
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface grafting of Fc-binding peptides as a simple platform to immobilize and identify antibodies that selectively capture circulating endothelial progenitor cells†
Omar S.
Bashth
 a,
Mohamed A.
Elkhodiry
a,
Gaétan
Laroche
b and
Corinne A.
Hoesli
*ac
a,
Mohamed A.
Elkhodiry
a,
Gaétan
Laroche
b and
Corinne A.
Hoesli
*ac
aDepartment of Chemical Engineering, McGill University, Canada. E-mail: corinne.hoesli@mcgill.ca
bCentre de Recherche du CHU de Québec & Département de Génie des Mines, des Matériaux et de la Métallurgie, Université Laval, Canada
cDepartment of Biomedical Engineering, McGill University, Canada
First published on 9th September 2020
Abstract
Antibody surface immobilization is a promising strategy to capture cells of interest from circulating fluids in vitro and in vivo. An application of particular interest in vascular interventions is to capture endothelial progenitor cells (EPCs) on the surface of stents to accelerate endothelialization. The clinical impact of EPC capture stents has been limited by the lack of efficient selective cell capture. Here, we describe a simple method to immobilize a variety of immunoglobulin G antibodies through their fragment crystallizable (Fc) regions via surface-conjugated RRGW peptides for cell capture applications. As an EPC capture model, peripheral blood endothelial colony-forming cells suspended in cell culture medium with up to 70% serum were captured by immobilized anti-CD144, anti-CD34 or anti-CD309 antibodies under laminar flow. The endothelial colony-forming cells were successfully enriched from a mixture with peripheral blood mononuclear cells using surfaces with anti-CD309 but not anti-CD45. This antibody immobilization approach holds great promise to engineer vascular biomaterials with improved EPC capture potential. The ease of immobilizing different antibodies using the same Fc-binding peptide surface grafting chemistry renders this platform suitable to screen antibodies that maximize cell capture efficiency and selectivity.
1 Introduction
Surface-immobilized antibodies are broadly applied to selectively capture target cells through cell surface antigen binding1,2 for applications ranging from diagnostics3 to immunotherapy.4–6 In the regenerative medicine field, antibody-functionalized surfaces have been used to enrich stem and progenitor cells for in situ tissue repair and regeneration.7,8 Endothelial progenitor cell (EPC) capture stents, for example, aim to selectively capture cells that accelerate endothelialization to reduce the incidence of restenosis.9 One cell type that can be used as an EPC capture model is endothelial colony-forming cells (ECFCs), which have high proliferative potential in vitro and can directly contribute to neovascularization in vivo.10,11 The Genous™ (OrbusNeich Medical Technologies Inc., FL, USA) stent, a stainless-steel stent coated with anti-CD34 antibodies, represents the first commercial implementation of this approach. Although previous human clinical trials have demonstrated a better early endothelialization on these stents compared with drug-eluting stents,12,13 other long-term studies have confirmed the formation of late neointimal hyperplasia and restenosis.14–16 This late in-stent restenosis could be due to poor ECFC selectivity by CD34 antibodies, as there are other circulating progenitor cells (CD34+) that could be captured on the surface.17 Certain CD34+ cells might differentiate into immune cells that mediate inflammatory responses and disturb the signaling and activation pathways of smooth muscle cells (SMCs), leading to intimal hyperplasia.18,19 Antibodies with higher specificity for ECFCs such as anti-CD144 antibodies were found to reduce the neointimal area observed in stainless steel stents compared to anti-CD34 antibodies.20Most of the antibody surface immobilization techniques available for in vitro studies or for in vivo cell capture applications have relied on adsorption, direct covalent conjugation or interactions with bio-affinity bacterial proteins. The simplest method to immobilize antibodies on surfaces is adsorption, but this method can lead to a reduction in antigen binding due to desorption and conformations with reduced availability of antigen-binding sites.21 Antibody conjugation via primary amine or carboxylic acid functional groups is less prone to desorption and it is a method currently used to modify commercially-available EPC capture stents.22 However, this technique suffers from a lack of control over antibody orientation on the surface due to the prevalence of these functional groups throughout the antibody structure.23 Directional antibody immobilization can be achieved via binding of the fragment crystallizable (Fc) region to surfaces grafted with bacterial products such as protein A or G.24,25 Disadvantages of this strategy include the immunogenicity of these bacterial proteins and their high affinity for albumin,24,26 which may increase fouling in the presence of biological fluids.
The disadvantages and limitations of such immobilization methods have led to the evolution and development of easily synthesized peptides that are able to both immobilize antibodies and control their orientation. For instance, Tsai et al.27 designed a short peptide sequence, RRGW, with a strong affinity toward the Fc region of mouse immunoglobulin G (IgG). With this and other short synthesized peptides, it becomes convenient to screen a large number of antibodies in a single experiment through an easy switch between different antibodies. The additional advantage of using these peptides to immobilize antibodies rather than proteins, such as protein A or G, is their small size that provides better control in grafting the molecule on the surface and preventing steric hindrance. These synthesized peptides have been designed and used for antibody purification purposes,28,29 and to fabricate biosensors chips.30 To our knowledge, no studies have tested the efficiency of peptide-based antibody immobilization to capture circulating cells. Here, we describe a method allowing controlled surface conjugation of RRGW peptides to immobilize and screen multiple antibodies aimed to capture and enrich ECFCs selectively from a heterogeneous mixture of cells.
2 Materials and methods
2.1 Antibody immobilization via RRGW peptides
Aminated polystyrene Petri dishes (#354732, Corning®, PureCoat™, USA) were cut into 2 cm × 3 cm slides using a hot wire (#K02B, Lompoc, USA) or – for flow chamber studies – into 2.95 cm × 2.51 cm slides using a Micro Mill (Datron Neo 3-axis CNC Mill, Cell imaging and analysis network, McGill University, Canada). The edges were surrounded by Teflon tape (PTFE, thread seal tape) to maintain the added solutions (90 μL cm−2) in each step. After each of the reaction steps, excess solutions were removed, and the slides were washed twice with 0.2 μm-filtered phosphate-buffered saline solution, pH 7.4 (PBS, #21600010, Thermo Fisher Scientific™). The slides were, first, incubated in 3 mg mL−1 of sulfo-succinimidyl-4-(p-maleimidophenyl)-butyrate (Sulfo-SMPB, #BC24, G-Biosciences) in PBS for 2 h in the dark with 90 rpm agitation (Ecotron, Infors HT). Next, 2 μL (around 0.64 μL mm−2) droplets of 30 μM to 5000 μM RRGW peptide (RRGW(PEG3)C; Biomatik, Cambridge, Canada) solutions in PBS were deposited onto the surface and incubated in static humid chambers in the dark for 2 h. Controls with adsorbed peptides were obtained in the same fashion, but omitting the sulfo-SMPB step. After blocking for 40 min in the dark at 90 rpm agitation using Dako serum-free protein block (#X0909, Agilent Dako, CA, USA), the slides were entirely covered with 10 μg mL−1 of primary antibodies resuspended in PBS and incubated for 1 h in the dark at 90 rpm agitation. The primary mouse anti-human antibodies used were CD105 (#323202), CD309 (#393002), CD14 (#367102), CD144 (#348502), all from Biolegend (San Diego, USA), and CD34 (#550760, BD Bioscience, ON, Canada). The negative control (no binding to anti-mouse secondary antibodies) consisted of slides functionalized with RRGW (300 μM) followed by immobilizing primary rabbit anti-human CD45 (#154885, Abcam, Canada). To detect the immobilized primary antibodies, surfaces were submerged in 10 μg mL−1 of Alexa Fluor 488 F(ab′)2-goat anti-mouse IgG (#A11017, Thermo Fisher Scientific™) in PBS. After 1 h incubation in the dark at 90 rpm agitation, the slides were washed twice in PBS and twice in RO water and then air-dried. The spots and surrounding background were imaged using a 10× objective of a confocal microscope (Zeiss LSM 5 Exciter, Germany).2.2 Stability of the immobilized antibodies in different serum concentrations
Mouse anti-human CD105 antibodies were immobilized on surfaces with spots of adsorbed (without sulfo-SMPB) or covalently grafted (with sulfo-SMPB) RRGW peptides (300 μM RRGW during the peptide grafting step). The following different aqueous conditions were prepared: (1) PBS; (2) endothelial cell growth medium-2 (EGM-2, supplement and growth factor kit #CC-4176 was added to endothelial cell growth basal medium – EBM-2 – #00190860, Lonza, Basel, Switzerland) with 10% fetal bovine serum (FBS, #SH3039603, Thermo Fisher Scientific™); (3) 70% FBS in EGM-2. Slides were then incubated in these solutions in a cell culture incubator (37 °C, 5% CO2) in static conditions or under flow (1.5 dyn cm−2 wall shear stress) using a parallel-plate flow chamber system previously described by Hoesli et al.31 After 1 h, slides were washed twice with PBS before adding Alexa Fluor 488 F(ab′)2-goat anti-mouse secondary antibodies, followed by imaging.2.3 PBMC isolation and ECFC formation
ECFCs were isolated from the peripheral blood of healthy donors as previously described.32 Briefly, blood samples were received from 6 healthy donors under informed consent following a protocol (Study No. A06-M33-15A) approved by the Ethics Institutional Review Board at McGill University. Next, a 6-well plate was coated with 50 μg mL−1 type 1 rat tail collagen (#A1048301, Thermo Fisher Scientific™) for 1 h, then washed with sterile Dulbecco's phosphate-buffered saline (DPBS, #14190250, Thermo Fisher Scientific™). Fresh blood samples were collected in 60 mL syringes coated with heparin (#02303086, Sandoz Canada Inc., Canada). The blood was then diluted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with sterile DPBS and added to SepMate™ tubes (#85450, Stem Cell Technologies Inc., Vancouver, British Columbia, Canada) filled with 15 mL of Histopaque® 1077 (#10771, Sigma-Aldrich®). The tubes were centrifuged with the break turned off, and the PBMC-containing top fraction was poured off and collected into 50 mL tubes. The collected PBMCs were washed twice with EBM-2, resuspended in EGM-2 with 10% FBS and seeded into the 6-well plate at a cell density of 12.5 million cell per mL. Screening for late colony formation was carried on for 30 days by using a phase-contrast of an inverted microscope (VWR International). Colonies were then trypsinized with TrypLE (#12604-021, Life Technologies, USA), resuspended in complete EGM-2 and expanded in collagen-coated cell culture flasks. Prior to any experiments, confluent cells were detached from the surface with TrypLE and enumerated using the inverted microscope after staining with Trypan blue (#15250061, Thermo Fisher Scientific™). ECFCs used in this study were between passages 4 and 7.
:1 ratio with sterile DPBS and added to SepMate™ tubes (#85450, Stem Cell Technologies Inc., Vancouver, British Columbia, Canada) filled with 15 mL of Histopaque® 1077 (#10771, Sigma-Aldrich®). The tubes were centrifuged with the break turned off, and the PBMC-containing top fraction was poured off and collected into 50 mL tubes. The collected PBMCs were washed twice with EBM-2, resuspended in EGM-2 with 10% FBS and seeded into the 6-well plate at a cell density of 12.5 million cell per mL. Screening for late colony formation was carried on for 30 days by using a phase-contrast of an inverted microscope (VWR International). Colonies were then trypsinized with TrypLE (#12604-021, Life Technologies, USA), resuspended in complete EGM-2 and expanded in collagen-coated cell culture flasks. Prior to any experiments, confluent cells were detached from the surface with TrypLE and enumerated using the inverted microscope after staining with Trypan blue (#15250061, Thermo Fisher Scientific™). ECFCs used in this study were between passages 4 and 7.
2.4 ECFC capture under flow
ECFC capture under flow was performed using a custom parallel-plate flow system with four independent chambers described previously by Hoesli et al.31 The flow system tubing was autoclaved while the flow chamber itself, as well as the antibody-modified slides, were incubated for 15 min in 100% ethanol and left to dry in a biosafety cabinet immediately before assembly. The following different cell culture media were prepared: (1) serum-free EGM-2 (EGM-2 without serum added from the kit); (2) 10% FBS (final concentration) in EGM-2; (3) 70% FBS in EGM-2. Before starting the flow, 15 mL of each solution was added to different 50 mL reservoirs, and the flow chamber system (parallel plate system, medium reservoirs and flow dampeners) was then placed inside a cell culture incubator (37 °C, 5% CO2) to equilibrate temperature and pH. ECFCs were harvested, resuspended in the corresponding media and sieved through a 40 μm mesh size cell strainer (#07201430, Thermo Fisher Scientific™) to remove cell aggregates before being injected into the medium reservoirs using a syringe and needle. The final average cell concentration in the reservoir was 1.2 ± 4 × 104 cells per mL. ECFCs were circulated in the flow chamber system at a wall shear stress of 1.5 dyn cm−2 using peristaltic pumps (Masterflex® model 7544-80 with Masterflex® L/S 2-channel easy-load II pump head using L/S 13 BPT tubing). After 1 h, the flow was stopped, surfaces were recovered from flow chambers, and the captured cells were fixed with 4% paraformaldehyde (4% PFA, #CAAAJ61899-AP, VWR International) for 10 min. The slides were washed and then stored in PBS for immunohistochemistry.2.5 The role of antigen-binding sites in cell capture
To block their fragment antigen-binding (Fab) regions, anti-human CD309 antibodies immobilized via covalently grafted RRGW were incubated with 20 μM of soluble CD309 proteins (#10012-H08H, Cedarlane, Canada) for 1 h in the dark at 90 rpm agitation. Slides were then washed twice with PBS and RO water. ECFCs were circulated over anti-human CD309 functionalized surfaces with or without CD309 blocking at 1.5 dyn cm−2 wall shear stress for 1 h, as described above. Surfaces recovered from flow chambers, and the captured cells were fixed with 4% PFA, washed and stored in PBS until performing immunohistochemistry.2.6 Selective capture of ECFCs from a cell mixture
ECFCs were stained with calcein acetoxymethyl (AM) according to the manufacturer's instruction (#C1430, Thermo Fisher Scientific™). Briefly, a volume of 7 mL of 1 μg mL−1 calcein-AM was added in each T-flask to mark ECFCs with a green fluorescent dye, and the flasks were incubated for 30 min inside cell culture incubator (37 °C, 5% CO2). Human PBMCs were isolated from fresh blood as described earlier and mixed at 1:1 ratio with the pre-labeled ECFCs harvested using TrypLE, as described above. The cell mixture was then resuspended in serum-free media and circulated for 1 h at a wall shear stress of 1.5 dyn cm−2 over surfaces modified with either mouse anti-human CD309 or mouse anti-human CD45. After 1 h, the captured cells were fixed for 10 min in 4% PFA and washed with PBS. Cell nuclei were then stained, and samples were imaged immediately.
2.7 Immunohistochemistry
Fixed samples were permeabilized with 0.1% Triton-X (#T8787, Sigma-Aldrich®) in PBS for 15 min, and slides were then washed with PBS. Cell nuclei were stained with 1 μg mL−1 of 4′,6-diamidino-2-phenylindole (DAPI, #D9542, Sigma-Aldrich®) resuspended in RO water for 10 min, followed by washing in RO water and then PBS. Using VECTASHIELD® HardSet™ antifade mounting medium (#H-1400, Vector laboratories Inc., USA), slides were mounted on coverslips and imaged using fluorescence microscopy (Olympus IX81, Tokyo, Japan) equipped with a programmable Prior® stage (Prior Scientific Inc., Rockland, MA). A total of 40 to 45 images – taken at pre-programmed positions – per each condition in every replicate were analyzed using the ImageJ program. Cells in each DAPI image were counted using “threshold” and “analyze particle” tools.2.8 Statistics
Data represent the mean values ± standard deviation (error bars) of at least 3 independent experimental replicates. In antibody detection experiments, each experimental replicate consisted of 3 spots with at least 6 images that were taken to cover each side of the spot. The mean intensity of each spot was quantified using CellProfiler™ software with the “measure object intensity” plug-in. For experiments with cells, each replicate was conducted with ECFCs from a different donor. In the proof-of-concept selective cell capture study, 2 experiments were conducted; each with a different PBMC donor but 4 independent ECFC donors to investigate surface capture selectivity of ECFCs without considering PBMCs between-donor variability. Unless otherwise mentioned, comparisons between multiple groups were performed using analysis of variance (ANOVA) followed by Tukey–Kramer HSD post-hoc tests. To compare various samples with the control group (Fig. 1D and 2), Dunnett's test was performed. Differences between groups were considered to be statistically significant at p < 0.05. Using Grubbs's test, an outlier point was determined and rejected from one replicate of the cell capture study in the 10% FBS condition (Fig. 4B). All statistical analyses were performed using JMP® (SAS Institute Inc., Cary, NC). | ||
| Fig. 1 Immobilization of mouse anti-human CD105 on conjugated or adsorbed RRGW spots on polystyrene substrates. (A) The chemical structure of the covalent conjugation of RRGW peptide into activated polystyrene surfaces with sulfo-SMPB. (B) Schematic representation of the experimental design to immobilize and detect the grafted antibodies on the surface. (C) Fluorescence imaging of immobilized antibodies on either adsorbed (without sulfo-SMPB) or conjugated (with sulfo-SMPB) spots of RRGW (300 μm during the conjugation step) through detection by AF488-F(ab′)2-goat anti-mouse. (D) Average of mean intensities of the detected antibodies on adsorbed or conjugated RRGW spots. The concentrations represent the amount of RRGW added during the conjugation or adsorption step, where 0 μm as a control represents the background regions without RRGW. Samples were compared to the control with **p < 0.01, *p < 0.05 and NS (not significant) >0.05, n = 3 experimental replicates (6 spots analyzed per experimental replicate). | ||
 | ||
| Fig. 2 Average of mean intensities of different mouse anti-human antibodies immobilized on conjugated spots of RRGW (300 μm during the conjugation step) on polystyrene substrates and detected by AF488-F(ab′)2-goat anti-mouse. The control is a rabbit anti-human CD45 immobilized on conjugated RRGW detected by the same anti-mouse secondary antibody. Samples were compared to the control with *p < 0.05 and **p < 0.01, n = 3 experimental replicates (6 spots analyzed per experimental replicate). | ||
3 Results
3.1 Directional and specific immobilization of antibodies on RRGW-modified surfaces
To maximize the availability of hypervariable antibody regions for cell surface antigen binding, antibodies were immobilized via interactions of the Fc region with covalently conjugated RRGW peptides, as shown in Fig. 1A. To determine the nitrogen and the amine content of the substrates used for conjugation, the commercially available PureCoat™ aminated surfaces were characterized using X-ray photoelectron spectroscopy (XPS) accompanied by chemical derivatization (see supplementary materials). As 4-(trifluoromethyl) benzaldehyde (TFBA) reacts with primary amines, the atomic nitrogen percentage decreased in samples with the chemical derivatization (20% ± 2%) compared with no chemical derivatization (22.0% ± 0.9%) as shown in Fig. S1.† The amine percentage in the PureCoat™ substrates was calculated to be 1.4% ± 0.2% with an average amine selectivity ([NH2]/N) of 0.06 ± 0.01. Sulfo-SMPB treatment and peptide grafting were both associated with an increase in the water contact angle, indicating a decrease in surface hydrophilicity after peptide grafting compared to the untreated PureCoat™ substrates (Fig. S2†).To confirm selective antibody immobilization only on regions functionalized with RRGW, peptide solutions were spotted onto surfaces that were washed before complete immersion in primary antibody followed by F(ab′)2 secondary antibody solutions for immunofluorescent detection as shown in Fig. 1B and C. The secondary antibody fragments of F(ab′)2 were used to eliminate signal from the interaction between the Fc region of the full secondary antibodies and RRGW peptides. As expected, the fluorescence signal detected via secondary antibodies was significantly higher in regions with covalently grafted RRGW spots in a range of concentrations from 40 μM to 500 μM compared to the background area where no peptides were grafted.
With covalent grafting of RRGW, the fluorescence intensity increased when concentrations of the peptide added in solution were raised from 30 μM to 300 μM. Above 300 μM concentration (which corresponds to 9 ± 2 × 109 total number of antibody molecules; Fig. 1D and S3†), a decrease in fluorescence signal was detected. This decrease could be due to peptide aggregation in solution hindering their surface grafting, or to fluorescence quenching of the secondary antibodies at higher densities.33 Contrary to surfaces with covalent peptide grafting, the fluorescence signal detected from adsorbed RRGW was not significantly different from the background. These results indicate that antibodies were specifically immobilized on RRGW spots, and that the antibody surface density was significantly higher with covalent RRGW grafting compared to adsorbed RRGW peptides. Based on these results, a concentration of 300 μM RRGW peptide was selected to be conjugated during the reaction scheme to maximize antibody surface density in subsequent experiments.
3.2 Successful immobilization of different antibodies on conjugated RRGW
A practical method of surface immobilization should be readily translatable to other antibodies, at least within the same immunoglobulin subclass. Different representative mouse IgG antibodies were successfully immobilized on PureCoat™ aminated surfaces treated with sulfo-SMPB and RRGW (spots of 300 μM solution), including anti-human CD34, CD31 (platelet endothelial cell adhesion molecule-1), CD144 (vascular endothelial cadherin), CD105 (endoglin), CD14 and CD309 (vascular endothelial growth factor receptor 2) antibodies. The detected signal from the different antibodies was significantly higher than the negative control (immobilized rabbit anti-human CD45, which should not be detected by anti-mouse secondary antibodies). This indicates that the detection method of the immobilized antibodies was specific and that the RRGW-conjugated method successfully immobilized several IgGs while maintaining similar grafting efficiency.3.3 Enhanced stability of immobilized antibodies on conjugated RRGW substrates
As this surface modification technique could eventually be applied in vivo, where the substrates would be in contact with biological fluids, the stability of the immobilized antibodies on conjugated RRGW was investigated. Under all aqueous conditions, the signals of the immobilized antibodies on the conjugated RRGW under flow were significantly higher than the background. As shown from the representative spot images in Fig. 3A, no significant differences in antibody retention on conjugated RRGW were observed between static and dynamic flow conditions. Conversely, a decrease in antibody surface density of high magnitude (∼30% loss of the signal after subtracting background signal; Fig. 3B) was observed for antibodies immobilized on surfaces with adsorbed RRGW peptides when exposed to flow. Although the difference between the signal detected before and after flow on surfaces with adsorbed RRGW did not reach statistical significance, the signal after flow exposure was not significantly different from the background (Fig. 3B). This suggests that adsorbed RRGW peptides and/or antibodies interacting with these peptides were washed away after 1 h of flow exposure.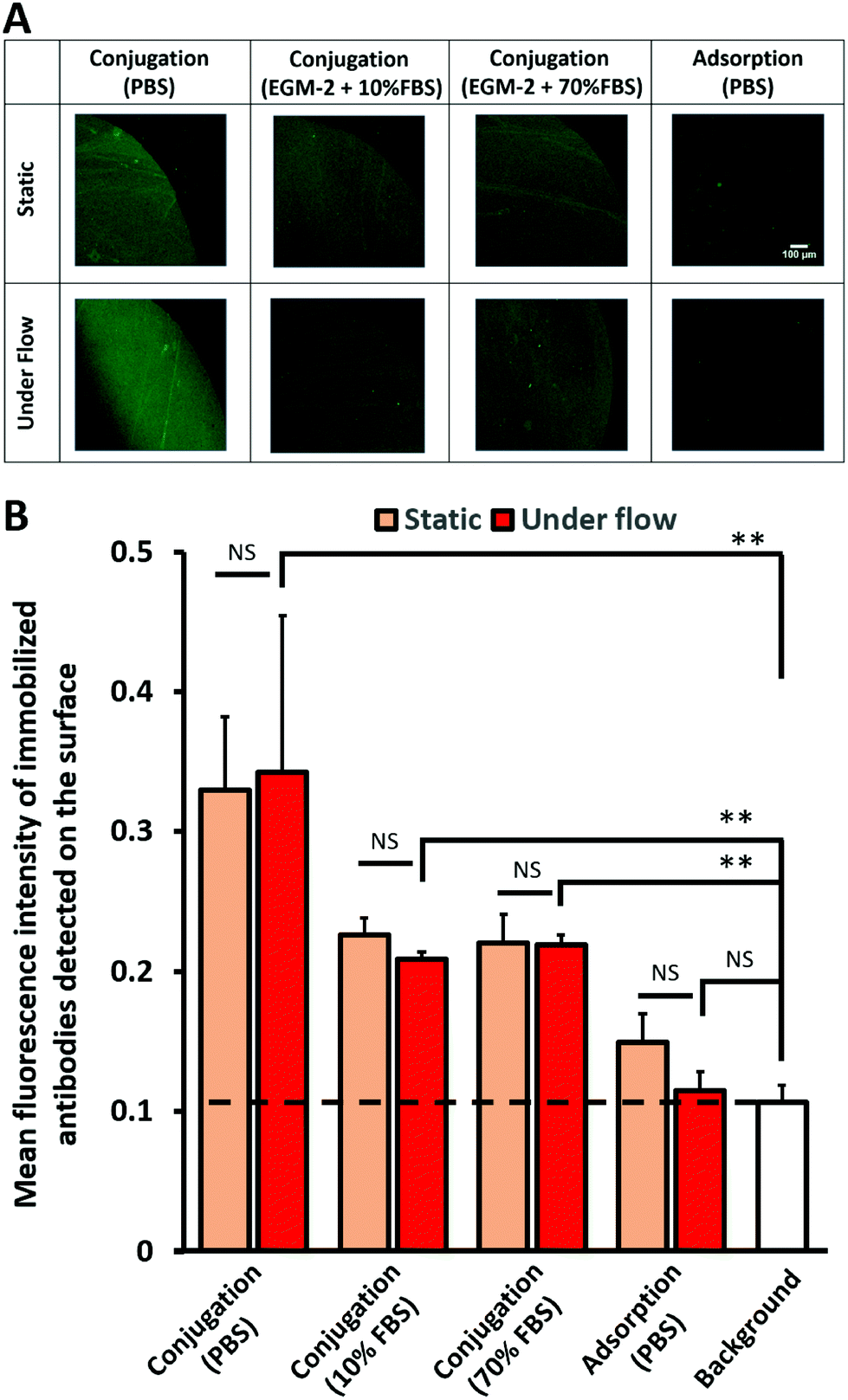 | ||
| Fig. 3 Stability study of immobilized mouse anti-human CD105 antibodies on conjugated or adsorbed spots of RRGW (300 μm during the conjugation step) incubated for 1 h in static conditions or under flow at 1.5 dyn cm−2 wall shear stress. Antibodies were detected by AF488-F(ab′)2-goat anti-mouse. (A) Fluorescence imaging of antibodies on either conjugated RRGW incubated in PBS, 10% and 70% FBS in EGM-2, or adsorbed RRGW incubated in PBS. (B) Average of mean intensities of the spot regions with the background (no RRGW) as a control. *p < 0.05, **p < 0.01 and NS (not significant) >0.05, n = 3 experimental replicates (6 spots analyzed per experimental replicate). | ||
Even though the dissociation constant of the RRGW and antibodies was reported in the literature to be low,27 there was a sharp decrease in the density of the immobilized antibodies between surfaces incubated in PBS (serum-free) and those incubated under serum conditions. Interestingly, the density and the amount of the immobilized antibodies after incubating the substrates at 10% and 70% serum were similar (1.4 ± 0.2 × 109 total number of antibody molecules; Fig. 3B and S3†). This indicates that the modified surfaces reached a stable state where the immobilized antibodies were not affected by the amount and the concentration of the present proteins in up to 70% serum. Overall, higher antibody retention was observed on surfaces with conjugated RRGW compared to adsorbed RRGW peptides, even in the presence of elevated serum concentrations.
3.4 Selective ECFC capture under flow
To assess the ability of the developed antibody immobilization method to capture circulating cells, ECFCs were resuspended in different serum concentrations and circulated for 1 h over surfaces modified with anti-CD14 (no expression expected in ECFCs), anti-CD34 (moderate expression in ECFCs), anti-CD144 or anti-CD309 (both with high expression in ECFCs).11,32 In serum-free EGM-2, the difference in the number of captured ECFCs per surface area was only significant between anti-CD309 and anti-CD14 (Fig. 4A and B). This difference was not statistically significant when anti-CD144 and anti-CD34 were compared with anti-CD14. In the presence of serum, all anti-endothelial cell antibodies (anti-CD34, anti-CD144, anti-CD309) captured significantly more cells compared with anti-CD14. | ||
| Fig. 4 Selective ECFC capture using parallel-plate flow chambers for 1 h at a wall shear stress of 1.5 dyn cm−2 over modified surfaces with antibodies immobilized on conjugated RRGW. (A) Representative images of nuclei stained with DAPI (blue). (B) Average of the number of captured ECFCs per mm2 surface area at different serum concentrations. At each serum condition, the antibodies recognizing endothelial cells were compared with anti-CD14 negative controls. *p < 0.05, **p < 0.01 and NS (not significant) >0.05, n = 6 experimental replicates. (C) Competitive binding study to demonstrate the role of available Fab regions in cell capture by blocking anti-CD309 antibodies with soluble CD309 proteins, n = 3 experimental replicates with *p < 0.05. | ||
Despite antibody losses observed under flow in the presence of serum (Fig. 3), the number of captured cells on the surface was significantly higher at 70% FBS compared to 10% FBS or serum-free conditions (Fig. 4B). The higher ECFC capture efficiency observed on surfaces with anti-endothelial cell antibodies compared to anti-monocyte (anti-CD14) suggests that the amount of the antibodies immobilized on the surface was sufficient to enhance cell recognition. Furthermore, the ECFC capture efficiency mediated by anti-CD309 surfaces was significantly reduced in the presence of soluble CD309 protein added to block the interactions between the Fab region and cell surface proteins (Fig. 4C).
3.5 Enhanced selectivity of capturing ECFCs from a heterogeneous mixture of cells by immobilized anti-CD309 antibodies
As a proof of the concept of selective ECFC capture from a heterogeneous mixture of cells, ECFCs were mixed 1:1 with PBMCs and then perfused over RRGW-conjugated surfaces presenting either anti-CD309 (expressed by ECFCs) or anti-CD45 (expressed by PBMCs but not ECFCs32,34) immobilized antibodies. In general, the total number of adhered PBMCs was significantly higher on the anti-CD45 surfaces, suggesting that these cells might have a higher adhesion propensity under flow compared to ECFCs (Fig. 5A). This could be due to the culture step required for ECFC expansion, which may lead to reduced expression of receptors required for adhesion under flow or due to higher inertial forces applied to the ECFCs resulting from their significantly larger diameter compared to the PBMCs. Importantly, significantly higher ECFC surface densities and concentrations were observed on the anti-CD309 compared to the anti-CD45 surfaces (Fig. 5B). Together with the dose-dependent effect of surface-immobilized antibodies via conjugated RRGW (Fig. 4C), these results provide substantial evidence of the roles of the surface-immobilized antibodies in promoting selective ECFC capture.
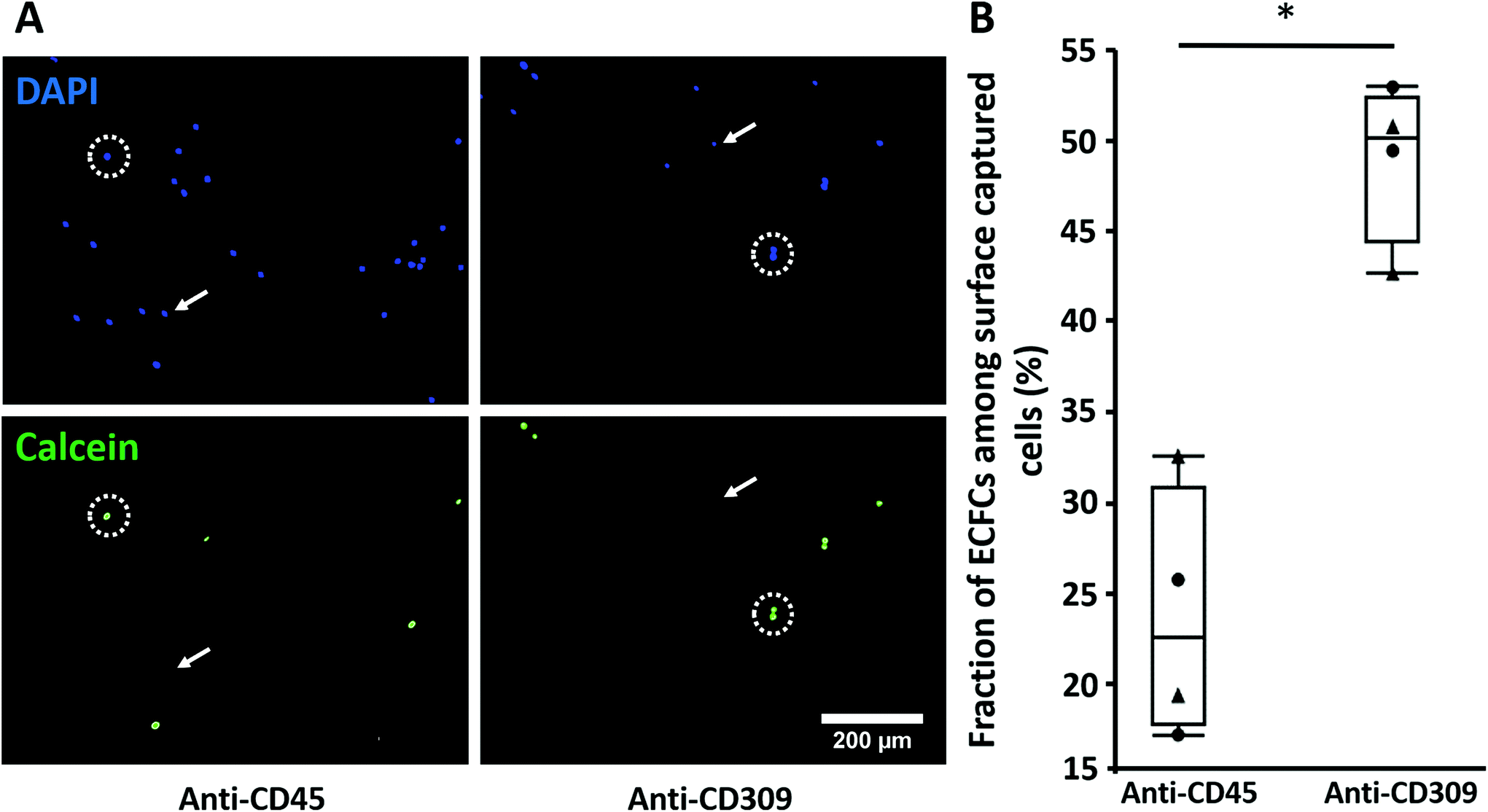 | ||
| Fig. 5 Selective capture of ECFCs from a mixture of cells. ECFCs were labeled with calcein (green fluorescent dye) and mixed at a ratio of 1:1 with PBMCs. The mixture was then resuspended in serum-free EGM-2 and perfused for 1 h at a wall shear stress of 1.5 dyn cm−2 using the parallel-plate flow chambers system. (A) Representative images of all cell types stained for nuclei with DAPI (blue), and the prelabelled ECFCs with calcein (green) on anti-CD45 and anti-CD309 modified substrates. White arrows indicate PBMCs (DAPI+ and calcein−) while white dashed circles indicate ECFCs (DAPI+ and calcein+). (B) The fraction of ECFCs among surface captured cells on modified substrates with either anti-CD45 or anti-CD309 antibodies. The triangle and the circle represent the different two independent PBMC donors that were mixed with ECFCs. *p < 0.05 with n = 4 experimental replicates. | ||
4 Discussion
In this work, the effect of RRGW-mediated immobilized antibodies on EPC capture was studied using ECFCs. Many previous in vitro studies have used HUVECs as a model cell type to develop EPC capture surfaces. However, HUVECs and other more mature endothelial cells display reduced in vitro angiogenesis capacity,35,36 in addition to being less representative of EPCs present in the circulation. The great challenge in developing ECFC capture stents is the presence of other circulating cells that might interfere with selective cell targeting. The selectivity of ECFC capture from a 1:1 mixture with PBMCs increased nearly 2-fold on anti-CD309 vs. anti-CD45 antibodies immobilized via RRGW peptides. The CD309-mediated ECFC capture was abrogated in the presence of soluble CD309 proteins, further demonstrating that ECFC capture was mediated by the immobilized antibodies via RRGW peptides (Fig. 4C). Similar observations were previously reported for HUVEC capture on protein G-immobilized anti-CD309 antibodies with or without blocking the antigen-binding sites.1
Previous studies of EPC capture technologies typically focused on one or two potential capture antibodies.1,2,20,37 To our knowledge, no single broad comparative study has been conducted between multiple EC-specific antibodies and non-EC specific controls. Here, a broad comparison between 3 antibodies recognizing ECFCs (anti-CD309, anti-CD34 and anti-CD144) and 2 antibodies that should not bind ECFCs (anti-CD14 and anti-CD45) was conducted in the same experimental system. Moreover, the effect of different serum concentrations on ECFC capture efficiency was investigated as antibodies and other suspended proteins may potentially outcompete and displace the grafted molecules. Antibody retention was reduced when surfaces were incubated in serum. This may be due to the desorption of adsorbed molecules present even in the “conjugated” conditions or to antibody displacement by antibodies present in serum.38,39 Given the lack of concentration-dependent effects of antibody displacement at different serum concentrations (Fig. 3) and the low dissociation constant of RRGW peptides,27 antibodies are expected to be retained in vivo for at least 1 h. Importantly, surfaces with conjugated RRGW retained significantly more antibodies than surfaces with adsorbed RRGW. Contrary to the physical adsorption of antibodies on the surface that was previously shown to be affected by salt concentrations,40 the developed antibody immobilization strategy via conjugated RRGW peptide is stable and preserved at up to 70% serum for at least 1 h under flow.
Despite lower antibody retention in conditions with 70% or 10% serum, ECFC capture efficiency was significantly enhanced at higher serum concentrations (Fig. 4). This could be due to the presence of proteins in the serum, which may enhance firm cell adhesion either directly (e.g., extracellular matrix protein adsorption41–43) or indirectly (e.g., growth factor receptor activation44,45). Moreover, antibodies recognizing ECs captured significantly more ECFCs than the anti-monocyte antibodies (anti-CD14) even at higher serum concentrations, supporting the feasibility of using the RRGW-conjugated surfaces for EPC capture applications.
The conjugation method applied in this study relies on an available single thiol (–SH) group from a terminal cysteine residue on the RRGW(PEG3)C peptide sequence (Fig. 1A). This approach permits not only directional antibody immobilization via Fc/RRGW interactions but also control over peptide orientation on surfaces – contrary to a previous method where RRGW-NH2 peptides were grafted onto surfaces with carboxylic acid groups using the 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide [EDC/NHS] chemistry.27 Another advantage of the peptide sequence used in this study is the integration of polyethylene glycol (PEG) within the sequence, which was previously proven to minimize non-desirable and non-specific protein adsorption.46–48 Furthermore, the conjugation scheme of the RRGW(PEG3)C sequence could readily be applied to other Fc-binding peptides in the literature such as KHRFNKD and HFRRHL.30,49 Thus, a screening platform including multiple Fc-binding peptides and cell capture antibodies could be envisioned. Mixtures of antibodies could also be applied to the RRGW-functionalized surfaces to maximize cell capture efficiency and selectivity. Finally, the peptide surface conjugation scheme could be applied to immobilize antibodies onto any substrate with available primary amines on the surface, including stents and bioresorbable vascular scaffolds. These stents and scaffolds, with immobilized antibodies on the surface, could then be implanted into animal models such as pigs to compare their capacity to capture ECFCs and promote endothelialization with commercially available stents such as the Genous™ technology.
Although this study focused on ECFC capture for vascular applications, the technique could be easily translated to target other cell types. Another anticipated application, for example, includes capturing circulating tumor cells for cancer diagnosis.50 The ease of immobilizing a variety of IgG antibodies (6 in total in this study) via the same initial Fc-binding peptide surface conjugation strategy facilitates screening of different cell capture antibodies. A large number of different antibodies and antibody combinations could readily be immobilized on surfaces to screen cell capture potential, particularly when combined with higher throughput flow systems using microfluidics or commercial systems with higher numbers of parallel flow paths compared to the 4-chamber custom flow system used here.
5 Conclusion
This study presents a method to selectively capture circulating cells with antibodies immobilized through surface conjugation of the Fc-binding peptide, RRGW. Antibody-modified surfaces captured circulating ECFCs suspended in serum-free or serum-containing medium. Anti-CD309 modified surfaces significantly increased the selective capture of ECFCs from a heterogeneous mixture of cells. The ease of immobilizing a variety of IgG antibodies without changing the underlying Fc-binding peptide surface grafting chemistry creates a platform that is highly versatile and amenable to screening applications. Furthermore, the chemistry used in this platform could be easily translated to variable stent materials – such as cobalt chromium alloys – with available amines on the surface introduced, for example, via plasma-based procedures.51 This platform holds great promise to engineer better cell capture biomaterials such as stents, biosensor platforms and other biomaterials for cellular therapy and regenerative medicine applications.Conflicts of interest
There are no conflicts that the authors would like to declare.Acknowledgements
The authors would like to thank Lisa Danielczak and Linda Peltier for their help in arranging the blood donation, and Jonathan Brassard and Julia Manalil for their help in reviewing the manuscript. We would also like to thank Gad Sabbatier for his help in performing the XPS analysis and Mariève Boulanger for analytical method development. This study was financially supported by the Canadian Institute for Health Research (CIHR, MOP 142285) and the Canadian Foundation for Innovation (CFI, project 35507). This work was supported via travel awards and networking opportunities offered by the Quebec Center for Advanced Materials (QCAM), the Quebec Network for Research on Protein Function (PROTEO), the McGill Regenerative Medicine (MRM) network, the Quebec Cell, Tissue and Gene Therapy Network (ThéCell) and the Quebec Cardiometabolic Health, Diabetes and Obesity Research Network – CMDO (ThéCell and CMDO are thematic networks supported by the Fonds de recherche du Québec–Santé).References
- B. D. Markway, O. J. McCarty, U. M. Marzec, D. W. Courtman, S. R. Hanson and M. T. Hinds, Tissue Eng., Part C, 2008, 14, 97–105 CrossRef CAS.
- K. Larsen, C. Cheng, D. Tempel, S. Parker, S. Yazdani, W. K. den Dekker, J. H. Houtgraaf, R. de Jong, S. Swager-ten Hoor, E. Ligtenberg, S. R. Hanson, S. Rowland, F. Kolodgie, P. W. Serruys, R. Virmani and H. J. Duckers, Eur. Heart J., 2012, 33, 120–128 CrossRef CAS.
- S. K. Vashist, E. Marion Schneider, E. Lam, S. Hrapovic and J. H. T. Luong, Sci. Rep., 2014, 4, 4407 CrossRef.
- L. H. Lambert, G. K. E. Goebrecht, S. E. De Leo, R. S. O'Connor, S. Nunez-Cruz, T.-D. Li, J. Yuan, M. C. Milone and L. C. Kam, Nano Lett., 2017, 17, 821–826 CrossRef CAS.
- L. Wu, E. Seung, L. Xu, E. Rao, D. M. Lord, R. R. Wei, V. Cortez-Retamozo, B. Ospina, V. Posternak, G. Ulinski, P. Piepenhagen, E. Francesconi, N. El-Murr, C. Beil, P. Kirby, A. Li, J. Fretland, R. Vicente, G. Deng, T. Dabdoubi, B. Cameron, T. Bertrand, P. Ferrari, S. Pouzieux, C. Lemoine, C. Prades, A. Park, H. Qiu, Z. Song, B. Zhang, F. Sun, M. Chiron, S. Rao, K. Radošević, Z.-Y. Yang and G. J. Nabel, Nat. Cancer, 2020, 1, 86–98 CrossRef.
- J. Matic, J. Deeg, A. Scheffold, I. Goldstein and J. P. Spatz, Nano Lett., 2013, 13, 5090–5097 CrossRef CAS.
- S. Pacelli, S. Basu, J. Whitlow, A. Chakravarti, F. Acosta, A. Varshney, S. Modaresi, C. Berkland and A. Paul, Adv. Drug Delivery Rev., 2017, 120, 50–70 CrossRef CAS.
- N. Charles, J. L. Liesveld and M. R. King, Biotechnol. Prog., 2007, 23, 1463–1472 CrossRef CAS.
- W. Wojakowski, A. Pyrlik, M. Król, P. Buszman, A. Ochała, K. Milewski, G. Smolka, D. Kawecki, A. Rudnik, T. Pawłowski, T. Jadczyk, R. Wyderka, W. Cybulski, S. Dworowy and M. Tendera, Minerva Cardioangiol., 2013, 61, 301–311 CAS.
- R. J. Medina, C. L. Barber, F. Sabatier, F. Dignat-George, J. M. Melero-Martin, K. Khosrotehrani, O. Ohneda, A. M. Randi, J. K. Y. Chan, T. Yamaguchi, V. W. M. Van Hinsbergh, M. C. Yoder and A. W. Stitt, Stem Cells Transl. Med., 2017, 6, 1316–1320 CrossRef.
- D. A. Ingram, L. E. Mead, H. Tanaka, V. Meade, A. Fenoglio, K. Mortell, K. Pollok, M. J. Ferkowicz, D. Gilley and M. C. Yoder, Blood, 2004, 104, 2752–2760 CrossRef CAS.
- M. A. M. Beijk, M. Klomp, N. J. W. Verouden, N. van Geloven, K. T. Koch, J. P. S. Henriques, J. Baan, M. M. Vis, E. Scheunhage, J. J. Piek, J. G. P. Tijssen and R. J. de Winter, Eur. Heart J., 2009, 31, 1055–1064 CrossRef.
- J. Aoki, P. W. Serruys, H. van Beusekom, A. T. L. Ong, E. P. McFadden, G. Sianos, W. J. van der Giessen, E. Regar, P. J. de Feyter, H. R. Davis, S. Rowland and M. J. B. Kutryk, J. Am. Coll. Cardiol., 2005, 45, 1574–1579 CrossRef CAS.
- W. K. den Dekker, J. H. Houtgraaf, Y. Onuma, E. Benit, R. J. de Winter, W. Wijns, M. Grisold, S. Verheye, S. Silber, E. Teiger, S. M. Rowland, E. Ligtenberg, J. Hill, M. Wiemer, P. den Heijer, B. J. Rensing, K. M. Channon, P. W. J. C. Serruys and H. J. Duckers, Atherosclerosis, 2011, 219, 245–252 CrossRef CAS.
- G.-A. Giurgea, A. Heuberger, J. Babayev, S. Winkler, O. Schlager, I. M. Lang and M. Gyöngyösi, PLoS One, 2018, 13, e0201416 CrossRef.
- H. J. Duckers, T. Soullié, P. den Heijer, B. Rensing, R. J. de Winter, M. Rau, H. Mudra, S. Silber, E. Benit, S. Verheye, W. Wijns and P. W. Serruys, EuroIntervention, 2007, 3, 350–358 CrossRef.
- A. Schober, R. Hoffmann, N. Oprée, S. Knarren, E. Iofina, G. Hutschenreuter, P. Hanrath and C. Weber, Am. J. Cardiol., 2005, 96, 1116–1122 CrossRef.
- P. van den Hoogen, M. M. H. Huibers, J. P. G. Sluijter and R. A. de Weger, J. Cardiovasc. Transl. Res., 2015, 8, 106–116 CrossRef.
- J.-L. Hillebrands, F. A. Klatter and J. Rozing, Arterioscler., Thromb., Vasc. Biol., 2003, 23, 380–387 CrossRef CAS.
- J. M. Lee, W. Choe, B.-K. Kim, W.-W. Seo, W.-H. Lim, C.-K. Kang, S. Kyeong, K. D. Eom, H.-J. Cho, Y.-C. Kim, J. Hur, H.-M. Yang, H.-J. Cho, Y.-S. Lee and H.-S. Kim, Biomaterials, 2012, 33, 8917–8927 CrossRef CAS.
- M. Shen, J. Rusling and C. K. Dixit, Methods, 2017, 116, 95–111 CrossRef CAS.
- M. J. B. Kutryk, R. J. Cottone and S. M. Rowland Jr., US Pat, 7803183B2, 2000 Search PubMed.
- H. Y. Song, X. Zhou, J. Hobley and X. Su, Langmuir, 2012, 28, 997–1004 CrossRef CAS.
- M. Linhult, H. K. Binz, M. Uhlén and S. Hober, Protein Sci., 2002, 11, 206–213 CrossRef CAS.
- N. G. Welch, J. A. Scoble, B. W. Muir and P. J. Pigram, Biointerphases, 2017, 12, 02D301 CrossRef.
- P. A. Nygren, M. Eliasson, L. Abrahmsén, M. Uhlén and E. Palmcrantz, J. Mol. Recognit., 1988, 1, 69–74 CrossRef CAS.
- C. W. Tsai, S. L. Jheng, W. Y. Chen and R. C. Ruaan, Anal. Chem., 2014, 86, 2931–2938 CrossRef CAS.
- X. Sun, J. Weaver, S. R. Wickramasinghe and X. Qian, Polymers, 2018, 10, 778 CrossRef.
- T. Sugita, M. Katayama, M. Okochi, R. Kato, T. Ichihara and H. Honda, Biochem. Eng. J., 2013, 79, 33–40 CrossRef CAS.
- R.-J. Yoo and S.-J. Choi, BioChip J., 2016, 10, 88–94 CrossRef CAS.
- C. A. Hoesli, C. Tremblay, P.-M. Juneau, M. D. Boulanger, A. V. Beland, S. D. Ling, B. Gaillet, C. Duchesne, J. Ruel, G. Laroche and A. Garnier, ACS Biomater. Sci. Eng., 2018, 4, 3779–3791 CrossRef CAS.
- M. A. Elkhodiry, M. D. Boulanger, O. Bashth, J.-F. Tanguay, G. Laroche and C. A. Hoesli, Biotechnol. Bioeng. Search PubMed.
- L. Dworak, A. J. Reuss, M. Zastrow, K. Rück-Braun and J. Wachtveitl, Nanoscale, 2014, 6, 14200–14203 RSC.
- A. Nakano, T. Harada, S. Morikawa and Y. Kato, Pathol. Int., 1990, 40, 107–115 CrossRef CAS.
- G. Finkenzeller, N. Torio-Padron, A. Momeni, A. T. Mehlhorn and G. B. Stark, Tissue Eng., 2007, 13, 1413–1420 CrossRef CAS.
- H. Bompais, J. Chagraoui, X. Canron, M. Crisan, X. H. Liu, A. Anjo, C. Tolla-Le Port, M. Leboeuf, P. Charbord, A. Bikfalvi and G. Uzan, Blood, 2004, 103, 2577–2584 CrossRef CAS.
- W.-H. Lim, W.-W. Seo, W. Choe, C.-K. Kang, J. Park, H.-J. Cho, S. Kyeong, J. Hur, H.-M. Yang, H.-J. Cho, Y.-S. Lee and H.-S. Kim, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 2798–2805 CrossRef CAS.
- J. Buijs, D. D. White and W. Norde, Colloids Surf., B, 1997, 8, 239–249 CrossRef CAS.
- A. Makaraviciute, A. Ramanavicius and A. Ramanaviciene, Anal. Methods, 2015, 7, 9875–9884 RSC.
- X. Zhao, F. Pan, L. Garcia-Gancedo, A. J. Flewitt, G. M. Ashley, J. Luo and J. R. Lu, J. R. Soc., Interface, 2012, 9, 2457–2467 CrossRef CAS.
- X. Lü, H. Zhang, Y. Huang and Y. Zhang, Regener. Biomater., 2018, 5, 261–273 CrossRef.
- J. L. Dewez, A. Doren, Y. J. Schneider and P. G. Rouxhet, Biomaterials, 1999, 20, 547–559 CrossRef CAS.
- A. J. García, Biomaterials, 2005, 26, 7525–7529 CrossRef.
- P. A. Underwood, J. M. Whitelock, P. A. Bean and J. G. Steele, J. Biomater. Sci., Polym. Ed., 2002, 13, 845–862 CrossRef CAS.
- C. Wilson, R. Clegg, D. Leavesley and M. Pearcy, Tissue Eng., 2005, 11, 1–18 CrossRef CAS.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Langmuir, 2005, 21, 12327–12332 CrossRef CAS.
- C. Bernhard, S. J. Roeters, J. Franz, T. Weidner, M. Bonn and G. Gonella, Phys. Chem. Chem. Phys., 2017, 19, 28182–28188 RSC.
- E. M. Davis, J. M. Platnich, R. T. Irvin and D. A. Muruve, ASAIO J., 2015, 61, 710–717 CrossRef CAS.
- S. Menegatti, B. G. Bobay, K. L. Ward, T. Islam, W. S. Kish, A. D. Naik and R. G. Carbonell, J. Chromatogr., A, 2016, 1445, 93–104 CrossRef CAS.
- P. Bankó, S. Y. Lee, V. Nagygyörgy, M. Zrínyi, C. H. Chae, D. H. Cho and A. Telekes, J. Hematol. Oncol., 2019, 12, 48 CrossRef.
- S. Diaz-Rodriguez, P. Chevallier, C. Paternoster, V. Montaño-Machado, C. L. Noël, L. Houssiau and D. Mantovani, RSC Adv., 2019, 9, 2292–2301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00650e |
| This journal is © The Royal Society of Chemistry 2020 |