
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring microfluidics as a tool to evaluate the biological properties of a titanium alloy under dynamic conditions†
Sarah-Sophia D.
Carter
 ,
Laurent
Barbe
,
Maria
Tenje
and
Gemma
Mestres
*
,
Laurent
Barbe
,
Maria
Tenje
and
Gemma
Mestres
*
Division of Microsystems Technology, Department of Materials Science and Engineering, Science for Life Laboratory, Uppsala University, 751 22 Uppsala, Sweden. E-mail: gemma.mestres@angstrom.uu.se; Tel: +46 18 471 3235
First published on 1st October 2020
Abstract
To bring novel biomaterials to clinical use, reliable in vitro models are imperative. The aim of this work was to develop a microfluidic tool to evaluate the biological properties of biomaterials for bone repair. Two approaches to embed medical grade titanium (Ti6Al4V) on-chip were explored. The first approach consisted of a polydimethylsiloxane microfluidic channel placed onto a titanium disc, held together by an additively manufactured fixture. In the second approach, a titanium disc was assembled onto a microscopic glass slide, using a double-sided tape microfluidic channel. Both approaches demonstrated potential for maintaining MC3T3-E1 preosteoblast-like cell cultures on-chip, as was shown by the vast majority of living cells after 1 day. In addition, the cells cultured on-chip showed a more elongated morphology compared to cells grown under static conditions and a tendency to align to the direction of the flow. For longer-term (i.e. 10 days) studies, the glass-based chip was selected. Assessment of cell viability showed a high number of living cells during the entire experimental period. Cell proliferation and differentiation studies revealed an increase in cell proliferation on-chip, suggesting that proliferation was the dominating process at the detriment of differentiation in this micrometric dynamic environment. These results illustrate the importance of optimizing in vitro cell culture conditions and how these may affect biomaterial testing outcomes. Overall, this work provides a step towards more in vivo-like microfluidic testing platforms, which are expected to provide more reliable in vitro screening of biomaterials.
1. Introduction
The longer life expectancy of the global population has increased the need for repairing bone injuries resulting from trauma or local diseases. A promising approach to restore such injuries involves the use of biomaterials.1 Over the past decades the biomaterial field has advanced tremendously, shifting from a focus on inert materials to bioactive materials that elicit biological responses and resorb over time, stimulating the formation of new bone.2 Nevertheless, despite the enormous research activities, only a fraction of potential novel biomaterials for bone repair reach clinical application.3 In order to reach the clinic, biomaterials need to be thoroughly evaluated, which requires reliable in vitro models. Currently used models for biomaterials for bone repair do however correlate poorly with in vivo results.3Although seemingly inert, bone is a dynamic tissue, which is continuously remodeled in order to repair damaged bone and adapt to functional demands, such as mechanical load. This process is coordinated by the activities of bone forming osteoblasts, bone resorbing osteoclasts and osteocytes, which are the cells involved in orchestrating the bone remodeling process.4 Previous studies have shown that pressure differences in the interstitial fluid, the fluid throughout the extracellular matrix, play a key role in the ability of bone cells to sense their mechanical environment.5–7 These changes in the interstitial fluid are thought to influence the shear stress acting on cell membranes and thereby influence the cellular response.
Whereas the traditional static cell culture vessels have provided significant insights into the biological properties of biomaterials, nowadays the added value of increased physiological relevance in in vitro testing is widely acknowledged.3 As recently highlighted by Mestres et al., microfluidic technology offers a promising tool for more accurate in vitro screening of biomaterials.8 By using a microfluidic approach, cells can be cultured confined in channels of only tens to hundreds of micrometers, thereby providing a more physiologically relevant microenvironment compared to classical macroscale cultures.9 Moreover, microfluidics enables controlled perfusion of cells that are adhered to the surface of a biomaterial, and offers the possibility to adjust relevant microenvironmental parameters, such as fluid shear stress, mechanical load, biochemical concentration gradients and environmental cues.9–12 For example, by varying flow rates or channel dimensions, multiple shear stress conditions can be recreated in single microfluidic devices.13 Moreover, low shear stress conditions can be achieved by shielding the cells from the flow, for instance by using a cell culture channel in combination with separate lateral flow channels.14 More complex and physiologically relevant systems can be built by combining multiple microenvironmental parameters simultaneously in one device. For example, combining mechanical stimulation with chemical gradients, combining environmental conditions (e.g. hypoxia) with a chemical gradient of a potential drug candidate or combining multiple cell types and different extracellular environments.15–17
Although the approach of evaluating biomaterials for bone repair on-chip is in its infancy, multiple studies have already highlighted the potential of this methodology.17–22 A relevant example is given by Stamp et al. who developed a microfluidic system to study the short-term (i.e. 60 minutes) effect of shear stress, temperature and pH on the adhesion of human Saos-2 osteosarcoma cells to a titanium alloy (Ti6Al4V ELI).23 More recently, biocompatible micropatterned polylactic acid was integrated on-chip, which allowed evaluation of the effect of biomaterial geometries on human MG-63 osteosarcoma cell morphology and distribution, both under perfusion and diffusion flow regimes.24
In this study, microfluidic chips integrating a biomaterial for bone repair were fabricated and subsequently assessed, using conventional methods such as fluorescent microscopy and colorimetric assays. As a model for medical implants, a well-studied biomaterial was chosen, namely medical grade titanium (Ti6Al4V, abbreviated as Ti), which is frequently used as a dental and orthopedic implant material.25 Two designs to embed Ti on-chip are proposed and discussed in terms of inertness of the materials used, fabrication and integration with standard biochemical assays to assess cell behavior. The most promising design was selected to study cell viability, proliferation and differentiation of MC3T3-E1 preosteoblast-like cells for a period of 10 days. The ultimate aim of this work was to explore microfluidic technology to provide a microfluidic-based tool that more closely resembles the in vivo conditions of bone and can be used to evaluate the biological properties of Ti.
2. Materials and methods
A medical grade (grade 5) titanium alloy (Ti6Al4V, referred to as Ti) was selected as a biomaterial model. To obtain reproducible surfaces, the Ti discs (∅ = 12 mm, height = 1 mm) were grinded and subsequently sonicated in isopropanol and distilled water for 10 min each. The average surface roughness value after the mechanical treatment was 0.08 ± 0.007 μm (ESI Fig. 1†).Two designs to embed Ti on-chip were explored. The first approach, later referred to as the Ti-polydimethylsiloxane (PDMS)-chip, comprised a PDMS microfluidic channel and Ti disc held together by an additively manufactured fixture. In the second approach, named the Ti-glass-chip, a Ti disc was attached to a microscopic glass slide using a double-sided tape microfluidic channel.
2.1. Fabrication of Ti-PDMS-chips
The PDMS microfluidic channel was prepared by soft lithography, using a SU-8 mold that was fabricated by photolithography. In short, a 4′′ silicon wafer was laminated with a 200 μm thick SUEX® resist layer (DJ Microlaminates) and subsequently UV patterned to obtain individual patterns (Karl Süss MA6 Mask Aligner) (Fig. 1A). PDMS (Dow Corning, SYLGARD™ 184) was prepared according to manufacturer's instructions in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 elastomer:curing agent ratio and poured over the SU-8 mold. After overnight (i.e. 16 hours) curing at 65 °C, the PDMS was peeled off from the mold and punched to release individual circular PDMS pieces (∅ = 12 mm), which accommodated the microfluidic channels.
:1 elastomer:curing agent ratio and poured over the SU-8 mold. After overnight (i.e. 16 hours) curing at 65 °C, the PDMS was peeled off from the mold and punched to release individual circular PDMS pieces (∅ = 12 mm), which accommodated the microfluidic channels.
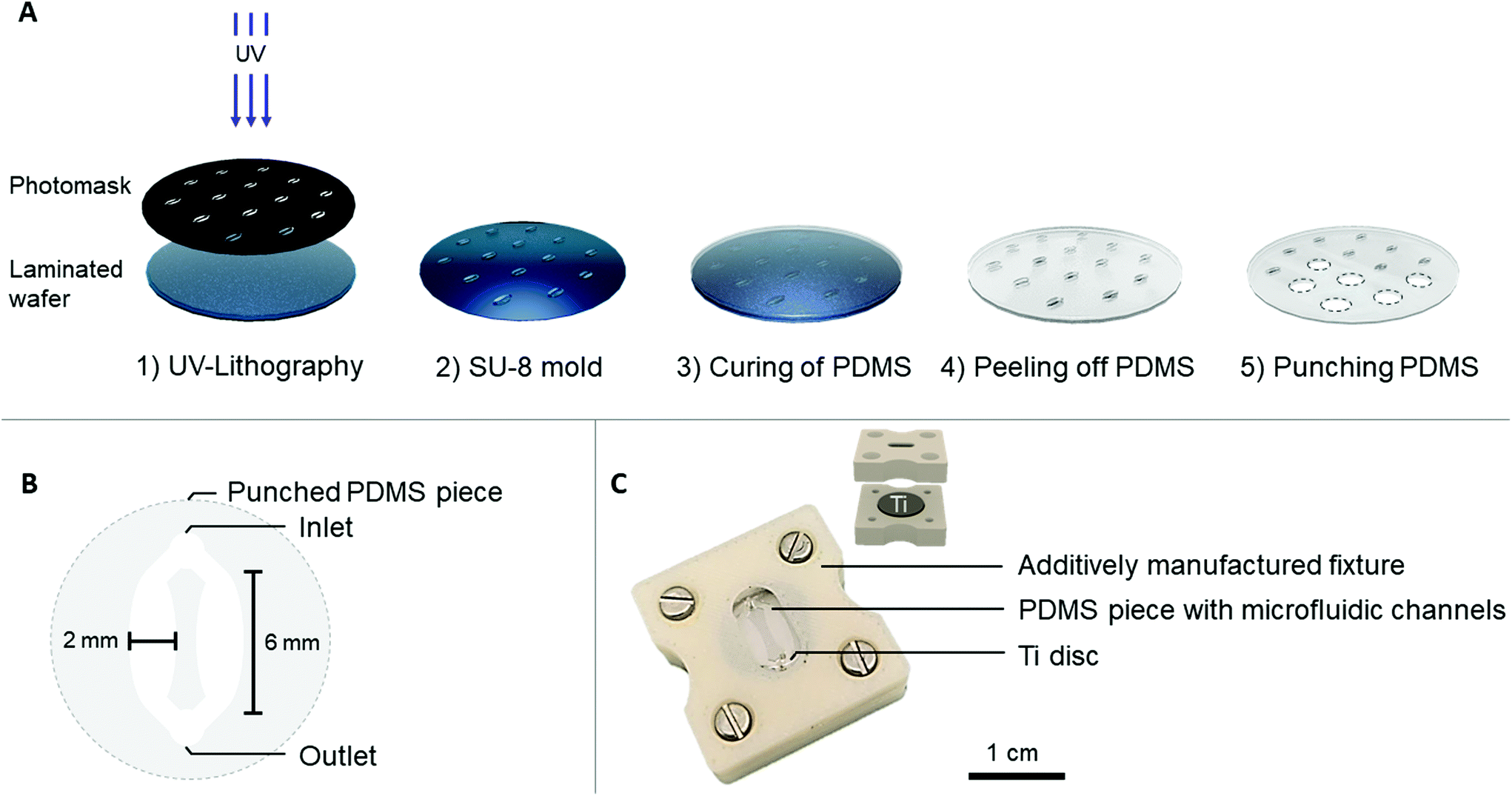 | ||
| Fig. 1 (A) Schematic of the process used to fabricate PDMS microfluidic channels for the Ti-PDMS-chip. (B) Schematic of one punched PDMS piece containing the two microfluidic channels. (C) Photograph of an assembled Ti-PDMS-chip and schematic of the opened chip to the side. | ||
Each PDMS piece contained one punched inlet and outlet (∅ = 1 mm), which bifurcated into two oval-shaped microfluidic channels (later referred to as cell culture areas) each with the following dimensions: l = 6 mm, w (largest) = 2 mm, h = 200 μm (Fig. 1B). This design with two cell culture areas was chosen to ensure the option of using conventional biochemical analysis, which are often optimized for macroscale cultures that usually have higher cell number and volume of reagents. To provide a tight seal between the PDMS microfluidic channel and the Ti disc, an acrylonitrile butadiene styrene (ABS) additively manufactured fixture was fabricated (Dimension Elite, Stratasys). This fixture consisted of an upper part, fitting the PDMS channel and a lower part, fitting the Ti disc (Fig. 1C). The upper part contained a window through which the PDMS and microfluidic channels were visible. The tubing (∅inner = 0.38 mm, ∅outer = 1.09 mm, Portex™ Fine Bore LDPE Tubing, Smiths Medical™) was directly plugged into the PDMS, connecting each chip to a peristaltic pump (LabV1-11 peristaltic pump, Shenchen). Prior to use, the PDMS channels and additively manufactured fixture were sterilized with 70% ethanol, the Ti discs were autoclaved and the chip was assembled under aseptic conditions. The cell culture medium was pre-equilibrated in typical cell culture conditions (i.e. 37 °C, 5% CO2, humidified atmosphere) before starting the perfusion.
2.2. Fabrication of Ti-glass-chips
The Ti-glass-chips consisted of a double-sided tape (468MP, 3M) microfluidic channel with dimensions of l = 26 mm, w (largest) = 10 mm, h = 130 μm (Fig. 2A), which was prepared using a cutting plotter (Craft ROBO Pro, Graphtech). The microfluidic channel was designed with a relatively narrow inlet that transitioned into a circular chamber-like structure on the Ti (later referred to as cell culture area) shaping back to a narrow channel at the outlet. The microfluidic channel, which covered the Ti almost entirely, was sandwiched between a conventional microscopic glass slide and a cover glass. The cover glass contained laser cut holes (AIO G, Östling Markingsystems), which served as a window to dock the Ti disc (∅hole = 12.05 mm) and the tubing (∅hole = 0.7 mm). A Ti disc was positioned in-plane with the cover glass and was sealed with an optical adhesive (NOA 76, Norland Products) (Fig. 2B). For the fluidic connections, silicone tubing jackets (∅inner = 1 mm, ∅outer = 3 mm, VWR, ref. no. 228-0701P) were glued onto the cover glass with silicone coating (ELASTOSIL® A07 Translucent, WACKER). All components were UV sterilized prior to use and mounted under aseptic conditions. The same tubing, peristaltic pump and set-up as described in section 2.1 were used. The cytotoxicity of the double-sided tape and optical adhesive were tested and showed no adverse effects on cell viability (ESI Fig. 2†).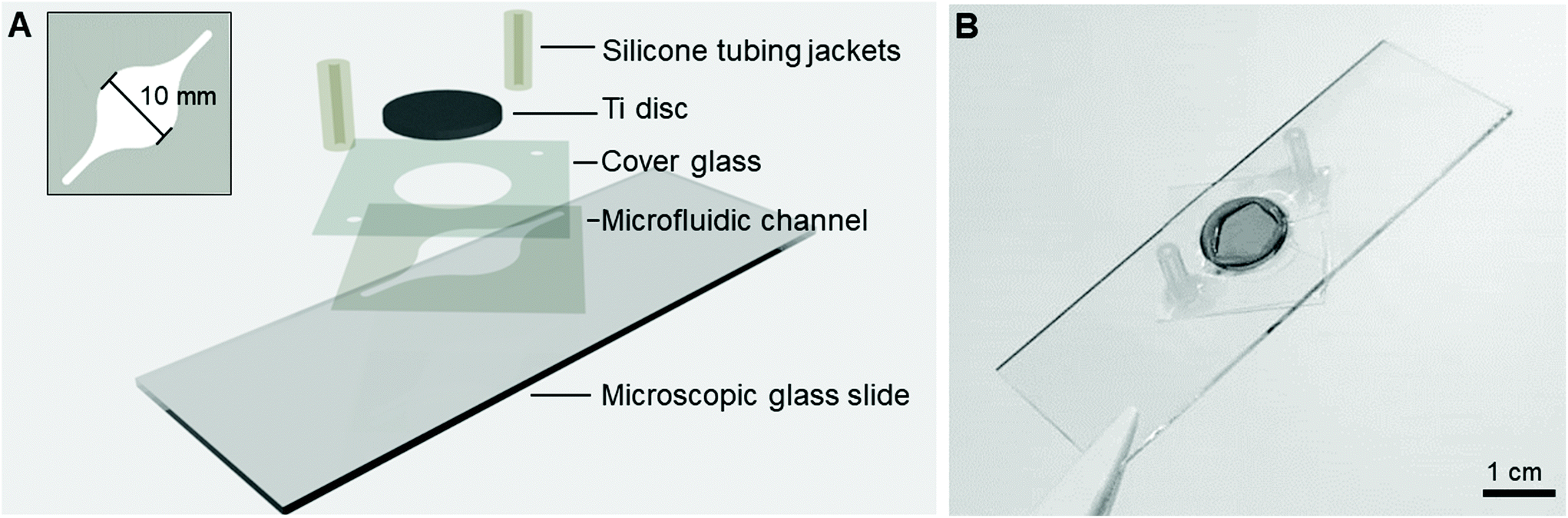 | ||
| Fig. 2 (A) Schematic of the different layers constituting the Ti-glass-chip and (B) a top-view photograph of the completed device. | ||
2.3. Prediction of wall shear stress
The wall shear stress in both chips was evaluated using COMSOL Multiphysics 5.5. software (COMSOL, Inc., Burlington, MA, USA), using the laminar flow module. The cell culture medium was modeled as an incompressible, homogenous, Newtonian fluid with a density of 1 g cm−3 and dynamic viscosity of 0.78 mPa s.26,27 The flow rate at the inlet was set to 2 μl min−1 and a zero pressure condition was applied to the outlet. No-slip boundary conditions were applied to the microchannel walls.2.4. Cell studies
For cell viability, proliferation and differentiation, MC3T3-E1 cells were cultured in MEM (HyClone™, ref. no. SH30265.01) further supplemented with 10 v/v % FBS, 1 v/v % penicillin/streptomycin, 50 μg ml−1 ascorbic acid (Sigma-Aldrich, ref. no. A7631) and 10 mM beta-glycerophosphate (Sigma-Aldrich, ref. no. G9422). For the Ti-on-chip samples, cells were seeded at 45000 cells per cm2 using a pipette. After seeding, the chips were flipped, meaning that the cells could sediment and adhere on top of the Ti. As controls, cells were seeded at the same cell density on Ti discs kept under static conditions in 24-well plates (Ti-static) and directly on polystyrene 48-well plates (PS-static). Both the chips and static samples were placed in the incubator for 4 hours to allow the cells to adhere. After this time, unidirectional perfusion was started through the chips at a flow rate of 2 μl min−1, which continued throughout the entire experiment. It should be noted that the flow rate was chosen to match previously performed studies and to fall within the disperse range of shear stress values reported in literature, as elaborated on in the discussion. This, in combination with practical considerations (i.e. consumption of cell culture medium), is the reason for choosing the flow rate of 2 μl min−1. The medium in the static samples was replaced every two days.
For image analysis, CellProfiler software (version 3.0.0) was used. To determine cell viability, the ratio between the dead cells and total amount of nuclei was quantified. For the evaluation of cell morphology and alignment, the cell nucleus was analyzed.28–31 Each cell nucleus was segmented and fitted to an ellipse in order to determine its eccentricity. Eccentricity is the ratio of the distance between the foci of the ellipse and its major axis length and ranges from 0 to 1, where 0 describes a circle and 1 a line segment. To determine cell alignment to the direction of the flow, the angle between the major axis of the fitted ellipse and the direction of the flow was calculated, ranging from −90° to 90°. However, for plotting, the absolute values were used. An angle of 0° refers to cell nuclei oriented in the direction of the flow, whereas an angle of 90° indicates a perpendicular orientation. To have the same sample size in all conditions, a threshold on the amount of analyzed nuclei (randomly selected) was set, based on the minimum amount of nuclei in the different samples. For quantification, two samples from each condition were analyzed at each time-point and at least three random fields were imaged for each sample.
After 1, 5 and 10 days of culture, Ti-static and PS-static were rinsed twice with PBS (Gibco™ 14200067) to remove detached cells and subsequently lysed using 400 μl of 0.1 v/v % Triton-X (Sigma-Aldrich, ref. no. T8787) for 50 minutes at 37 °C. The same procedure was followed for Ti-glass-chip, but to account for the differences in volume between the static conditions and on-chip, after lysing, the extracted solution from the chip was diluted to 400 μl using the same cell lysis buffer. Subsequently, a 50 μl aliquot was taken from each sample and incubated with 100 μL LDH assay reagents in a 96-well plate. After 25 minutes of incubation at room temperature protected from light, LDH activity was determined by measuring the absorbance at 490 nm and background absorbance at 690 nm (TECAN, Spark®). To account for the differences in surface area available for the cells on-chip and on the static samples, the absorbance values were normalized for the surface area, which were 0.82 cm2 and 1.1 cm2, respectively. The experiments were performed three times, using three samples per condition at each time-point in each experiment, unless mentioned otherwise. The results are presented as mean ± standard deviation from one representative experiment.
The total protein concentration was determined using a micro BCA protein assay kit (Fisher Scientific, ref. no. 23227), following manufacturer's instructions. In short, 30 μl aliquots of the prepared cell lysates were taken and combined with microBCA working solution in a 1:8 sample:working solution ratio in a 48-well plate. After 30 minutes incubation at 37 °C, the absorbance was read at 562 nm (TECAN, Spark®). The experiments were performed three times, using three samples per condition at each time-point in each experiment, unless mentioned otherwise. The results are presented as mean ± standard deviation from one representative experiment.
2.5. Statistical analysis
Statistical analysis was performed using Minitab version 18. The data was evaluated by one-way analysis of variance (ANOVA), two-sided, at a significance level of α = 0.05. Post-hoc Tukey test was performed to investigate differences between samples. A Levene's test was used to assess homogeneity of variances between groups. When significant, Welch's ANOVA, with post-hoc Games-Howell test was used to assess differences between groups.3. Results
3.1. Modelling wall shear stress on-chip
The wall shear stress in the area of both Ti-on-chips was modelled using COMSOL. This simulation showed that the wall shear stress in cell culture area was between ∼2.5 × 10−3–2.5 × 10 −2 dyne per cm2 in both the Ti-PDMS-chip and Ti-glass chip (Fig. 3A and B). The exact wall shear stress distribution along the cell culture areas can be found in ESI Fig. 3 and 4.†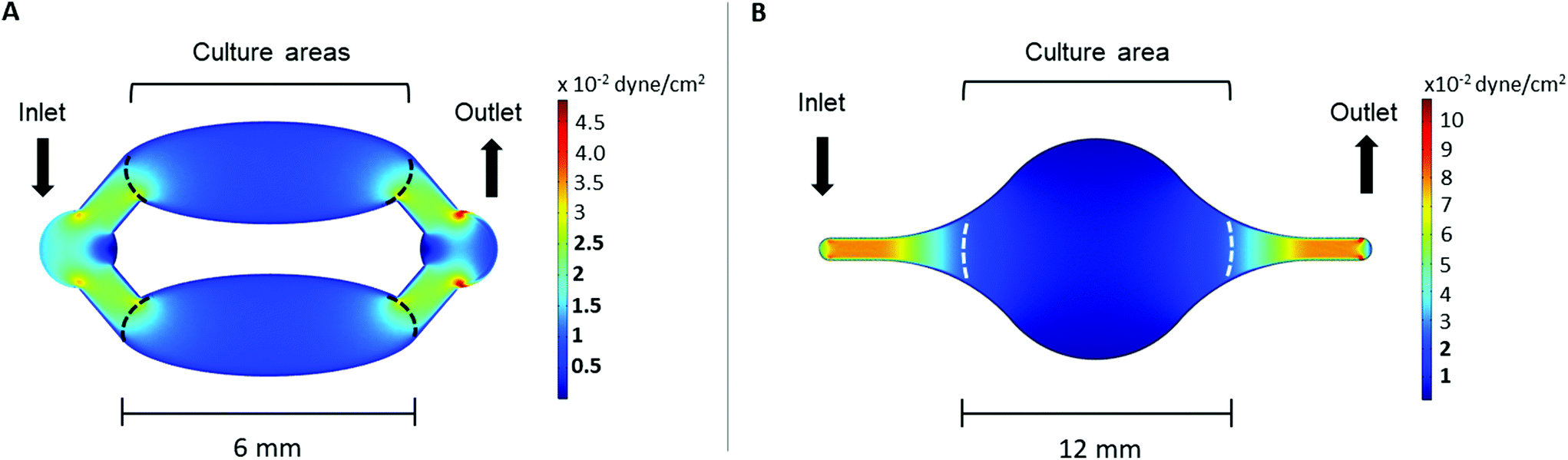 | ||
| Fig. 3 COMSOL simulations of the wall shear stress in the (A) Ti-PDMS-chip and (B) Ti-glass-chip, in which the areas marked by the dashed lines indicate the border of the area designed for cell culture. The shear stress values marked in bold in the legend indicate the wall shear stress levels relevant to the cell culture areas. | ||
3.2. Cell studies
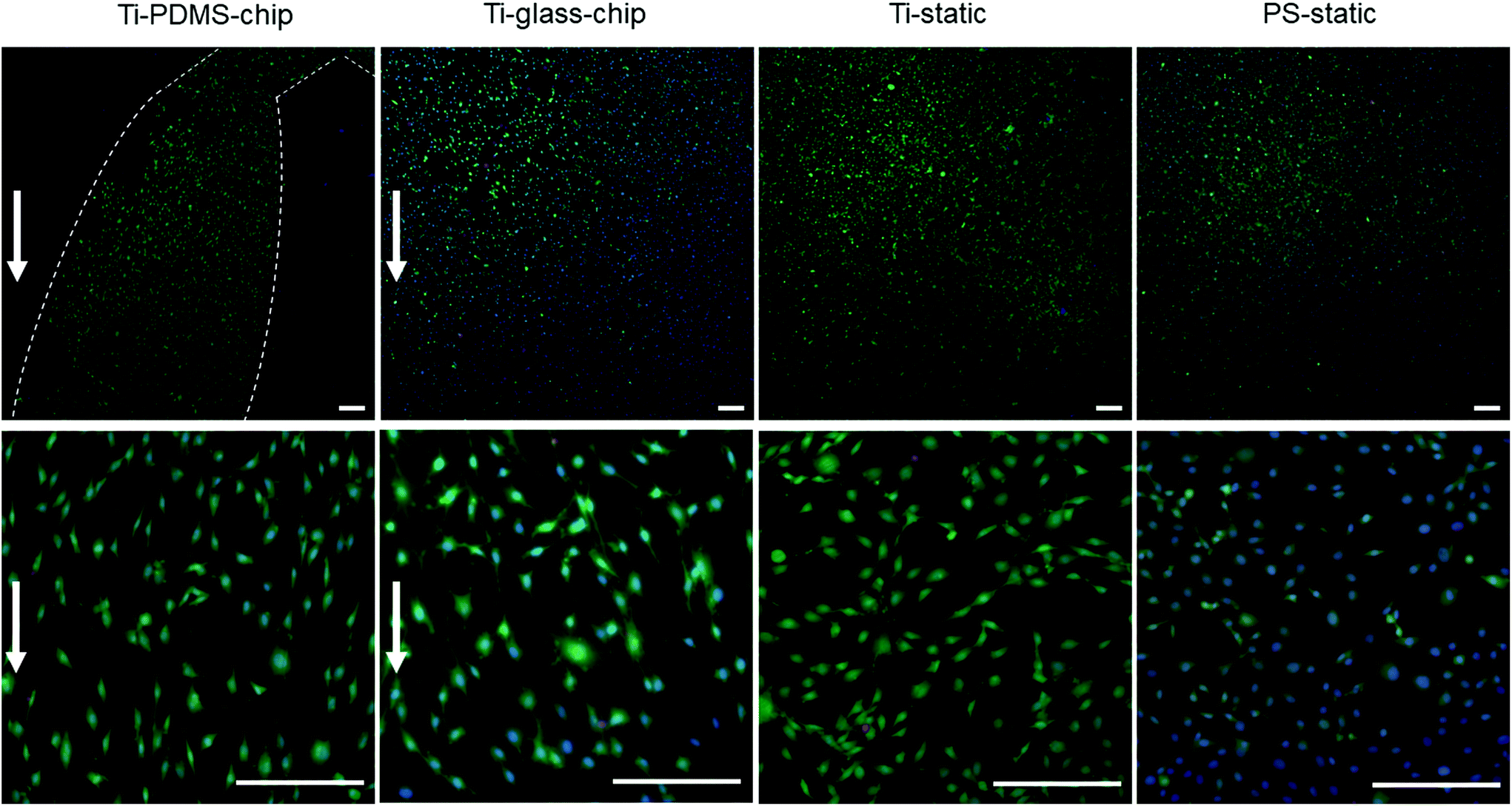 | ||
| Fig. 4 MC3T3-E1 cell viability and morphology determined by calcein-AM/propidium iodide/Hoechst staining after 1 day of culture on the Ti-PDMS-chip, Ti-glass-chip, Ti-static and PS-static. For the Ti-PDMS-chip sample, the outline of the microfluidic channel is marked with dotted lines and for both chips, the direction of the flow is indicated with an arrow. Scale bar corresponds to 200 μm. | ||
Compared to both static conditions, cells grown on Ti-PDMS-chip showed a more elongated morphology, as is indicated by eccentricity values closer to 1 (p < 0.0005 compared to both static conditions) (Fig. 5A). A similar trend was seen for the Ti-glass-chip (p < 0.0005 compared to both static conditions). In addition, significant differences were observed between the two chips, showing higher eccentricity values for cells grown on Ti-PDMS-chip (p < 0.0005). Regarding cell orientation, compared to both static conditions, on which the cells were oriented into all directions, the majority of the cells grown on both chips revealed to be orientated along the direction of the flow (i.e. 0°) (p < 0.0005 for both chips, compared to both static conditions) (Fig. 5B). In addition, significant differences were observed between Ti-PDMS-chip and Ti-glass-chip, showing enhanced tendency to align to the direction of the flow for cells grown on Ti-PDMS-chip (p < 0.0005). No significant differences in cell viability, morphology and orientation were observed between the two static samples.
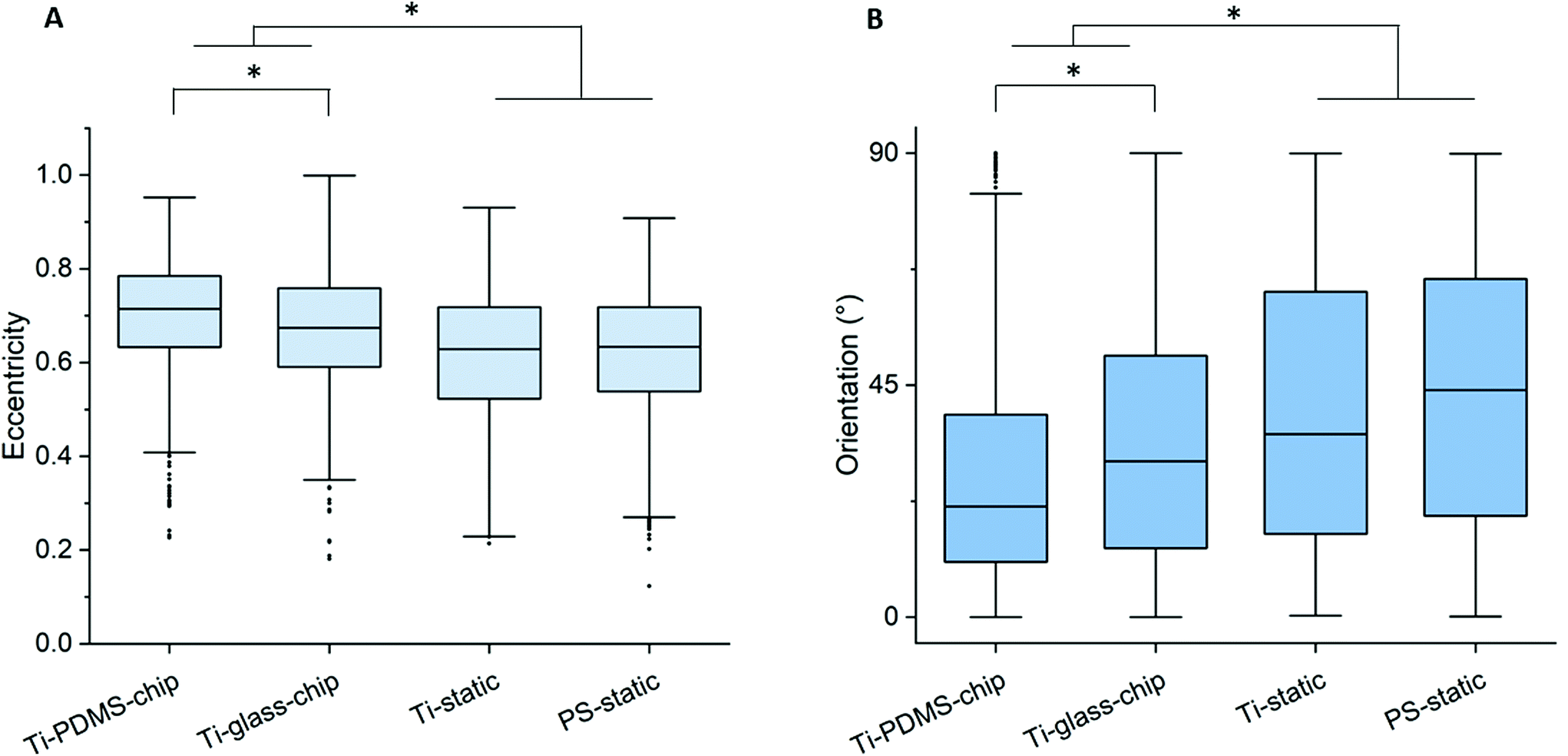 | ||
| Fig. 5 (A) Eccentricity of MC3T3-E1 cell nuclei on Ti-PDMS-chip, Ti-static and PS-static after 1 day, in which 0 describes a circle and 1 a line segment. (B) Nuclear orientation of MC3T3-E1 cells on Ti-PDMS-chip, Ti-static and PS-static after 1 day, in which an angle of 0° refers to cell nuclei oriented in the direction of the flow. Each dot represents one nucleus with n = 750. | ||
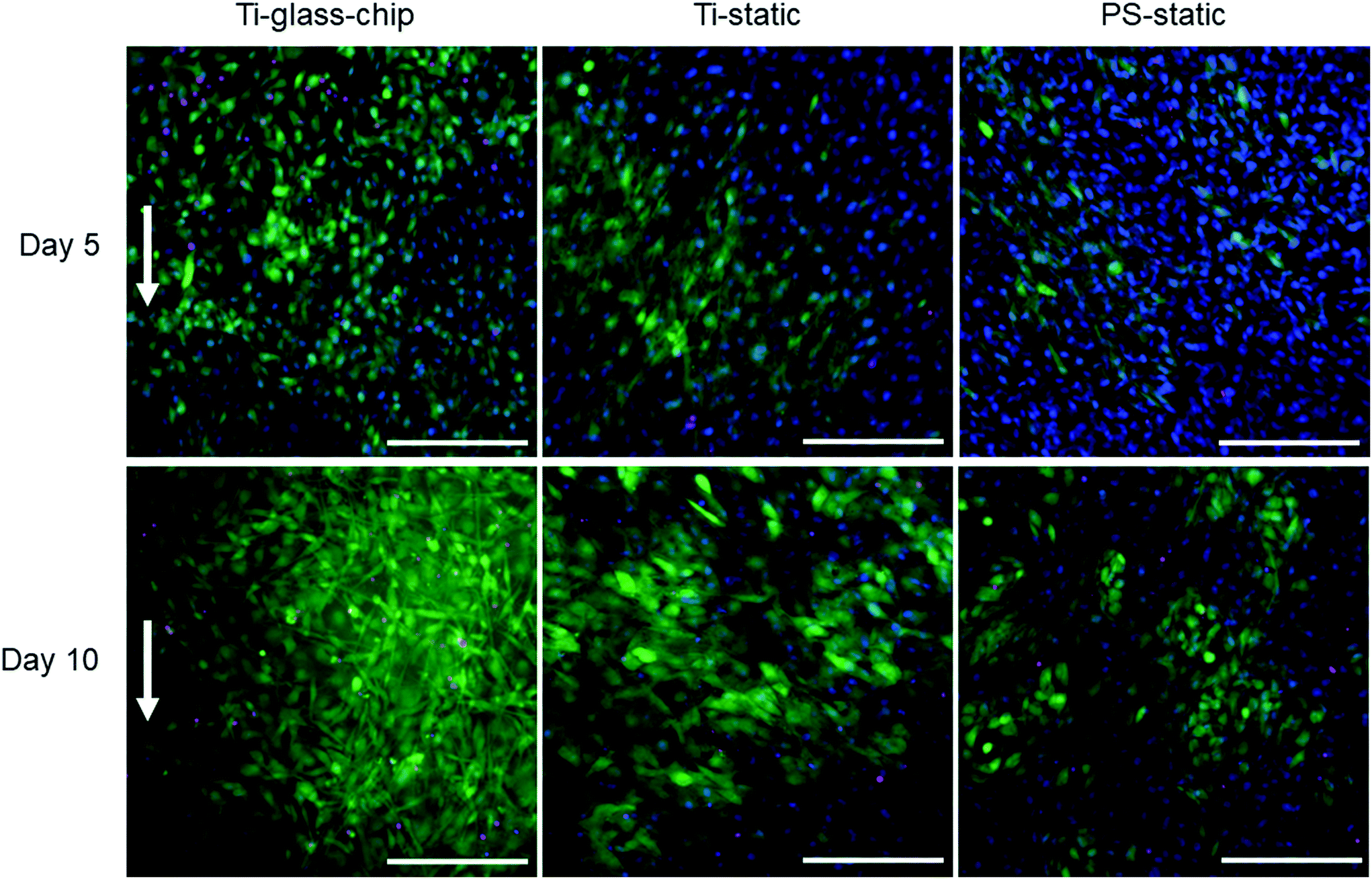 | ||
| Fig. 6 MC3T3-E1 cell viability and morphology determined by calcein-AM/propidium iodide/Hoechst staining after 5 days and 10 days of culture on the Ti-glass-chip, Ti-static and PS-static. The direction of the flow is indicated with an arrow. Scale bar corresponds to 200 μm. | ||
To quantitate the biological properties of Ti under dynamic conditions, cell proliferation and differentiation of MC3T3-E1 cells cultured on the Ti-glass-chip were assessed for a period of 10 days using the colorimetric LDH and ALP assays, respectively. As observed in Fig. 7A, cell proliferation increased noticeably over the first 5 days for Ti-glass-chip, Ti-static and PS-static and the cell number stagnated afterwards (p-values indicated in ESI Table 1†). After 1 day of culture, no significant differences were observed in terms of cell proliferation between cells grown on-chip and in either of the static conditions. Starting from day 5, cells that had grown on-chip showed a statistically significant increase in cell proliferation compared to both static controls (p < 0.0005 for both). This trend continued to day 10, on which cell proliferation was statistically higher for cells cultured on Ti-glass-chip compared to cells cultured on either Ti-static or PS-static (p = 0.003 for both).
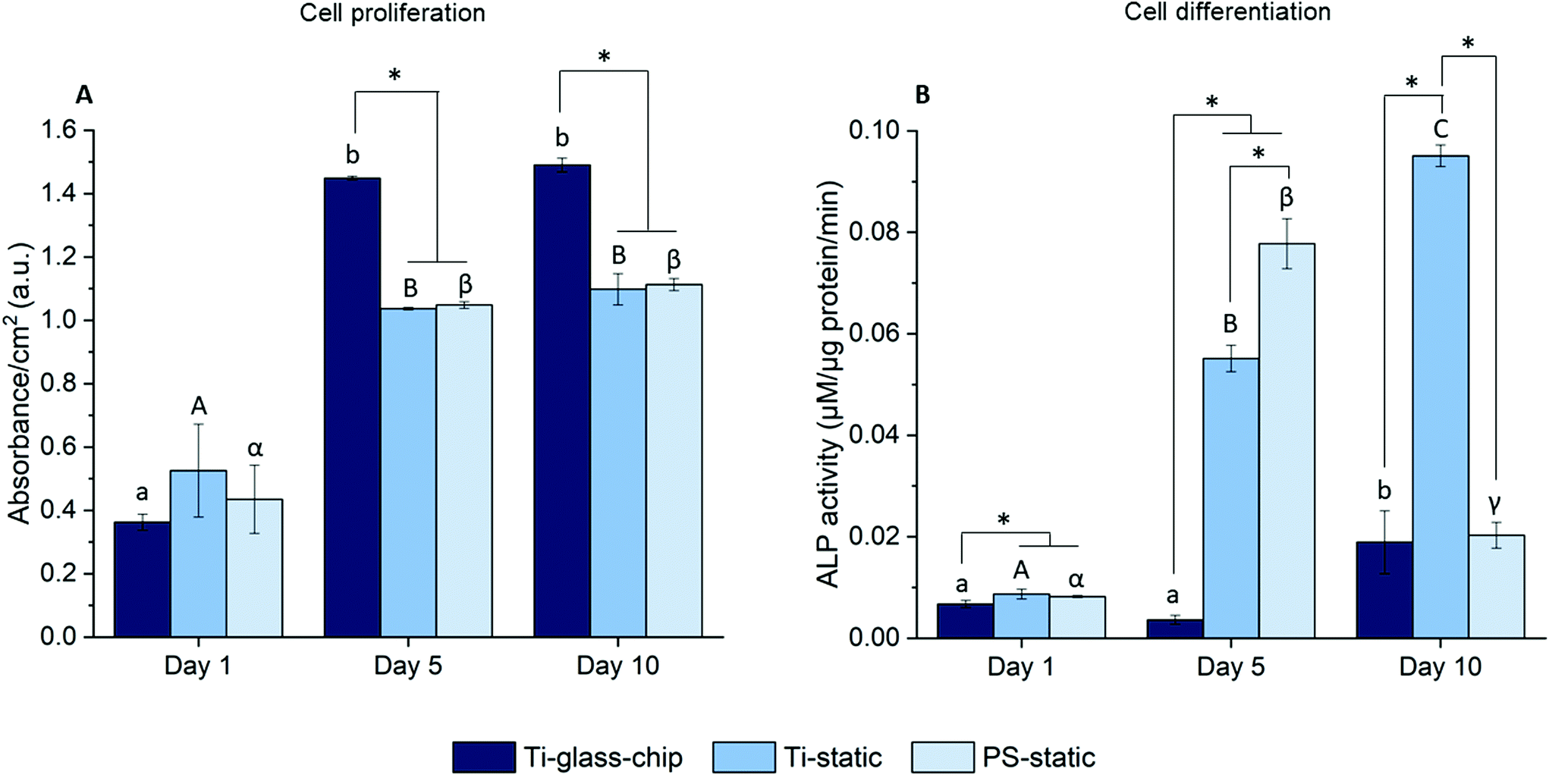 | ||
| Fig. 7 (A) Cell proliferation and (B) differentiation of MC3T3-E1 cells after 1 day, 5 days and 10 days of culture on the Ti-glass-chip, Ti-static and PS-static. Bars labeled with different letters indicate statistically significant differences between subsequent time-points within each condition (p < 0.05). *corresponds to significant differences between the samples at each time-point (p < 0.05). n = 2 for Ti-glass-chip on day 5 and 10. | ||
Regarding cell differentiation, different trends were observed for the different samples over time. For cells grown on Ti-glass-chip, from day 1 to day 5 no increase in ALP activity was observed. However, from day 5 to day 10, ALP activity increased. For cells grown on Ti-static, ALP activity already increased drastically from day 1 to day 5, which continued to day 10. For PS-static, ALP activity peaked on day 5 and decreased on day 10 (Fig. 7B, p-values indicated in ESI Table 2†). Already on day 1, significant differences were observed between the samples, showing lower levels in ALP activity for cells grown on Ti-glass-chip compared to cells grown on Ti-static (p = 0.003) and on PS-static (p = 0.008). At this time point, no differences in ALP activity were detected between cells growing on the static samples. On day 5 and 10, larger levels in ALP activity were observed for cells grown on Ti-static and PS-static in comparison to Ti-glass-chip. On day 5 in particular, compared to the cells grown on-chip, ∼15 and 21 times higher levels in ALP activity were shown for Ti-static (p = 0.001) and PS-static (p < 0.0005), respectively. When comparing the static samples, the cells directly grown on PS-static showed significantly higher levels of ALP activity (p = 0.01) than the Ti-static samples. On day 10, cells growing on Ti-static still showed significantly higher levels of ALP activity compared to the cells grown on Ti-glass-chip (p = 0.001). In addition, ALP activity levels from cells grown on PS-static dropped, reaching similar levels in ALP activity as for the cells grown on Ti-glass-chip, showing much lower levels compared to Ti-static (p = 0.001).
3.3. Comparison of Ti-PDMS-chip and Ti-glass-chip
Two approaches to integrate Ti discs were explored. Although both the Ti-PDMS-chip and Ti-glass-chip allowed for viable cultures on-chip, each approach comes with its own advantages and disadvantages. To allow larger-scale use of the devices and successful on-chip cultures, certain requirements need to be considered. These include, but are not limited to, the inertness of the materials, robustness of the fabrication processes, and the integration with standard biochemical assays to assess cell behavior. Table 1 briefly describes the advantages and challenges of the Ti-PDMS-chip and the Ti-glass-chip.| Ti-PDMS-chip | Ti-Glass-chip | ||||
|---|---|---|---|---|---|
| Advantage | Disadvantage or challenge | Advantage | Disadvantage or challenge | ||
| Materials | Handling and inertness of the materials in contact with the cells | PDMS is easy to prepare and readily moldable into a large variety of channel geometries.32–34 | PDMS may absorb small hydrophobic molecules,35,36 it may release PDMS compounds affecting cell behavior37,38 and medium may evaporate since PDMS is gas permeable.39 | Mostly standard materials used, among which glass is inert. Double-sided tape has shown to be compatible with on-chip biological applications40,41 and both double-sided tape and optical adhesive were considered suitable for our application, as no adverse effects on cell viability were observed during the cytotoxicity studies and over the period of 10 days on-chip. | N/A |
| Costs | Both PDMS and the ABS polymer used for the additively manufactured fixture have low material costs. | The fabrication of the SU-8/silicon master mold is relatively expensive.41,42 Costs could be reduced by molding the PDMS off a 3D printed master. | Relatively inexpensive materials.41,42 | N/A | |
| Fabrication | Equipment needed | Fabrication of the additively manufactured fixture is done almost fully automatically by the machine, reducing labor-intensive work. | Fabrication of the SU-8/silicon master mold relies on photolithography, which requires specialized equipment, a cleanroom environment and trained personnel. | The cutter plotter is a relatively affordable machine, easy to use and offers a good option for prototyping for microfluidic applications.40,41,43 | The laser cutter is a relatively specialized equipment only available in some facilities. |
| Fabrication process and results | Soft lithography allows for high resolution microfluidic channels. Once the silicon master mold is prepared, it can be reused. This also applies for the additively manufactured fixture. | It takes multiple fabrication steps upon completion of the silicon master mold (∼3.5 hours) and additionally ∼2.5 hours for molding and preparing one set of 14 PDMS microfluidic channels (i.e. each silicon master mold contained 14 patterns). | The fabrication does not require a clean room environment. Since the chip is made from (semi-) transparent materials, the Ti and microfluidic channel can be visualized directly, which allows for quality control during the entire experiment. | Delicate process, requires multiple manual aligning steps with long (e.g. 24 hours to cure silicone coating) waiting times. Compared to the Ti-PDMS-chip, the steps are time consuming (∼4.5 hours to create 14 chips), of which none of the components can be re-used. In addition, the edges of the channels may be less sharp compared to the Ti-PDMS-chip, which may be of extra importance when working with small channel dimensions. | |
| Integration of Ti discs | The additively manufactured fixture allows for accurate and convenient alignment of a microfluidic channel onto a Ti disc. | The additively manufactured fixture needs to be carefully closed, ensuring a tight seal between PDMS and a Ti disc, without creating back-pressure in the channel. | N/A | Compared to the Ti-PDMS-chip, the integration of Ti is more time consuming, since each disc is glued one-by-one. | |
| Integration with standard biochemical assays | Microscopy and plate reader assays | The additively manufactured fixture can be opened and a Ti disc can be easily transferred to e.g. a well plate, which facilitates integration with standard biochemical assays. | Given the thickness of the full chip, for high quality imaging, the Ti disc needs to be removed from the additively manufactured fixture. This adds another step and potential risk of damaging the cell surface. | The chip format (i.e. standard microscopic slide) and its dimensions (i.e. thin) allow for direct imaging under the microscope (i.e. within the working distance). | The Ti disc cannot be removed from the chip and therefore requires optimization of standard biochemical assays to assess cell behavior. |
4. Discussion
Thorough evaluation of biomaterials is a key step in the selection of successful bone implants. Current in vitro testing of novel biomaterials is largely driven by protocols provided by the International Organization of Standardization (ISO), which focus on biocompatibility and cytotoxicity. However, these approaches have shown to be inadequate to accurately predict in vivo outcomes, highlighting the need for alternative and more reliable in vitro testing platforms.3 The aim of this work was to develop a microfluidic tool that could be used to study the biological properties of clinically relevant biomaterials for bone repair. This was achieved by integrating medical grade titanium in a microenvironment that more closely resembles the in vivo bone niche, particularly by providing fluid flow and confining the cells in micrometric channels.8Two approaches to integrate medical grade titanium (Ti6Al4V; referred to as Ti) were explored using two different main materials (PDMS or glass) and two different channel designs (with similar shear stress in the culture areas). Both culture areas were designed to be large enough to allow a sufficient amount of cells for accurate biochemical analysis, this being possible with two smaller channels in the case of Ti-PDMS-chip and with a larger chamber-like channel in the Ti-glass chip. These two approaches were compared in terms of the handling and inertness of the materials, fabrication processes and integration with standard methods used to evaluate cell behavior. Although both approaches have their own benefits and challenges, given the overall findings (Table 1), the Ti-glass-chip was selected for longer-term cell viability, proliferation and differentiation studies. The rationale for choosing the Ti-glass-chip approach was based on the material properties and accessibility of the approach (i.e. access to materials and equipment needed to fabricate the chip).
In the PDMS chip, the microfluidic channel consists of a PDMS roof and sidewalls, placed onto the Ti discs. Although PDMS is a commonly used material in the field, controversy exists on its biological inertness.34,37 Particularly, in our recent work it was shown that PDMS oligomers enhanced the differentiation of MC3T3-E1 preosteoblast-like cells.38 Whereas the Ti-PDMS-chip comes with the advantage of reusing several of the fabricated components (i.e. silicon master mold and additively manufactured fixture), specialized equipment and a cleanroom setting are required. Although the Ti-glass-chip contains only single use components and involves a more delicate fabrication process, most of the components of the Ti-glass-chip are available in regular laboratories, making this approach much more accessible. Moreover, even though images could be obtained on both types of chips, the Ti disc integrated in the Ti-PDMS-chip had to be transferred to a well plate to ensure sharp images. Whereas this brings the advantage of using standard off-chip biochemical assays and protocols, it requires an additional step and risk of damaging the cell culture area. In contrast, the microscope slide format and optical properties of the Ti-glass-chip allowed for direct imaging. However, the fact that it is not possible to remove the Ti disc from the Ti-glass-chip, could be considered challenging because conventional biochemical methods are often optimized for macroscale cultures that usually have higher cell number and volume of reagents than on-chip platforms. Nevertheless, in the current study, in order to perfuse the majority of the Ti, relatively large channel dimensions were chosen, compromising between scalability and measurability when using colorimetric methods. This means that only a slight modification of the standard biochemical methods to evaluate cell behaviour was required, namely further dilution of the cell lysate (needed for ALP, BCA and LDH assays) on-chip to match the volume in the well plates.
Evaluating biomaterials for bone repair on-chip is still in early stage. To the best of our knowledge, only two other studies have assessed osteoblast behaviour on a titanium alloy using a microfluidic approach. The first study involved a co-culture approach to assess the effect of prokaryotic cells present on Ti surfaces on MC3T3-E1 cell adhesion and viability.44 In the second study, Stamp et al. used a microfluidic system (previously developed20) to study the effect of shear stress, temperature and pH on the adhesion of Saos-2 osteosarcoma cells to a titanium alloy.23 The cells were seeded at a predetermined temperature and pH and subsequently exposed to flow, which was induced by acoustic streaming. The results showed that the cells were more affected (i.e. higher cell detachment) by extreme temperatures and pH when exposed to the flow regime than when maintained under static conditions. Noteworthy, in this device, the titanium alloy served as the lid of the flow chamber, meaning that the cells were facing the bottom of the flow chamber. In static experiments performed over 60 minutes, the authors reported on gravity-induced detachment of Saos-2 osteosarcoma cells from the titanium alloy. Although this study provides valuable insights into cell–biomaterial interactions under dynamic conditions, cell behavior was only assessed after a short period of time. In addition, apart from cell attachment, no additional characteristics were evaluated. Based on their findings, our chips were flipped in such a manner that the Ti disc was positioned at the bottom of the chip and the cells were on top of the Ti.
MC3T3-E1 cells were successfully grown on Ti integrated on-chip, both in PDMS-based and glass-based chips (Fig. 4 and 6). When assessing cell morphology on the Ti-PDMS-chip after 1 day of culture, a clear difference was observed between cells cultured on-chip, compared to cells cultured on either of the static conditions. On-chip the cells showed a more elongated morphology and orientation along the direction of the flow, compared to the cells grown on either of the static conditions (Fig. 5A and B). Cell morphology is key in cellular function and it is well known that osteoblast-like cells respond to fluid induced shear stress. In fact, multiple studies have already reported on similar results in morphological changes and orientation due to fluid flow.45,46 Even though the overall shear stress levels experienced by the cells in the cell culture areas of both chip designs were similar, cells grown on the Ti-PDMS-chip showed this tendency of cell elongation and alignment to a larger extent than cells grown on the Ti-glass-chip. A possible explanation for these observations may be related to the microfluidic channel design and resulting shear stress distribution within each of the culture areas in the Ti-PDMS-chip and Ti-glass-chip. Since the dimensions of the cell culture area of the Ti-glass-chip were larger than that of the Ti-PDMS-chip, the change in shear stress was more gradual along the entire cell culture area (ESI Fig. 3 and 4†). In contrast, the more abrupt change in shear stress experienced by the cells cultured in the Ti-PDMS-chip, resulting in the majority of the cells being exposed to the same shear stress value, could have caused the more defined morphology and orientation of cells along the flow of cell culture media. In addition, in the Ti-glass-chip the rather narrow inlet widens towards a relatively wide chamber-like structure, meaning that the areas surrounding the center of the cell culture area are shielded from the original direction of the flow.
Noteworthy, previously it has been shown that osteoblast morphology and alignment can also be affected by topographical characteristics of the substrate, particularly when these features exhibit similar dimensions as the cellular microenvironment.47,48 However, since no differences in cell morphology were observed between cells grown on Ti-static and PS-static for 24 hours (Fig. 6A), the changes in cell morphology and alignment were likely affected by the fluid flow, rather than due to contact guidance by the biomaterial surface.
For more in-depth evaluations, the Ti-glass-chip was selected to study cell viability, proliferation and differentiation over a period of 10 days. Both cell viability and cell proliferation studies confirmed successful culture of MC3T3-E1 cells on-chip (Fig. 6 and 7). Whereas cell proliferation on-chip became significantly elevated compared to both static conditions from day 5 onwards, ALP activity reached much higher levels in the static conditions, indicating that cell proliferation is the dominating process on this chip. These findings are in agreement with the general consensus that distinct proliferation and differentiation stages exist during osteoblast maturation and in line with previous work, which reported enhanced cell proliferation of osteogenic cell types when exposed to dynamic culture conditions.49–51 A potential explanation for this observation is an enhanced supply of nutrients and oxygen to the cells and more effective waste removal from the cells when being continuously perfused (i.e. complete medium renewal approximately every 20 minutes).
Physical stimulations, such as shear stress are often mentioned as key factors for the differentiation of osteogenic cells. Whereas the physiological shear stress in bone is reported to fall between 0.06–30 dyne per cm2, a larger range in shear stress values are reported to impact osteogenic response.5,7 For example, when MC3T3-E1 cells were cultured in a glass microfluidic device (coated with poly-L-lysine and matrigel) for a period of 10 days at a shear stress level of 7 × 10−2 dyne per cm2, elevated levels in ALP activity were shown when compared to the static control (i.e. well plate). However, this work had two major experimental differences with our study, including the modification of MC3T3-E1 cells with a green fluorescent protein and the addition of other osteogenic factors such as dexamethasone and bone morphogenetic protein 2 (BMP-2) in the medium.52 In another study, in which a PDMS-glass microfluidic system was fabricated to evaluate the effect of low fluid-flow induced stress (i.e. 1.5 × 10−5–5 × 10−4 dyne per cm2) on proliferation and differentiation of MC3T3-E1 cells it was shown that not only proliferation, but also differentiation was promoted, however only in this range of low fluid-induced stress.53 More recently, Babaliari reported on a glass-polymethyl methacrylate microfluidic system to assess MC3T3-E1 cell behavior on collagen matrices. In their work, in which the estimated shear stress exerted on the cells was either 0.3 or 0.5 dyne per cm2, both cell proliferation and differentiation were enhanced after 7 days when compared to the static control (i.e. collagen-coated glass). Interestingly, for cell proliferation, the only significant differences were observed for cells cultured at 0.5 dyne per cm2 and for cell differentiation for cells cultured at 0.3 dyne per cm2.46 In the current study, the cell culture area of the Ti-glass-chip had a maximum wall shear stress of 2.5 × 10−2 dyne per cm2, according to COMSOL simulations when a flow rate of 2 μl min−1 was maintained. This shear stress value falls within the same order of magnitude as multiple of the above-mentioned studies and does therefore at first glance not seem likely to have attributed to the results obtained in our device. However, comparing our findings to work from Yu et al., who showed that values over 4 × 10−3 dyne per cm2 inhibited proliferation and differentiation, suggests that no conclusive statements regarding the effect of shear stress on the proliferation and differentiation can yet be made.53
Apart from enhanced transport of nutrients and waste removal, continuous perfusion may also cause depletion of autocrine and paracrine factors that support differentiation. Various cell-secreted factors have been reported to be involved in the regulation of osteoblast function.54,55 For MC3T3-E1 cells in particular, a number of cytokines and hormones linked to cell differentiation are known to be secreted, among these being insulin-like growth factor (IGF) I, IGF-II and bone morphogenetic proteins (BMPs) BMP-2 and BMP-4.56–58 In line with this, previous works have suggested that a depletion of autocrine and paracrine factors can affect cell survival and differentiation of mouse embryonic stem cells in microfluidic systems.59–61 This effect of autocrine and paracrine signaling could be further explored by decreasing the flow rate and/or recirculating the cell culture medium.
Although the selected designs and conditions are not directly translatable to the in vivo bone environment, our findings demonstrate the power of in vitro cell culture conditions and how these may drastically affect testing outcomes. However, at present the approach of integrating biomaterials in microfluidic systems is still in its infancy and for the field to advance, validation studies in which the biological evaluation of biomaterials on-chip is compared to standard in vitro and in vivo methods are imperative.
5. Conclusions
In this study, microfluidic chips integrating medical grade titanium (Ti6Al4V) were developed and assessed for their potential to study the biological properties of this biomaterial under dynamic conditions (i.e. continuous perfusion). Both a Ti-PDMS-chip and Ti-glass-chip were fabricated and showed to be promising for on-chip evaluation of medical grade titanium. Short-term culture on both chips revealed high cell viability and a more elongated morphology for cells cultured under dynamic conditions, with a clear tendency to align to the direction of the flow. After considering the inertness of the materials, robustness of the fabrication processes, the accessibility of the approach in terms of materials and equipment and lastly the integration with standard biochemical assays, the Ti-glass-chip was selected for longer-term cell experiments. These studies showed successful culture of MC3T3-E1 cells over the 10 days course of the experiments and revealed that cell proliferation was the dominating process over cell differentiation on this chip. Overall, this study provides a step towards the development of more complex in vitro methods, which could potentially offer a more reliable option to screen the biological properties of biomaterials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Milena De Albuquerque Moreira and Hugo Nguyen for their assistance with the chip fabrication processes and Anna Blasi Romero for the valuable discussions. GM acknowledges the Swedish Council Formas (#2016-00781), Swedish Council Vetenskapsrådet (#2017-05051) and Göran Gustafsson's Foundation (#1841) for funding this research. MT acknowledges funding from the Knut and Alice Wallenberg Foundation (#2016-0112).References
- J. Henkel, M. A. Woodruff, D. R. Epari, R. Steck, V. Glatt, I. C. Dickinson, P. F. M. Choong, M. A. Schuetz and D. W. Hutmacher, Bone Res., 2013, 1, 216–248 CrossRef CAS.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS.
- G. Hulsart-Billström, J. I. Dawson, S. Hofmann, R. Müller, M. J. Stoddart, M. Alini, H. Redl, A. El Haj, R. Brown, V. Salih, J. Hilborn, S. Larsson and R. O. C. Oreffo, Eur. Cells Mater., 2016, 31, 312–322 CrossRef.
- U. H. Lerner, Semin. Orthod., 2012, 18, 237–248 CrossRef.
- S. Weinbaum, S. C. Cowin and Y. Zeng, J. Biomech., 1994, 27, 339–360 CrossRef CAS.
- I. Owan, D. B. Burr, C. H. Turner, J. Qiu, Y. Tu, J. E. Onyia and R. L. Duncan, Am. J. Physiol., 1997, 273, C810–C815 CrossRef CAS.
- L. Wang, S. P. Fritton, S. Weinbaum and S. C. Cowin, J. Biomech., 2003, 36, 1439–1451 CrossRef.
- G. Mestres, R. A. Perez, N. L. D'Elia and L. Barbe, Biomed. Phys. Eng. Express, 2019, 5, 032001 CrossRef.
- M. Domenech, H. Yu, J. Warrick, N. M. Badders, I. Meyvantsson, C. M. Alexander and D. J. Beebe, Integr. Biol., 2009, 1, 267–274 CrossRef CAS.
- N. Tandon, D. Marolt, E. Cimetta and G. Vunjak-Novakovic, Biotechnol. Adv., 2013, 31, 1020–1031 CrossRef CAS.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- B. D. Riehl and J. Y. Lim, Cells, 2012, 1, 1225–1245 CrossRef CAS.
- S. Kou, L. Pan, D. van Noort, G. Meng, X. Wu, H. Sun, J. Xu and I. Lee, Biochem. Biophys. Res. Commun., 2011, 408, 350–355 CrossRef CAS.
- B. Harink, S. Le Gac, D. Barata, C. Van Blitterswijk and P. Habibovic, Lab Chip, 2014, 14, 1816–1820 RSC.
- K. Middleton, S. Al-Dujaili, X. Mei, A. Günther and L. You, J. Biomech., 2017, 59, 35–42 CrossRef CAS.
- B. Harink, S. Le Gac, D. Barata, C. van Blitterswijk and P. Habibovic, Electrophoresis, 2015, 36, 475–484 CrossRef CAS.
- N. Jusoh, S. Oh, S. Kim, J. Kim and N. L. Jeon, Lab Chip, 2015, 15, 3984–3988 RSC.
- J. A. Burdick, A. Khademhosseini and R. Langer, Langmuir, 2004, 20, 5153–5156 CrossRef CAS.
- C. Moraes, G. Wang, Y. Sun and C. A. Simmons, Biomaterials, 2010, 31, 577–584 CrossRef CAS.
- A. Hartmann, M. Stamp, R. Kmeth, S. Buchegger, B. Stritzker, B. Saldamli, R. Burgkart, M. F. Schneider and A. Wixforth, Lab Chip, 2014, 14, 542–546 RSC.
- F. Abeille, F. Mittler, P. Obeid, M. Huet, F. Kermarrec, M. E. Dolega, F. Navarro, P. Pouteau, B. Icard, X. Gidrol, V. Agache and N. Picollet-D'Hahan, Lab Chip, 2014, 14, 3510–3518 RSC.
- B. Zhou, X. Gao, C. Wang, Z. Ye, Y. Gao, J. Xie, X. Wu and W. Wen, ACS Appl. Mater. Interfaces, 2015, 7, 17181–17187 CrossRef CAS.
- M. E. M. Stamp, A. M. Jötten, P. W. Kudella, D. Breyer, F. G. Strobl, T. M. Geislinger, A. Wixforth and C. Westerhausen, Diagnostics, 2016, 6, 38 CrossRef CAS.
- D. Barata, E. Provaggi, C. Van Blitterswijk and P. Habibovic, Lab Chip, 2017, 17, 4134–4147 RSC.
- M. Kaur and K. Singh, Mater. Sci. Eng., C, 2019, 102, 844–862 CrossRef CAS.
- P. M. Hinderliter, K. R. Minard, G. Orr, W. B. Chrisler, B. D. Thrall, J. G. Pounds and J. G. Teeguarden, Part. Fibre Toxicol., 2010, 7, 36 CrossRef CAS.
- C. Wang, B. M. Baker, C. S. Chen and M. A. Schwartz, Arterioscler. Thromb. Vasc. Biol., 2013, 33, 2130–2136 CrossRef CAS.
- J. H. C. Wang, E. S. Grood, J. Florer and R. Wenstrup, J. Biomech., 2000, 33, 729–735 CrossRef CAS.
- A. Khakbaznejad, B. Chehroudi and D. M. Brunette, J. Biomed. Mater. Res., Part A, 2004, 70, 206–218 CrossRef CAS.
- A. W. Feinberg, W. R. Wilkerson, C. A. Seegert, A. L. Gibson, L. Hoipkemeier-Wilson and A. B. Brennan, J. Biomed. Mater. Res., Part A, 2008, 86, 522–534 CrossRef.
- M. Versaevel, T. Grevesse and S. Gabriele, Nat. Commun., 2012, 3, 1–11 Search PubMed.
- D. C. Duffy, J. C. Mcdonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. Ijzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482, 323–328 CrossRef CAS.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- S.-S. D. Carter, A.-R. Atif, S. Kadekar, I. Lanekoff, H. Engqvist, O. P. Varghese, M. Tenje and G. Mestres, Organs-on-a-Chip, 2020, 2, 100004 CrossRef.
- Y. S. Heo, L. M. Cabrera, J. W. Song, N. Futai, Y.-C. Tung, G. D. Smith and S. Takayama, Anal. Chem., 2007, 79, 1126–1134 CrossRef CAS.
- P. Nath, D. Fung, Y. A. Kunde, A. Zeytun, B. Branch and G. Goddard, Lab Chip, 2010, 10, 2286–2291 RSC.
- T. E. Winkler, M. Feil, E. F. G. J. Stronkman, I. Matthiesen and A. Herland, Lab Chip, 2020, 20, 1212–1226 RSC.
- D. I. Walsh III, D. S. Kong, S. K. Murthy and P. A. Carr, Trends Biotechnol., 2017, 35, 383–392 CrossRef.
- S. R. A. Kratz, C. Eilenberger, P. Schuller, B. Bachmann, S. Spitz, P. Ertl and M. Rothbauer, Sci. Rep., 2019, 9, 9287 CrossRef CAS.
- J. H. Lee, H. Wang, J. B. Kaplan and W. Y. Lee, Acta Biomater., 2010, 6, 4422–4429 CrossRef CAS.
- S. M. Ponik, J. W. Triplett and F. M. Pavalko, J. Cell. Biochem., 2007, 100, 794–807 CrossRef CAS.
- E. Babaliari, G. Petekidis and M. Chatzinikolaidou, Bioengineering, 2018, 5, 66 CrossRef CAS.
- B. Zhu, Q. Lu, J. Yin, J. Hu and Z. Wang, Tissue Eng., 2005, 11, 825–834 CrossRef CAS.
- C. Wu, M. Chen, T. Zheng and X. Yang, Biomed. Mater. Eng., 2015, 26, S155–S164 Search PubMed.
- L. D. Quarles, D. A. Yohay, L. W. Lever, R. Caton and R. J. Wenstrup, J. Bone Miner. Res., 1992, 7, 683–692 CrossRef CAS.
- S. Sinlapabodin, P. Amornsudthiwat, S. Damrongsakkul and S. Kanokpanont, Mater. Sci. Eng., C, 2016, 58, 960–970 CrossRef CAS.
- J. van den Dolder, G. N. Bancroft, V. I. Sikavitsas, P. H. M. Spauwen, J. A. Jansen and A. G. Mikos, J. Biomed. Mater. Res., Part A, 2003, 64, 235–241 CrossRef.
- K. Jang, K. Sato, K. Igawa, U. Chung and T. Kitamori, Anal. Bioanal. Chem., 2008, 390, 825–832 CrossRef CAS.
- W. Yu, H. Qu, G. Hu, Q. Zhang, K. Song, H. Guan, T. Liu and J. Qin, PLoS One, 2014, 9, e89966 CrossRef.
- F. J. Hughes, W. Turner, G. Belibasakis and G. Martuscelli, Periodontol. 2000, 2006, 41, 48–72 CrossRef.
- Y. Han, X. You, W. Xing, Z. Zhang and W. Zou, Bone Res., 2018, 6, 1–11 CrossRef CAS.
- S. Amarnani, H. L. Merriman, T. A. Linkhart, D. J. Baylink and S. Mohan, J. Bone Miner. Res., 1993, 8, 157–165 CrossRef CAS.
- M. Suzawa, Y. Takeuchi, S. Fukumoto, S. Kato, N. Ueno, K. Miyazono, T. Matsumoto and T. Fujita, Endocrinology, 1999, 140, 2125–2133 CrossRef CAS.
- K. M. Thrailkill, S. R. Siddhanti, J. L. Fowlkes and L. D. Quarles, Bone, 1995, 17, 307–313 CrossRef CAS.
- D. Ellison, A. Munden and A. Levchenko, Mol. Biosyst., 2009, 5, 1004–1012 RSC.
- K. Blagovic, L. Y. Kim and J. Voldman, PLoS One, 2011, 6, e22892 CrossRef CAS.
- E. L. Moreno, S. Hachi, K. Hemmer, S. J. Trietsch, A. S. Baumuratov, T. Hankemeier, P. Vulto, J. C. Schwamborn and R. M. T. Fleming, Lab Chip, 2015, 15, 2419–2428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00964d |
| This journal is © The Royal Society of Chemistry 2020 |