
One-step synthesis of composite hydrogel capsules to support liver organoid generation from hiPSCs†
Yaqing
Wang‡
abc,
Haitao
Liu‡
bc,
Min
Zhang
bc,
Hui
Wang
bc,
Wenwen
Chen
bc and
Jianhua
Qin
 *bcde
*bcde
aSchool of Chemistry, Dalian University of Technology, Dalian 116024, P.R. China. E-mail: jhqin@dicp.ac.cn
bCAS Key Laboratory of Separation Science for Analytical Chemistry, DalianInstitute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, P.R. China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, P.R. China
dInstitute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beijing, 100101, P.R. China
eCAS Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, 200031, P.R. China
First published on 15th August 2020
Abstract
Advances in biomaterials, especially in hydrogels, have offered great opportunities for stem cell organoid engineering with higher controllability and fidelity. Here, we propose a novel strategy for one-step synthesis of composite hydrogel capsules (CHCs) that enable engineering liver organoids from human induced pluripotent stem cells (hiPSCs) in an oil-free droplet microfluidic system. The CHCs composed of a fibrin hydrogel core and an alginate–chitosan composite shell are synthesized by an enzymatic crosslinking reaction and electrostatic complexation within stable aqueous emulsions. The proposed CHCs exhibit high uniformity with biocompatibility, stability and high-throughput properties, as well as defined compositions. Moreover, the established system enables 3D culture, differentiation and self-organized formation of liver organoids in a continuous process by encapsulating hepatocyte-like cells derived from hiPSCs. The encapsulated liver organoids consisting of hepatocyte- and cholangiocyte-like cells show favorable cell viability and growth with consistent size. Furthermore, they maintain proper liver-specific functions including urea synthesis and albumin secretion, replicating the key features of the human liver. By combining stem cell biology, defined hydrogels and the droplet microfluidic technique, the proposed system is easy-to-operate, scalable and stable to engineer stem cell organoids, which may offer a robust platform to advance organoid research and translational applications.
1. Introduction
Building 3D organ models in vitro is a formidable goal to recapitulate human physiology with various biomedical applications including disease modeling, regenerative medicine and tissue engineering. Organoids, referring to 3D multicellular tissues derived from human embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), tissue-resident adult stem cells (ASCs) or progenitor cells by the processes of self-organization, recapitulate the key architectural and functional features of in vivo organs.1–4 They represent a major technological breakthrough of organ models for applications in biomedicine. Progress in the organoid field has enabled successful generation of various organoid types from human pluripotent stem cells (hPSCs) for applications in developmental studies, disease modeling and drug screening.5–7 Generally, most organoids are generated in Petri dishes, mainly relying on self-organization of stem cells in animal-derived extracellular matrices (e.g., Matrigel).2,5,8 However, these organoid systems are often limited by the high variability of tissue composition and architecture due to the lack of defined 3D matrices. Moreover, current protocols often required labor-intensive multistep manual operations, possibly resulting in the risk of contamination and marked variability among organoids.Advances in biomaterials, especially in hydrogels, have provided great opportunities to improve the construction of organoids with higher controllability and fidelity.4 Hydrogels, including natural9,10 (e.g., fibrin and alginate) and synthetic11 (e.g., polyethylene glycol, PEG) polymers, have been preliminarily utilized as extracellular matrix (ECM) scaffolds to instruct the organization and generation of stem cell organoids. These chemically defined matrices can mimic the native ECM microenvironment and improve the physiological relevance of organoids by control over their biochemical and physical properties, which may reduce the variability of organoids. Additionally, hydrogels with flexible structures, including porous scaffolds,12 micropatterns,13 and spheroids14 at the micro to millimeter scale, are utilized to guide the formation of organoids with specific architectures using different engineering methods. But there are still some limitations in engineering organoids with high fidelity and reproducibility.
Hydrogel capsules are recognized as ideal scaffolds for 3D culture of cellular spheroids, like organoids, with high uniformity due to the well-defined morphologies and a high surface area.15,16 They are currently fabricated by chemical reactions or physical interaction in water-in-oil (W/O) droplet systems. The reagents used in these systems, such as the oil phase and/or surfactants, may reduce the biocompatibility of hydrogel capsules, leading to potential protein or enzyme denaturation, inhibition of cell growth, and limited mass transfer in subsequent cell culture.17–19 Alternatively, the aqueous two-phase system (ATPS) has provided an oil-free manner in droplet microfluidic systems for the fabrication of biomimetic and/or cell-laden hydrogel scaffolds.20–25 Hydrogel capsules generated in such systems can be solidified under moderate conditions, and are potential scaffolds for cell assembly and 3D culture.
In this work, we present a novel strategy for one-step fabrication of composite hydrogel capsules (CHCs) that allow the 3D culture and formation of liver organoids from hiPSCs in an oil-free droplet microfluidic system. The capsules are fabricated based on the interfacial complexation of the oppositely charged Na-alginate (NaA) and chitosan (CS), as well as the enzymatic reaction of fibrinogen and thrombin in this system. The characteristics of capsules, such as morphology and composition, are investigated. In addition, the size and inner hydrogel components are scalable and adjustable by changing the flow rates and the hydrogel precursors within core flow. Then, hiPSC-derived hepatic cells are self-organized into liver organoids with a uniform size in an array format by encapsulation in CHCs. The cell viability and expression of liver specific markers, as well as liver-specific functions of organoids in CHCs, are examined. This integrative approach paves a way for engineering organoids using hydrogel scaffolds and might extend their potential applications in drug testing and translational research.
2. Materials and methods
2.1. Materials
Dextran (500 kDa, GE), PEG (20 kDa, Aladdin), Na-alginate (55 cps, Qingdao Hyzlin Biology Development Co., Ltd), chitosan (100–200 cps, Aladdin), glacial acetic acid (Hengxing Chemical Reagent), NaCl (Tianjin Damao Chemical Reagents Factory), fibrinogen (Sigma), thrombin (Sigma), fluorescein isothiocyanate isomer I (FITC) (95%, J&K), polydimethylsiloxane (PDMS, SYLGARD 184) and the curing agent (Dow Corning Corporation), and photoresist (SU-8 3035, MicroChem Corporation) are used. All reagents are used as received.2.2. Microfluidic chip fabrication
The layered microfluidic chip was fabricated via standard soft lithography and micromolding methods. Briefly, the PDMS at a weight ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was molded from photoresist templates after being cured at 80 °C for 2 h in an oven to form structured top and middle layers of the chip. Subsequently, the top layer was bonded to the middle layer that was bonded to a plain PDMS block after punching with a diameter punch of 1.5 mm and oxygen plasma treatment. Both the height and width of the microchannels in the top layer are 200 μm, while both the height and width of the microchannels in the middle layer are 100 μm. In addition, the thickness and diameter of the pneumatic valve are 100 μm and 1 mm, respectively, which could control the generation of droplet templates.
:1 was molded from photoresist templates after being cured at 80 °C for 2 h in an oven to form structured top and middle layers of the chip. Subsequently, the top layer was bonded to the middle layer that was bonded to a plain PDMS block after punching with a diameter punch of 1.5 mm and oxygen plasma treatment. Both the height and width of the microchannels in the top layer are 200 μm, while both the height and width of the microchannels in the middle layer are 100 μm. In addition, the thickness and diameter of the pneumatic valve are 100 μm and 1 mm, respectively, which could control the generation of droplet templates.
2.3. Capsule fabrication
The oil-free droplet microfluidic system was composed of 17% (w/w) PEG and 15% (w/w) dextran (DEX), which were dissolved in deionized water. 1% (w/w) NaA and 5 mg mL−1 fibrinogen were dissolved in the DEX solution to form the core flow solution. And 1% (w/w) CS and 2.5 U mL−1 thrombin were dissolved in the PEG solution with pH = 5 adjusted by acetic acid to form the shell flow solution. A pure 17% PEG solution was used as the middle flow solution. The core, middle and shell flow solutions were pumped into their inlets of the microfluidic chip by using syringe pumps (11 Picro Plus, Harvard). In the droplet generation unit, the pneumatic valve was pushed and pulled periodically as the compressed air in and out, thus leading to a droplet template generation at the end of a switch cycle. Then, droplet templates were delivered into shell flow by the PEG phase at the second junction, wherein the electrostatic complexation of NaA and CS, as well as the enzymatic crosslinking reaction of fibrinogen and thrombin, occurred. The generated capsules were collected in the collection tank with physiological saline/culture medium containing 2.5 U mL−1 thrombin for further gelation of fibrinogen for 10 min.2.4. ATR–FTIR analysis
The composition of the capsule shell (synthesized without fibrinogen/thrombin) was studied using a FTIR spectrometer (Bruker Tensor II, Germany) and compared with that of unreacted Na-alginate/chitosan. Here, the samples were initially suspended in deionized (DI) water and then freeze-dried for subsequent analysis.2.5. Scanning electron microscopy (SEM) analysis
Hydrous hybrid capsules were freeze-dried in a lyophilizer (SCIENTZ-10ND, China) overnight. Then they were coated with a layer of gold for 220 s with a sputter coater (SBC-12, KYKY) after being fixed on the aluminum stub using conductive adhesive tape to improve the electrical conductivity of capsules. The size and surface morphology of the capsules were characterized using a SEM (Hitachi TM3000, Japan) at 15 kV.2.6. FITC-labeled chitosan synthesis
To label the CS layer within the capsules, FITC was chosen to tag the CS component. The FITC-labeled CS (FITC-CS) was synthesized using the reaction between the –NH2 group of CS and –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS group of FITC.26 Briefly, 0.5 g CS was dissolved in 50 mL acetic acid (0.1 M) before adding 50 mL methanol and 25 mL methanol containing FITC (2 mg mL−1). After reaction in the dark at room temperature for 3 h, FITC-CS was precipitated by adding 0.2 M NaOH until the pH increased to 9–10. Unreacted FITC was washed away with a mixture of methanol and water in a 7:3 volume ratio separated by centrifugation (17800g, 10 min) until no fluorescence could be detected in the supernatant. The FITC-CS was then dialyzed with a dialysis bag (cut-off MW = 7000) in 3 L of DI water for 3 days in the dark. The DI water was replaced with fresh water daily. The dialyzed FITC-CS was freeze-dried in a lyophilizer for 3 days for the subsequent experiments. The fluorescence images of FITC labeled capsules were acquired with a fluorescence microscope (Olympus IX71, Japan).
CS group of FITC.26 Briefly, 0.5 g CS was dissolved in 50 mL acetic acid (0.1 M) before adding 50 mL methanol and 25 mL methanol containing FITC (2 mg mL−1). After reaction in the dark at room temperature for 3 h, FITC-CS was precipitated by adding 0.2 M NaOH until the pH increased to 9–10. Unreacted FITC was washed away with a mixture of methanol and water in a 7:3 volume ratio separated by centrifugation (17800g, 10 min) until no fluorescence could be detected in the supernatant. The FITC-CS was then dialyzed with a dialysis bag (cut-off MW = 7000) in 3 L of DI water for 3 days in the dark. The DI water was replaced with fresh water daily. The dialyzed FITC-CS was freeze-dried in a lyophilizer for 3 days for the subsequent experiments. The fluorescence images of FITC labeled capsules were acquired with a fluorescence microscope (Olympus IX71, Japan).
2.7. Cell culture and differentiation
Human liver cell line (HepaRG) was purchased from the Cell Bank of the Chinese Academy of Sciences (Kunming, China). The HepaRG cells were cultured using Dulbecco's Modified Eagle's Medium (DMEM, high glucose, Gibco), which was supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% streptomycin and penicillin (Biyotime). They were cultured in a humidified incubator composed of 5% CO2 at 37 °C and passaged until achieving 80%–90% confluence.Human iPSCs (donated by Dr Ning Sun) were cultured using mTeSR1 medium (StemCell) on Matrigel-coated six-well plates (1:50 dilution, BD) under a humidified atmosphere with 5% CO2.27–29 The culture medium was changed daily. Prior to generating liver organoids, iPSCs were initially induced into hepatocyte-like cells by sequentially adding specific growth factors into the medium on Petri dishes. The differentiation protocol was modified according to previous studies.30 For definitive endoderm (DE) differentiation, iPSCs were incubated in RPMI-1640 medium (Invitrogen) supplemented with 1% B27 supplement (Invitrogen), 1% KSR (Invitrogen), 1% GlutaMAX (Invitrogen), 1% penicillin–streptomycin (Sigma), and 100 ng mL−1 activin-A (PeproTech) for 5 days. For hepatic progenitor differentiation and expansion, 20 ng mL−1 HGF (PeproTech) and 10 ng mL−1 bFGF (PeproTech) were added to the medium from days 5 to 10. Then, the medium was replaced by HCM medium (ScienCell) supplemented with 10−7 M dexamethasone (Sigma-Aldrich) and 10 ng mL−1 oncostatin M (OSM, R&D) to facilitate hepatocyte differentiation for additional 5 days. On day 14 of differentiation, hepatic cells were ready for the encapsulation experiments. After day 15, the cells were cultured without OSM for long term.
2.8. Cell encapsulation
HepaRG cells and hepatocyte-like cells at day 14 of differentiation from iPSCs were dissociated by 0.25% trypsin-EDTA (Gibco), and then resuspended in core flow solution with a density of 4 × 106 cells per mL (HepaRG) and 3–5 × 107 cells per mL (hepatocyte-like cells) for encapsulation experiments, respectively. Here, the core flow solution was prepared by mixing cell suspensions with a concentrated NaA solution (22.5% (w/w) DEX, 1.5% (w/w) NaA and 7.5 mg mL−1 fibrinogen dissolved in physiological saline) in a 1:2 ratio. The final solution contained 15% (w/w) DEX, 1% (w/w) NaA and 5 mg mL−1 fibrinogen. 17% (w/w) PEG and 17% (w/w) PEG with 1% (w/w) CS/2.5 U mL−1 thrombin were also dissolved in physiological saline as the middle and shell flow, respectively. All these solutions were filtered with a 0.22 μm syringe filter (Millex Syringe Filter) before being pumped into the microfluidic chip device. The generated cell-laden capsules were collected in physiological saline/culture medium containing 2.5 U mL−1 thrombin for further gelation of fibrinogen for 10 min. The cells in capsules were finally transferred to cell culture medium within a 24-well culture plate (Guangzhou Jet Bio-Filtration Co., Ltd) and cultured in a humidified incubator with 5% CO2 at 37 °C with the fresh medium being changed daily.
2.9. Size measurement and distribution statistics
The images of cell-free capsules were acquired in the physiological saline bath with an inverted microscope (Olympus IX-71, Japan). The images of cell-laden capsules were acquired in the cell culture medium with an inverted microscope (Leica DMi8, Germany). The size measurement and distribution statistics of capsules/cellular spheroids are implemented with Image J software (http://rsb.info.nih.gov/ij/).2.10. Cell viability analysis
Cell viability of the encapsulated cells was evaluated by using live/dead assay. The cell-laden capsules were incubated in cell culture medium with ethidium homodimer-1 (red, dead, 1:500) and calcein-AM (green, live, 1:1000) (LIVE/DEAD viability/cytotoxicity assay kit, Gibco) at 37 °C for 25 min. Then, they were rinsed with physiological saline before imaging under a fluorescence microscope (Leica DMI3000 B, Germany). The viability percentage of cells within capsules was determined by measuring the area of live cells (green fluorescence), which was divided by the total area of live + dead cells (red fluorescence) at a given focal plane.
2.11. Real-time quantitative PCR
The whole mRNA was extracted from iPSCs, 2D liver cells in Petri dishes, and liver organoids in capsules with a Trizol reagent (TAKARA) to detect the associated gene expression level. The final concentration of mRNA was adjusted to 250 ng mL−1. The cDNA was then synthesized by reverse transcription polymerase chain reaction (RT-PCR) (TAKARA). Subsequently, real-time quantitative PCR was implemented with a SYBR Green kit (TAKARA) under the following reaction conditions (40 cycles): denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s. The primer pairs were as follows: AFP forward: 5′-CTT TGG GCT GCT CGC TAT GA-3′, reverse: 5′-GCA TGT TGA TTT AAC AAG CTG CT-3′; ALB forward: 5′-GCC TTT GCT CAG TAT CTT-3′, reverse: 5′-AGG TTT GGG TTG TCA TCT-3′; RBP4 forward: 5′-CCA GAA GCG CAG AAG ATT G-3′, reverse: 5′-TTT CTT TCT GAT CTG CCA TCG-3′; CYP3A4 forward: 5′-TTC AGC AAG AAG AAC AAG GAC AA-3′, reverse: 5′-GGT TGA AGA AGT CCT CCT AAG C-3′; CK7 forward: 5′-AAG AAC CAG CGT GCC AAG TT-3′, reverse: 5′-CAC GCT CAT GAG TTC CTG GT-3′; CK19 forward: 5′-TCC GAA CCA AGT TTG AGA CG-3′, reverse: 5′-CCC TCA GCG TAC TGA TTT CCT-3′; GAPDH forward: 5′-CAA TGA CCC CTT CAT TGA CC-3′, reverse: 5′-GAC AAG CTT CCC GTT CTC AG-3′. Quantification was performed using GAPDH as the reference gene.2.12. Fluorescence immunohistochemistry
HepaRG cells in capsules were soaked in 4% paraformaldehyde (Tianjin Damao Chemical Reagents Factory) for 15 min to be immobilized after cultivation for 10 days. Then they were permeabilized with 0.2% Triton X-100 (Amresco) for 10 min and blocked in a blocking solution containing 10% goat serum for 2 h at room temperature, successively. Next, these cells were incubated at 4 °C overnight with primary goat antihuman albumin (diluted 1:400, Beyotime Company), which were then exposed to secondary antibodies (cy3 labeled donkey goats IgG (H + L) (diluted 1:200, Beyotime Company) for 1 h at room temperature. The cell nuclei were stained with DAPI (1:4000, CST 4083) for 10 min at room temperature. For the encapsulated liver organoids, they were collected after encapsulation for 7 days. Then, they were fixed, permeabilized, blocked, and incubated with primary antibodies as described above. Here, the primary antibodies were as follows: albumin (goat, 1:1000, Bethyl A80-129A), CYP3A4 (mouse, 1:500, Absin abs132219), CK7 (rabbit, 1:8000, Abcam ab181598) and CK19 (mouse, 1:250, Abcam ab7754). The organoids were then incubated with Cy3 (1:500, Beyotime) and Alexa Fluor 488- or 594- (1:200, Cell Signaling) conjugated secondary antibodies at room temperature for 1 h. DAPI (1:4000) was used to stain the cell nuclei. All the images were acquired with a confocal microscope (Olympus IX81, Japan).
2.13. Urea synthesis and albumin secretion
For quantitative analysis of the urea synthesis and albumin secretion from the encapsulated cells cultured in 24-well plates, the supernatant of culture medium was collected punctually. Here, the supernatants were collected on days 1, 3, 5, 7 and 9 for the encapsulated HepaRG cells and on days 2, 4, 6 and 8 for the encapsulated liver organoids, which were kept at −80 °C before being used. A Urea Assay Kit (DIUR-100) was used to measure urea concentrations. The samples held the same volume as the standard sample and the blank control. Each well of the 96-well plate contained 100 μL mixture of the urea standard, the medium and the samples. After incubating the mixture in the dark (1 h, 25 °C), the absorbance at 430 nm was read using a microplate reader. The results were expressed as μg mL−1. Similarly, the concentration of secreted albumin in the medium was measured using a human Albumin ELISA kit (Bethyl Laboratories) according to the instructions. The concentration of albumin secretion was determined by the measuring the absorbance at 450 nm and expressed as μg mL−1.2.14. Statistical analysis
In this work, all the experiments were performed at least in triplicate. All the data were presented as mean ± standard deviation (SD), unless stated differently. Statistical significance was declared when *p < 0.05, **p < 0.01 and ***p < 0.001 as determined by one-way ANOVA and Student's t-test.3. Results and discussion
3.1. Design and manipulation of the oil-free droplet microfluidic platform
The existing methods for organoid generation mostly rely on the self-organization of stem cells within 3D Matrigel in low-adhesion Petri dishes. The undefined compositions and unrestricted structures of the 3D matrices might lead to organoid formation with higher variation in size and morphology. In order to generate human organoids from hiPSCs with a uniform size and morphology, we designed and established an oil-free droplet microfluidic system to fabricate hydrogel capsules in a mild and controllable fashion. Dextran (DEX) solution with NaA/fibrinogen and PEG solution are pumped into the device serving as core and middle flows, respectively, for the formation of droplet templates (Fig. 1A). The compressed air driven by a solenoid valve controller is utilized to activate the pneumatic valve for controlling the generation manners of droplet templates, which could partially address the inherent limitation of low interfacial tension in ATPS31 due to partial conversion of mechanical energy provided by the pneumatic valve into surface energy.32 As shown in Fig. 1B, the pneumatic valve is pushed and pulled periodically as the compressed air in and out, thus leading to the deformation of the core channel and a droplet generation at the end of a switch cycle. Also, the mechanical valves within the microfluidic device can facilitate the stable and controllable generation of droplet templates in an all-in-water microenvironment, avoiding the utilization of the oil phase that may lead to potential cytotoxicity.17–19 In addition, compared to the passive microfluidic flow focusing platform,33 our valve-based platform can significantly increase the uniformity and throughput of water-in-water droplets. This improvement is beneficial for the controllable and massive generation of organoids from hiPSCs. Moreover, the PEG solution with CS/thrombin (shell flow) is used to react with core flow by diffusion of CS/thrombin to the droplet templates for the generation of CHCs (Fig. 1D).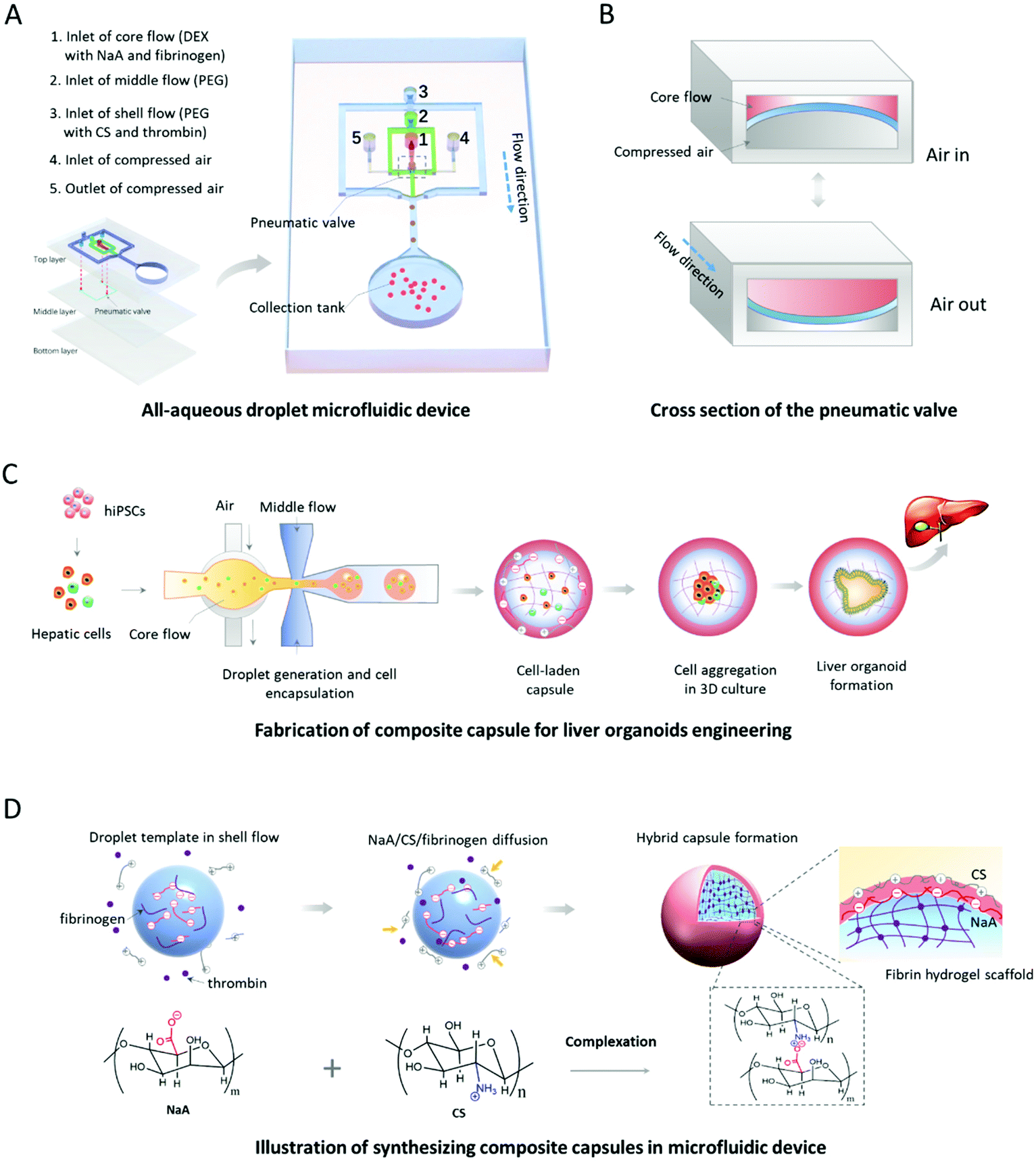 | ||
| Fig. 1 Schematic diagram of the oil-free droplet microfluidic system to fabricate composite hydrogel capsules (CHCs) that enable the engineering of liver organoids. (A) The configuration of the droplet microfluidic device, including shell flow, middle flow, core flow, compressed air, pneumatic valve, and collection tank. (B) The procedure of the pneumatic valve for the generation of droplet templates. (C) The flow chart of engineering human liver organoids from hiPSCs with the defined capsules. (D) The illustration of the synthesis of defined hydrogel capsules in the microfluidic device. | ||
The fabrication of hydrogel capsules is mainly based on the controllable diffusion of four reagents (NaA, CS, fibrinogen, thrombin), the fast complexation of oppositely charged NaA and CS, and the mild enzymatic reaction of fibrinogen and thrombin in the system. Specifically, droplet templates generated at the pneumatic valve, containing NaA and fibrinogen, are delivered into the shell flow by immiscible middle flow, where the NaA and fibrinogen molecules diffuse to the middle flow due to the concentration gradient. Similarly, CS and thrombin molecules within the shell flow can diffuse to the middle flow. When all four reactants meet at a spherical interface, the complexation of oppositely charged NaA and CS and the enzymatic reaction of fibrinogen and thrombin occur to form a binary complex and fibrin hydrogel, respectively. Since the binary complex of NaA and CS, serving as a semipermeable membrane, can stop the further diffusion of macromolecules (e.g., NaA, CS, fibrinogen), the well-defined binary shell and hydrogel core are maintained in the capsules. Herein, the reaction time (including diffusion and complexation processes) between NaA and CS is only 1.9–4.7 s. The very short time is sufficient to ensure the synthesis of core–shell CHCs without agglomeration occurrence during the subsequent collection process in the proposed chip. In addition, hundreds to thousands of capsules can be produced within one minute using mechanical valves in the system (Fig. S1 and Movie S1, ESI†), indicating the efficiency of our system for generating microdroplets with well-controllability and high speed. It also suggested the potential of this system for fabricating cell-loaded 3D scaffolds in a high-throughput manner. In addition, the speed of microdroplet generation can be easily increased by fabricating more parallel units in the microfluidic device without losing the homogeneity of droplets, which has been proved in the previous work.20 The microfluidic chip is actually composed of three layers of polydimethylsiloxane (PDMS) bonded together (Fig. 1A and Fig. S2†). The core, middle and shell flows were directly introduced into the microchannels of the top layer, while the compressed air was injected into the middle layer via the inlets of the top layer. As such, hydrogel capsules can be produced in this system, which allows 3D culture and self-organized formation of human liver organoids in a continuous process by encapsulating hiPSC-derived hepatic cells (Fig. 1C).
3.2. The controllable synthesis of composite hydrogel capsules (CHCs)
The process of organoid formation in traditional approaches is often spontaneous and uncontrollable, which would largely affect their reproducibility. To generate organoids with consistent and controllable dimension using hydrogel capsules, the oil-free droplet system was optimized in the following experiments. To explore the key parameters that affect the capsule size, we measured the diameter of the capsules generated under various conditions. Herein, three factors, including rates of multi-phase flows, valve switch cycle, and concentrations of NaA/CS, were studied systematically. Flow rates are crucial factors in the system to affect the size of droplet templates and then the size of capsules. The effects of the core flow were initially observed, which was varied from 0.1 to 0.4 μL min−1. The diameter of CHCs increased with the increase of the core flow rate significantly (***p < 0.001) (Fig. 2A and B). This result was obviously in accordance with that in the general droplet microfluidic system due to the correlation between the dispersed phase flow rate and droplet size within a certain range.34 However, we do not observe regular effects of middle/shell flow rates on the diameter of CHCs (Fig. S3†). The regular changes of the diameter of CHCs were mainly attributed to the core flow rate rather than middle/shell flow rates. This is in agreement with previous reports that the size of droplets was mainly regulated by the flow rate of core stream under the jetting regime.35,36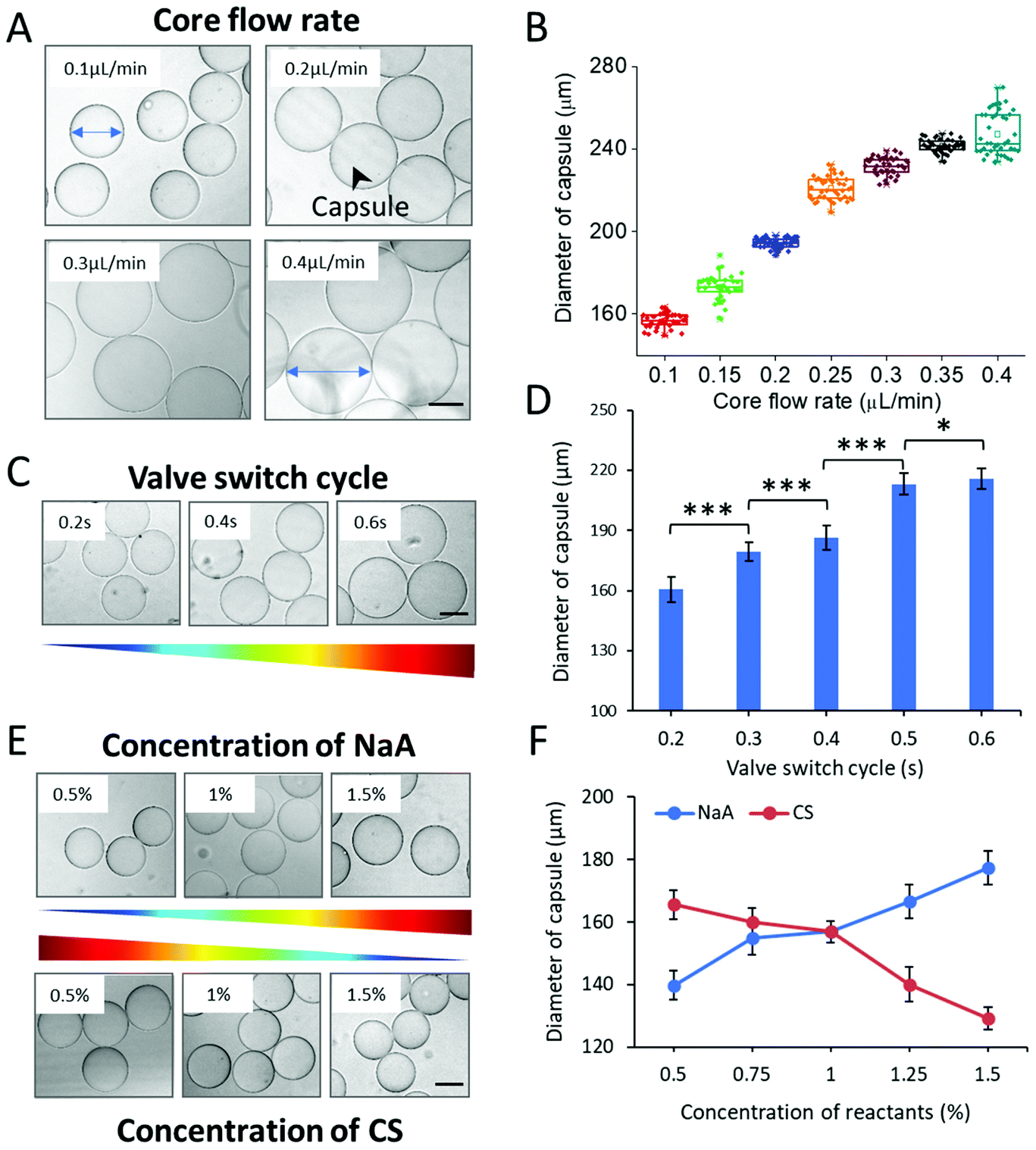 | ||
| Fig. 2 Effects of different fluid parameters on the diameter of capsules. (A) Bright-field images of hydrogel capsules synthesized at different core flow rates, ranging from 0.1 to 0.4 μL min−1. (B) The plot of capsule diameter at different core flow rates (0.1 to 0.4 μL min−1). (C) Bright-field images of capsules synthesized under different valve switch cycles, ranging from 0.2 to 0.6 s. (D) Diameter profiles of capsules obtained under different valve switch cycles. Data are shown as mean ± SD. Student's t-tests were performed. *p < 0.05, ***p < 0.001. (E) The images of capsules synthesized with different concentrations of NaA/CS. (F) The diameter of capsules as a function of the concentrations of NaA/CS. Quantitative analysis of the diameter of capsules were performed on at least 30 capsules. Scale bars: 100 μm. | ||
Furthermore, we measured the size distribution of CHCs generated in different experimental groups to evaluate the polydispersity and stability of our proposed method. As shown in Fig. 2B and Fig. S2,† these capsules exhibited uniform features and exhibited a relatively small coefficient of variation (CV < 5%), leading to the potential of CHCs to constrain the shape and size of loaded cargoes.
As above, the droplet templates were generated with the control of pneumatic valve, of which switch cycle was also an important parameter that affected the CHC diameter. Herein, the valve switch cycle was defined as the total time of valve being pushed and pulled. We measured the size of hydrogel capsules generated with different valve cycles, ranging from 0.2 to 0.6 s. In this case, the size of CHCs was significantly increased even with a small increment of the valve cycle (e.g., 0.1 s) (Fig. 2C and D). According to our previous work, the valve switch cycle influenced the size of droplet templates directly due to mass conservation,20 thus resulting in the variation of capsules. In addition, the binary shells of capsules were formed by the diffusion of NaA and CS in this system. Herein, the driving force of the diffusion was the concentration gradient according to Fick's first law
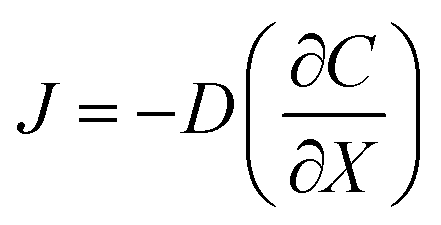 | (1) |
3.3. Structural and componential characterization of CHCs
The internal structure, spatial distribution, and well-defined compositions are the key features of hydrogel capsules, making them suitable as scaffolds for 3D cell culture and tissue growth. To evaluate these features of CHCs, capsules under hydrous and freeze-dried conditions were investigated with an optical microscope and a scanning electron microscope (SEM). Initially, the hydrous capsules, synthesized with fluorescein isothiocyanate-labeled chitosan (FITC-CS) and without fibrinogen/thrombin, were investigated under an optical microscope. Such capsules exhibited a transparent circular section, of which the edge was darker than the center (Fig. 3A). Then, it was proved from the fluorescence images that the FITC-CS was located at the dark edge of CHCs (Fig. 3B). As mentioned in section 2.1, the binary complex of NaA and CS can serve as a semipermeable membrane to stop the further diffusion of CS into the capsule under the fabrication conditions, leading to the deposition of FITC-CS only at the surface of the capsule. To further demonstrate the hollow structure of these capsules, the freeze-dried CHCs were prepared and characterized subsequently. The thin shell is presented in the SEM images, of which the thickness was about 200 nm (Fig. 3D).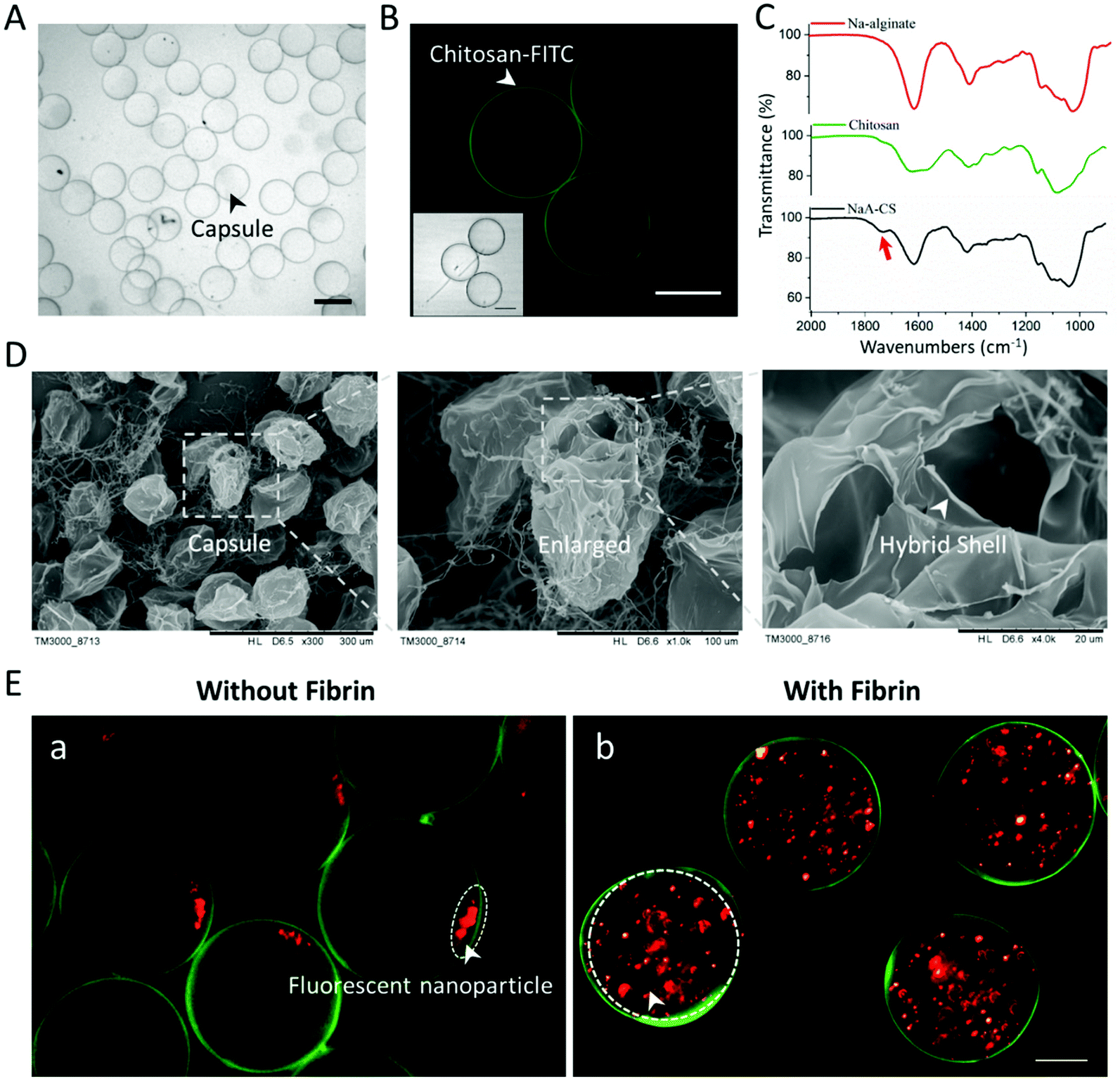 | ||
| Fig. 3 Characterization of the structure and composition of hydrogel capsules. (A) Bright-field image of massively produced hydrogel capsules. Scale bar: 200 μm. (B) Fluorescence image of hydrogel capsules synthesized with FITC-chitosan. Scale bar: 100 μm. (C) The FTIR spectra of Na-alginate, chitosan and NaA–CS binary capsules. (D) SEM images of freeze-dried capsules. (E) Fluorescence image of capsules filled with fluorescent nanoparticles. Scale bar: 50 μm. For (A to E), all the capsules were fabricated under the same conditions (rates of core, middle and shell flow: 0.15, 2, and 4 μL min−1; valve cycle: 0.4 s; concentrations of NaA/CS: 1%). For E-b, capsules were fabricated under the aforementioned flow conditions, but with the existence of fibrinogen and thrombin in the core and shell flows, respectively. | ||
In addition, the complexation of oppositely charged NaA and CS in the shell and the enzymatic reaction of fibrinogen and thrombin in the core were also evaluated. The composition of the CHC shell (synthesized without fibrinogen/thrombin) was studied using a FTIR spectrometer and compared with that of unreacted NaA/CS. As shown in Fig. 3C, the peaks in the region of 950–1200 cm−1 exhibited in all the spectra are a result of various vibrations of the carbohydrate rings that are the building blocks of both NaA and CS. The characteristic peaks of NaA are at 1618 (asymmetric COO– stretching) and 1415 cm−1 (symmetric COO– stretching).37 Also, the typical peaks of CS are at 1630 (amide I band), 1590 (N–H bending of amine and amide II band), and 1385 cm−1 (amide III band).37,38 The detailed peak assignments are shown in Fig. S4 and Table S1.†
When the NaA–CS capsules were formed, the peaks in the spectra changed significantly. The asymmetric and symmetric COO– stretching of NaA slightly shifted from 1618 to 1613 cm−1 and 1415 to 1413 cm−1, respectively, and became broader due to the decrease of carboxyl charge density.39 As for CS, the amide I and amide II bands shifted into a singlet band at 1613 cm−1, and the N–H bending of amine at 1590 cm−1 disappeared because of the reduction of free amino groups.38,40 In addition, a new peak (arrow) at 1730 cm−1 appeared on spectra of the NaA–CS capsule, which belonged to the asymmetric stretching of COO–.41 All these results indicated that the electrostatic complexation occurred between negatively charged NaA and positively charged CS within the capsules. To demonstrate the successful reaction of fibrinogen and thrombin in the CHCs, fluorescent nanoparticles (red) were added in the core flow to form the capsules without/with fibrinogen and thrombin. As shown in Fig. 3Ea, the fluorescent nanoparticles deposited at a corner in the CHCs due to gravity after 12 h. In contrast, the fluorescent nanoparticles maintained homogeneous distribution in the capsules with the existence of fibrin. As such, the CHCs holding well-defined spatial distribution and compositions were produced in our system.
3.4. Evaluation of capsules as scaffolds for 3D cell culture
As above, the fabricated capsules in this system exhibited high uniformity and defined compositions, which might provide promising scaffolds for cell encapsulation and scalable 3D culture. In order to assess the possibility of CHCs for encapsulation of cells, human liver (HepaRG) cells were initially resuspended in the core flow to settle in CHCs. Here, these capsules could maintain their structural integrity for at least 25 days under cell culture conditions, demonstrating the stability of hydrogel capsules for cell culture (Fig. S5†). After encapsulation, the cell-laden capsules with fibrinogen in the core were immersed in thrombin solution to further facilitate the production of the fibrin matrix in the capsules by the enzymatic reaction. As shown in Fig. S6A,† HepaRG cells aggregated and spontaneously formed small spheroids in CHCs after encapsulation at day 1. These cell spheroids kept growing until filling up the whole capsules during 10 days of culture. The sizes distribution of the spheroids at days 1 and 10 was also analyzed, which showed a relatively narrow particle size distribution at both days (Fig. S6B†). These data indicate that the CHCs can constrain the shape and size of the 3D tissue with uniform morphology.Compared to the cellular spheroids produced using other methodologies, such as micro-wells42 and hanging drop,43 these spheroids are more promising in their reproducible generation and show higher uniformity and potential of cell therapy due to the inherent advantages of droplet microfluidics and the presence of capsules. Moreover, the hepatic spheroids showed favorable cell viability demonstrated by liver/dead assay over time (Fig. S6C†). The proliferation ability of hepatic spheroids was quantitatively evaluated at days 1, 4, 7, and 10 after cell encapsulation, suggesting that the liver tissues grew well in CHCs (Fig. S6D†). As seen from the SEM images, the hepatocytes within the spheroids exhibited tight junctions (Fig. S6E†). These data further confirmed the proper survival and proliferation properties of liver cells and high biocompatibility of this system. In addition, the liver-specific function was identified by immunostaining for albumin in liver tissues on day 10. As shown in Fig. S6F,† the liver spheroids within CHCs displayed a positive expression of albumin. To assess the liver-specific functions of liver tissues in CHCs over an extended period quantitatively, we measured the urea synthesis. The level of urea production by the liver tissues gradually increased from day 1 to day 9 (Fig. S6G†). These results indicate that the CHC system is beneficial to HepaRG cell self-assembly and proliferation after encapsulation, as well as facilitate proper viability, prolonged 3D culture and liver-specific function of the tissues.
3.5. Generation and 3D culture of human liver organoids in CHCs
The hiPSCs possess a powerful capability of self-renewal and self-organization, which allows the generation of organoids in the 3D culture system. At present, liver organoids derived from hiPSCs are a new type of in vitro liver model for disease modeling and drug testing.30,44 In the following experiment, we investigated the feasibility of CHCs as 3D scaffolds for engineering liver organoids from hiPSCs. The hiPSCs are initially differentiated into definitive endoderm and hepatic progenitors with addition of sequential chemical factors under 2D culture conditions. Then, the differentiated hepatic cells are resuspended in the core flow with fibrinogen and encapsulated into capsules. As shown in Fig. 4A, the encapsulated cells were dispersedly distributed within the core fibrin scaffold of capsules on day 0. By day 2, the hepatic cells are found to self-organize into single complete spheroids with a clear edge in CHCs. Furthermore, the spheroids grew well and rapidly expanded into large organoids from day 2 to day 6, demonstrating the efficient development and generation of liver organoids in CHCs.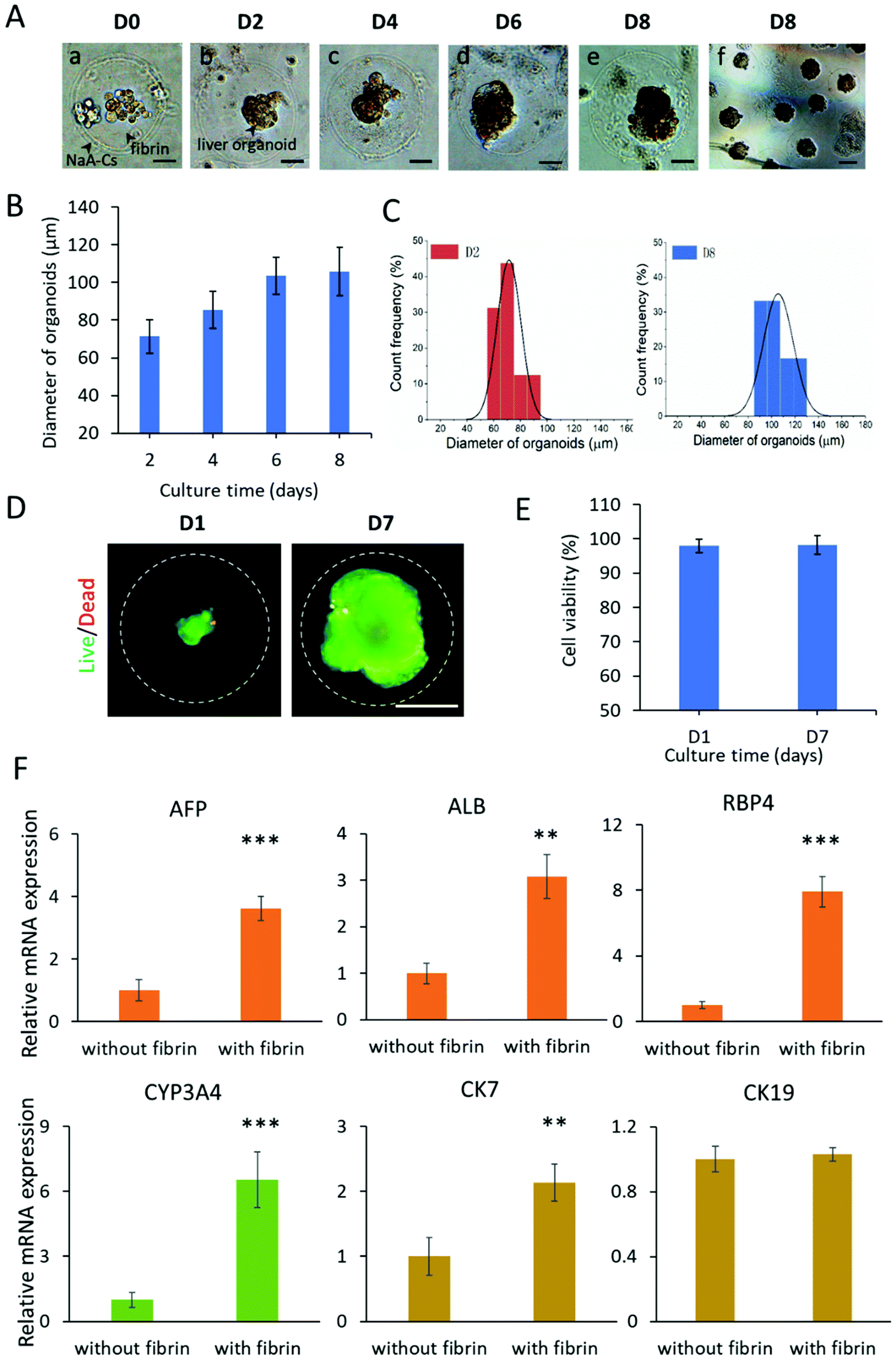 | ||
| Fig. 4 The encapsulation and formation of hiPSC-derived liver organoids in CHCs. (A) Bright-field images of liver organoids after encapsulation in capsules for 0, 2, 4, 6 and 8 days. Scale bars for a–e: 50 μm; f: 100 μm. (B) Diameter profiles of liver organoids in capsules on days 2, 4, 6 and 8. Quantitative analysis of the diameter of liver organoids was performed on at least 15 organoids. Data are shown as mean ± SD. (C) Size distribution was assessed by the diameter of cellular spheroids in capsules on days 2 and 8. (D and E) Cell viability of liver organoids was evaluated at days 1 and 7 of culture in capsules by live/dead staining and fluorescence quantitative analysis. The green and red fluorescence represents live and dead cells, respectively. Scale bar: 50 μm. (F) The expressions of hepatocyte-associated genes (AFP, ALB, RBP4 and CYP3A4) and cholangiocyte-associated genes (CK7, CK19) were examined in liver organoids with or without fibrin scaffolds in CHCs using real-time PCR. The expression values were normalized to GAPDH. Three independent experiments were performed. Data are shown as mean ± SD. **p < 0.01, ***p < 0.001. | ||
To quantitatively evaluate the growth of liver organoids in capsules, the size distribution of organoids from 2 days to 8 days of culture was measured by the average diameters. As shown in Fig. 4B, the liver organoids exhibited increased size as development proceeded. The liver organoids can grow up to about 106 μm in size after culturing for 8 days in CHCs. Finally, massive liver organoids in CHCs with consistent size and morphology were obtained (Fig. 4A). Also, these organoids exhibit a relatively narrow particle size distribution at days 2 and 8 (Fig. 4C). In addition, the cell viability of organoids was examined on days 1 and 7, showing the favorable viability (over 95%) of liver organoids in capsules (Fig. 4D and E). These data indicate that the CHC system is highly biocompatible for cell encapsulation, growth, organization and organoid formation due to the mild all-aqueous liquid environment. Moreover, the uniform capsules are beneficial to facilitate the production of organoids with consistent morphology and size, which may reduce the variability of organoids. This flexible system may also be utilized as scaffolds for engineering other types of 3D microtissues in vitro, thus providing a new approach to build biomimetic 3D tissue models.
3.6. Identification of the differentiation and functions of liver organoids in CHCs
Liver organoids derived from hiPSCs are multicellular tissues including hepatocytes and cholangiocytes. To identify the differentiation features of liver organoids in CHCs, the gene expression profiles related to hepatic lineages were analyzed by real-time PCR (Fig. S7†). The gene expression in hiPSC acted as the control. As expected, the mature hepatocyte (ALB, RBP4), cytochrome P450 gene (CYP3A4), and cholangiocyte (CK7, CK19) markers were examined at increased levels in liver organoids in CHCs. Notably, the organoids in CHCs showed significantly higher expression of the ALB, RBP4, CYP3A4 and CK7 genes compared to the 2D liver cells at the same stage, demonstrating the enhanced differentiation of liver-specific genes in 3D liver organoids in capsules.In vivo, the physical and chemical microenvironment factors are crucial for the liver organogenesis, such as 3D matrix. Fibrin is a biocompatible hydrogel, which could serve as an ECM scaffold to offer an appropriate environment for 3D cell culture, differentiation and formation of organoids.45,46 To examine the effect of fibrin scaffolds on the differentiation of liver organoids, we performed real-time PCR analysis of hepatic lineage genes. As shown in Fig. 4F, both the hepatocyte (AFP, ALB, RBP4, CYP3A4) and cholangiocyte (CK7) related genes showed markedly higher expression with fibrin compared to that without fibrin during the process of organoid differentiation in capsules (Fig. 4F). The data indicated that defined 3D fibrin scaffold could facilitate the differentiation and maturation of liver organoids, further confirming the importance of a controlled favorable ECM microenvironment on engineering stem cell organoids. Overall, these results demonstrate the feasibility of engineering liver organoids from hiPSCs using the CHC system.
Next, we further examined the protein expression and distribution of hepatic lineage identities in liver organoids by immunohistochemical analysis. As shown in Fig. 5A, the organoids abundantly expressed the hepatic-specific markers CYP3A4 and ALB. Moreover, they showed both positive expression of cholangiocyte markers CK7 and CK19 within the luminal structures, resembling bile ducts (Fig. 5B). The data revealed the cellular heterogeneity of liver organoids composed of hepatocytes and cholangiocytes. To further examine the functional properties of liver organoids in CHCs, we assessed the liver-specific functions of organoids during the culture period in capsules, including urea synthesis and albumin secretion (Fig. 5C and D). The liver organoids maintained a stable level of albumin and urea production in CHCs. Overall, this CHC system may offer an appropriate cellular microenvironment by integrating the 3D matrix and cell–cell interactions, which could facilitate the generation of functional liver organoids in a controlled manner. In future, this organoid-CHC system with well-defined properties and high biocompatibility may provide a potential platform for preliminary drug testing and serve as effective carriers for immune isolation to translate into clinical studies.
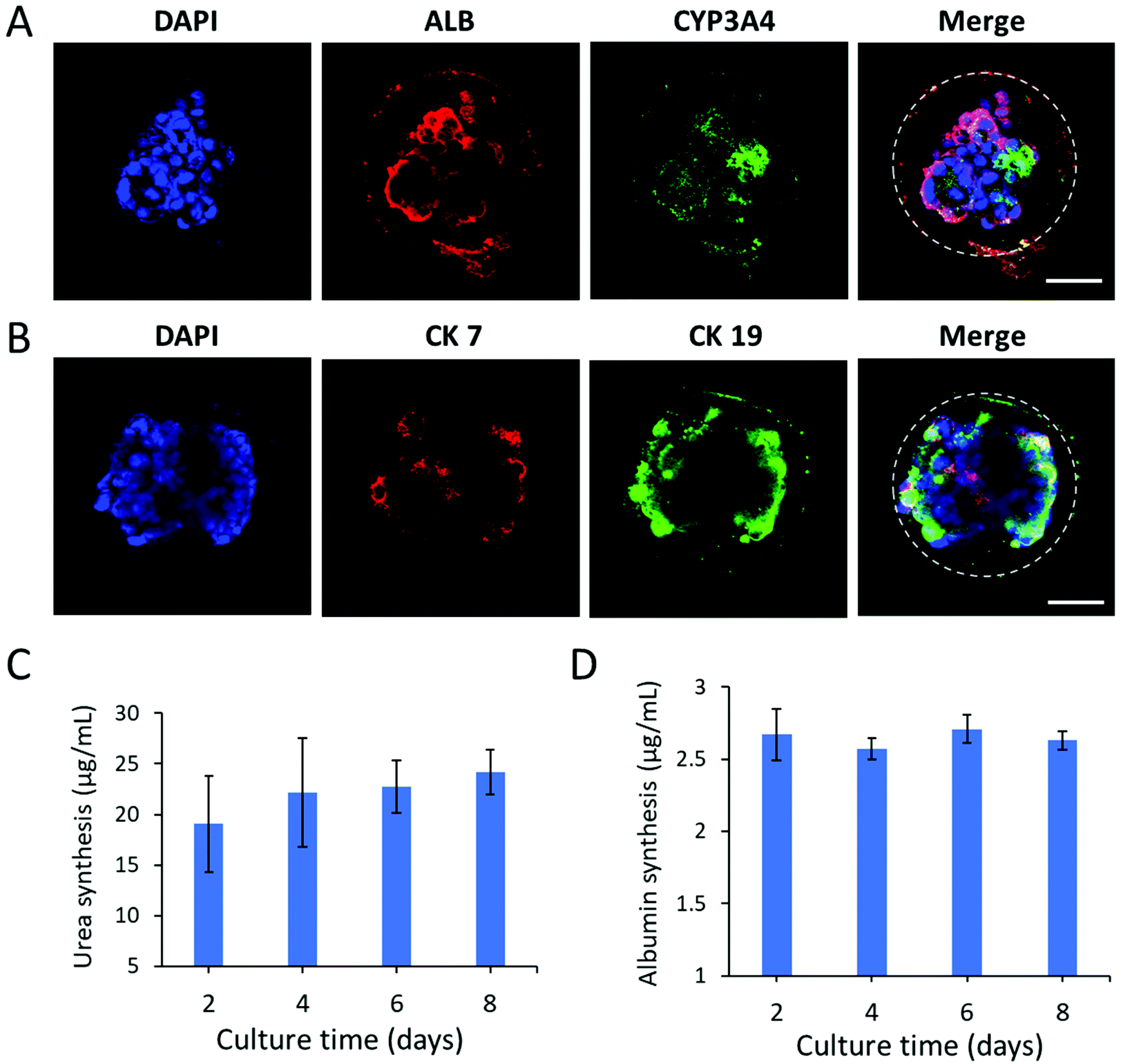 | ||
| Fig. 5 Identification of differentiation and hepatic functions of liver organoids in CHCs. Immunohistochemical staining of (A) hepatocyte markers (ALB, CYP3A4) and (B) cholangiocyte markers (CK7, CK19) in liver organoids after 7 days of encapsulation in capsules. DAPI stained the nuclei (blue). Scale bars: 50 μm. Albumin secretion (C) and urea synthesis (D) in liver organoids after encapsulation of 2, 4, 6 and 8 days. | ||
4. Conclusions
In summary, we proposed a new approach for one-step fabrication of defined composite hydrogel capsules for engineering stem cell organoids in an oil-free droplet microfluidic system. This system enables 3D culture, differentiation and generation of functional liver organoids from hiPSCs in a continuous process. The synthetic CHCs are characterized by high biocompatibility, stability, uniformity and high-throughput in a mild all-in-water microenvironment, which is beneficial for 3D culture, assembly and formation of organoids within the defined hydrogel scaffolds in a controlled manner. The produced liver organoids in CHCs showed favorable growth and uniform size, as well as liver-specific functions. The established CHC system provides a controllable and stable 3D scaffold for organoid generation in a high-throughput and reproducible way, partially addressing the potential limitations in conventional methods. By combining stem cell biology, defined hydrogels and droplet microfluidics, this work provides a proof-of-concept for organoid engineering using an integrative strategy. It also offers a robust platform to advance organoid applications in drug testing, disease modeling and regenerative medicine.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA16020900, XDB32030200, and XDB29050301), National Science and Technology Major Project (No. 2018ZX09201017-001-001), National Nature Science Foundation of China (No. 81573394 and 31971373), and Innovation Program of Science and Research from the DICP, CAS (DICP I201934).References
- T. Sato, R. G. Vries, H. J. Snippert, M. van de Wetering, N. Barker, D. E. Stange, J. H. van Es, A. Abo, P. Kujala, P. J. Peters and H. Clevers, Nature, 2009, 459, 262–265 CrossRef CAS.
- M. A. Lancaster and J. A. Knoblich, Science, 2014, 345, 1247125 CrossRef.
- G. Rossi, A. Manfrin and M. P. Lutolf, Nat. Rev. Genet., 2018, 19, 671–687 CrossRef CAS.
- H. T. Liu, Y. Q. Wang, K. L. Cui, Y. Q. Guo, X. Zhang and J. H. Qin, Adv. Mater., 2019, 1902042 CrossRef CAS.
- H. Clevers, Cell, 2016, 165, 1586–1597 CrossRef CAS.
- A. Fatehullah, S. H. Tan and N. Barker, Nat. Cell Biol., 2016, 18, 246–254 CrossRef.
- T. Takahashi, Annual Reviews, in Annual Review of Pharmacology and Toxicology, ed. P. A. Insel, Palo Alto, 2019, vol. 59, pp. 447–462 Search PubMed.
- A. L. Bredenoord, H. Clevers and J. A. Knoblich, Science, 2017, 355, eaaf9414 CrossRef.
- N. Broguiere, L. Isenmann, C. Hirt, T. Ringel, S. Placzek, E. Cavalli, F. Ringnalda, L. Villiger, R. Zullig, R. Lehmann, G. Rogler, M. H. Heim, J. Schuler, M. Zenobi-Wong and G. Schwank, Adv. Mater., 2018, 30, 12 CrossRef.
- M. M. Capeling, M. Czerwinski, S. Huang, Y. H. Tsai, A. Wu, M. S. Nagy, B. Juliar, N. Sundaram, Y. Song, W. M. Han, S. Takayama, E. Alsberg, A. J. Garcia, M. Helmrath, A. J. Putnam and J. R. Spence, Stem Cell Rep., 2019, 12, 381–394 CrossRef CAS.
- N. Glorevski, N. Sachs, A. Manfrin, S. Giger, M. E. Bragina, P. Ordonez-Moran, H. Clevers and M. P. Lutolf, Nature, 2016, 539, 560–564 CrossRef.
- S. S. Ng, K. Saeb-Parsy, S. J. I. Blackford, J. M. Segal, M. P. Serra, M. Horcas-Lopez, D. Y. No, S. Mastoridis, W. Jassem, C. W. Frank, N. J. Cho, H. Nakauchi, J. S. Glenn and S. T. Rashid, Biomaterials, 2018, 182, 299–311 CrossRef CAS.
- Z. Ma, J. Wang, P. Loskill, N. Huebsch, S. Koo, F. L. Svedlund, N. C. Marks, E. W. Hua, C. P. Grigoropoulos, B. R. Conklin and K. E. Healy, Nat. Commun., 2015, 6, 7413 CrossRef CAS.
- M. Xie, Q. Gao, H. Zhao, J. Nie, Z. Fu, H. Wang, L. Chen, L. Shao, J. Fu, Z. Chen and Y. He, Small, 2019, 15, e1804216 CrossRef.
- D. W. Scharp and P. Marchetti, Adv. Drug Delivery Rev., 2014, 67–68, 35–73 CrossRef CAS.
- G. Orive, E. Santos, D. Poncelet, R. M. Hernandez, J. L. Pedraz, L. U. Wahlberg, P. De Vos and D. Emerich, Trends Pharmacol. Sci., 2015, 36, 537–546 CrossRef CAS.
- C. Liu, W. Zheng, R. Xie, Y. Liu, Z. Liang, G. Luo, M. Ding and Q. Liang, Chin. Chem. Lett., 2019, 30, 457–460 CrossRef CAS.
- M. Mastiani, N. Firoozi, N. Petrozzi, S. Seo and M. Kim, Sci. Rep., 2019, 9, 15561 CrossRef.
- Y. Chao and H. C. Shum, Chem. Soc. Rev., 2020, 49, 114–142 RSC.
- H. T. Liu, H. Wang, W. B. Wei, H. Liu, L. Jiang and J. H. Qin, Small, 2018, 14, 1801095 CrossRef.
- K. Zhu, Y. Yu, Y. Cheng, C. Tian, G. Zhao and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 4826–4832 CrossRef CAS.
- M. Navi, N. Abbasi, M. Jeyhani, V. Gnyawali and S. S. H. Tsai, Lab Chip, 2018, 18, 3361–3370 RSC.
- C. G. Liu, W. C. Zheng, R. X. Xie, Y. P. Liu, Z. Liang, G. A. Luo, M. Y. Ding and Q. L. Liang, Chin. Chem. Lett., 2019, 30, 457–460 CrossRef CAS.
- C. Shao, Y. Liu, J. Chi, J. Wang, Z. Zhao and Y. Zhao, Research, 2019, 2019, 9783793 CrossRef.
- H. Wang, Z. Zhao, Y. Liu, C. Shao, F. Bian and Y. Zhao, Sci. Adv., 2018, 4, eaat2816 CrossRef.
- M. Huang, E. Khor and L. Y. Lim, Pharm. Res., 2004, 21, 344–353 CrossRef.
- N. Sun, N. J. Panetta, D. M. Gupta, K. D. Wilson, A. Lee, F. Jia, S. Hu, A. M. Cherry, R. C. Robbins, M. T. Longaker and J. C. Wu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15720–15725 CrossRef.
- L. Wang, C. Xu, Y. J. Zhu, Y. Yu, N. Sun, X. Q. Zhang, K. Feng and J. H. Qin, Lab Chip, 2015, 15, 4283–4290 RSC.
- Q. J. Wang, H. Yang, A. B. Bai, W. Jiang, X. Y. Li, X. H. Wang, Y. S. Mao, C. Lu, R. Z. Qian, F. Guo, T. L. Ding, H. Y. Chen, S. F. Chen, J. Y. Zhang, C. Liu and N. Sun, Biomaterials, 2016, 105, 52–65 CrossRef CAS.
- Y. Q. Wang, H. Wang, P. W. Deng, W. W. Chen, Y. Q. Guo, T. T. Tao and J. H. Qin, Lab Chip, 2018, 18, 3606–3616 RSC.
- I. Ziemecka, V. van Steijn, G. J. Koper, M. Rosso, A. M. Brizard, J. H. van Esch and M. T. Kreutzer, Lab Chip, 2011, 11, 620–624 RSC.
- Z. Z. Chong, S. H. Tan, A. M. Ganan-Calvo, S. B. Tor, N. H. Loh and N. T. Nguyen, Lab Chip, 2016, 16, 35–58 RSC.
- B. U. Moon, N. Abbasi, S. G. Jones, D. K. Hwang and S. S. H. Tsai, Anal. Chem., 2016, 88, 3982–3989 CrossRef CAS.
- D. M. Headen, G. Aubry, H. Lu and A. J. Garcia, Adv. Mater., 2014, 26, 3003–3008 CrossRef CAS.
- P. Guillot, A. Ajdari, J. Goyon, M. Joanicot and A. Colin, C. R. Chim., 2009, 12, 247–257 CrossRef CAS.
- A. Sauret and H. C. Shum, Int. J. Nonlinear Sci. Numer. Simul., 2012, 13, 351–362 Search PubMed.
- M. Y. Ji, X. Y. Sun, X. B. Guo, W. J. Zhu, J. L. Wu, L. Chen, J. H. Wang, M. M. Chen, C. Cheng and Q. Q. Zhang, Food Hydrocolloids, 2019, 90, 515–522 CrossRef CAS.
- M. G. Sankalia, R. C. Mashru, J. M. Sankalia and V. B. Sutariya, Eur. J. Pharm. Biopharm., 2007, 65, 215–232 CrossRef.
- H. X. Liu, F. Liu, Y. Ma, H. D. Goff and F. Zhong, Carbohydr. Polym., 2020, 236, 11 Search PubMed.
- B. Sarmento, D. Ferreira, F. Veiga and A. Ribeiro, Carbohydr. Polym., 2006, 66, 1–7 CrossRef.
- B. Smitha, S. Sridhar and A. A. Khan, Eur. Polym. J., 2005, 41, 1859–1866 CrossRef.
- S. F. Wong, D. Y. No, Y. Y. Choi, D. S. Kim, B. G. Chung and S. H. Lee, Biomaterials, 2011, 32, 8087–8096 CrossRef.
- T. J. Bartosh, J. H. Ylostalo, A. Mohammadipoor, N. Bazhanov, K. Coble, K. Claypool, R. H. Lee, H. Choi and D. J. Prockop, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13724–13729 CrossRef CAS.
- S. Akbari, G. G. Sevinc, N. Ersoy, O. Basak, K. Kaplan, K. Sevinc, E. Ozel, B. Sengun, E. Enustun, B. Ozcimen, A. Bagriyanik, N. Arslan, T. T. Onder and E. Erdal, Stem Cell Rep., 2019, 13, 627–641 CrossRef CAS.
- H. Bruns, U. Kneser, S. Holzhuter, B. Roth, J. Kluth, P. M. Kaufmann, D. Kluth and H. C. Fiegel, Tissue Eng., 2005, 11, 1718–1726 CrossRef CAS.
- M. Jaramillo, S. S. Singh, S. Velankar, P. N. Kumta and I. Banerjee, J. Tissue Eng. Regener. Med., 2015, 9, 1–12 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01085e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |