
Dramatic differences in the fluorescence of AIEgen-doped micro- and macrophase separated systems†
Zehua
Song
a,
Xiaolin
Lv
b,
Longcheng
Gao
 *a and
Lei
Jiang
ac
*a and
Lei
Jiang
ac
aLaboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, Key Laboratory of Beijing Energy, School of Chemistry and Environment, Beihang University, Beijing 100191, P. R. China. E-mail: lcgao@buaa.edu.cn
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, P. R. China
cKey Laboratory of Bio-inspired Materials and Interfacial Science, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 1st December 2017
Abstract
A block copolymer (BCP) and a polymer blend containing small amounts of fluorogenic molecules with aggregation-induced emission (AIE) characteristics exhibited dramatic differences in the fluorescence properties. In particular, AIEgens with different polarities were doped in BCP polystyrene-b-polylactide (PS-b-PLA) and the PS/PLA blend. For the BCP, whether the AIEgens were selectively located in one phase or not, the intramolecular motions of the phenyl rings in AIEgens were affected by both the PS and PLA segment motions, due to the covalent bonds between the constitutive blocks. Therefore, the fluorescence was affected by the segment movements of PS and PLA. In contrast, the AIEgen-doped polymer blend exhibited location dependent fluorescence. The fluorescence was affected by the segment movements where the AIEgens were located, because there were no connections between the PS and PLA chains. This visualization method will provide new insights into monitoring the chain motions of macromolecules with more complex structures.
Introduction
Different polymers combined into a single material, either chemically or physically, lead to a wide range of phase behaviours. For example, binary homopolymer blends and block copolymers (BCP) exhibit macrophase and microphase separation, respectively. For blends, the interfacial tension favors a reduction in the surface area that leads to macroscopic segregation.1 BCPs are polymers consisting of two or more blocks of mutually immiscible segments, chemically connected end-to-end. They tend to form separated phases, but macroscopic phase separation is prevented because of the chemical connectivity. Instead, a microphase separated structure is formed.2,3 The morphologies directly influence the physical properties and ultimately the applications. For instance, the macrophase separated structures enable the polymer blends in the fields like bulk-heterojunction solar cells,4 plastics and rubbers.5,6 And well-defined nanostructures of BCPs have been extensively studied and applied in the fields like lithography,7,8 drug delivery,9,10 interface materials,11–14 energy harvest,15 and so on.Many techniques have been used to detect the morphologies of the phase separated systems, including TEM, SAXS, etc. Besides aggregatochromic probes are exploited for monitoring the conformation transition and morphology visualization.16–19 The fluorescent probes with aggregation-induced emission (AIE) characteristics are promising candidates, due to the distinct light emission behaviors between molecularly-dispersed and aggregate states.20–24 The AIEgens are very sensitive to the changes in the micro-/nanostructures where they are placed, and are able to detect the subtle details of the microenvironments. After doping with AIEgens, the polymer segment motions will interfere with the intramolecular motions of the AIEgens.25 For example, the rheological properties vary abruptly before and after glass transition. The intramolecular motions of AIEgens can be tuned from the restriction of intramolecular rotations (RIR) to free rotation.26,27 The variations can be monitored by the fluorescent probes.
Herein, we discovered that the AIEgen-doped micro- and macrophase separated systems exhibited dramatically different fluorescence properties, corresponding to the essential differences between the BCPs and the polymer blends.28 In the segregations formed by the simplest A and B linear BCPs, the block–block joints are localized at the interfaces, and the chains are stretched, resulting in the loss of system entropy.1 Thus, the segment movements in the A phase interfere with the movements in the adjacent B phase through the covalent bonds. If the fluorescent probes are selectively doped in the A phase, the transitions of the constitutive A and B blocks in a separated phase are likely to be monitored. In contrast, for the segregations formed by the A/B blends, the interfacial tension tends to be minimized. The polymer motions in the A phase and the adjacent B phase are relatively independent. In contrast with the BCP systems, only the transitions of the polymer phase that is selectively doped with the fluorescent probes can be monitored.
Experimental
Materials
Tetraphenylethene (TPE) and dicarboxyl tetraphenylethylene (TPE-2COOH) were synthesized according to the literature.29,30 The block copolymer PS-b-PLA (Mn = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000, PDI = 1.09, Polymer Source Inc.), homopolymer PLA (Mn = 60000, Energy Chemical Inc.), and Tetrakis(4-carboxylphenyl)ethylene (TPE-4COOH, Alfa chemical, 97%) were used as received. The PS homopolymer was polymerized in our lab by radical polymerization (Mn = 26000, PDI = 1.21). The NMR results are given in Fig. S1 and S2 (ESI†).
000–14000, PDI = 1.09, Polymer Source Inc.), homopolymer PLA (Mn = 60000, Energy Chemical Inc.), and Tetrakis(4-carboxylphenyl)ethylene (TPE-4COOH, Alfa chemical, 97%) were used as received. The PS homopolymer was polymerized in our lab by radical polymerization (Mn = 26000, PDI = 1.21). The NMR results are given in Fig. S1 and S2 (ESI†).
Results and discussion
We chose polystyrene (PS)/polylactic acid (PLA) as the model system. PS and PLA are chemically immiscible, due to the contrast in the solubility parameters.31 Tetraphenylethene (TPE) based fluorescent molecules were doped into the polymer matrix. The selective location of the AIEgens in one phase is a key factor. The polarity of the TPE derivatives is tuned by modifying different numbers of carboxyl groups (Scheme 1). The probes show increased polarity with the increase of acidic groups, resulting in the decrease of the Rf values on a TLC plate using dichloromethane/n-hexane (1:2) as the developing solvent. TPE, with no carboxyl group, shows the lowest polarity, and tends to be selectively dissolved in the PS domains either in PS-b-PLA or in the PS/PLA blend. Tetracarboxyl substituted TPE-4COOH, with the highest polarity, tends to selectively dissolve in the PLA domains. Dicarboxyl substituted TPE-2COOH, with moderate polarity, tends to dissolve in both the PS and PLA domains. The different locations of the fluorescent probes are expected to reflect the microstructure differences from the BCP and the polymer blend.
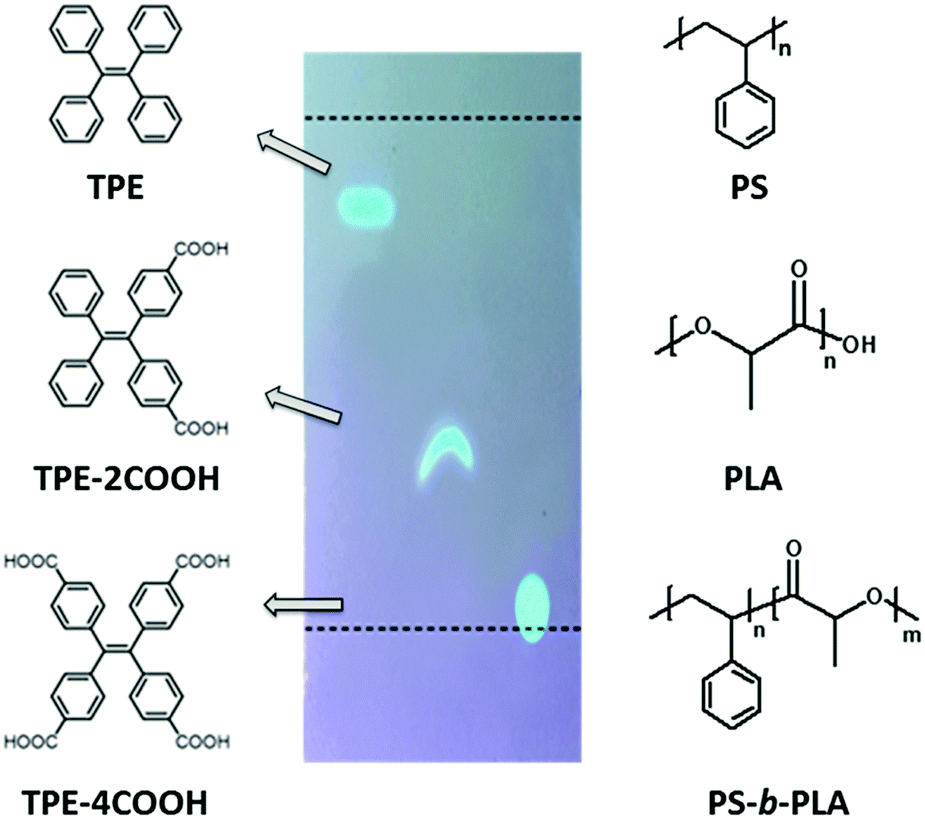 | ||
| Scheme 1 The molecular structures of TPE, TPE-2COOH, TPE-4COOH, polystyrene (PS), polylactic acid (PLA) and PS-b-PLA. The fluorescent probes show increased polarity with the increase of acidic groups, resulting in the decrease of the Rf values on a TLC plate (1:2 of dichloromethane/n-hexane). | ||
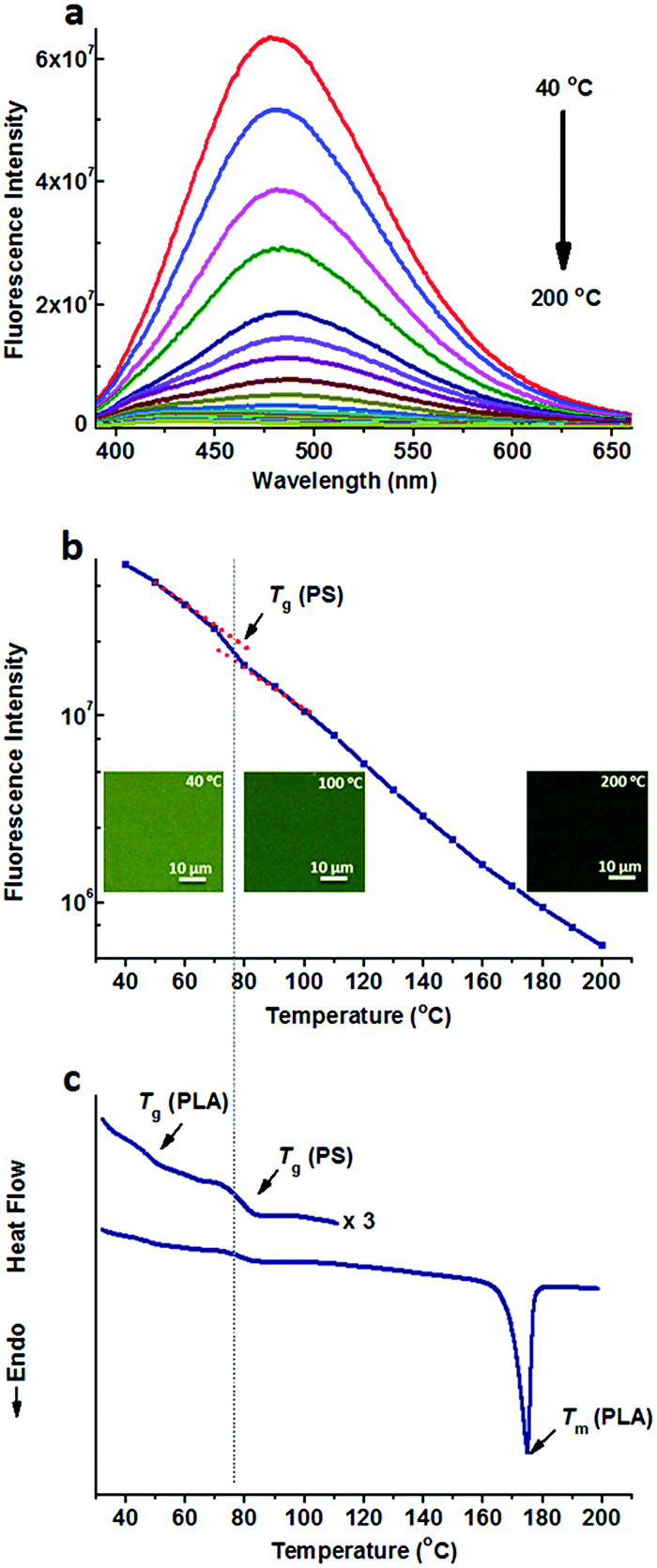
Fig. 1a shows the fluorescence spectra of TPE@PS-b-PLA at various temperatures. At low temperatures, TPE@PS-b-PLA emitted strong blue light (485 nm). The fluorescence intensity decreased with the increase of temperature. The logarithm of the peak fluorescence intensity was plotted as a function of temperature (Fig. 1b). We can see that the fluorescence intensity drops drastically with the increase of temperature. At low temperature, the intramolecular motions of phenyl rings of TPE, including rotation, vibration, torsion, and bending, were restricted to some extent. TPE emitted efficiently because the excited state energy is annihilated through radiation decay. As the temperature increased, free volume in the PS nanodomains increased and the movements of PS segments were enhanced. Thus, the intramolecular motions of TPE were activated and more energy of the excited state was consumed. This led to the weak fluorescence emission at higher temperatures. More importantly, three transitions around 56 °C, 97 °C, and 167 °C were corresponded to the Tg of PLA, Tg of PS, and Tm of PLA, respectively. The transitions were confirmed by the differential scanning calorimetry (DSC) curve of the PS-b-PLA (Fig. 1c) and the fluorescence properties of the corresponding homopolymers (Fig. S3–S8, ESI†). Even though TPE was selectively located in the PS nanodomains, the PLA segment movement actuated the motions of the PS segment, and sequentially induced the intramolecular motions of TPE. It was the covalent bond between the two blocks that played the key role. The temperature dependent fluorescence properties of TPE@PS-b-PLA were confirmed by the fluorescence microscopy results, as shown in Fig. S9 (ESI†).
 | ||
| Fig. 1 (a) Fluorescence spectra of TPE@PS-b-PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicate three transitions as the temperature increased. (c) The DSC curve of PS-b-PLA exhibited three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. The inset shows the fluorescent microscopy images at different temperatures. | ||
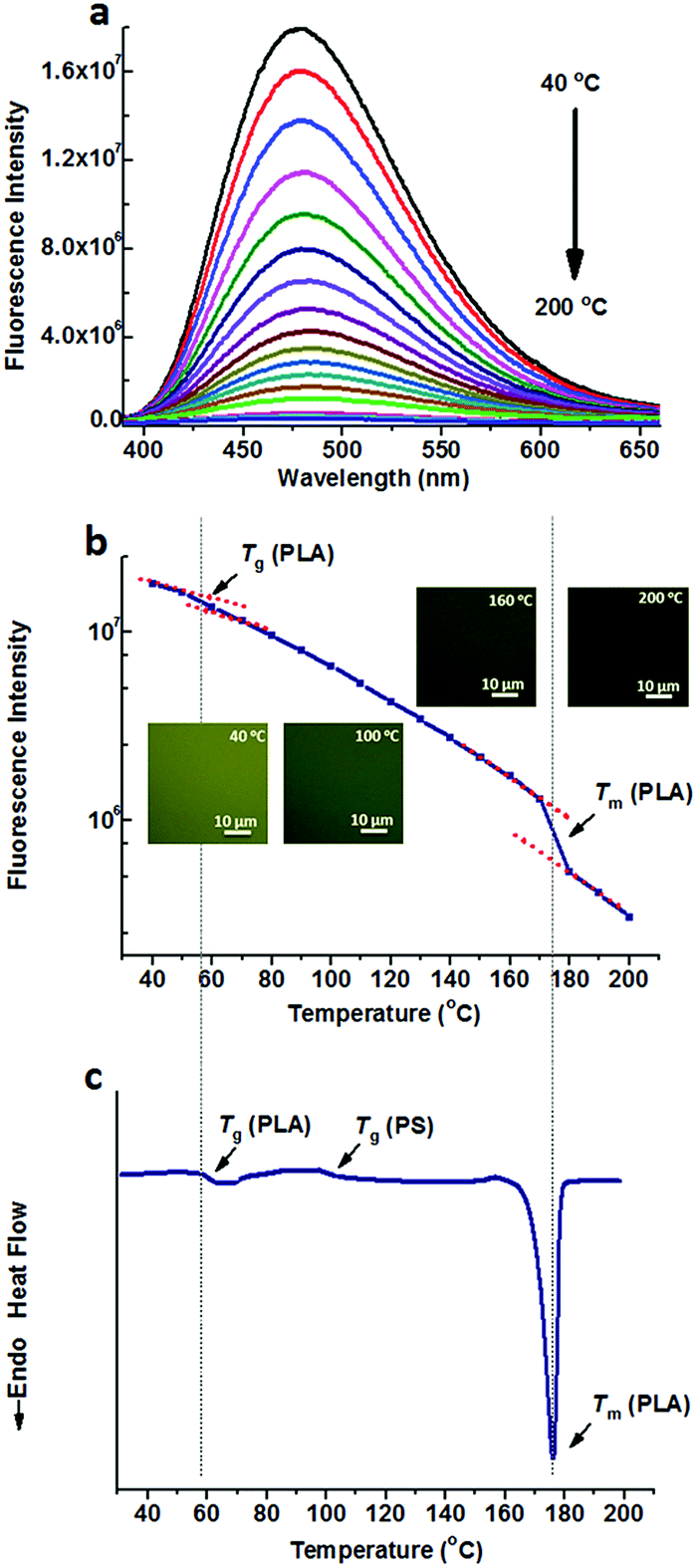
On the other hand, TPE was doped into the PS/PLA blend. Similar to that in the PS-b-PLA system, TPE was selectively located in the PS domains. Temperature dependent fluorescence spectra of TPE@PS/PLA are shown in Fig. 2a. The fluorescence intensity decreased as the temperature increased. Meanwhile, the fluorescence microscopy results showed that the emission decayed with the increase in temperature (Fig. S10, ESI†). Fig. 2b shows the plots of the peak intensity as a function of temperature. Different from that of TPE@PS-b-PLA, only one transition at around 78 °C was observed, corresponding to the Tg of PS. The transitions of PLA, both Tg and Tm, were absent on the plot. The DSC curve of the PS/PLA blend showed the Tg of PLA (50 °C), Tg of PS (78 °C), and Tm of PLA (175 °C) in Fig. 2c. That is to say, the movements of PS segments induced the intramolecular motions of TPE, while those of the PLA could not. The molecular weight of the PS block in BCP was close to that of the PS homopolymer (Fig. S3 and S4, ESI†) which was one component in the polymer blend PS/PLA, the increase of the Tg compared to that of the PS homopolymer was related to its connection with the hard PLA block.32
 | ||
| Fig. 2 (a) Fluorescence spectra of TPE@PS/PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicate the Tg of PS. (c) The DSC curve of the PS/PLA blend exhibits three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. The inset shows the fluorescence microscopy images at different temperatures. | ||
The dramatically different fluorescence properties of the TPE doped BCP and blend systems reflected the microstructure information. The BCP formed a microphase separated structure, as shown in Fig. S11 (ESI†). TPE was selectively dissolved in the PS domains. The intramolecular motions of TPE were not only activated by the PS segment movements, but were also influenced by the movements of the PLA in the adjacent domains, through the connections between the two blocks. Thus, the TPE fluorescence monitored the segment movements of the two blocks. However, for the blend, PS and PLA formed macroscopic separate phases, as shown in Fig. S12 (ESI†). The correlation between the adjacent phases was weak. Because most TPE molecules were located in the PS domains, the intramolecular motions of TPE were interfered by only PS segment movements rather than PLA.
Compared to the nonpolar TPE, TPE-4COOH with a much higher polarity can be selectively dissolved in the PLA domains either in PS-b-PLA or the PS/PLA blend, due to the strong hydrogen bond interaction. Fig. 3a shows the fluorescence spectra of TPE-4COOH@PS-b-PLA at various temperatures. Similar to those of TPE doped systems, at low temperatures, TPE-4COOH@PS-b-PLA emitted strong blue light. The fluorescence intensity decreased with the increase of temperature. Also, the fluorescence microscopy results showed the trend (Fig. S13, ESI†). The logarithm of the maximum fluorescence intensity was plotted as a function of temperature (Fig. 3b). We can see three transitions at 56 °C, 96 °C, and 167 °C, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA, respectively. The transitions were in accordance to the DSC result in Fig. 3c. Even though TPE-4COOH was selectively located in the PLA nanodomains, the PS segment movement actuated the motions of PLA segments, and sequentially induced the intramolecular motions of TPE-4COOH.
 | ||
| Fig. 3 (a) Fluorescence spectra of TPE-4COOH@PS-b-PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicate three transitions as the temperature increases. The inset shows the fluorescence microscopy images at different temperatures. (c) The DSC curve of TPE-4COOH@PS-b-PLA exhibits three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. | ||
When TPE-4COOH was doped into the PS/PLA blend, it was selectively located in the PLA domains. Temperature dependent fluorescence spectra of TPE-4COOH@PS/PLA are shown in Fig. 4a. The fluorescence intensity decreased as the temperature increased. We also performed the fluorescence microscopy test, which exhibited the same trend (Fig. S14, ESI†). Fig. 4b shows the plots of maximum intensity as a function of temperature. Different from that of TPE-4COOH@PS-b-PLA, transitions at around 54 °C and 172 °C were observed, corresponding to the Tg and Tm of PLA. The Tg of PS was absent on the plot. The DSC curve of TPE-4COOH@PS/PLA showed the Tg of PLA (60 °C), Tg of PS (97 °C), and Tm of PLA (172 °C) in Fig. 4c. That is to say, the movements of PLA segments induced the intramolecular motions of TPE, while the PS could not.
 | ||
| Fig. 4 (a) Fluorescence spectra of TPE-4COOH@PS/PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicating two transitions as the temperature increases, corresponding to the Tg and Tm of PLA, respectively. The Tg of PS was not detected in this situation. The inset shows the fluorescence microscopy images at different temperatures. (c) The DSC curve of TPE-4COOH@PS/PLA exhibits three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. | ||
Either TPE or TPE-4COOH was selectively dissolved in one phase. They could distinguish PS-b-PLA from PS/PLA directly. Dicarboxyl substituted TPE-2COOH shows moderate polarity. It tends to dissolve in the PLA and PS domains, or maybe at the interface of the PS and PLA domains, due to the amphiphilic character of TPE-2COOH. Fig. 5a shows the fluorescence spectra of TPE-2COOH@PS-b-PLA at various temperatures. The fluorescence intensity decreased with the increase of temperature, as also confirmed by the fluorescence microscopy results (Fig. S15, ESI†). Meanwhile, on the plot of the peak fluorescence intensity as a function of temperature (Fig. 5b), we can also see three transitions around 58 °C, 97 °C, and 165 °C, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA, respectively. The DSC curve of TPE-4COOH@PS-b-PLA showed the Tg of PLA (60 °C), Tg of PS (98 °C), and Tm of PLA (165 °C) in Fig. 5c. It was a quite normal phenomenon, because TPE-2COOH was non-selectively located in the PLA and PS domains, where the intramolecular motions of TPE-2COOH can be in situ actuated.
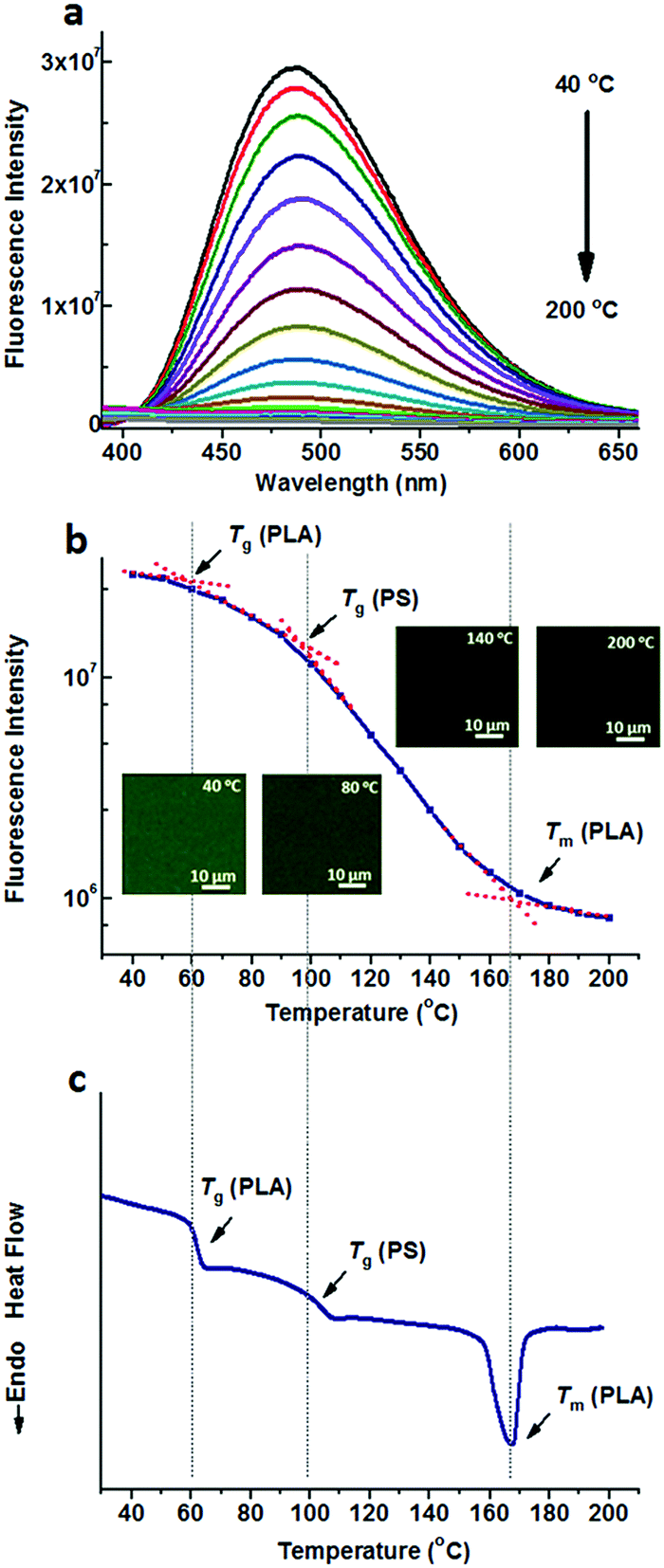 | ||
| Fig. 5 (a) Fluorescence spectra of TPE-2COOH@PS-b-PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicate three transitions as the temperature increases. The inset shows the fluorescence microscopy images at different temperatures. (c) The DSC curve of TPE-2COOH@PS-b-PLA exhibits three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. | ||
TPE-2COOH was doped into the PS/PLA blend. Temperature dependent fluorescence spectra of TPE-2COOH@PS/PLA are shown in Fig. 6a and b shows the plots of maximum intensity as a function of temperature, as well as the temperature dependent fluorescence microscopy experiments (Fig. S16, ESI†). Similar to that of TPE-2COOH@PS-b-PLA, transitions at around 53 °C and 172 °C were observed, corresponding to the Tg and Tm of PLA, as well as the Tg of PS at 97 °C. The DSC curve of TPE-2COOH@PS/PLA showed the Tg of PLA (60 °C), Tg of PS (98 °C), and Tm of PLA (170 °C) in Fig. 6c. That is to say, the movements of both PLA and PS segments affected the intramolecular motions of TPE-2COOH. This was totally different from those of TPE and TPE-4COOH doped PS/PLA blends.
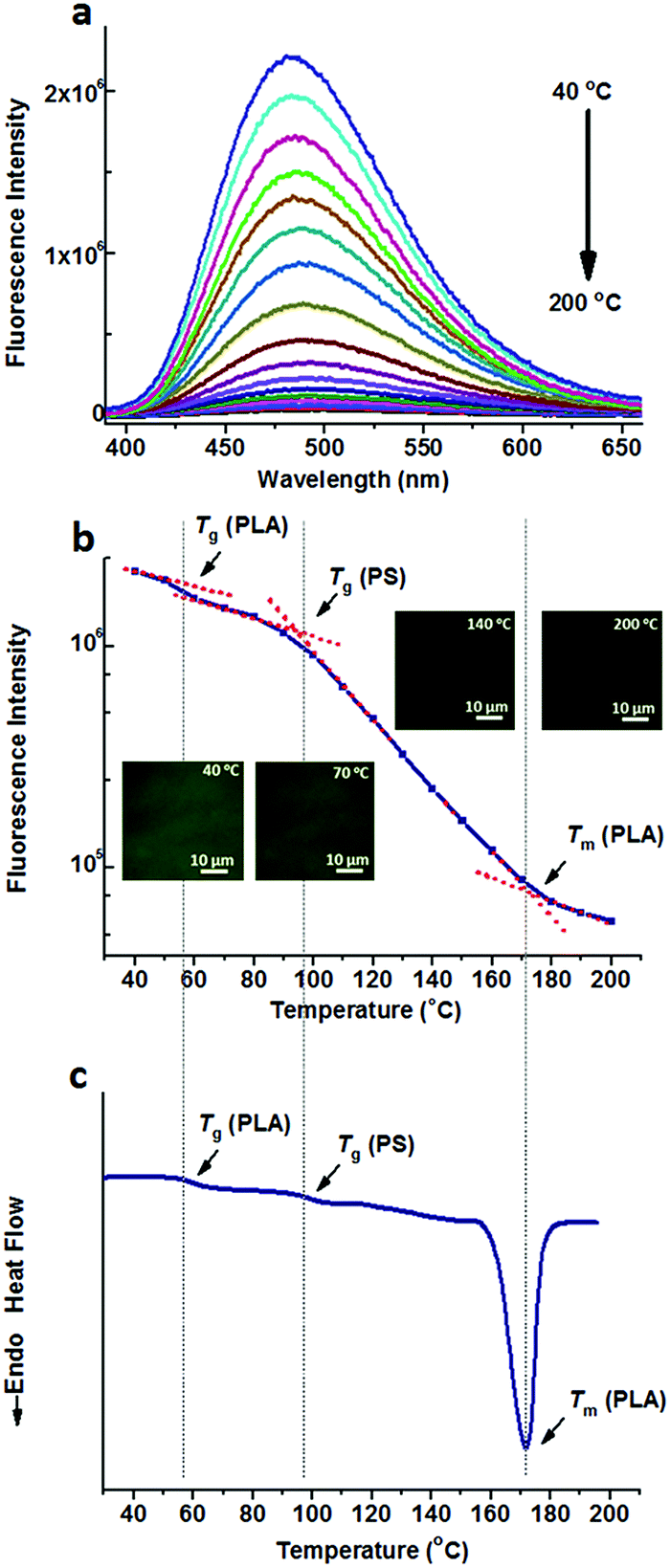 | ||
| Fig. 6 (a) Fluorescence spectra of TPE-2COOH@PS/PLA at different temperatures ranging from 40 °C to 200 °C, and (b) the fluorescence intensity as a function of temperature, indicate three transitions as the temperature increases. The inset shows the fluorescence microscopy images at different temperatures. (c) The DSC curve of TPE-2COOH@PS/PLA exhibits three transitions, corresponding to the Tg of PLA, Tg of PS, and Tm of PLA. | ||
The fluorescent probes reflected the chain movements where the probes were located. As shown in Fig. 7, when doped with selective fluorescent probes, the BCP and blend systems exhibited different fluorescence properties. The chain movements of the constitutive blocks in the BCP could affect the fluorescent probes, due to the connection of the covalent bonds. In contrast, in the blend system, the chain movements of polymers without fluorescent probes cannot affect the fluorescent probes. For non-selective fluorescent probes, no remarkable differences can be detected between the BCP and blend systems, because all the chain movements nondistinctively influence the fluorescent probes, whether in BCP or blend systems. It needs to say that the selective location of TPE and TPE-4COOH in the BCP and blend systems is deduced from the like-dissolves-like rule. The wide polarity range of the fluorescent probes facilitates selective dissolution in the polymers with matching solubility parameters.
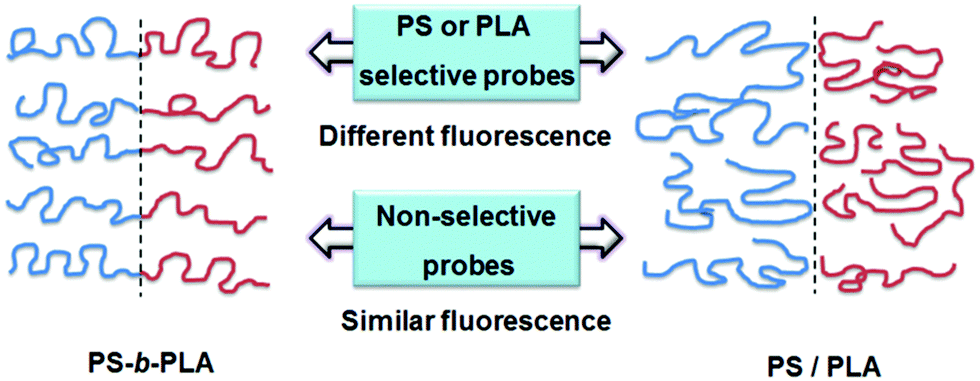 | ||
| Fig. 7 Schematic diagram of the tendency of different fluorescent probes doped in the BCP and blend systems. When doped with selective probes, they exhibit different fluorescence properties. Thus, the BCP and the blend can be distinguished using the fluorescence probe method. While doped with nonselective probes, they exhibit similar fluorescence properties. | ||
However, the direct experimental detection of the location is hard to realize in the current situation, due to the low content of the fluorescent probes. In return, the high sensitivity of the fluorescent probes provides us a visualization method for monitoring the segment motions of complex systems.
Conclusion
In conclusion, we discovered the dramatic differences in the fluorescence of AIEgen-doped between BCPs and polymer blends by selectively doping small amounts of fluorescent probes with different polarities. When the probes were selectively dissolved into one microphase of the BCP, the temperature ranges of the transitions were strongly dependent on the polymers where the probes were located, as well as the covalently bonded polymers. The movements of the blended polymers in the adjacent macrophases hardly interfered with the intramolecular motions of probes. Fluorescence probes make it possible to distinguish BCPs and polymer blends, and also provide a real-time, on-site visualization method for monitoring the segment motions of macromolecules with complex structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Haoke Zhang at the Hong Kong University of Science and Technology for helpful discussion. The work was supported by the National Key Research and Development Program of China (2017YFA0206904, 2017YFA0206900) and the Fundamental Research Funds for the Central Universities.Notes and references
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- S. Hobbs, M. Dekkers and V. Watkins, Polymer, 1988, 29, 1598–1602 CrossRef CAS.
- F. Bruder and R. Brenn, Phys. Rev. Lett., 1992, 69, 624–627 CrossRef CAS PubMed.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2009, 110, 146–177 CrossRef PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- B. L. Xue, L. C. Gao, Y. P. Hou, Z. W. Liu and L. Jiang, Adv. Mater., 2013, 25, 273–277 CrossRef CAS PubMed.
- X. Sui, Z. Zhang, Z. Zhang, Z. Wang, C. Li, H. Yuan, L. Gao, L. Wen, X. Fan and L. Yang, Angew. Chem., Int. Ed., 2016, 55, 13056–13060 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Z. Geng, S. Guan, H. M. Jiang, L. C. Gao, Z. W. Liu and L. Jiang, Chin. J. Polym. Sci., 2014, 32, 92–97 CrossRef CAS.
- Z. Zhang, X. Sui, P. Li, G. Xie, X. Y. Kong, K. Xiao, L. Gao, L. Wen and L. Jiang, J. Am. Chem. Soc., 2017, 139, 8905–8914 CrossRef CAS PubMed.
- G. Iasilli, A. Battisti, F. Tantussi, F. Fuso, M. Allegrini, G. Ruggeri and A. Pucci, Macromol. Chem. Phys., 2014, 215, 499–506 CrossRef CAS.
- C. Zhu, S. Pang, J. Xu, L. Jia, F. Xu, J. Mei, A. Qin, J. Sun, J. Ji and B. Tang, Analyst, 2011, 136, 3343–3348 RSC.
- Q. Zhu, L. Huang, J. Su and S. Liu, Chem. Commun., 2014, 50, 1107–1109 RSC.
- R.-H. Chien, C.-T. Lai and J.-L. Hong, J. Phys. Chem. C, 2011, 115, 20732–20739 CAS.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2010, 133, 660–663 CrossRef PubMed.
- J. Yang, J. Huang, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663–2684 RSC.
- I. Giuseppe, B. Antonella, T. Francesco, F. Francesco, A. Maria, R. Giacomo and P. Andrea, Macromol. Chem. Phys., 2014, 215, 499–506 CrossRef.
- H. Shi, J. Liu, J. Geng, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 9569–9572 CrossRef CAS PubMed.
- Y. L. Liu, Z. K. Wang, W. Qin, Q. L. Hu and B. Z. Tang, Chin. J. Polym. Sci., 2017, 35, 365–371 CrossRef CAS.
- K. Binder, Adv. Polym. Sci., 1994, 112, 181–299 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- M. Nakamura, T. Sanji and M. Tanaka, Chemistry, 2011, 17, 5344 CrossRef CAS PubMed.
- I. M. D. Arenaza, E. Meaurio, B. Coto and J. R. Sarasua, Polymer, 2010, 51, 4431–4438 CrossRef.
- D. S. Kaplan, J. Appl. Polym. Sci., 1976, 20, 2615–2629 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc04771a |
| This journal is © The Royal Society of Chemistry 2018 |