
Probing enantioselectivity in rhodium-catalyzed Si–C bond cleavage to construct silicon-stereocenters: a theoretical study†
Zhaoyuan
Yu‡
 b,
Tao
Zhang‡
b,
Ruopeng
Bai
b and
Yu
Lan
*ab
b,
Tao
Zhang‡
b,
Ruopeng
Bai
b and
Yu
Lan
*ab
aDepartment College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450001, China. E-mail: lanyu@cqu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400030, China
First published on 14th December 2018
Abstract
The rhodium-catalyzed asymmetric synthesis of dibenzooxasilines developed by Hayashi and co-workers provides an efficient method to construct tetraorganosilicon stereocenters. In the present study, density functional theory (DFT) calculations were performed to investigate the mechanism and enantioselectivity of this reaction. Theoretical calculations indicate that the mechanism involves the initial formation of an aryloxorhodium complex followed by Rh–Si exchange to afford an arylrhodium complex. The favorable oxidative addition/reductive elimination to cleave one Si–C(phenyl) bond from the arylrhodium complex determines the enantioselectivity. The enantioselectivity originates from the silyl moiety extruding from the phenyl ring on the rhodium atom in the reductive elimination transition state.
Introduction
Organosilicon compounds generally have high thermal and chemical stabilities and are readily available, relatively inexpensive and non-toxic; thus, as organometallic nucleophiles, they possess many advantages over other organometallic compounds.1 Compared with other organometallic nucleophiles, organosilicon compounds are weak nucleophiles because the carbon-silicon bond is only weakly polarized.2 Because of the above-mentioned special properties, organosilicon compounds are widely used in various transition-metal (e.g., nickel,3 copper,4 iridium,5 palladium,6 rhodium,7 ruthenium,8 iron9)-catalyzed organic reactions. For example, Chatani and co-workers reported rhodium-catalyzed intermolecular coupling of 2-trimethylsilylphenyl boronic acids with alkynes by the cleavage of an inactive C(sp3)–Si bond.10 Xi and coworkers developed an efficient process involving Rh-catalyzed selective cleavage of a C(sp3)–Si bond and consequent cis-addition to a C–C unsaturated bond, affording silatricyclic compounds.11 Murakami and coworkers presented a unique palladium-catalyzed intermolecular σ-bond exchange reaction between C–C and C–Si σ-bonds.12Various methods are available for the preparation of carbon-stereogenic compounds.13 In contrast, there are very limited reports on the synthesis of organosilicon compounds containing tetraorganosilicon stereocenters.14 It is well known that chiral silicon compounds do not exist in nature. Therefore, broadening the scope of accessible chiral organosilicon compounds is highly desirable. The pioneering work on the asymmetric construction of chiral silicon centers was reported by the Corriu15 and Kumada groups in the 1970s.16 Since then, a number of catalytic systems have been developed for the synthesis of chiral silicon compounds.17 For example, Nishihara and coworkers reported palladium-catalyzed enantioselective arylation of secondary silanes.18 Takai and coworkers reported asymmetric rhodium-catalyzed Si–H silylation to synthesize spirosilabifluorene derivatives.19
Recently, Hayashi and coworkers reported a straightforward process for the formation of silicon-stereogenic organosilanes using rhodium-catalyzed enantioselective transmetallation of prochiral organosilicon compounds (Scheme 1).20 A combination of [Rh(OH)(coe)2]2 and the (S,S)-Me-Duphos ligand afforded the product in 89% yield and 91% ee. However, the origin of the enantioselectivity was unclear. From the reaction shown in Scheme 1, formation of the chiral silicon center involves S–C bond cleavage. As we will see, how the Si–C bond is cleaved and how the cleavage is related to the formation of the chiral silicon center are especially interesting. In order to elucidate the key factors in the formation of the chiral silicon center, we carried out density functional theory (DFT) calculations to explore the reaction mechanisms and origins of the enantioselectivity of the reaction shown in Scheme 1.
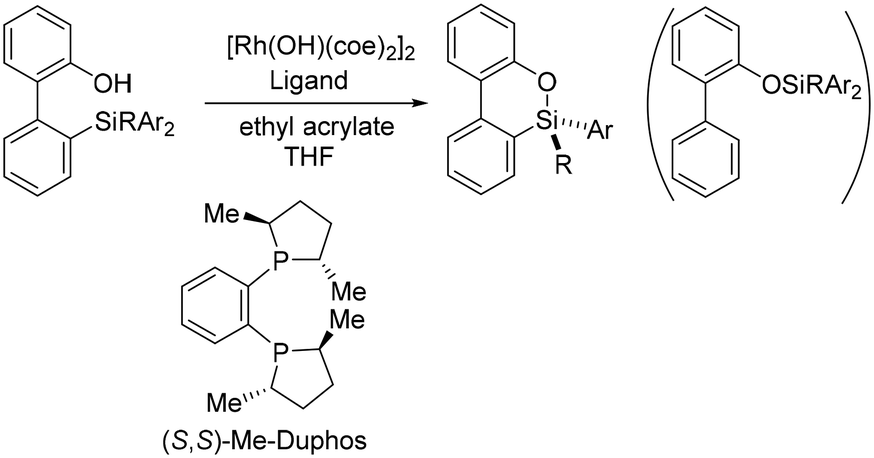 | ||
| Scheme 1 Rhodium-catalyzed asymmetric synthesis of dibenzooxasilines. | ||
Computational methods
All the DFT calculations were carried out with the GAUSSIAN 09 program package.21 Density functional theory at the level of B3LYP22 with the standard 6-31G(d) basis set23 (LanL2DZ basis set24 for rhodium atom) was used for geometry optimizations. Harmonic frequency calculations were performed at the same level of theory for all the stationary points to confirm them as local minima or transition structures and to derive the thermochemical corrections for the enthalpies and Gibbs free energies.The recently developed MN12-L functional25 was used to calculate the single point energies. Solvent (tetrahydrofuran) effects were also considered in the single point energy calculations based on the gas-phase optimized stationary points using the SMD solvation model.26 The larger basis set 6-311+G(d,p) (LANL08f basis set27 for the rhodium atom) was used in the single point energy calculations. The energies given and discussed in this paper are the solvation-corrected MN12-L calculated Gibbs free energies. The NCIPLOT analyses were conducted with Multiwfn28 and VMD.29 The 3D images of the calculated structures were prepared using CYLview.30
Results and discussion
From the reaction shown in Scheme 1, it is convenient to assume that the reaction mechanism involves O-coordination of a substrate molecule to Rh followed by oxidative addition (OA) involving Si–C bond cleavage and then reductive elimination to form an Si–O bond, affording the product molecule. In order to achieve this, two possible pathways must be considered.31Scheme 2 shows the two possible catalytic cycles. Both cycles start with A, a species generated from transmetallation between a monomeric Rh complex and the substrate. In path a (blue line), an Si–C(phenyl) bond is oxidatively added to the Rh(I) metal center of A to afford the Rh(III) intermediate B, which then undergoes Si–O bond-forming reductive elimination, producing the phenylrhodium species C and the dibenzooxasiline product. In path b, an alternative OA, which cleaves the Si–C(biphenyl) bond instead, occurs to afford a different Rh(III) intermediate (D), which undergoes Si–O bond-forming reductive elimination to provide E. The transformation of A to E is in fact achieved via a formal metathesis between the relevant Si–C and Rh–O bonds. From E, oxidative addition of one Si–C(phenyl) bond affords F, followed by reductive elimination, again yielding the phenylrhodium species C and the product. The two pathways join at the phenylrhodium species C. From C, insertion of ethyl acrylate into the Rh–phenyl bond takes place to afford the square-planar intermediate G, from which subsequent protonolysis with another substrate molecule regenerates the active aryloxorhodium species A and completes the catalytic cycle.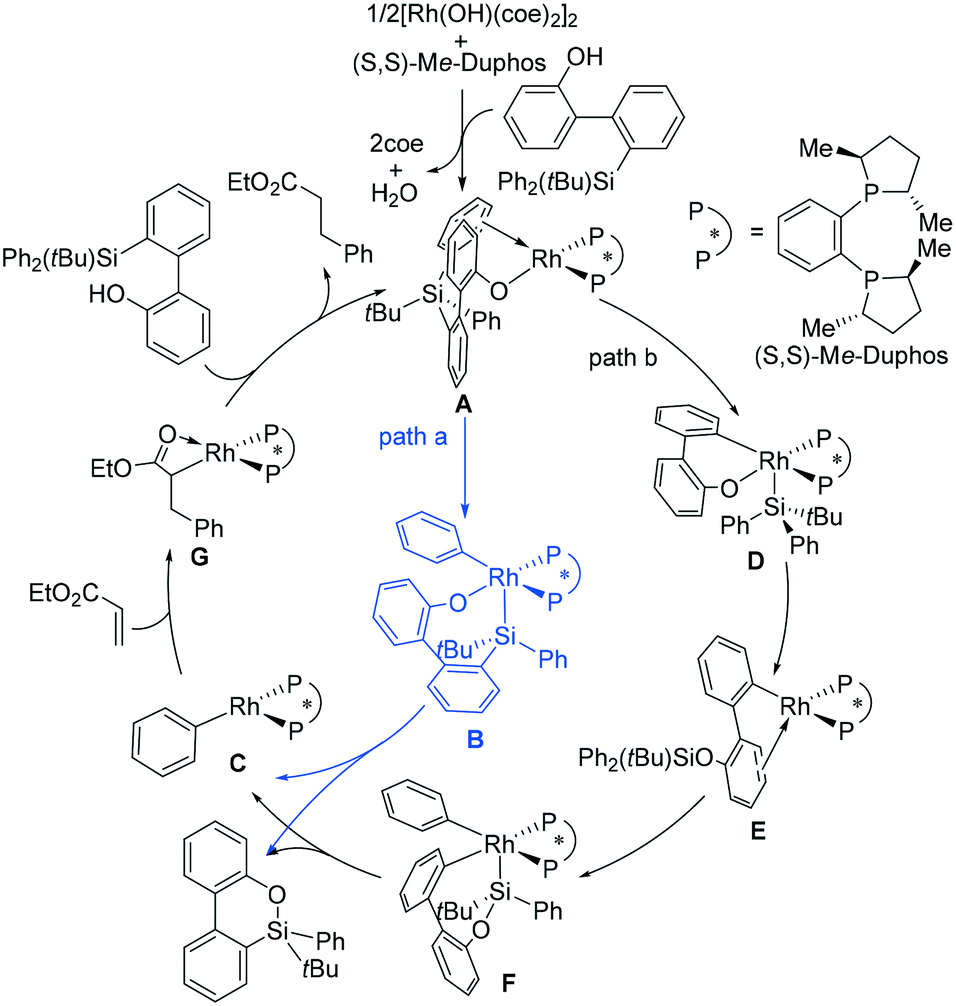 | ||
| Scheme 2 Proposed mechanisms for the Rh-catalyzed synthesis of dibenzooxasilines. | ||
From Scheme 2, the active aryloxorhodium species A is formed by reacting the precatalyst with the substrate in the presence of the chiral ligand (S,S)-Me-Duphos. The formation of A from the precatalyst is indeed very thermodynamically favorable (see Fig. S1 in the ESI†), supporting the mechanistic hypothesis presented in Scheme 2.
Formation of the dibenzooxasiline from aryloxorhodium species A
On the basis of the mechanisms shown in Scheme 2, we first consider the oxidative addition (A → B) and reductive elimination (B → C) processes, as shown in Fig. 1. Experimentally, the (S,S)-Me-Duphos ligand affords the product in 89% yield and 91% ee; therefore, in our calculations, we used (S,S)-Me-Duphos as the ligand. Starting from active species A, in which one of the phenyl rings on the silicon atom coordinates with the Rh(I) center, intramolecular oxidative addition of the Si–C(phenyl) bond can take place via the transition state TSA-B(R) with a 21.2 kcal mol−1 barrier. The square pyramidal Rh(III) intermediate B-R is then formed and is endothermic by 15.3 kcal mol−1. The subsequent reductive elimination occurs via the transition state TSB-C(R) with a free energy barrier of 7.2 kcal mol−1 relative to B-R and releases the (R)-configuration dibenzooxasiline product. Alternatively, the dibenzooxasiline product can be formed from complex Avia a direct transmetallation transition state TSA-C(R) with a 22.4 kcal mol−1 barrier.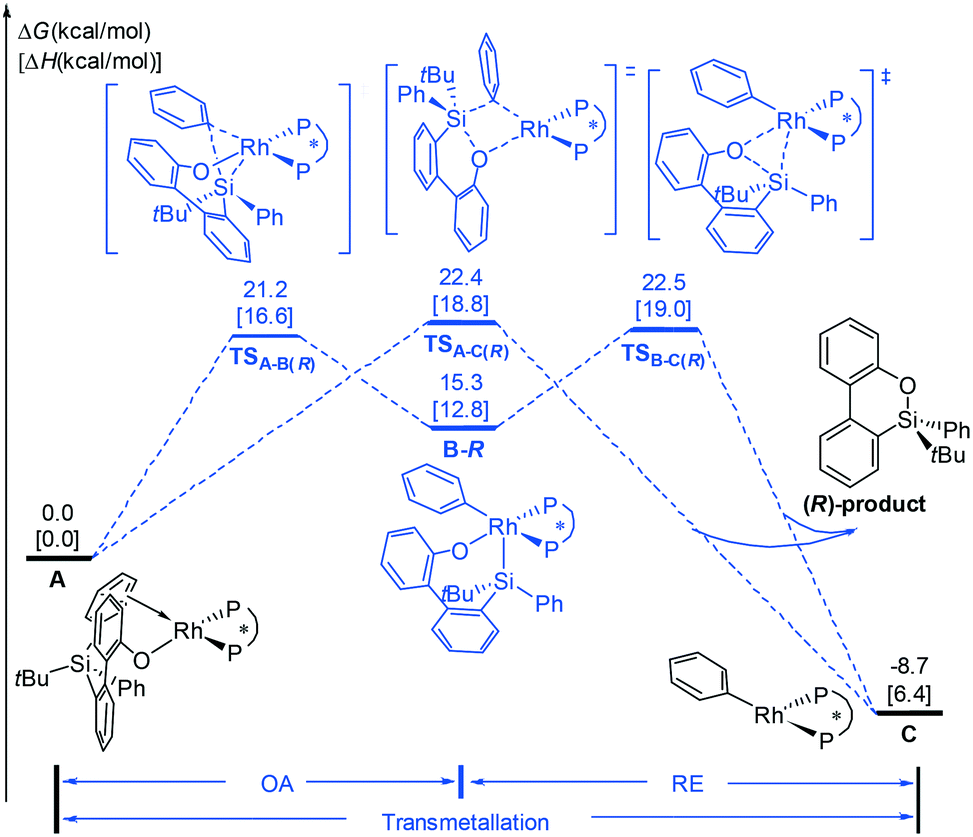 | ||
| Fig. 1 The free energy profiles calculated for the formation of the (R)-configuration dibenzooxasiline product starting from intermediate A. | ||
Rh–Si exchange to form arylrhodium species E followed by transmetallation
According to previous experimental studies32 and computational studies,33 Rh–Si exchange may also occur. In the following sections, our main objective is to obtain deep insight into the cleavage of the Si–C(biphenyl) bond versus the Si–C(phenyl) bond. The calculated energy profiles to form arylrhodium complex E are shown in Fig. 2. The calculation results reveal that Si–C(biphenyl) bond cleavage viaTSA-D requires an activation free energy of 16.7 kcal mol−1 to afford the Rh(III) intermediate D. The generated intermediate D adopts a square pyramidal geometry and is endergonic by 3.0 kcal mol−1. The subsequent reductive elimination process requires an overall barrier of 19.2 kcal mol−1 (from A to TSD-E). On the other hand, the transmetallation pathway via transition state TSA-E to afford the arylrhodium E is 5.3 kcal mol−1 lower in energy than the oxidative addition/reductive elimination pathway. The above results suggest that the Rh–Si exchange is an energy-downhill process and occurs directly via a transmetallation transition state.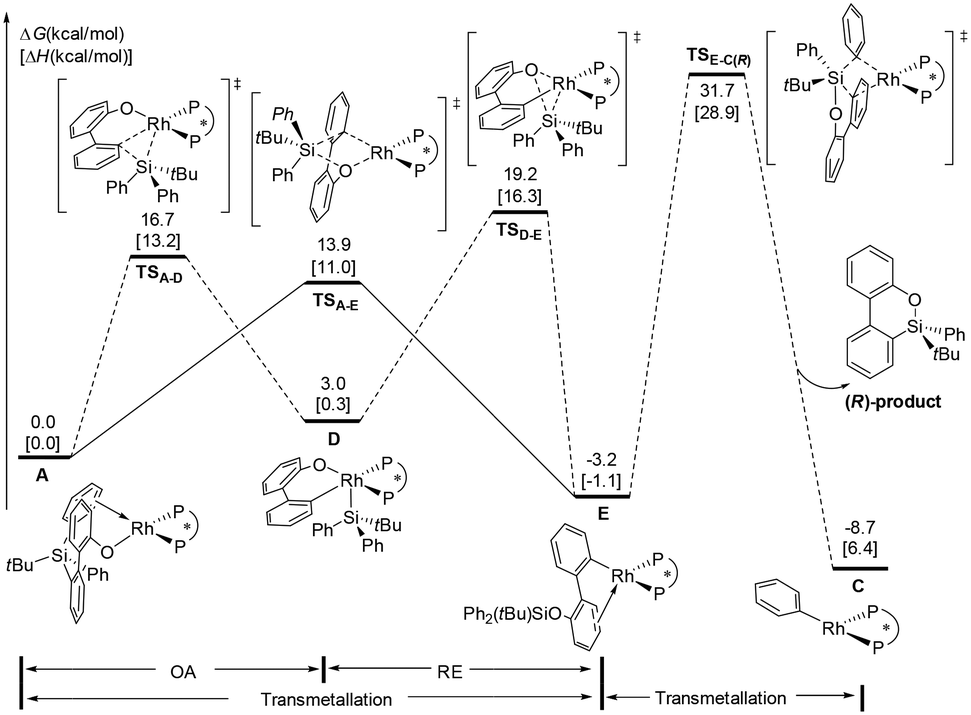 | ||
| Fig. 2 The free energy profiles for Rh–Si exchange/transmetallation from intermediate A. | ||
From the π-coordinated arylrhodium complex E (Fig. 3), transmetallation of one of the two Si–C(phenyl) bonds via transition state TSE-C(R) occurs to afford the (R)-configuration product and phenylrhodium complex C with a very high barrier of 34.9 kcal mol−1. Clearly, this pathway is very energetically unfavorable. The energy profiles calculated for the formation of the byproduct from complex E are presented in the ESI† (Fig. S2).
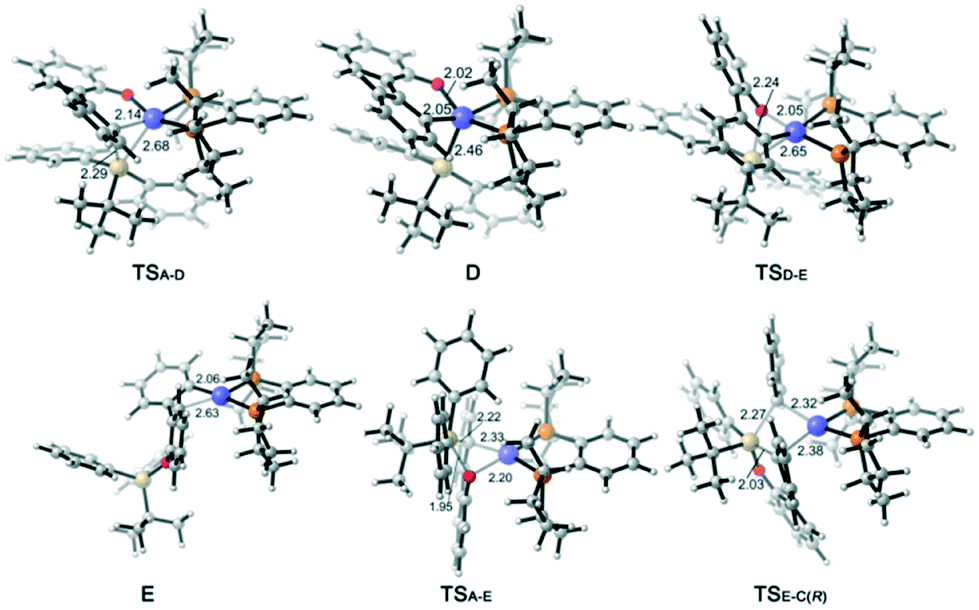 | ||
| Fig. 3 Optimized structures of selected intermediates and transition states; distances are given in angstroms and angles in degrees. | ||
Oxidative addition of the Si–C(phenyl) bond of arylrhodium species E
In addition to the transmetallation pathway, the alternative oxidative addition/reductive elimination pathway was explored. As shown in Fig. 4, a facile oxidative addition process can occur through transition state TSE-F(R) to form intermediate F-R; subsequent reductive elimination affords the phenylrhodium C and the product. This stepwise pathway requires a 14.5 kcal mol−1 overall barrier from intermediate E. Therefore, our calculation results indicate that formation of the (R)-configuration product is favored via oxidative addition/reductive elimination of arylrhodium complex E, and reductive elimination of the Si–C(biphenyl) bond is the enantioselectivity-determining step.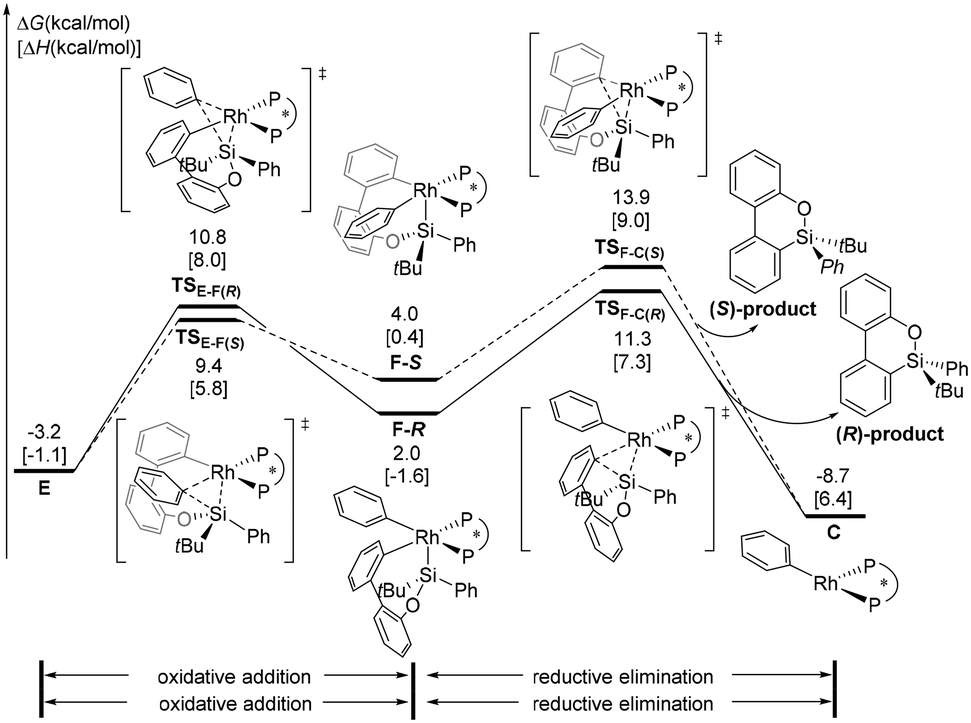 | ||
| Fig. 4 The free energy profiles for the oxidative addition and reductive elimination mechanisms. | ||
The next issue that must be addressed is the origin of enantioselectivity. The free energy profile leading to the (S)-configuration product via the oxidative addition/reductive elimination pathway from arylrhodium complex E is illustrated in Fig. 4. The formation of the Rh(III) intermediate F-Svia transition state TSE-F(S) is slightly more favorable than that by TSE-F(R). However, the reductive elimination transition state TSF-C(S) is 2.6 kcal mol−1 less stable than TSF-C(R). The above results suggest that oxidative addition/reductive elimination leading to the observed (R)-configuration product is more favourable, and the value of ee (96%) predicted based on the energy difference between TSF-C(R) and TSF-C(S) is in good agreement with the experimental result (91%). From phenylrhodium C, the calculated energy profile to regenerate the active species complex A is given in the ESI† (Fig. S3).
In order to obtain details of the origins of the enantioselectivity of this reaction, all possible transition states that result in different chiral products were computed. The chiral environment created by the (S,S)-Me-Duphos ligand can be described according to the quadrant analysis (Fig. 5a).34 The NE and SW quadrants are both occupied by methyl groups on the ligand; thus, these two quadrants are sterically more encumbered than the NW and SE quadrants (where the two hydrogen atoms are oriented toward the Rh center). These chiral pockets created by the ligand are expected to affect the orientations of the substrates. As shown in Fig. 5b, both F-R and F-S adopt square pyramidal geometries, and the silyl group occupies the vertex position. However, in the transition state TSF-C(R), the Si–C(biphenyl) bond forms in the SE quadrant (less steric repulsion quadrant). In contrast, in TSF-C(R), the Si–C(biphenyl) bond forms in the methyl group-blocked SW quadrant. Moreover, in the process of Si–C(biphenyl) bond formation, the silicon atom will approach the carbon atom of the Rh–C(biphenyl) bond accompanied by the silyl moiety, which tends to squeeze the phenyl ring (green phenyl group on the rhodium atom). In the structure of TSF-C(R), the dihedral angle of C1-Rh-P-C2 is 170.7°, which indicates that the phenyl ring on the rhodium is tilted slightly toward the NW quadrant. However, in the transition state TSF-C(S), the silyl group will extrude the phenyl group on the rhodium atom and tilt it toward the NE quadrant (methyl group-blocked quadrant). Consequently, the phenyl group is considerably distorted from the square-planar geometry to minimize steric interaction (the dihedral angle of C1-Rh-P-C2 in TSF-C(S) is 109.6°).
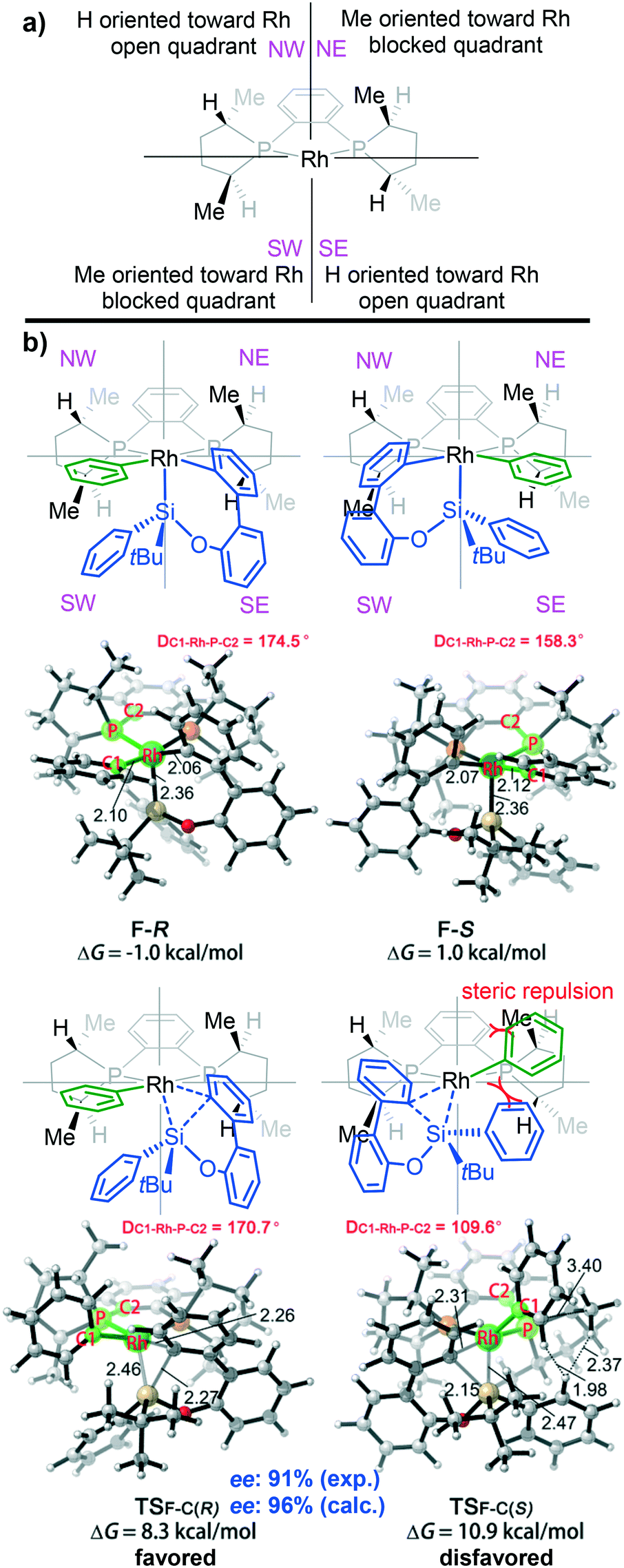 | ||
| Fig. 5 (a) Quadrant analysis of the chiral environment created by the (S,S)-Me-Duphos ligand. (b) Optimized structures of F-R, F-S, TSF-C(R) and TSF-C(S). Distances are given in angstroms and angles in degrees. | ||
To obtain a clearer view of the steric repulsion induced by the spatial environment, NCIPLOT analysis was performed to describe the steric repulsion. As shown in Fig. 6, the higher activation energy barrier of transition state TSF-C(S) can be rationalized by the labelled steric repulsion between the phenyl groups on the silicon atom and the phosphine ligand.
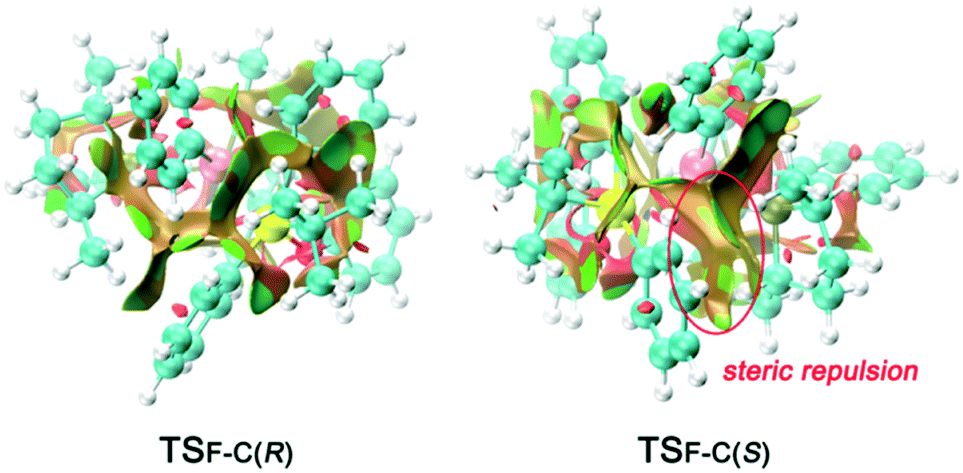 | ||
| Fig. 6 NCIPLOTs of TSF-C(R) and TSF-C(S). The s = 0.6 au isosurface is colored according to a BGR scheme over the range of −0.02 < sign(λ2)ρ < 0.02 au. Blue indicates strong attractions, green indicates very weak interactions, and red indicates strong repulsion. | ||
Conclusions
In summary, the detailed mechanism of the rhodium-catalyzed asymmetric synthesis of dibenzooxasilines has been investigated with DFT calculations. The calculation results revealed that the mechanism consists of the following steps: (i) the hydroxorhodium complex initially undergoes deprotonation with the substrate to generate the aryloxorhodium intermediate; (ii) Rh–Si exchange occurs to afford the arylrhodium complex; (iii) oxidative addition/reductive elimination generates the dibenzooxasiline product; (iv) the active aryloxorhodium species A is regenerated with the aid of ethyl acrylate.The related enantioselectivity of this reaction has also been investigated. The results indicate that the enantioselectivity is determined during the step of oxidative addition/reductive elimination from the arylrhodium complex. The origin of the enantioselectivity has been elucidated by quadrant analysis and NCIPLOT analysis. It was found that in the reductive elimination transition state TSF-C(S), the silyl group will squeeze the phenyl ring on the rhodium atom to tilt toward the methyl group-blocked quadrant, which causes significant steric repulsion and eventually leads to formation of the (R)-configuration product.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Helpful discussions with Prof. Zhenyang Lin of The Hong Kong University of Science and Technology are gratefully acknowledged. This project was supported by the National Science Foundation of China (Grants 21822303 and 21772020) and Fundamental Research Funds for the Central Universities (Chongqing University) (No. 2018CDXZ0002; 2018CDPTCG0001/4).References
- (a) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed; (b) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835–846 CrossRef CAS; (c) T. Hiyama and E. Shirakawa, Top. Curr. Chem., 2002, 219, 61–85 CrossRef CAS; (d) L. Li, Y. Zhang, L. Gao and Z. Song, Tetrahedron Lett., 2015, 56, 1466–1473 CrossRef CAS.
- (a) T. H. Chan and D. Wang, Chem. Rev., 1992, 92, 995–1006 CrossRef CAS; (b) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS; (c) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC; (d) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (e) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- (a) K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2006, 8, 483–485 CrossRef CAS PubMed; (b) K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 6094–6095 CrossRef CAS PubMed; (c) J. Terao, H. Watabe, H. Watanabe and N. Kambe, Adv. Synth. Catal., 2004, 346, 1674–1678 CrossRef CAS.
- (a) S. K. Kang, T. H. Kim and S. J. Pyun, J. Chem. Soc., Perkin Trans. 1, 1997, 6, 797–798 RSC; (b) Y. Nishihara, K. Ikegashira, K. Hirabayashi, J.-i. Ando, A. Mori and T. Hiyama, J. Org. Chem., 2000, 65, 1780–1787 CrossRef CAS PubMed; (c) H. Taguchi, K. Ghoroku, M. Tadaki, A. Tsubouchi and T. Takeda, J. Org. Chem., 2002, 67, 8450–8456 CrossRef CAS PubMed.
- B. Su, T. G. Zhou, X. W. Li, X. R. Shao, P. L. Xu, W. L. Wu, J. F. Hartwig and Z. J. Shi, Angew. Chem., Int. Ed., 2017, 56, 1092–1096 CrossRef CAS PubMed.
- (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS; (b) Y. Hatanaka and T. Hiyama, Tetrahedron Lett., 1990, 31, 2719–2722 CrossRef CAS; (c) K. Itami, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2001, 123, 5600–5601 CrossRef CAS PubMed; (d) Y. Liang, W. Geng, J. Wei and Z. Xi, Angew. Chem., Int. Ed., 2012, 51, 1934–1937 CrossRef CAS PubMed; (e) Y. Liang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2011, 133, 9204–9207 CrossRef CAS PubMed; (f) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef; (g) J.-i. Yoshida, K. Tamao, M. Takahashi and M. Kumada, Tetrahedron Lett., 1978, 19, 2161–2164 CrossRef.
- (a) T.-S. Huang and C.-J. Li, Chem. Commun., 2001, 2348–2349 RSC; (b) T. Matsuda and Y. Ichioka, Org. Biomol. Chem., 2012, 10, 3175–3177 RSC; (c) C. Walter and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 3818–3820 CrossRef CAS PubMed; (d) M. Yu and X. Fu, J. Am. Chem. Soc., 2011, 133, 15926–15929 CrossRef CAS PubMed; (e) Q. W. Zhang, K. An and W. He, Angew. Chem., Int. Ed., 2014, 53, 5667–5671 CrossRef CAS PubMed.
- F. Delpech, J. Mansas, H. Leuser, S. Sabo-Etienne and B. Chaudret, Organometallics, 2000, 19, 5750–5757 CrossRef CAS.
- (a) H. Hashimoto, A. Matsuda and H. Tobita, Organometallics, 2006, 25, 472–476 CrossRef CAS; (b) H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004–4006 RSC; (c) E. Shirakawa, S. Masui, R. Narui, R. Watabe, D. Ikeda and T. Hayashi, Chem. Commun., 2011, 47, 9714–9716 RSC.
- M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477–19488 CrossRef CAS PubMed.
- Q. Yang, L. Liu, Y. Chi, W. Hao, W.-X. Zhang and Z. Xi, Org. Chem. Front., 2018, 5, 860–863 RSC.
- S. Okumura, F. Sun, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2017, 139, 12414–12417 CrossRef CAS PubMed.
- (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (b) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (d) E. Negishi, Acc. Chem. Res., 2002, 15, 340–348 CrossRef; (e) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (f) A. Suzuki, Acc. Chem. Res., 2002, 15, 178–184 CrossRef.
- (a) X. F. Bai, J. F. Zou, M. Y. Chen, Z. Xu, L. Li, Y. M. Cui, Z. J. Zheng and L. W. Xu, Chem. - Asian J., 2017, 12, 1730–1735 CrossRef CAS PubMed; (b) J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 720–724 CrossRef CAS PubMed; (c) J. O. Bauer and C. Strohmann, Eur. J. Inorg. Chem., 2016, 2016, 2868–2881 CrossRef CAS; (d) K. Igawa, D. Yoshihiro, Y. Abe and K. Tomooka, Angew. Chem., Int. Ed., 2016, 55, 5814–5818 CrossRef CAS PubMed; (e) P.-W. Long, X.-F. Bai, F. Ye, L. Li, Z. Xu, K.-F. Yang, Y.-M. Cui, Z.-J. Zheng and L.-W. Xu, Adv. Synth. Catal., 2018, 360, 2825–2830 CrossRef CAS; (f) R. Shintani, Asian J. Org. Chem., 2015, 4, 510–514 CrossRef CAS; (g) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616–1620 CrossRef CAS PubMed; (h) H. Wen, X. Wan and Z. Huang, Angew. Chem., Int. Ed., 2018, 57, 6319–6323 CrossRef CAS PubMed; (i) G. Zhan, H. L. Teng, Y. Luo, S. J. Lou, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2018, 57, 12342–12346 CrossRef CAS; (j) Q.-W. Zhang, K. An, L.-C. Liu, Q. Zhang, H. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125–1129 CrossRef CAS PubMed; (k) W.-T. Zhao, Z.-Q. Lu, H. Zheng, X.-S. Xue and D. Zhao, ACS Catal., 2018, 8, 7997–8005 CrossRef CAS.
- (a) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1974, 64, C51–C54 CrossRef CAS; (b) R. J. P. Corriu and J. J. E. Moreau, Tetrahedron Lett., 1973, 14, 4469–4472 CrossRef.
- T. Hayashi, K. Yamamoto and M. Kumada, Tetrahedron Lett., 1974, 15, 331–334 CrossRef.
- (a) R. Shintani, K. Moriya and T. Hayashi, J. Am. Chem. Soc., 2011, 133, 16440–16443 CrossRef CAS PubMed; (b) R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305–7308 CrossRef CAS PubMed; (c) R. Shintani, R. Takano and K. Nozaki, Chem. Sci., 2016, 7, 1205–1211 RSC.
- Y. Kurihara, M. Nishikawa, Y. Yamanoi and H. Nishihara, Chem. Commun., 2012, 48, 11564–11566 RSC.
- (a) Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520–1522 CrossRef CAS PubMed; (b) M. Murai, Y. Takeuchi, K. Yamauchi, Y. Kuninobu and K. Takai, Chem. - Eur. J., 2016, 22, 6048–6058 CrossRef CAS.
- R. Shintani, E. E. Maciver, F. Tamakuni and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 16955–16958 CrossRef CAS PubMed.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- (a) J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921–2923 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS; (c) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS; (c) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 13171–13174 RSC.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (b) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed.
- (a) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS; (b) L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed; (c) Z. Yu, Z. Jin, M. Duan, R. Bai, Y. Lu and Y. Lan, J. Org. Chem., 2018, 83, 9729–9740 CrossRef CAS PubMed.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, (http://www.cylview.org) Search PubMed.
- (a) W. J. Chen and Z. Lin, Dalton Trans., 2014, 43, 11138–11144 RSC; (b) H. Xie, H. Zhang and Z. Lin, New J. Chem., 2013, 37, 2856 RSC; (c) J. Zhang, J. Z. Xu, Z. J. Zheng, Z. Xu, Y. M. Cui, J. Cao and L. W. Xu, Chem. - Asian J., 2016, 11, 2867–2875 CrossRef CAS PubMed; (d) T. Zhou, L. Xu and Y. Xia, Org. Lett., 2013, 15, 6074–6077 CrossRef CAS PubMed; (e) T. Zhou and Y. Xia, Organometallics, 2014, 33, 4230–4239 CrossRef CAS.
- T. Seiser and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 10163–10167 CrossRef CAS PubMed.
- Z. Yu and Y. Lan, J. Org. Chem., 2013, 78, 11501–11507 CrossRef CAS PubMed.
- (a) S. E. Denmark, W. T. Chang, K. N. Houk and P. Liu, J. Org. Chem., 2015, 80, 313–366 CrossRef CAS PubMed; (b) C. Zheng, C. X. Zhuo and S. L. You, J. Am. Chem. Soc., 2014, 136, 16251–16259 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02261e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |