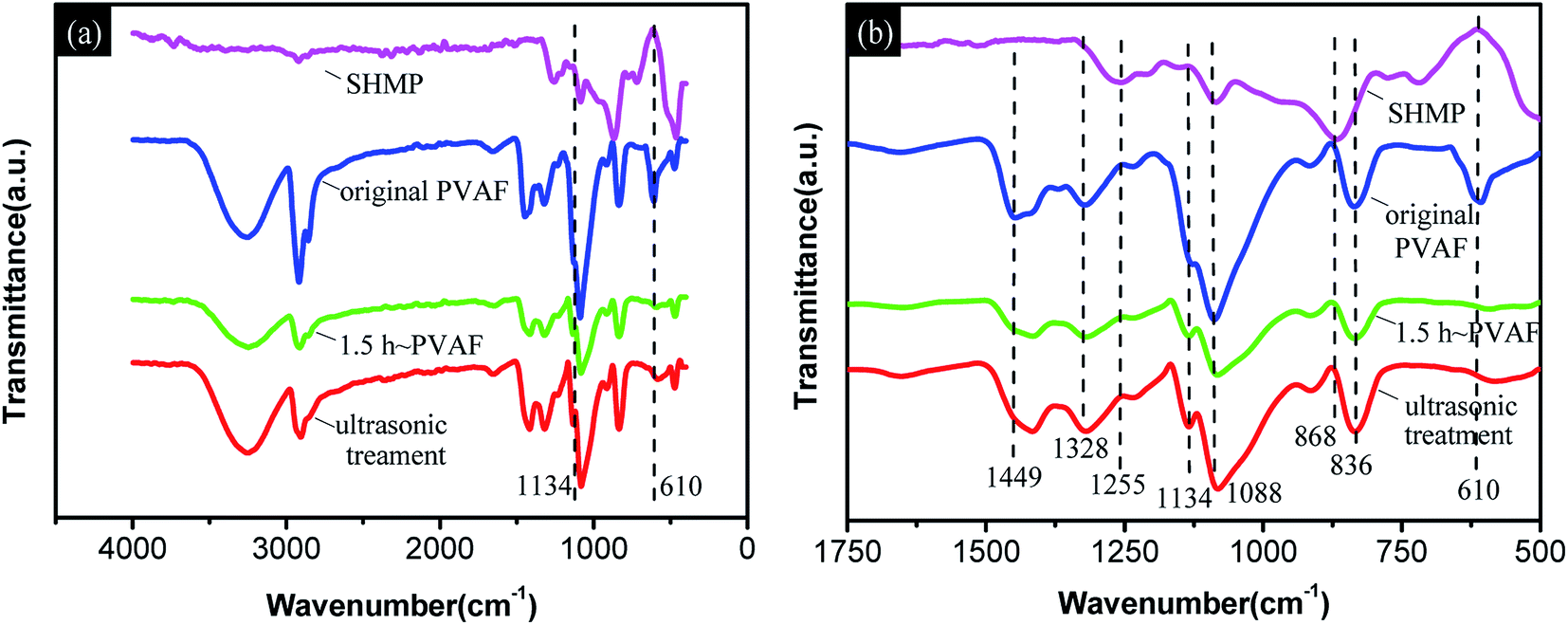 | ||
| Fig. 3 (a) Infrared spectra in the range of 4000–400 cm−1 for SHMP, original PVAF, PVAF soaked for 1.5 h, and pre-soaked PVAF with further ultrasonic vibration treatment; (b) expansion of the spectra in (a) over the range of 1750–500 cm−1. | ||
Comparing the spectra for the PVAF before and after soaking for 1.5 h, the newly developed C–O–P stretching vibration band at 1134 cm−1 and disappearance of the O–H out-of-plane bending vibration at 610 cm−1 are most likely due to the reaction of the hydroxyl groups of PVAF with the phosphate groups of SHMP molecules during the pre-soaking process. Moreover, the spectra for the soaked PVAF before and after sonication have similar absorption bands, particularly at 1134 cm−1, indicating that the chemical bond connection between the PVAF and SHMP was formed in the modified pre-soaking process.
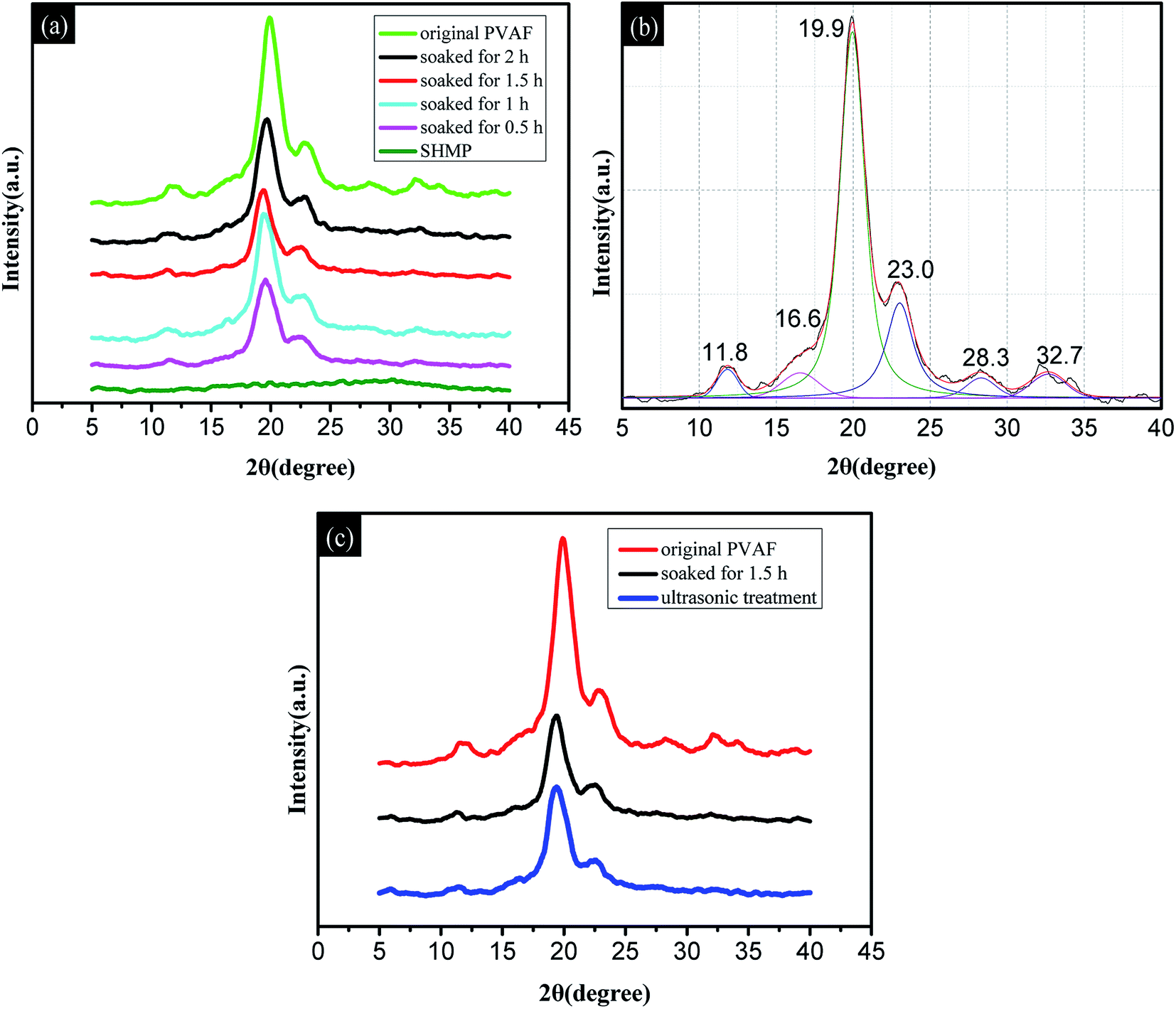 | ||
| Fig. 4 Wide-angle X-ray diffraction patterns: (a) SHMP, original PVAF, and PVAF soaked for varying amounts of time; (b) typical fitting curve for the original PVAF; (c) comparison of the original PVAF, PVAF soaked for 1.5 h, and pre-soaked PVAF with further ultrasonic treatment. | ||
| 2θ (°) | Original PVAF | 0.5 h | 1.0 h | 1.5 h | 2.0 h | Ultrasonic treatment |
|---|---|---|---|---|---|---|
| 11.8 | 23.46 | 7.85 | 13.49 | 5.41 | 11.85 | 6.47 |
| 16.6 | 38.68 | 17.00 | 40.08 | 18.23 | 28.19 | 19.46 |
| 23.0 | 144.02 | 77.91 | 123.88 | 92.43 | 108.00 | 80.24 |
| 28.3 | 25.92 | 15.62 | 28.69 | 14.42 | 52.63 | 22.85 |
| 32.7 | 32.71 | 5.87 | 16.67 | 23.31 | 29.42 | 7.77 |
| 19.9 | 460.76 | 218.46 | 304.44 | 207.76 | 299.26 | 216.54 |
Fig. 4c shows the X-ray diffraction patterns for the original PVAF and the PVAF soaked for 1.5 h before and after ultrasonic vibration treatment. It is clear that the soaked PVAF samples have similar diffraction patterns before and after ultrasonic vibration treatment, as well as similar peak areas mainly at 19.9° and 23.0° in Table 1. This provides further confirmation that the chemical bond connection between the PVAF and SHMP was formed in the modified pre-soaking process. These results are also in good agreement with the FT-IR spectra.
3.2. Mechanical properties of composite materials
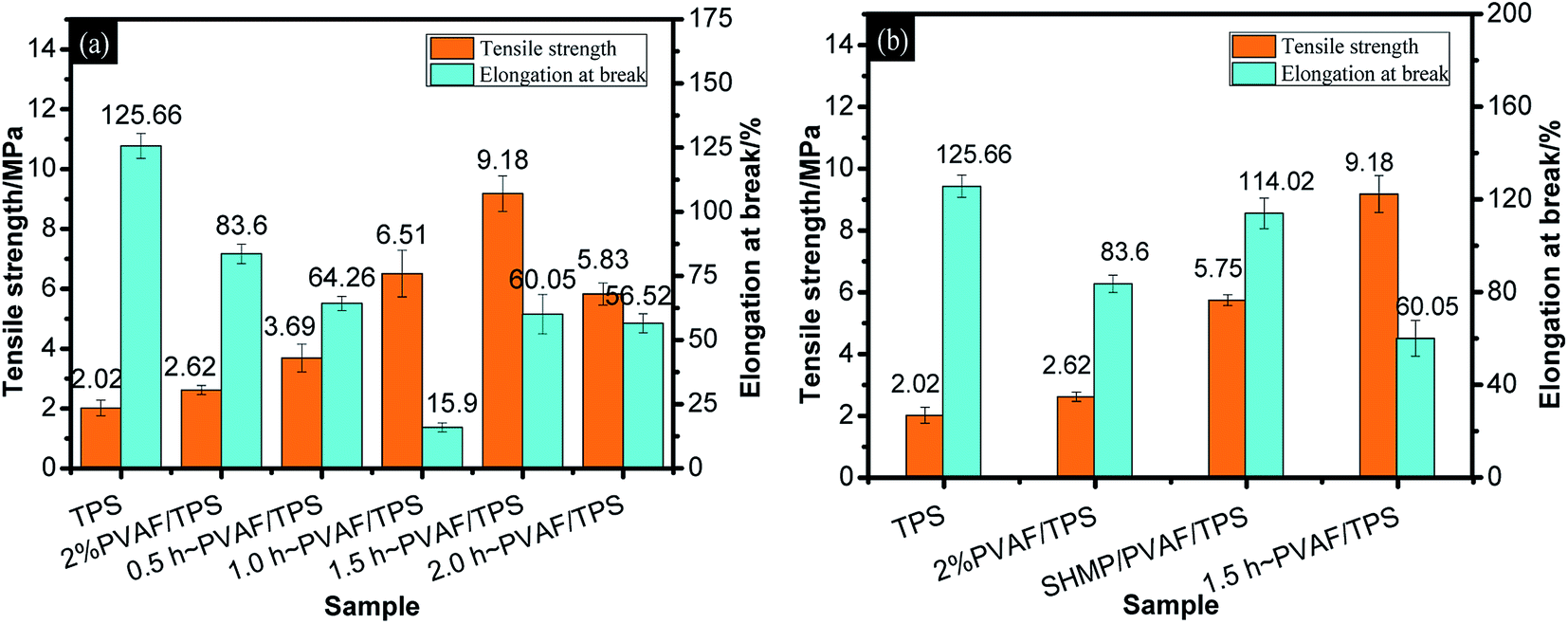 | ||
| Fig. 5 Mechanical properties of samples: (a) TPS, PVAF, and pre-soaked PVAF-reinforced TPS composites with different soaking times; (b) TPS, PVAF, TPS reinforced with PVAF pre-soaked for 1.5 h, and SHMP-loaded PVAF/TPS. | ||
To verify that the increase in tensile strength was a consequence of the reinforcing effect of the modified PVAF rather than a pure SHMP crosslinking effect with the starch macromolecules, as shown in Fig. 5b, we provide a comparison of the mechanical properties between the PVAF/TPS composites with 1.5 h of pre-soaking and the corresponding SHMP-loaded PVAF/TPS comprising the same PVAF and SHMP contents. It is clear that the reinforcing effect of modified PVAF with a strength of 9.18 MPa is larger than the corresponding SHMP-loaded PVAF/TPS strength of 5.75 MPa. Further confirmation can be obtained through the SEM images of the fractured cross sections of these samples after the tensile tests, as shown in Fig. 6.
 | ||
| Fig. 6 SEM images of fractured cross-sections after the tensile tests: (a) TPS; (b) 2% PVAF-reinforced TPS; (c) SHMP-loaded PVAF/TPS; (d) TPS reinforced with PVAF pre-soaked for 1.5 h. | ||
As observed in Fig. 6, the fractured cross-sections of TPS and PVAF/TPS composites exhibited different morphologies. TPS featured a homogeneously smooth surface, as shown in Fig. 6a, which is consistent with previous reports in the literature.21 The fractured cross-section of the 2 wt% PVAF-reinforced TPS still retained a smooth surface, with the protruding fibers dispersed homogenously, which is an important factor on mechanical properties, as shown in Fig. 6b. However, the SHMP-loaded PVAF/TPS composite exhibited a rougher cross-section, as shown in Fig. 6c, mainly resulting from the strong interaction between SHMP and the starch macromolecule matrix, which may indicate a brittle fracture. In contrast, a smooth and flat fractured cross-section was observed for reinforced PVAF/TPS composites treated with pre-soaked PVAF, as shown in Fig. 6d; no obvious protruding fibers can be observed on the fractured surface, which suggests that the pre-soaked PVAFs were chemically connected to the starch macromolecule matrix by SHMP in the composites. Therefore, the pre-soaked PVAFs can transfer stress effectively during the tensile process, and play a key role in enhancing the mechanical properties of PVAF/TPS composites. The SEM images of the fracture cross-sections are also in good agreement with the observed mechanical properties.
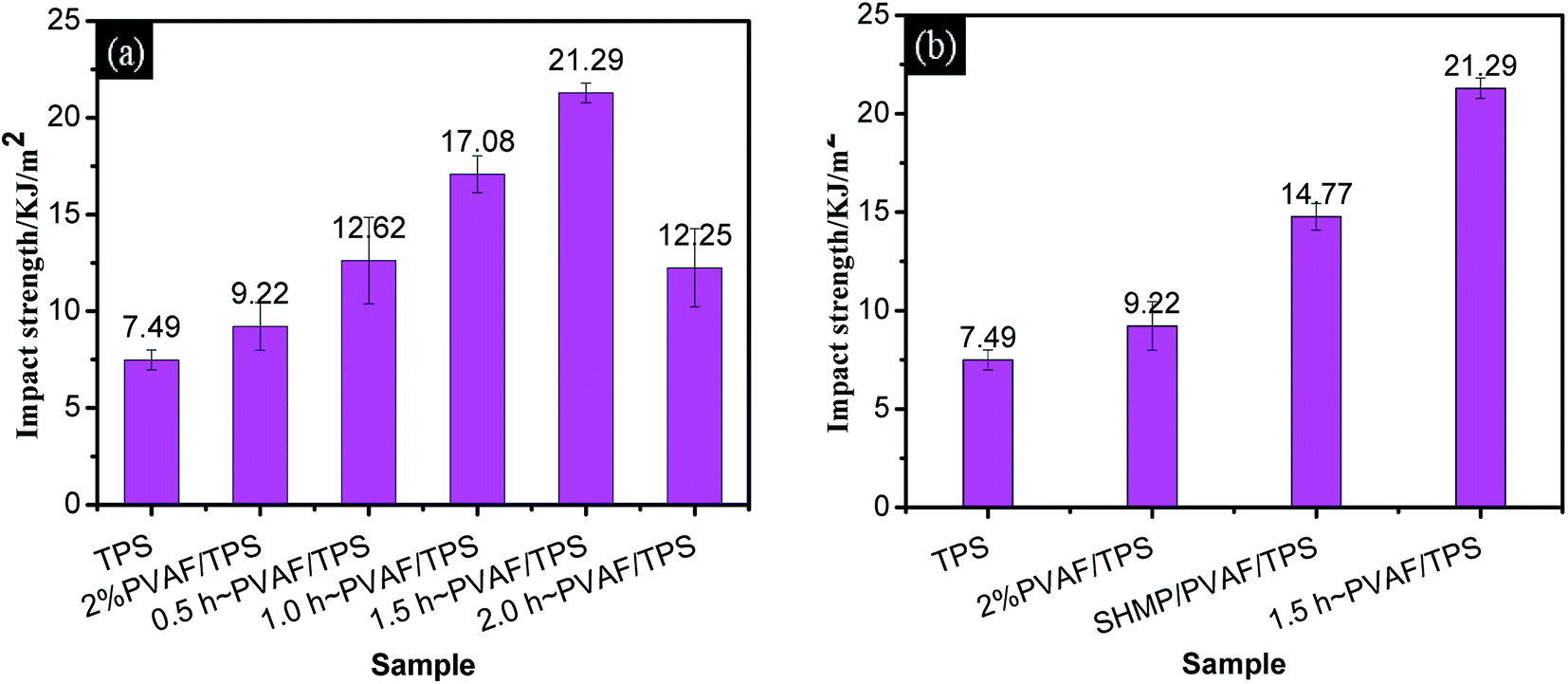 | ||
| Fig. 7 Impact performance: (a) TPS, PVAF, and pre-soaked PVAF-reinforced TPS composites with different soaking times; (b) TPS, PVAF, TPS reinforced with PVAF pre-soaked for 1.5 h, and SHMP-loaded PVAF/TPS. | ||
Fig. 7b provides a comparison of the impact strengths for the PVAF/TPS composites pre-soaked for 1.5 h and the corresponding SHMP-loaded PVAF/TPS comprising the same PVAF and SHMP contents. It is clear that the modified PVAF impact strength of 21.29 kJ m−2 is larger than the corresponding SHMP-loaded PVAF/TPS strength of 14.77 kJ m−2. Therefore, the SHMP and PVAF co-reinforced crosslinked structure also resulted in an increased ability to absorb impact energy.
3.3. Dynamic mechanical thermal properties
Dynamic mechanical analysis (DMA) curves for the starch composites were obtained by scanning the temperature from −90 °C to 120 °C at four frequencies: 1, 3.33, 5, and 10 Hz. Fig. 8a and b show the storage modulus curve and loss factor curve, respectively, for the PVAF/TPS system at a frequency of 5 Hz.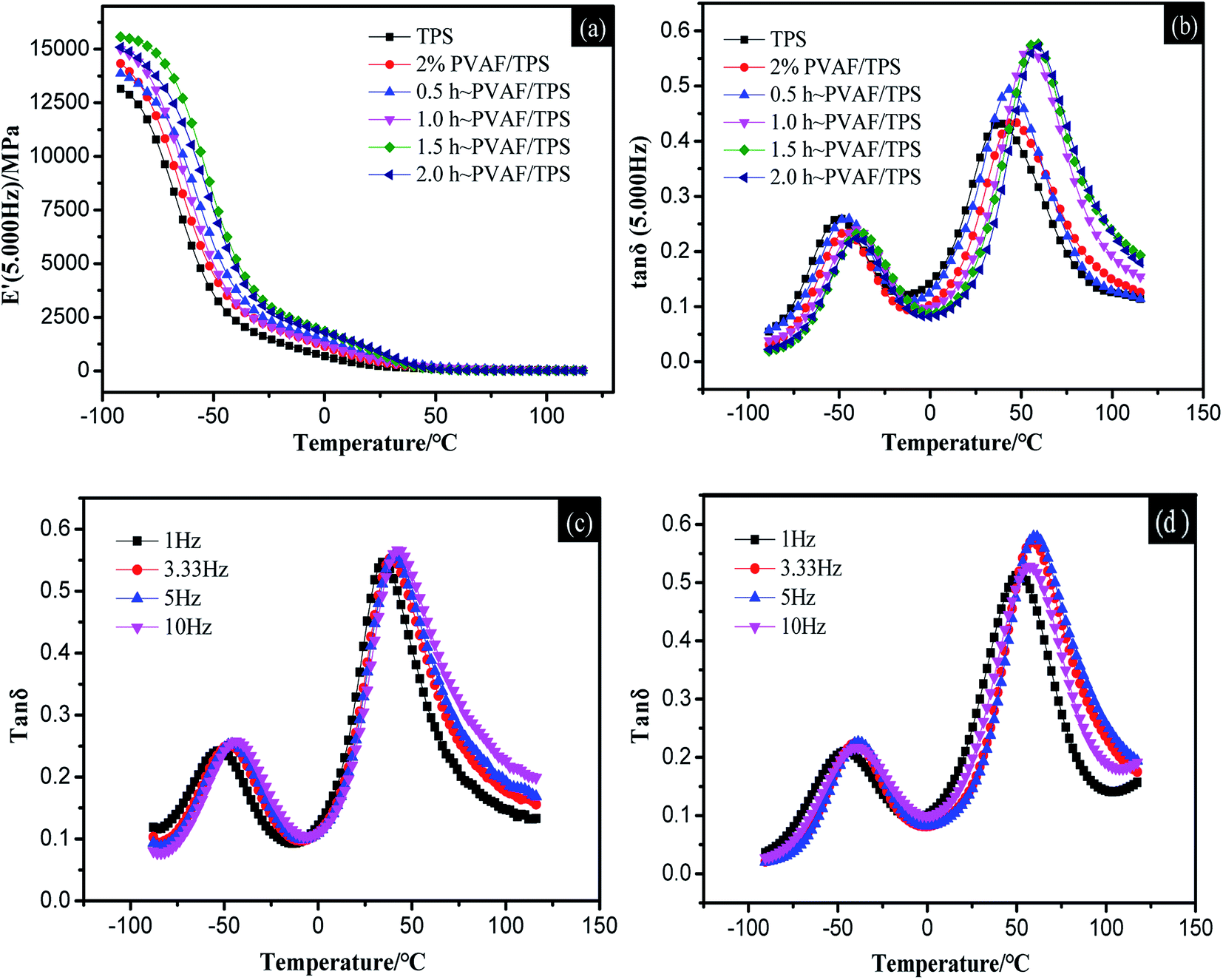 | ||
| Fig. 8 DMA curves for TPS and different PVAF/TPS composites at 5 Hz: (a) storage modulus vs. temperature, (b) loss factor vs. temperature; loss factor vs. temperature curves at different frequencies: (c) TPS, (d) 1.5 h-PVAF/TPS. | ||
By performing a temperature scan at a series of frequencies, the loss factor curves at different frequencies for typical samples of TPS and 1.5 h-2% PVAF/TPS composite were obtained, as shown in Fig. 8c and d, respectively. From the relationship between the peak temperature, Tα (K), and the frequency (Hz) of the tangent (tanδ), the apparent activation energy, ΔE, can be obtained with the following equation:
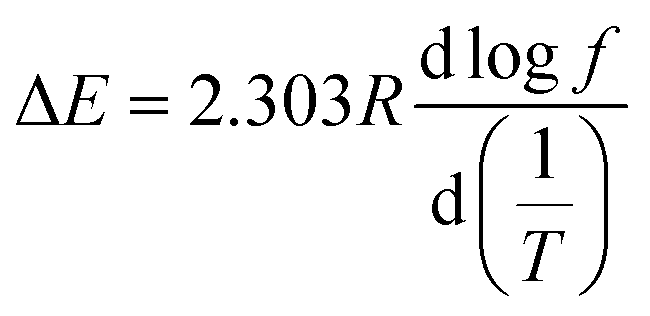 f with respect to 1/T.46
f with respect to 1/T.46
To investigate the interaction between the PVAFs and starch matrix further, the thermomechanical behavior of PVAF/TPS and the modified PVAF/TPS composites were investigated using the DMA technique. Fig. 8a and b illustrate the thermomechanical responses (storage modulus (E′) and loss factor (tanδ), respectively) of the PVAF/TPS and modified PVAF/TPS systems at a range of temperatures from −90 °C to 120 °C. As shown in Fig. 8a, the storage modulus of TPS, PVAF/TPS, and the modified PVAF/TPS composites decreased with increasing temperature. At the same time, the storage modulus of the modified PVAF/TPS composites was higher than that of the PVAF/TPS and TPS at the same temperature. With increasing immersion time of the PVAFs in SHMP solution, the storage modulus of the modified PVAF/TPS composites also increased. This is attributed to the higher mechanical strength of PVAFs than starch, followed by the SHMP solution surface treatment of PVAFs that can directly form a crosslinked network structure with the starch matrix, blocking the movement of the starch molecular chain, resulting in an increased storage modulus.
Fig. 8b shows the loss factor curves for TPS, PVAF/TPS, and the modified PVAF/TPS composite. Several authors16,17 have reported that starch–glycerol mixtures are partially miscible systems, giving rise to a starch-rich phase and a glycerol-rich phase. As shown by the observation of two relaxation peaks in Fig. 8b, the relaxation occurring at a lower temperature (Tβ) was associated with the Tg of the glycerol-rich phase, while the relaxation at a higher temperature (Tα) was associated with the Tg of the starch-rich phase. Fig. 8b and Table 2 indicate that the Tα and Tβ of TPS were lower than the transition temperatures of PVAF/TPS, and the Tα and Tβ of the modified PVAF/TPS system were further improved with increasing soaking time. Owing to the formation of the crosslinked network structure between PVAFs as well as between the PVAFs and starch matrix, the molecular chain movement of the starch was blocked, leading to an increased transition temperature of PVAF/TPS. The activation energies for the samples in Table 2 indicate that the activation energy of the composite with a soaking time of 1.5 h was the highest, which suggests that the relaxation energy of the starch macromolecules in the composite was higher.
| Sample | Tβ (°C) | Tα (°C) | Activation energy (kJ mol−1) |
|---|---|---|---|
| TPS | −52.63 | 37.36 | 173.182 |
| 2% PVAF/TPS | −46.63 | 43.76 | 224.616 |
| 0.5 h ∼ PVAF/TPS | −44.62 | 45.37 | 233.304 |
| 1.0 h ∼ PVAF/TPS | −40.59 | 50.32 | 280.574 |
| 1.5 h ∼ PVAF/TPS | −38.45 | 55.41 | 349.944 |
| 2.0 h ∼ PVAF/TPS | −36.29 | 55.38 | 336.318 |
3.4. Water wettability
The contact angles of TPS and the PVAF/TPS composites are shown in Fig. 9. TPS exhibited the lowest contact angle of 46.36°, whereas the surface of PVAF/TPS composites had higher contact angles. In particular, the contact angle of the composite reinforced with PVAF pre-soaked for 1.5 h exhibited the minimal water wettability, with an associated contact angle of 66.25°, which is nearly 65% higher than that of the original TPS (46.36°). This indicates that the pre-soaking treatment of PVAF in SHMP can form an effective chemical connection between the PVAFs and starch macromolecules, thus destroying some of the hydroxyl groups in the PVAFs and starch macromolecules, and thereby slightly reducing the water wettability of the PVAF/TPS composite. The contact angle of the sample soaked for 1.5 h was nearly 65% higher than that of the original TPS of 46.36°. Therefore, the surface water wettability of the PVAF/TPS composites is mainly related to the degree of cross-linking between the PVAFs and starch macromolecule through SHMP, and these properties are also in good agreement with the observed mechanical properties.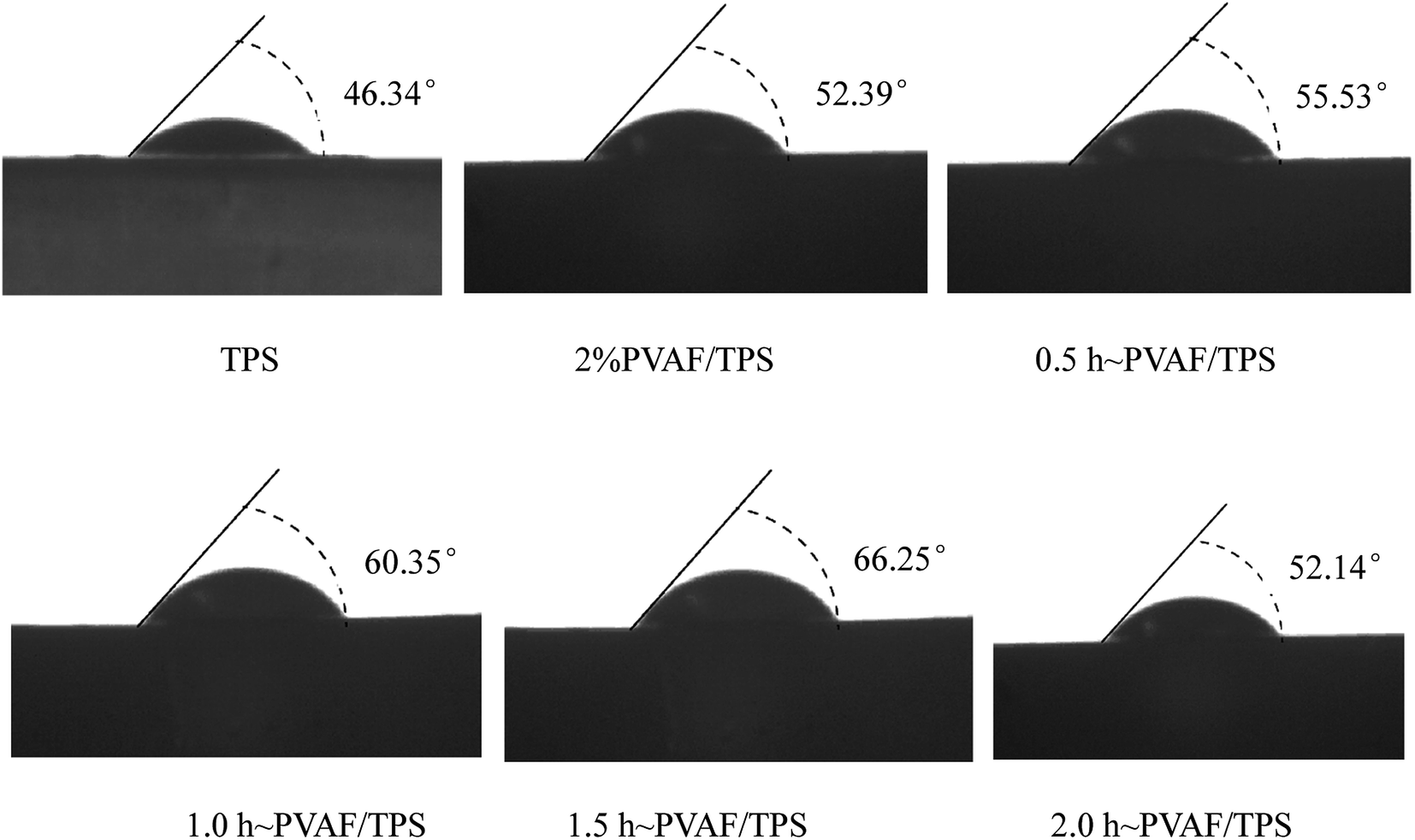 | ||
| Fig. 9 Images of the contact angles of TPS, PVAF, and composites of TPS reinforced with PVAF pre-soaked for different times. | ||
3.5. Reaction mechanism
To clarify the mechanism for the reinforcing effect of the modified PVAF, a schematic illustration of the typical fabrication process for the PVAF/TPS composites is shown in Fig. 10. The change of mechanical properties could be brought about by a combination of factors: (1) increased interfacial adhesion between the filler and the matrix, in this case, the local grafting SHMP around PVA fibers after pre-soaking reacts further with the starch macromolecule matrix, forms the main chemical attachment in the PVAF/TPS composites, and we also could take it as “crosslinking between PVAF and starch” in Fig. 10, which plays a key role in enhancing the mechanical properties based on our work; (2) the possible cross-linking between the fibers or the matrix macromolecules, in this case, the grafted PVAFs by SHMP can react with different PVAFs, which is shown in Fig. 10 as “crosslinking between PVAFs”; in addition, some free SHMPs maybe react with different starch macromolecules, and named “crosslinking between starch chains”,however, these possible reactions could not contribute significantly to the enhanced mechanical properties. Therefore, the effective crosslinked structure between PVAFs and the starch macromolecule matrix is the key mechanism for the reinforcing effect of pre-soaked PVAF in the PVAF/TPS composites.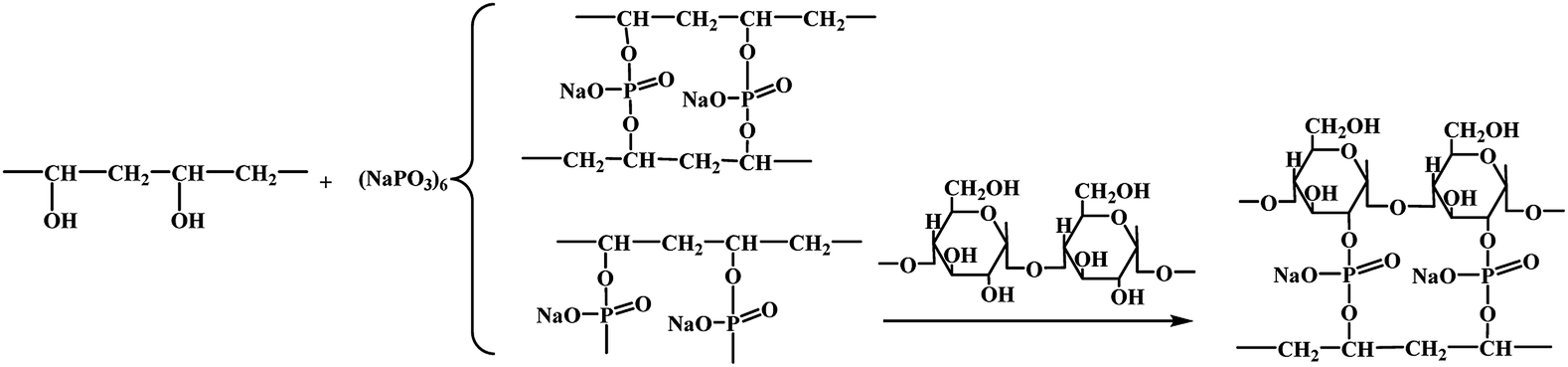 | ||
| Fig. 10 Mechanism of cross-linking reaction of modified-PVAF and TPS. | ||
4. Conclusions
In this study, the effect of SHMP-modified PVAFs on TPS is investigated. When the PVAFs are soaked in an 8% SHMP solution for 1.5 h, the tensile strength of the PVAF/TPS composites reaches 9.18 MPa, approached with the 11.88 MPa tested under the same condition of the commercial LDPE plastic produced by Sinopec Yangzi Petrochemical Co. Ltd. (Nanjing, China), indicating improved mechanical properties from the inclusion of SHMP-modified PVAF. In addition, the storage modulus of the 1.5 h ∼ PVAF/TPS sample is the highest at a given temperature, and its glass transition temperature and apparent activation energy increase from 37.36 °C and 173.182 kJ mol−1 to a maximum of 55.41 °C and 349.944 kJ mol−1 because the SHMP is mostly coated on the surface of the PVAFs, which changes the surface structure of the fibers and contributes to the crosslinked structure between the fibers and the starch matrix. This study demonstrates that is possible to produce pre-soaked PVAF/TPS composites through traditional extrusion and injection molding with better mechanical properties and processability on a large scale. It is also possible for these composites to be used for packaging and catering, including for trays, lids, cutlery, cups, and straws.Conflicts of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.Acknowledgements
The authors thank the Higher school in Jiangsu province college students' practice innovation training programs (201910298025Z), Jiangsu Government Scholarship for overseas studies, Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_1108), and the Natural Science Foundation of Jiangsu Province (BK20140967).References
- X. H. Ma, W. L. Zhang and M. J. Zhang, Research status of corn starch, Liangshi yu Youzhi, 2019, 32, 4–6 Search PubMed.
- J. Prachayawarakorn, L. Hommanee, D. Phosee and P. Chairapaksatien, Property improvement of thermoplastic mung bean starch using cotton fiber and low-density polyethylene, Starch/Staerke, 2010, 62, 435–443 CrossRef CAS.
- O. V. López, L. A. Castillo, M. A. García, M. A. Villara and S. E. Barbosa, Food packaging bags based on thermoplastic corn starch reinforced with talc nanoparticles, Food Hydrocolloids, 2015, 43, 18–24 CrossRef.
- T. P. Mohan and K. Kanny, Thermoforming studies of corn starch-derived biopolymer film filled with nanoclays, J. Plast. Film Sheeting, 2016, 32, 163–188 CrossRef CAS.
- N. Follain, C. Joly, P. Dole and C. Bliard, Properties of starch based blends. Part 2. Influence of poly vinyl alcohol addition and photocrosslinking on starch based materials mechanical properties, Carbohydr. Polym., 2005, 60, 185–192 CrossRef CAS.
- G. J. He, Q. Liu and M. R. Thompson, Characterization of structure and properties of thermoplastic potato starch film surface cross-linked by UV irradiation, Starch/Staerke, 2013, 65, 304–311 CrossRef CAS.
- P. Yin, D. D. Zha, B. Guo, B. G. Li and P. X. Li, Effect of wheat straw oxidation on thermoplastic starch composites: mechanical, thermal, and rheological process behaviors, J. Thermoplast. Compos. Mater., 2020, 33, 646–658 CrossRef.
- B. F. Bergel, S. Dias Osorio, L. M. da Luz and R. M. C. Santana, Effects of hydrophobized starches on thermoplastic starch foams made from potato starch, Carbohydr. Polym., 2018, 200, 106–114 CrossRef CAS PubMed.
- A. H. M. Zain, M. K. Ab Wahab and H. Ismail, Biodegradation Behaviour of Thermoplastic Starch: The Roles of Carboxylic Acids on Cassava Starch, J. Polym. Environ., 2018, 26, 691–700 CrossRef CAS.
- Q. J. Sun, H. R. Fan and L. Xiong, Preparation and characterization of starch nanoparticles through ultrasonic-assisted oxidation methods, Carbohydr. Polym., 2014, 106, 359–364 CrossRef CAS PubMed.
- D. Gumul, M. Krystyjan, K. Buksa, R. Ziobro and T. Zięba, The influence of oxidation, extrusion and oxidation/extrusion on physico-chemical properties of potato starch, Starch/Staerke, 2014, 66, 190–198 CrossRef CAS.
- H. Y. Kim, J. L. Jane and B. Lamsal, Hydroxypropylation improves film properties of high amylose corn starch, Ind. Crops Prod., 2017, 95, 175–183 CrossRef CAS.
- C. M. Müller, J. B. Laurindo and F. Yamashita, Composites of thermoplastic starch and nanoclays produced by extrusion and thermopressing, Carbohydr. Polym., 2012, 89, 504–510 CrossRef PubMed.
- C. A. Romero-Bastida, D. R. Tapia-Blácido, G. Méndez-Montealvo, L. A. Bello-Pérez, G. Velázquez and J. Alvarez-Ramirez, Effect of amylose content and nanoclay incorporation order in physicochemical properties of starch/montmorillonite composites, Carbohydr. Polym., 2016, 152, 351–360 CrossRef CAS PubMed.
- N. F. Magalhaes and C. T. Andrade, Calcium bentonite as reinforcing nanofiller for thermoplastic starch, J. Braz. Chem. Soc., 2010, 21, 202–208 CrossRef CAS.
- L. Lendvai, I. Sajó and J. Karger-Kocsis, Effect of storage time on the structure and mechanical properties of starch/bentonite nanocomposites, Starch/Staerke, 2019, 71, 1800123 CrossRef.
- K. Kaewtatip and V. Tanrattanakul, Structure and properties of pregelatinized cassava starch/kaolin composites, Mater. Des., 2012, 37, 423–428 CrossRef CAS.
- J. A. Mbey, S. Hoppe and F. Thomas, Cassava starch–kaolinite composite film. Effect of clay content and clay modification on film properties, Carbohydr. Polym., 2012, 88, 213–222 CrossRef CAS.
- W. Sanhawong, P. Banhalee, S. Boonsang and S. Kaewpirom, Effect of concentrated natural rubber latex on the properties and degradation behavior of cot-ton-fiber-reinforced cassava starch biofoam, Ind. Crops Prod., 2017, 108, 756–766 CrossRef CAS.
- J. S. Santana, J. M. Do Rosário, C. C. Pola, C. G. Otoni, N. F. Ferreira Soares, G. P. Camilloto and R. S. Cruz, Cassava starch-based nanocomposites reinforced with cellulose nanofibers extracted from sisal, J. Appl. Polym. Sci., 2017, 134, 44637–44645 CrossRef.
- B. Guo, L. J. Wang, P. Yin, B. G. Li and P. X. Li, Ultra-high molecular weight polyethylene fiber-reinforced thermoplastic corn starch composite, J. Thermoplast. Compos. Mater., 2017, 30, 564–577 CrossRef CAS.
- B. Guo, D. D. Zha, C. Xue, P. Yin and P. X. Li, Effect of Polylactic Acid Fiber on the Properties of Thermoplastic Starch Plastics, Chin. J. Polym. Sci., 2018, 31, 261–266 CrossRef CAS.
- H. Tang, H. T. Jiang, L. J. Wang, L. Fan, B. Guo, P. X. Li and Q. S. Zhang, Study on the Polyvinyl Alcohol Fiber Reinforced Thermoplastic Starch, China Plast. Ind., 2013, 41, 110–113 CAS.
- M. Rico, S. Rodríguez-Llamazares, L. Barral, R. Bouza and B. Montero, Processing and characterization of polyols plasticized-starch reinforced with microcrystalline cellulose, Carbohydr. Polym., 2016, 149, 83–93 CrossRef CAS PubMed.
- J. Chen, F. S. Chen, Y. H. Meng, S. F. Wang and Z. Long, Oxidized microcrystalline cellulose improve thermoplastic starch-based composite films: thermal, mechanical and water-solubility properties, Polymer, 2019, 168, 228–235 CrossRef CAS.
- S. Karimi, P. M. Tahir, A. Dufresne, A. Karimi and A. A. Abdulkhani, Comparative study on characteristics of nanocellulose reinforced thermoplastic starch biofilms prepared with different techniques, Nord. Pulp Pap Res. J., 2014, 29, 41–45 CAS.
- Q. F. Chen, Y. Y. Liu and G. X. Chen, A comparative study on the starch-based biocomposite films reinforced by nanocellulose prepared from different non-wood fibers, Cellulose, 2019, 26, 2425–2435 CrossRef CAS.
- Z. Q. Liu, M. Jiang, X. Bai, X. G. Dong, J. Tong and J. Zhou, Effect of postcrosslinking modification with glutaraldehyde on the properties of thermoplastic starch/poly(vinyl alcohol) blend films, J. Appl. Polym. Sci., 2012, 124, 3774–3781 CrossRef CAS.
- K. El-Tahlawy, R. A. Venditti and J. J. Pawlak, Aspects of the preparation of starch microcellular foam particles crosslinked with glutaraldehyde using a solvent exchange technique, Carbohydr. Polym., 2007, 67, 319–331 CrossRef CAS.
- B. Sreedhar, D. K. Chattopadhyay, M. S. H. Karunakar and A. R. K. Sastry, Thermal and surface characterization of plasticized starch polyvinyl alcohol blends crosslinked with epichlorohydrin, J. Appl. Polym. Sci., 2006, 101, 25–34 CrossRef CAS.
- L. Y. Zhou, G. Y. Zhao and W. Jiang, Mechanical properties of biodegradable polylactide/poly(ether-blockamide)/thermoplastic starch blends: effect of the crosslinking of starch, J. Appl. Polym. Sci., 2016, 133, 42297–42304 Search PubMed.
- Y. Qin, W. T. Wang, H. Zhang, Y. Y. Dai, H. X. Hou and H. Z. Dong, Effects of Citric Acid on Structures and Properties of Thermoplastic Hydroxypropyl Amylomaize Starch Films, Materials, 2019, 12, 1565 CrossRef CAS PubMed.
- K. Bahram, B. K. N. Muhammad, J. Zaib, F. Wasif, R. N. Salman, A. Majid, A. Israr and H. Arshad, Effect of ultra-violet cross-linking on the properties of boric acid and glycerol co-plasticized thermoplastic starch films, Food Packag. Shelf Life, 2019, 19, 184–192 CrossRef.
- Y. Yin, J. Li, Y. Liu and Z. Li, Starch crosslinked with poly(vinyl alcohol) by boric acid, J. Appl. Polym. Sci., 2005, 96, 1394–1397 CrossRef CAS.
- Z. Q. Liu, Y. Dong, H. T. Men, M. Jiang, J. Tong and J. Zhou, Post-crosslinking modification of thermoplastic starch/PVA blend films by using sodium hexametaphosphate, Carbohydr. Polym., 2012, 89, 473–477 CrossRef CAS PubMed.
- D. W. Wang, Y. J. Xu, X. Li, C. M. Huang, K. S. Huang, C. K. Wang and J. T. Yeh, Mechanical Retention and Waterproof Properties of Bacterial Cellulose-Reinforced Thermoplastic Starch Biocomposites Modified with Sodium Hexametaphosphate, Materials, 2015, 8, 3168–3194 CrossRef CAS.
- J. G. Yu, N. Wang and X. F. Ma, The effects of citric acid on the properties of thermoplastic starch plasticized by glycerol, Starch/Staerke, 2005, 57, 494–504 CrossRef CAS.
- X. F. Sun, H. Wang, Q. H. Zeng and F. F. Zhang, Study on the preparation of edible cross-linked wheat starch by sodium hexametaphosphate, Liangshi yu Youzhi, 2016, 29, 39–41 Search PubMed.
- C. Xue, P. Yin, Z. D. Y. Jiang, B. Guo and P. X. Li, Crosslinking Modification of Thermoplastic Starch by Using Sodium Hexametaphosphate, Suliao, 2017, 46, 46–48 Search PubMed.
- D. Aritra, U. Ramagopal and D. Chandan, Feasibility of poly-vinyl alcohol/starch/glycerol/citric acid composite films for wound dressing applications, Int. J. Biol. Macromol., 2019, 131, 998–1007 CrossRef PubMed.
- W. A. Silva, J. Pereira, C. W. P. Carvalho and F. Q. Ferrua, Determination of color, topographic superficial image and contact angle of the biofilms of different starch sources, Cienc. Agrotecnol., 2007, 31, 154–163 CrossRef.
- T. X. Tao, Z. C. Wu, X. Q. Wang, M. S. Li and J. H. Zhang, Synthesis and spectra of compleses involving polyvinyl alcohol fiber ligands, Acta Polym. Sin., 2006, 3, 387–390 CrossRef.
- A. Bouzidi, W. Jilani, H. Guermazi, I. S. Yahia, H. Y. Zahran and G. B. Sakr, The effect of zinc iodide on the physicochemical properties of highly flexible transparent poly (vinyl alcohol) based polymeric composite films: opto electrical performance, J. Mater. Sci. Mater. Electron., 2019, 30, 11799–11806 CrossRef CAS.
- M. Kurumova, D. Lopez, R. Benavente and C. Mijangos, Effect of crosslinking on the mechanical and thermal properties of poly(vinyl alcohol), Polymer, 2000, 41, 9265–9272 CrossRef.
- O. C. Wokadala, N. M. Emmambux and S. S. Ray, Inducing PLA/starch compatibility through butyl-etherification of waxy and high amylose starch, Carbohydr. Polym., 2014, 112, 216–224 CrossRef CAS PubMed.
- N. Anousheh, F. O. Godey and A. Soldera, Unveiling the Impact of Regioisomerism Defects in the Glass Transition Temperature of PVDF by the Mean of the Activation Energy, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 419–426 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |