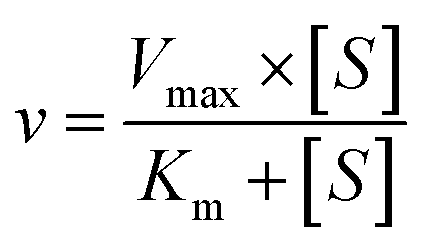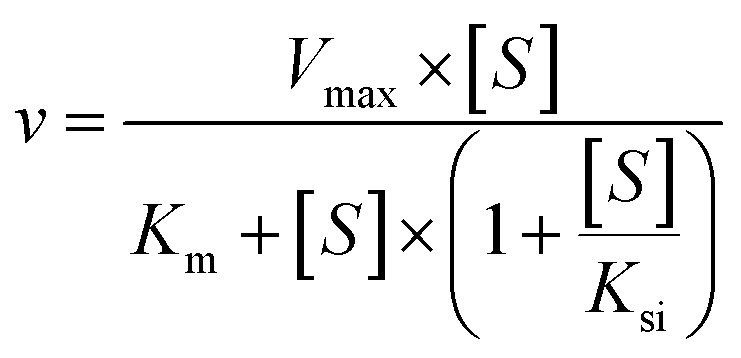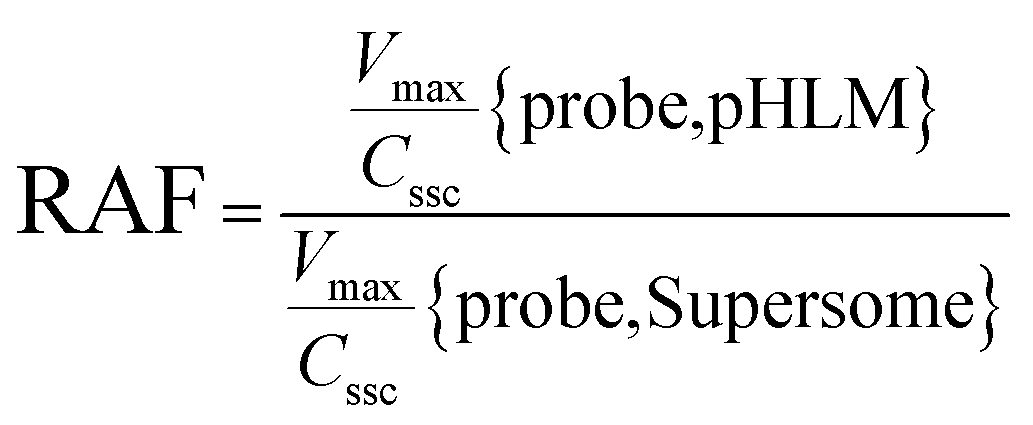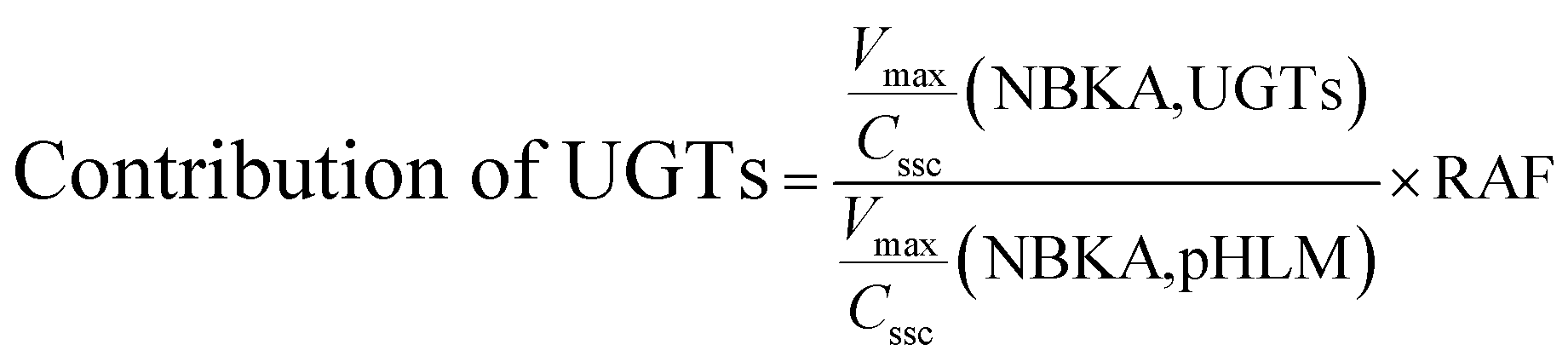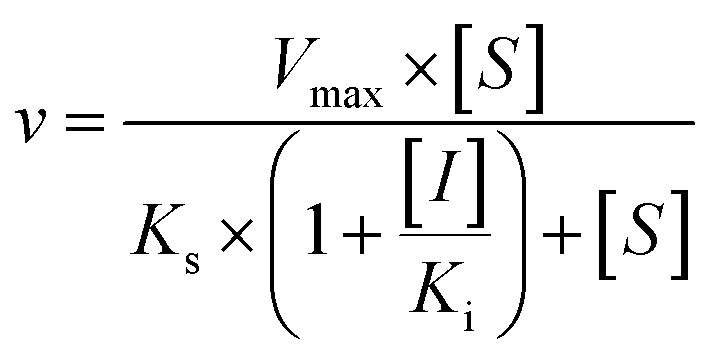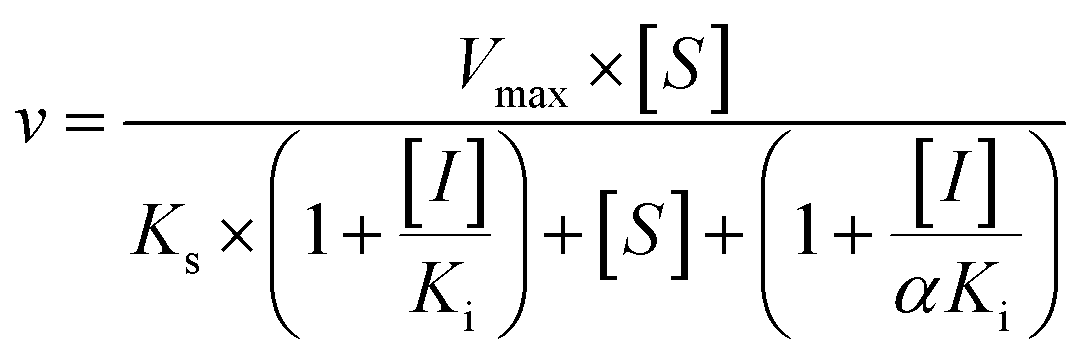DOI:
10.1039/C7RA10376J
(Paper)
RSC Adv., 2017,
7, 52661-52671
Characterization of human UDP-glucuronosyltransferases responsible for glucuronidation and inhibition of norbakuchinic acid, a primary metabolite of hepatotoxicity and nephrotoxicity component bakuchiol in Psoralea corylifolia L.†
Received
18th September 2017
, Accepted 28th October 2017
First published on 15th November 2017
Abstract
Norbakuchinic acid (NBKA) is the most abundant metabolite of bakuchiol (a hepatotoxicity and nephrotoxicity component in Psoralea corylifolia L.) in plasma and urine. The present study aimed to identify human UDP-glucuronosyltransferase (UGT) isoforms involved in NBKA glucuronidation, and to assess the inhibition potential of NBKA against recombinant UGTs. NBKA glucuronidation was determined using human liver microsomes (HLM) and human intestine microsomes (HIM) as well as expressed UGTs. The mechanisms of NBKA-UGT interactions were explored through kinetic characterization and modeling. As a result, two glucuronides (G1 and G2) were detected by HLM and HIM. G2 was dominantly produced with CLint values of 3.21 and 0.96 μL min−1 mg−1 in HLM and HIM, respectively. Furthermore, based on the relative activity factor (RAF) approach, UGT1A9 and 2B7 represented 5.98 and 46.16% of G2 in HLM, respectively. In addition, G2 was both significantly correlated with propofol-glucuronidation (r = 0.681, p = 0.015) and AZT-glucuronidation (r = 0.832, p = 0.0008). Also, NBKA glucuronidation showed marked species differences. Moreover, NBKA displayed broad-spectrum inhibition against human UGTs. Kinetic analyses showed that inhibition of these UGTs followed competitive (UGT1A1), mixed (UGT1A3, 1A6, 1A10, 2B7, 2B15 and 2B17) and non-competitive (UGT1A4, 1A7, 1A8 and 1A9) mechanisms, with Ki values ranging from 2.48 to 20.31 μM. Among them, NBKA exhibited moderate inhibitory effects against UGT1A1, 1A3, 1A4, 1A7, 1A8, 2B15 and 2B17 (both IC50 and Ki values lower than 8 μM). In contrast, the effect of NBKA was activating for UGT2B10. Taken altogether, these findings together demonstrate that NBKA has a potent and broad-spectrum inhibitory effect against most human UGTs and thus suggest that much caution should be exercised when high-dose BK is co-administered with UGT substrates.
1. Introduction
Bakuchiol (BK) is a phenolic compound isolated from the seeds of Psoralea corylifolia L., which has been broadly used as a kidney tonifying agent for the treatment of asthma, diarrhea and osteoporosis osteoporosis.1 Meanwhile, BK is the most abundant active compound, which accounts for 6.24% weight of the dried seeds.2 And also, it has attracted increasing interest for various biological activities including reduction of bone loss, antitumor and anti-inflammatory.3–6 However, high doses of BK could induce kidney toxicity in mice, and also showed toxicity in HepG2 and HK-2 cells in vitro.7–9 In addition, BK has poor bioavailability (3.2% in rats) due to significant first-pass metabolism.9–13 In our previous study, BK was mainly metabolized and transformed to norbakuchinic acid (NBKA), an oxidation product of BK.7,14 Further bioactivity evaluation suggested that NBKA exhibited no cytotoxicity on HKC-8 cells even at the highest concentration 50 μM.7 Moreover, NBKA was also present in the highest exposure in rat plasma, and then underwent conjugation through UDP-glucuronosyltransferases (UGTs) pathway to produce NBKA-O-glucuronides after oral administration of BK or Psoralea corylifolia L. extracts.7,14 However, the UGT isoforms involved in the glucuronidation of NBKA, and the interactions between NBKA and human UGTs still remain unknown. In consideration of the wide use of Psoralea corylifolia L., the frequency of the co-administration of BK with other clinical drugs is relatively high. Thus, it is very necessary to evaluate the potential risks of the herb–drug interactions between NBKA and other therapeutic drugs.
Phase II enzymes catalyze the conjugation of a hydrophilic moiety (mainly glucuronic acid and sulfonate group) to the drugs, which make drugs more water soluble and excretable.15 Human UGTs are involved in the metabolism of various endogenous substance and xenobiotic drugs by transferring a glucuronic acid from its cofactor uridine diphosphoglucuronic acid (UDPGA) to substrates.16 It is illustrated that human UGTs are responsible for approximately 35% of drugs metabolized by phase II enzymes.16 So far, human UGTs are mainly divided into four subfamilies, namely UGT1, UGT2, UGT3, and UGT8, based on their amino acid sequence.17 Most human UGTs are expressed in the liver, but several isoforms (UGT1A7, 1A8, 1A10, etc.) are predominantly expressed in these tissues such as the digestive tract (mainly intestine), kidney, lung, prostate, mammary and adrenal.18 Human intestinal UGTs are also considered as important determinants in intestinal metabolism, especially for those oral phenolic compounds.19 Therefore, the glucuronidation and the inhibition against human UGTs in the liver and gastrointestinal tract both play an important role in the oral bioavailability of many phenolic drugs or natural products.
The aim of this present study is to identify the main UGT enzymes involved in NBKA glucuronidation as published study previously,20 and to investigate the inhibitory effects of NBKA against human UGTs based on seven probe substrates.21,22 Hopefully, this study will be useful in disclosing new potential toxicities resulting from lowering activities of human UGTs when there is a co-administration of Psoralea corylifolia L. with other drugs.
2. Experimental
2.1. Chemicals and reagents
Alamethicin, uridine diphosphate glucuronic acid (UDPGA), D-saccharic-1,4-lactone and magnesium chloride (MgCl2) were provided from Sigma-Aldrich (St Louis, MO). Pooled human liver microsomes (HLM), human intestinal microsomes (HIM), monkey liver microsomes (MkLM), rat liver microsomes (RLM), mice liver microsomes (MLM), dog liver microsomes (DLM), guinea pig liver microsomes (GpLM), rabbit liver microsomes (RaLM), and recombinant expressed human UGT Supersomes™ (UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15 and 2B17) were all obtained from Corning Biosciences (New York, USA). All experiments were approved by the Ethics Committee of Jinan University in Guangzhou of China (Ethical Review No. 20130301003). All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). Norbakuchinic acid with purity over 98% were prepared and identified in our laboratory.7 Androsterone, 4-methylumbelliferone (4-MU), amitriptyline, β-estradiol, magnolol, propofol, suberoylanilide hydroxamic acid (SAHA), trifluoperazine (TFP) and zidovudine (AZT) were purchased from Aladdin Chemicals (Shanghai, China). All other chemicals and reagents were of analytical grade or the highest grade commercially available.
2.2. Glucuronidation assay
NBKA (0.25–100 μM) was incubated with HLM, HIM, MkLM, RLM, MLM, DLM, GpLM and RaLM and thirteen expressed UGTs enzymes to determine the rates of glucuronidation as published previously.20 Briefly, the incubation mixture (200 μL, 37 °C) mainly contained 50 mM Tris-hydrochloric acid buffer (pH = 7.4), and MgCl2, alamethicin, saccharolactone and UDPGA with the final concentration of 0.88 mM, 22 μg mL−1, 4.4 mM and 3.5 mM, respectively. The reaction was terminated by adding equal volume of ice-cold acetonitrile. The samples were vortexed and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g for 10 min. The supernatant was subjected to UPLC/Q-TOF-MS analysis. Incubation without UDPGA served as negative control to confirm the metabolites produced were UDPGA-dependent. All experiments were performed in triplicate. Preliminary experiments were performed to ensure that the rates of glucuronidation were determined under linear conditions with respect to the incubation time and protein concentration.
800g for 10 min. The supernatant was subjected to UPLC/Q-TOF-MS analysis. Incubation without UDPGA served as negative control to confirm the metabolites produced were UDPGA-dependent. All experiments were performed in triplicate. Preliminary experiments were performed to ensure that the rates of glucuronidation were determined under linear conditions with respect to the incubation time and protein concentration.
2.3. Chemical inhibition studies
Glucuronidation of NKBA in pooled HLM was measured in the absence or presence of an inhibitor (androsterone, magnolol and amitriptyline). Both androsterone and magnolol are the selective inhibitors for UGT1A9.23 Androsterone and amitriptyline are both reported to be a selective inhibitor of UGT2B7.24 NKBA (10 μM) were incubated in the absence or presence of three concentrations of androsterone (0.1, 1 and 10 μM), magnolol (0.1, 1 and 10 μM) and amitriptyline (0.1, 1 and 10 μM) for the glucuronidation. The incubations conditions were same as these of glucuronidation assays.
2.4. Enzymes kinetic evaluation
The kinetic models Michaelis–Menten equation and substrate inhibition equation were fitted to the data of metabolic rates versus substrate concentrations and displayed in eqn (1) and (2), respectively. Appropriate models were selected by visual inspection of the Eadie–Hofstee plot.25
The parameter were as follow. V is the formation rate of product. Vmax is the maximal velocity. Km is the Michaelis constant and [S] is the substrate. Ksi is the substrate inhibition constant. The intrinsic clearance (CLint) was obtained using the maximal reaction rates at the saturated substrate concentration (Vmax/Cssc) for Michaelis–Menten and substrate inhibition models.
| |
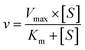 | (1) |
| |
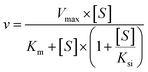 | (2) |
2.5. Activity correlation analysis
According to the glucuronidation assay protocol as described previously,20 the metabolic activities of individual HLMs (n = 12) toward NBKA, propofol (a probe substrate for UGT1A9) and zidovudine (a probe substrate for UGT2B7) were determined. NBKA (4 and 10 μM), propofol (500 μM) and zidovdin (1.25 mM) were separately incubated with UDPGA-supplemented individual HLM (1.0 mg mL−1) for 120 min. Correlation analyses were performed between NBKA glucuronidation (G2) and propofol glucuronidation, and between NBKA glucuronidation (G2) and zidovdin glucuronidation. Correlation (Pearson) analyses was performed using GraphPad Prism V5 software.
2.6. Contribution of UGT isoforms
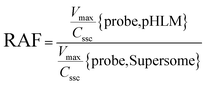
The contribution of individual expressed UGT enzymes to NBKA glucuronidation in HLM was evaluated by the relative activity factor (RAF) approach. The relative activity factor was defined as the activity ratio of HLM to an expressed UGT enzyme (Supersome) toward a probe substrate for this enzyme using eqn (3). The relative amount of NBKA glucuronidation in HLM attributed to an expressed UGT enzyme was estimated by multiplying the glucuronidation activity derived with this enzyme by the corresponding RAF. The RAFs were derived for UGT1A9 and 2B7 using the well-recognized probe substrates propofol and AZT, respectively. The contribution of individual UGT enzyme were calculated according to eqn (4).| |
 | (3) |
| |
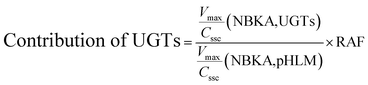 | (4) |
2.7. Assessment of inhibitory effects of NBKA against human UGTs
4-MU was used as the non-selective substrate for UGT1A3, 1A6, 1A7, 1A8, 1A10, 2B4 and 2B15.21 β-estradiol, TFP, propofol, AZT, amitriptyline and SAHA were typically used as the specific probe substrate for UGT1A1, 1A4, 1A9, 2B7, 2B10 and 2B17, respectively.22 In this study, the above probe substrates were incubated with the corresponding UGTs in both absence and presence of varying NBKA concentrations (1, 10 and 100 μM). Corresponding to the reported values of Km for each UGT isoform, the concentrations of these probe substrates were as follows: 60 μM β-estradiol for UGT1A1, 1200 μM 4-MU for UGT1A3, 40 μM TFP for UGT1A4, 100 μM 4-MU for UGT1A6, 20 μM 4-MU for UGT1A7, 800 μM 4-MU for UGT1A8, 40 μM propofol for UGT1A9, 30 μM 4-MU for UGT1A10, 1000 μM 4-MU for UGT2B4, 500 μM AZT for UGT2B7, 10 μM amitriptyline for UGT2B10, 250 μM 4-MU for UGT2B15 and 300 μM SAHA for UGT2B17.21,22 The incubation mixture were same as these in glucuronidation assay above.
2.8. Kinetic analyses for NBKA-associated inhibition on human UGTs
Half maximal inhibitory concentration (IC50) was determined using various concentrations of NBKA. The percent rate of control (in the absence of the NBKA) was calculated to assess whether NBKA had inhibition or activation effects or both. Inhibition kinetic parameters (Ki) were determined by using various concentrations of β-estradiol, TFP, propofol, AZT, amitriptyline, SAHA and 4-MU in the presence or absence of varying concentrations of NBKA.26 The inhibition kinetic type was evaluated through determining the intersection point in the Dixon plot. The reversible inhibitory type can be categorized into competitive inhibition, non-competitive inhibition, or mixed inhibition.27
Three kinetic models were used to calculate the Ki values by nonlinear regression using the eqn (5), eqn (6) and (7) for competitive inhibition, noncompetitive inhibition and mixed inhibition, respectively. The parameter were as follow. V is the velocity of the reaction; [S] and [I] are the substrate and inhibitor concentrations, respectively; Ki is the constant describing the affinity of the inhibitor to the enzyme; αKi describes the affinity of the inhibitor to the complex of enzyme and substrate; Ks is the constant describing the affinity of the substrate to the enzyme. Goodness of fit was determined by R2.
| |
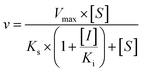 | (5) |
| |
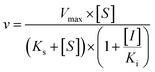 | (6) |
| |
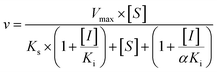 | (7) |
2.9. Analytical conditions
Metabolite screening of NBKA glucuronide was performed using a UPLC-Q-TOF/MS system (Waters Corporation, Manchester, U.K.). Chromatographic separation was achieved on a BEH C18 column (2.1 mm × 50 mm, 1.7 μm) maintained at 35 °C. The mobile phase consisted of water (A) and acetonitrile (B) (both including 0.1% formic acid, v/v), and the flow rate was 0.5 mL min−1. The gradient elution program was as follows: 10–50% B from 0 to 2.0 min, 50–100% B from 2.0 to 3.0 min. After holding 100% B for next 0.2 min, the column was returned to its starting condition. The detection wavelength was set at 254 nm and the injection volume was 8 μL.
The UPLC system was coupled to a hybrid quadrupole orthogonal time-of-flight (Q-TOF) tandem mass spectrometer (SYNAPT™ G2 HDMS, Waters, Manchester, U.K.) equipped with electrospray ionization (ESI). The operating parameters were as follow: capillary voltage of −2.5 kV (ESI−); Sample cone voltage 40 V (ESI−); ramp trap collision energy of 40–60 V (ESI−); extraction cone voltage of 4 V, source temperature of 100 °C, desolvation temperature 300 °C, cone gas flow of 50 L h−1 and desolvation gas flow of 800 L h−1. Argon was used as collision gas for CID in both MSE and MS2 mode. To ensure mass accuracy and reproducibility, the mass spectrometer was calibrated over a range of 50–1500 Da using solution of sodium formate. Leucine-enkephalin (m/z 554.2615 in negative ion mode) was used as external reference of LockSpray™ infused at a constant flow of 5 μL min−1 and data were centroid during acquisition.
2.10. Statistical analysis
Data are expressed as the mean ± SD (standard deviation). Model fitting and parameter estimation were performed by Graphpad Prism V5 software (SanDiego, CA). Mean differences between treatment and control groups were analyzed by two-tailed student's t test. The level of significance was set at p < 0.05 (*), p < 0.01 (**) or p < 0.001 (***).
3. Results
3.1. Structural identification of NKBA metabolites
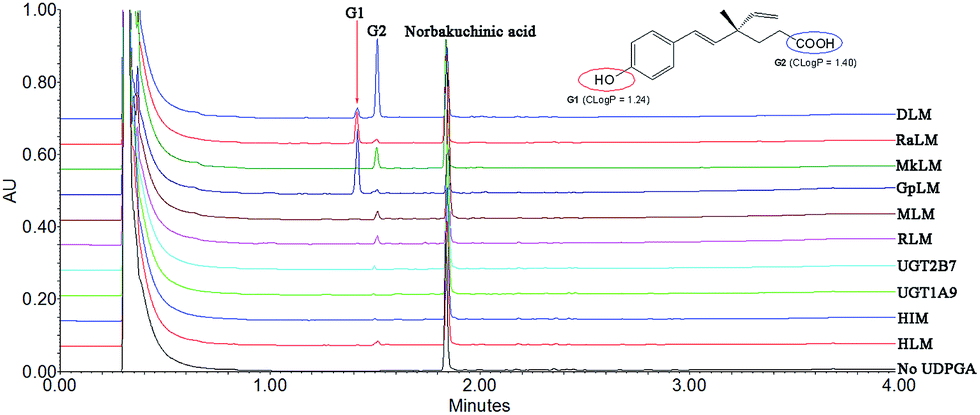
After incubation of NBKA with UDPGA-supplemented HLM, two additional peaks (tR = 1.41 and 1.53 min) which have a similar UV absorption profile in addition to NBKA were detected by UPLC-DAD analysis (Fig. 1). NBKA exhibited a typical [M + H]− ion at m/z 245.1171, and two main daughter ions at m/z 201.1365 and 173.0843 produced by losing a successive neutral fragment of CO2 and C2H4, respectively (Fig. S1†). The two metabolites had the same [M + H]− ion at m/z 421.1507, which was 176.0325 Da higher than that of NBKA (Fig. S1†). Meanwhile, the diagnostic fragment ions at m/z 175.0225 and 113.0208 were detected in MS/MS spectra, which indicated that they were characterized as mono-glucuronide of NBKA (Fig. S1†). Their chemical structures (G1 and G2) were shown in Fig. 1.
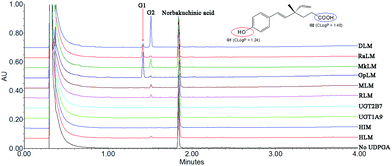 |
| | Fig. 1 Ultra-performance liquid chromatography analysis and chemical structures of norbakuchinic acid and its two glucuronides (G1 and G2). | |
3.2. Reaction phenotyping and chemical inhibitors assays
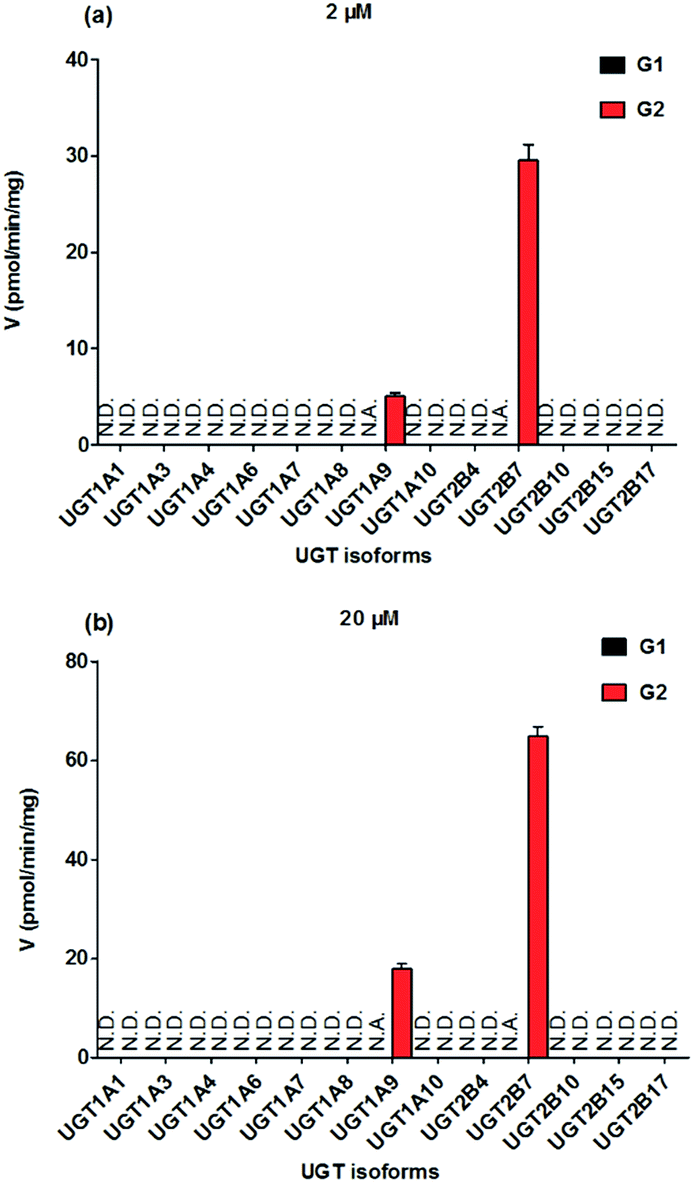
To identify the enzymes involving in the glucuronidation of NKBA, thirteen expressed UGT enzymes were analyzed for their catalysis activities (expressed as pmol min−1 mg−1 protein) at the substrate concentrations of 2 μM (Fig. 2a) and 20 μM (Fig. 2b). Among thirteen expressed UGT enzymes, only UGT1A9 and 2B7 involved in the glucuronidation of NBKA (Fig. 2a and b). Due to the concentration under the limit of quantification, it was unable to determine the kinetic parameters of G1 by UGT1A9 and 2B7.
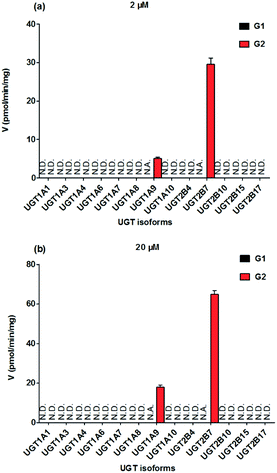 |
| | Fig. 2 Comparisons of glucuronidation rates of NBKA by thirteen expressed UGT enzymes at the substrate concentrations of 2 μM (a) and 20 μM (b). All experiments were performed in triplicate. N.D.: not detected; N.A.: due to the concentration under the limit of quantification, it was unavailable to determine the kinetic parameters of G1 by UGT1A9 and 2B7. | |
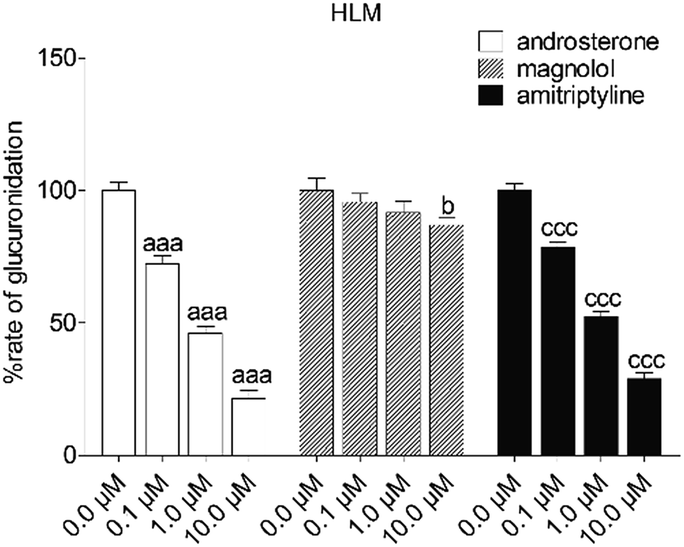
To reveal the roles of UGT1A9 and 2B7 in glucuronidation (G2) of NKBA, a series of chemical inhibition assays was performed by using highly selective UGT1A9 inhibitors (androsterone and magnolol) and UGT2B7 inhibitors (androsterone and amitriptyline). As shown in Fig. 3, androsterone (10 μM) exhibited the potent inhibition in HLM, with the remaining activity of 21.5% of the control values (Fig. 3). Furthermore, magnolol (10 μM) exhibited weak inhibition in HLM, decreasing the activity to be 86.9% of the control values (Fig. 3), whereas NBKA glucuronidation activity by HLM was decreased to be 28.9% of the control values in the presence of amitriptyline (10 μM) (Fig. 3).
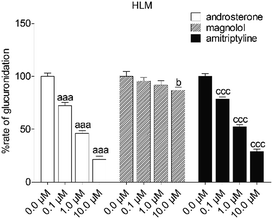 |
| | Fig. 3 Inhibitory effects of androsterone, magnolol and amitriptyline on the formation of NBKA-glucuronidated metabolites in HLM. All experiments were performed in triplicate. (a, b, c Compared with the control values of NBKA-glucuronidated metabolites in HLM, ap < 0.05, aap < 0.01, aaap < 0.001; bp < 0.05, bbp < 0.01, bbbp < 0.001; cp < 0.05, ccp < 0.01, cccp < 0.001). | |
3.3. Glucuronidation of NBKA in HLM, HIM and expressed UGTs
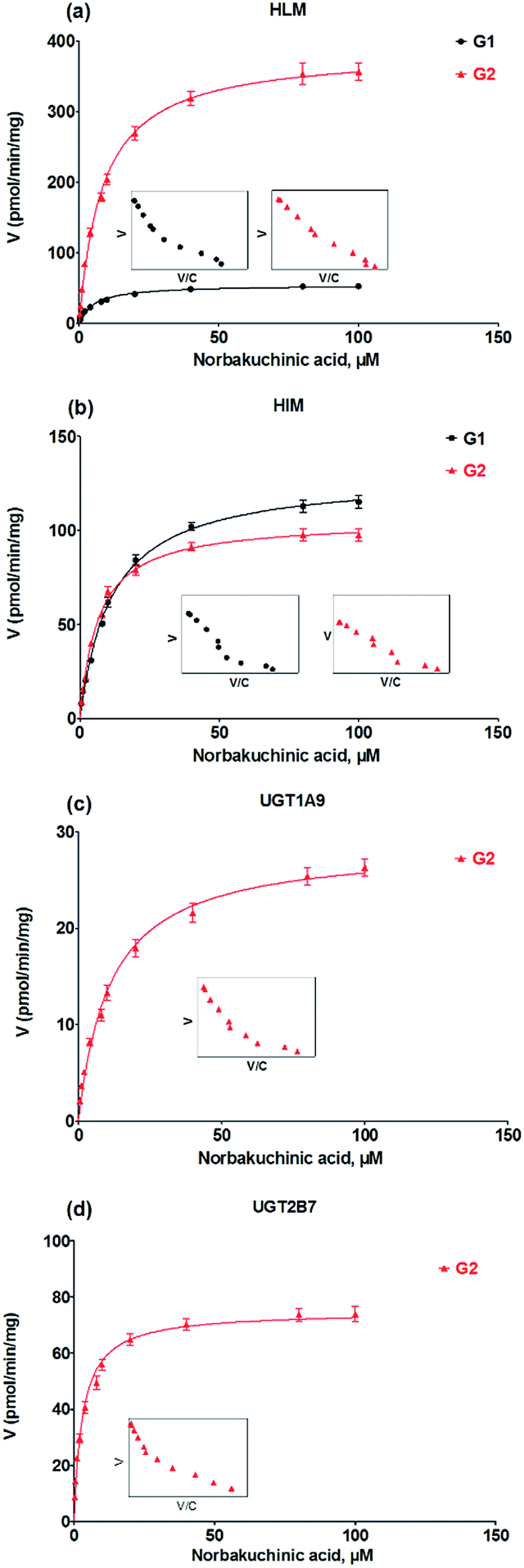
Kinetic profiling revealed that formation of G1 and G2 in HLM well modeled by Michaelis–Menten kinetics (Fig. 4a), whereas G1 and G2 by HIM followed the substrate inhibition equation and classical Michaelis–Menten kinetics, respectively (Fig. 4b). Obviously, G2 was the main metabolites by HLM and HIM with CLint values of 3.21 and 0.96 μL min−1 mg−1, respectively (Table 1). For G2, UGT1A9 (Fig. 4c) displayed weaker glucuronidation activity with the CLint value of 0.24 μL min−1 mg−1, while UGT2B7 (Fig. 4d) showed the highest glucuronidation activity (CLint = 0.70 μL min−1 mg−1) (Table 1).
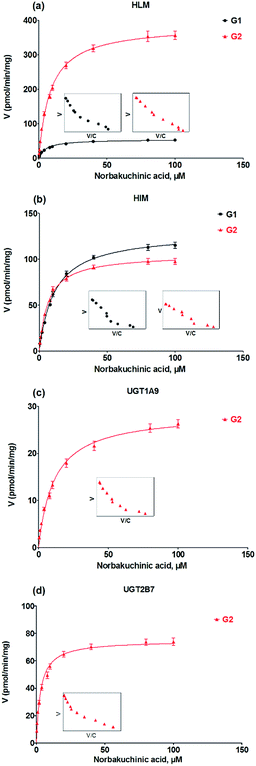 |
| | Fig. 4 Kinetic profiles for glucuronidation of norbakuchinic acid. (a) Kinetic profiles by pooled human liver microsomes (HLM); (b) kinetic profiles by pooled human liver microsomes (HIM); (c) kinetic profiles by expressed UGT1A9; (d) kinetic profiles by expressed UGT2B7. All experiments were performed in triplicate. | |
Table 1 Kinetic parameters derived for the two glucuronides (G1 and G2) of norbakuchinic acid by HLM, HIM, expressed UGT enzymes, MkLM, RLM, MLM, DLM, GpLM and RaLM (Mean ± SD). All experiments were performed in triplicatea
| Enzyme |
Metabolite |
Vmax (pmol min−1 mg−1) |
Km (μM) |
Ki (μM) |
CLint (μL min−1 mg−1) |
Model |
| G1 and G2 were the two glucuronides of norbakuchinic acid. N.A.: not available; +: under the limit of quantification; ++: unable to determine the kinetic parameters in the absence of a full kinetic profile; HLM: human liver microsomes; HIM: human intestine microsomes; MkLM: monkey liver microsomes; RLM: rat liver microsomes; MLM: mice liver microsomes; DLM: dog liver microsomes; GpLM: guinea pig liver microsomes; RaLM: rabbit liver microsomes; SI: substrate inhibition model; MM: Michaelis–Menten model. |
| HLM |
G1 |
54.11 ± 1.53 |
5.33 ± 0.59 |
N.A. |
0.53 ± 0.04 |
MM |
| G2 |
385.7 ± 5.06 |
8.47 ± 0.39 |
N.A. |
3.21 ± 0.16 |
MM |
| HIM |
G1 |
129.3 ± 2.33 |
11.36 ± 0.67 |
N.A. |
1.08 ± 0.07 |
MM |
| G2 |
105.3 ± 1.66 |
6.49 ± 0.38 |
N.A. |
0.96 ± 0.06 |
MM |
| UGT1A9 |
G1 |
+ |
+ |
+ |
+ |
+ |
| G2 |
28.63 ± 0.81 |
11.3 ± 1.05 |
N.A. |
0.24 ± 0.02 |
MM |
| UGT2B7 |
G1 |
++ |
++ |
++ |
++ |
++ |
| G2 |
73.73 ± 2.64 |
3.42 ± 0.53 |
N.A. |
0.70 ± 0.11 |
MM |
| MkLM |
G1 |
55.55 ± 1.74 |
9.61 ± 1.03 |
N.A. |
0.54 ± 0.06 |
MM |
| G2 |
936.2 ± 24.02 |
14.23 ± 1.14 |
N.A. |
7.20 ± 0.61 |
MM |
| RLM |
G1 |
98.42 ± 5.55 |
5.38 ± 1.19 |
N.A. |
0.96 ± 0.22 |
MM |
| G2 |
811.8 ± 19.26 |
8.13 ± 0.69 |
N.A. |
6.71 ± 0.59 |
MM |
| MLM |
G1 |
84.42 ± 2.89 |
8.63 ± 1.03 |
N.A. |
0.82 ± 0.10 |
MM |
| G2 |
1453.0 ± 137.1 |
14.86 ± 2.42 |
173.0 ± 48.94 |
36.33 ± 6.91 |
SI |
| DLM |
G1 |
803.2 ± 9.79 |
14.9 ± 0.56 |
N.A. |
7.44 ± 0.29 |
MM |
| G2 |
4988.0 ± 45.56 |
11.72 ± 0.35 |
N.A. |
35.63 ± 1.11 |
MM |
| GpLM |
G1 |
7001.0 ± 96.93 |
18.45 ± 0.75 |
N.A. |
48.28 ± 2.09 |
MM |
| G2 |
423.2 ± 9.91 |
17.99 ± 1.24 |
N.A. |
4.03 ± 0.29 |
MM |
| RaLM |
G1 |
2974.0 ± 48.17 |
15.18 ± 0.75 |
N.A. |
20.51 ± 1.08 |
MM |
| G2 |
347.0 ± 5.36 |
16.74 ± 0.77 |
N.A. |
3.30 ± 0.16 |
MM |
3.4. Activity correlation analysis
As mentioned, glucuronidation activity of propofol and AZT in HLM is a well-accepted functional marker for UGT1A9 and 2B7, respectively.20 It was shown that G2 was significantly correlated with propofol-glucuronidation (r = 0.681, p = 0.015) (Fig. 5a), whereas G2 were strongly correlated with AZT-glucuronidation with correlation factors (r = 0.832, p = 0.0008) (Fig. 5b), which the results indicated that UGT1A9 and 2B7 enzymes both played a critical role in NBKA glucuronidation.
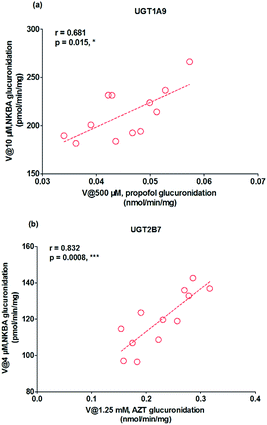 |
| | Fig. 5 Correlation analysis for glucuronidation of norbakuchinic acid. (a) Correlation analysis between NBKA-glucuronidation and propofol-glucuronidation in a bank of individual human liver microsomes (n = 12); (b) correlation analysis between NBKA-glucuronidation and AZT glucuronidation in a bank of individual human liver microsomes (n = 12); all experiments were performed in triplicate. (*p < 0.05, **p < 0.01, ***p < 0.001). | |
3.5. Contribution of UGT1A9 and 2B7 to NBKA glucuronidation in HLM
As prescribed in previous study, the RAF values for UGT1A9 and 2B7 were 0.80 and 2.11, respectively.20 The scaled Vmax/Cssc values of G2 was 0.192 (=0.24 × 0.80) μL min−1 mg−1 for UGT1A9 that represented 5.98% of the Vmax/Cssc values (3.21 μL min−1 mg−1) in HLM. The scaled Vmax/Cssc values of G2 was 1.48 (=0.70 × 2.11) ml min−1 mg−1 for UGT2B7 that represented 46.16% of the total glucuronidation activity in HLM. Clearly, UGT2B7 was the main UGT isoforms for the NBKA glucuronidation. These results also indicated that some other UGT isoforms were also responsible for the NBKA glucuronidation.
3.6. Glucuronidation of NBKA by DLM, RLM, MLM, MkLM, RaLM and GpLM
The apparent Vmax and Km values were determined for G1 and G2 by animal liver microsomes (Table 1). As shown in Fig. 6, human and these animal liver microsomes all could catalyze NBKA to produce the glucuronides with the CLint values of 0.53–48.28 μL min−1 mg−1 and 3.21–36.33 μL min−1 mg−1 for G1 and G2, respectively (Table 1). The formation of G1 and G2 by these animal liver microsomes were well modeled by the classical Michaelis–Menten kinetics (Fig. S2a∼e†) except G2 by MLM followed the substrate inhibition equation (Fig. S2f†). Clearly, there was a marked species difference (up to 91.1-fold) in hepatic glucuronidation of NBKA. Furthermore, since G2 was the main metabolite of NBKA, guinea pig and rabbits were probably the appropriate models for the studies on NBKA glucuronidation in humans due to its appropriate kinetic parameters (Table 1).
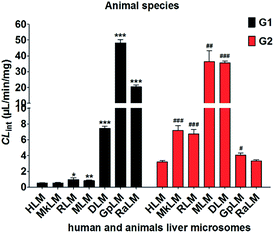 |
| | Fig. 6 Comparison of the CLint values of HLM and six animals microsomes. All experiments were performed in triplicate. (* compared with the CLint value of G1 in HLM, *p < 0.05, **p < 0.01, ***p < 0.001; # compared with the CLint value of G2 in HLM, #p < 0.05, ##p < 0.01, ###p < 0.001). | |
3.7. Effects of NBKA on the activities of recombinant human UGT enzymes
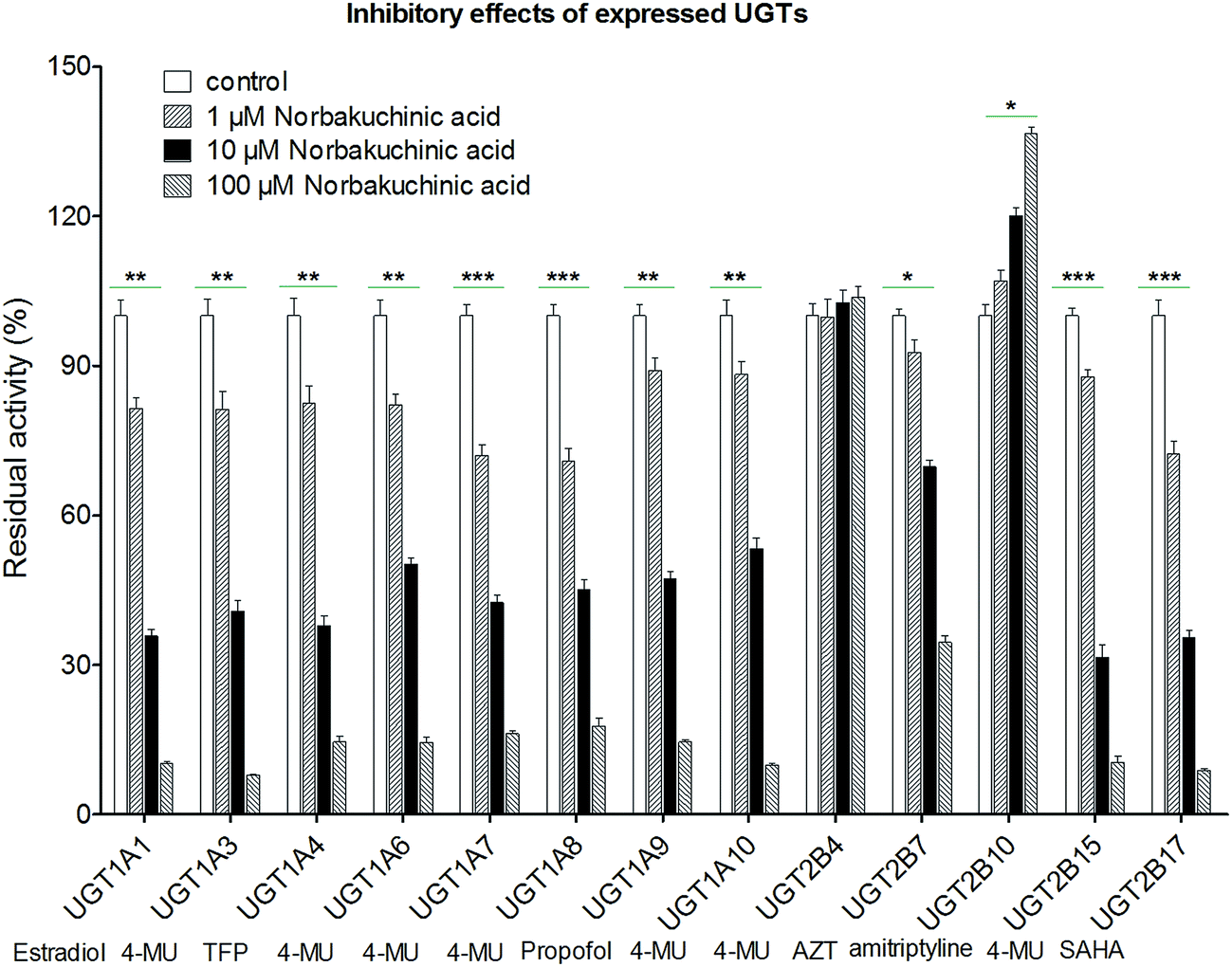
Modulation of recombinant human UGT activity by NBKA was determined using thirteen expressed UGT enzymes with commercial availability. The activities of human UGT enzymes were probed using the known substrates (β-estradiol, 4-MU, TFP, propofol, AZT, amitriptyline and SAHA) at a concentration around the Km value.21,22 As shown in Fig. 7, upon addition of NBKA (100 μM, final concentration), the catalytic activities of most human UGTs were strongly inhibited. Specifically, the residual activities of UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15 and 2B17 were 6.2%, 6.0%, 14.6%, 14.5%, 16.5%, 17.8%, 14.6%, 9.9%, 34.6%, 10.5%, and 8.9%, respectively, of their null controls. By contrast, NBKA showed activation effects on UGT2B10 (Fig. 7). Besides, UGT2B4 were not altered at three concentrations (Fig. 7).
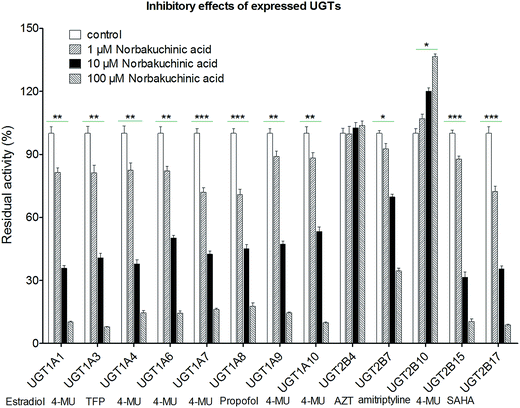 |
| | Fig. 7 Effects of NBKA on the activities of various UGT enzymes. The substrates selected in the activity assay for UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15, and 2B17 were β-estradiol (60 μM), 4-MU (4-methylumbelliferone, 1200 μM), TFP (trifluoperazine, 40 μM), 4-MU (100 μM), 4-MU (20 μM), 4-MU (800 μM), propofol (40 μM), 4-MU (30 μM), 4-MU (1000 μM), AZT (zidovudine, 500 μM), amitriptyline (10 μM), 4-MU (250 μM) and SAHA (suberoylanilide hydroxamic acid, 300 μM), respectively.* The glucuronidation rates in the presence of NBKA showed statistical difference compared to that in the absence of NBKA (*p < 0.05; **p < 0.01; p < 0.001). Each column represents the mean of triplicate determinations. 4-MU: 4-methylumbelliferone; AZT: zidovudine; SAHA: suberoylanilide hydroxamic acid; TFP: trifluoperazine. | |
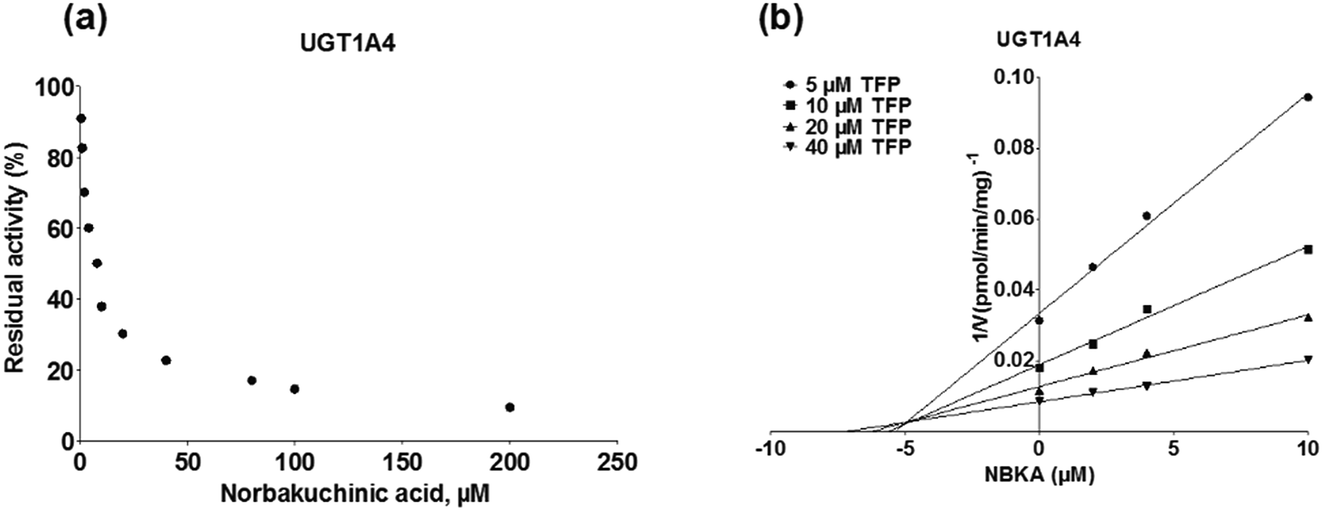
To further characterize the inhibitory effects of NBKA against human UGTs activities, dose-dependent inhibition curves were plotted using different NBKA concentrations. It could be seen from Fig. 8–10 and S3–S10† that NBKA inhibited these human UGTs in a concentration dependent manner. The calculated IC50 values were 5.55, 6.75, 7.06, 10.59, 6.47, 7.50, 10.36, 11.96, 39.07, 4.80 and 4.59 μM for UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15 and 2B17, respectively (Table 2). As have already been reported, the IC50 value of nilotinib against UGT1A1 was determined as 0.10 μM,28 while magnolol showed a strong inhibitory effect against UGT1A9 with the IC50 value of 0.08 μM,29 and androsterone inhibited UGT2B7 activity with the IC50 value of 12.58 μM.30 Compared with these three well-known UGT inhibitors, NBKA is found to be a strong inhibitor against UGT1A1, 1A3, 1A4, 1A7, 1A8, 2B15 and 2B17 (IC50 < 10 μM) while serving as a moderate inhibitor against UGT1A6, 1A9, 1A10 and 2B7 (IC50 range from 10 to 40 μM). These findings prompted us to further investigate the inhibition kinetic types and the corresponding inhibition parameters of NBKA, specifically for UGT1A1, 1A3, 1A4, 1A7, 1A8, 2B15 and 2B17.
Table 2 Inhibition kinetic parameters of norbakuchinic acid towards expressed human UGTsa
| Enzyme |
Substrate |
IC50 (μM) |
Ki (μM) |
α |
Type of inhibition |
Goodness of fit (R2) |
| 4-MU: 4-methylumbelliferone; AZT: zidovudine; SAHA: suberoylanilide hydroxamic acid; TFP: trifluoperazine. N.A.: not available. |
| UGT1A1 |
Estradiol |
5.55 ± 0.29 |
3.28 ± 0.16 |
N.A. |
Competitive |
0.9916 |
| UGT1A3 |
4-MU |
6.75 ± 0.40 |
2.67 ± 0.28 |
8.36 ± 0.25 |
Mixed |
0.9937 |
| UGT1A4 |
TFP |
7.06 ± 0.53 |
5.62 ± 0.67 |
N.A. |
Non-competitive |
0.9954 |
| UGT1A6 |
4-MU |
10.59 ± 0.49 |
13.58 ± 0.62 |
2.21 ± 0.17 |
Mixed |
0.9815 |
| UGT1A7 |
4-MU |
6.47 ± 0.38 |
7.28 ± 0.39 |
N.A. |
Non-competitive |
0.9946 |
| UGT1A8 |
4-MU |
7.50 ± 0.56 |
7.35 ± 0.40 |
N.A. |
Non-competitive |
0.9677 |
| UGT1A9 |
Propofol |
10.36 ± 0.75 |
13.56 ± 0.86 |
N.A. |
Non-competitive |
0.9928 |
| UGT1A10 |
4-MU |
11.96 ± 0.92 |
8.00 ± 0.38 |
2.11 ± 0.27 |
Mixed |
0.9949 |
| UGT2B7 |
AZT |
39.07 ± 2.67 |
20.31 ± 1.28 |
2.05 ± 0.19 |
Mixed |
0.9987 |
| UGT2B15 |
4-MU |
4.80 ± 0.37 |
3.72 ± 0.29 |
2.22 ± 0.24 |
Mixed |
0.9971 |
| UGT2B17 |
SAHA |
4.59 ± 0.50 |
2.48 ± 0.31 |
9.38 ± 0.59 |
Mixed |
0.9639 |
3.8. Kinetic analysis for inhibition of NBKA on recombinant human UGTs
To assess the possible mechanisms of NBKA inhibition against recombinant human UGT isoforms, inhibition kinetic analysis were carried out. The glucuronidation kinetics for UGTs (specific substrates) in the presence of NBKA was characterized using the Dixon plot based on the nonlinear regression analysis. In the Dixon plots, straight lines at different fixed substrate concentrations intersected in the second quadrant, indicating that inhibition of UGTs by NBKA followed the competitive or mixed-type mechanism.27
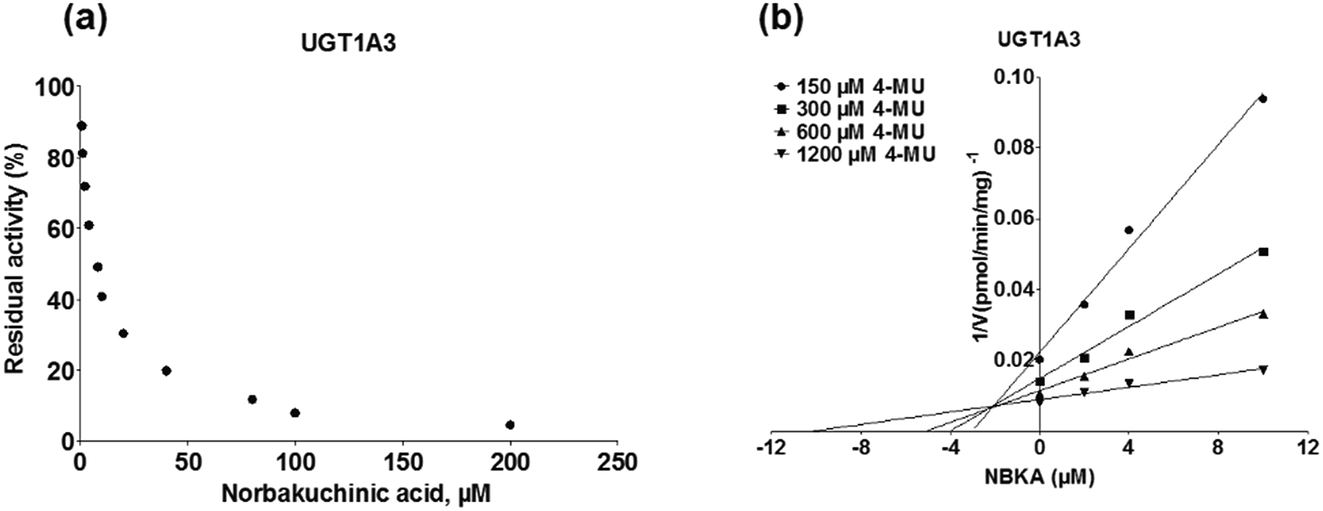
As shown in Fig. 8, Dixon plots (Fig. 8b) demonstrates that NBKA is a competitive inhibitor for UGT1A1-mediated β-estradiol-O-glucuronidation. Based on the nonlinear regression analysis, NBKA showed competitive inhibition against UGT1A1-catalyzed β-estradiol-3-glucuronidation, with a Ki value of 3.28 μM (Table 2). Likewise, Fig. 9 shows the mixed inhibition mechanism of NBKA against UGT1A3-mediated 4-MU-O-glucuronidation. The Ki value of NBKA against UGT1A3 in Fig. 9b was determined as 2.67 μM (Table 2). The inhibitory effects of UGT1A6, 1A10, 2B7, 2B15 and 2B17 exhibited the same manner as well as these of UGT1A3. The Ki values were 13.58, 8.00, 20.31, 3.72 and 2.48 μM for UGT1A6, 1A10, 2B7, 2B15 and 2B17, respectively (Table 2). By contrast, in the Dixon plot for inhibition of UGT1A4 metabolism (TFP as the substrate), the straight lines intersected on the x-axis, an indicator of noncompetitive mechanism (Fig. 10b). Similarly, the Ki values of NBKA against UGT1A4, 1A7, 1A8 and 1A9 were 5.62, 7.28, 7.35, 13.56 μM, respectively (Table 2). The concentration-dependent plots and Dixon plots of NBKA against UGT1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15 and 2B17 were shown in Fig. S3–S10,† respectively. The inhibition types and Ki values for these UGTs are listed in Table 1. These findings suggested that NBKA is a strong and non-selective inhibitor of most members from the UGT1A and UGT2B subfamilies.
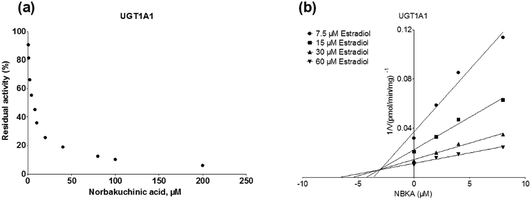 |
| | Fig. 8 Inhibition evaluation of NBKA toward recombinant UGT1A1-catalyzed β-estradiol glucuronidation. (a) NBKA exhibited concentration-dependent inhibition toward recombinant UGT1A1-catalyzed β-estradiol glucuronidation; (b) Dixon plot of NBKA's inhibition toward recombinant UGT1A1-catalyzed β-estradiol glucuronidation; all experiments were performed in triplicate. | |
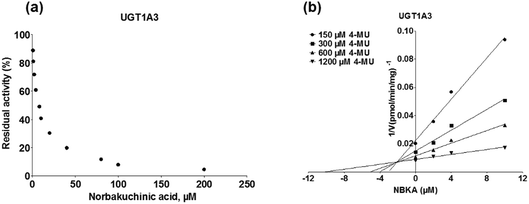 |
| | Fig. 9 Inhibition evaluation of NBKA toward expressed UGT1A3-catalyzed 4-MU-glucuronidation. Concentration-dependent plot (a) and Dixon plot (b) of NBKA's inhibition toward expressed UGT1A3-catalyzed 4-MU-glucuronidation. All experiments were performed in triplicate. 4-MU: 4-methylumbelliferone. | |
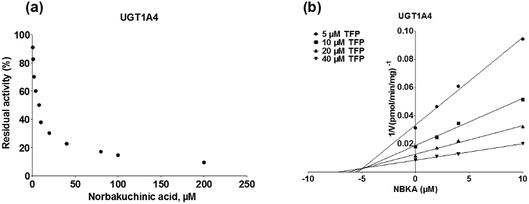 |
| | Fig. 10 Inhibition evaluation of NBKA toward expressed UGT1A4-catalyzed TFP-glucuronidation. Concentration-dependent plot (a) and Dixon plot (b) of NBKA's inhibition toward expressed UGT1A4-catalyzed TFP-glucuronidation. All experiments were performed in triplicate. TFP: trifluoperazine. | |
4. Discussion
NBKA, the most abundant primary metabolite of BK in rat plasma and urine after oral administration of BK or Psoralea corylifolia extracts, mainly underwent the glucuronidation metabolism to be excreted from the body.7,14 And also, NBKA exhibited no cytotoxicity on HKC-8 cells even at the highest concentration 50 μM.7 However, so far there is no published report regarding the glucuronidation of NBKA against human UGTs. In the present study, it was shown for the first time that NBKA was efficiently metabolized in the liver via the glucuronidation pathway. In addition, the finding that UGT2B7 was the contributor to glucuronidation of NBKA was strongly supported by three lines of evidence. First, of all hepatic UGT enzymes, UGT1A9 (Fig. 4c) and 2B7 (Fig. 4d) showed predominant activities towards NBKA with CLint values of 0.24 and 0.70 μL min−1 mg−1 protein for G2, respectively (Table 1). Furthermore, chemical inhibitor, magnolol (10 μM) inhibited the activities of UGT1A9, reducing NBKA glucuronidation activities of to be 86.9% of the control (Fig. 3), while the remaining activities of UGT2B7 for G2 were 21.5 and 28.9% in the presence of androsterone (10 μM) and amitriptyline (10 μM), respectively (Fig. 3). Second, as shown in Fig. 5a and b, NBKA glucuronidation was significantly correlated with propofol-glucuronidation (r = 0.681, p = 0.015) and AZT-glucuronidation (r = 0.832, p = 0.0008), respectively. Third, about 5.98% and 46.16% of NBKA glucuronidation (G2) in HLM was attributed to UGT1A9 and 2B7 based on the RAF approach. Therefore, the role of UGT enzymes in determining the body exposure (bioavailability) and elimination of this compound should not be underestimated.
To explore further the metabolic mechanism, the glucuronidation activity was obtained by kinetic profiling and modeling at a series of NBKA concentrations. The relative activities of different expressed UGT enzymes toward NBKA glucuronidation were evaluated by the derived CLint values. Use of CLint (=Vmax/Cssc) as an indicator of UGTs enzymes activity was advantageous, because (1) CLint represents the catalytic efficiency of the UGT enzyme and is independent of the substrate concentration; (2) compared with other kinetic parameters such as Cssc and Vmax, CLint is more relevant in an attempt to predict hepatic clearance in vivo.31 In addition, the CLint values of NBKA glucuronidation generated in HLM indicated a first-pass metabolism in the liver after oral administration, and the following hepatic glucuronidation should lead to a rapid elimination of NBKA from human body. Moreover, UGT1A9 and 2B7 are highly polymorphic enzymes.32,33 Since NBKA was predominantly metabolized by these two hepatic enzymes, a large pharmacokinetic variability was expected to exist among individuals with different UGT1A9 and 2B7 genotypes.
On the other hand, herb–drug interactions (HDIs) are significant clinical safety concerns. Many studies have reported that herbs can interact with several clinical drugs with narrow therapeutic indices, including warfarin, digoxin, docetaxel, irinotecan and so on.34 Hence, evaluations of the inhibitory potentials of herbs have essential implications for drug development. As the most abundant metabolite in rats, greater attention should be given to avoiding the potential risks of drug–herb interactions following the oral administration of BK or Psoralea corylifolia extracts by considering the potent inhibitory effects of NBKA on several human UGTs. Thus, the inhibitory effects of NBKA against human UGTs were investigated for the first time, and NBKA displayed potent inhibitory effects against several UGTs including UGT1A1, 1A3, 1A4, 1A7, 1A8, 2B15 and 2B17, with low Ki values ranging from 2.48 to 7.35 μM (Table 2). Compared with the inhibitory effects of BK against UGT1A6 (Ki = 5.3 μM), 1A8 (Ki = 1.8 μM), 1A10 (Ki = 92.6 μM)35 and UGT2B4 (Ki = 20.7 μM),36 this study showed that NBKA were with lower Ki values and also was a potent and broad-spectrum inhibitor of human UGTs (Fig. 7). However, to the regret, so far, there are no researches on the plasma concentration of norbakuchinic acid, the drug–drug interactions (DDI) potential in vivo for norbakuchinic acid via the inhibition against human UGTs enzymes were not predicted. Therefore, a research of NKBA pharmacokinetics in vivo is indispensable.
Recently, an increasing number of traditional herbs have attracted more attention for their potential toxic effects on human.37 Psoralea corylifolia has been widely used as adjuvant therapy in several countries, mainly for the treatment of bone diseases.3 Furthermore, it is recognized as one of the emerging hepatotoxins, and several cases on remarkably elevated bilirubin and acute liver injury after exposure to Psoralea corylifolia and its related proprietary medicine have been reported.38–40 It seems possible that the chemical compounds and xenobiotics in vivo of Psoralea corylifolia may be responsible for the hepatotoxicity. Previous studies demonstrated that the ethanol extracts of Psoralea corylifolia displayed strong inhibitory effect on UGT1A1, while several major compounds (such as bavachin, neobavaisoflavone, isobavachalcone, bavachinin, corylifol A and bakuchiol) were identified as strong inhibitors of UGT1A1, 1A6, 1A8, 1A10 and 2B4.35,36,41
It is also well known that the marketed drugs mainly metabolized by UGT1A1 (15%), 1A4 (20%) and 2B7 (35%).42 UGT1A1 attracted more particular attention, due to this conjugating enzyme plays a key role in the metabolism of a wide variety of clinical drugs (such as morphine and SN-38) and some important endogenous toxicants (like bilirubin and bile acid).30,42,43 So far, UGT1A1 is the only physiologically relevant UGT isoform involved in the metabolic clearance of endobiotics bilirubin, which is a toxic waste formed from heme degradation. Likewise, UGT1A4 expressed in liver is the main contributor for the metabolism of imipramine and trifluoperazine, whereas AZT and morphine are the main substrates of UGT2B7.30,42 The inhibition of the catalytic activity of human UGTs by drugs or xenobiotics may not only trigger clinical adverse herb–drug interactions, but also result in metabolic disorders of endogenous substances. In fact, there are numerous clinically relevant therapeutic drugs causing elevated unconjugated bilirubin in blood via UGT1A1 inhibition. For example, the mechanism of indinavir-induced hyperbilirubinemia has been revealed as the direct competitive inhibitory effects on bilirubin conjugating activity rather than suppression of enzyme expression.44 Inhibition of UGT1A1-mediated bilirubin clearance may also be an important reason for the nilotinib-induced elevated level of unconjugated bilirubin in serum.45 Of importance, UGT2B15 and UGT2B17 are both regulated at the transcriptional level by their own androgen substrates through the androgen receptor.46 In this study, our results clearly demonstrated that NBKA moderately inhibited the activities of UGT1A1, 1A3, 2B15 and 2B17 with Ki values below 5 μM. These findings indicate that NBKA might induce the clinically significant interactions with drugs that are primarily metabolized by these human UGTs, especially UGT1A1.
Notably, the possibility of in vivo inhibition of NBKA against human UGTs is complicated by many factors, such as genetic polymorphisms.32,33,47 It is also well known that some polymorphic expression of certain UGTs will dramatically reduce the expression and function of the corresponding enzyme. This may leads to underestimation of inhibitory effects for variants that possessing low catalytic activity. UGT1A1 is a highly polymorphic enzyme with high clinical significance.33 In factor, up to 42% African and South Asian populations possessing TA-repeat polymorphism (UGT1A1*28, commonly diagnosed as Gilbert's syndrome) manifest impaired bilirubin-conjugating activity.41 Thus, it is easily conceivable that individuals with UGT1A1*28 might be expected to be more susceptible to hyperbilirubinemia and subsequently hepatic dysfunction as a consequence of NBKA mediated inhibition of human UGT1A1. Similarly, polymorphisms in UGT1A7, UGT1A9 and UGT1A10, especially for UGT1A7*3, UGT1A9*3 and UGT1A10 T202I, will also influence the inhibitory effects in vivo.48 Moreover, the herb–drug interaction possibility of in vivo inhibition of NBKA against human UGTs needs to be fully considered.
5. Conclusions
In conclusion, NBKA, as the abundant metabolite in plasma and urine, were dominantly metabolized to produce two glucuronides (G1 and G2) in HLM and HIM. G2 was the main metabolites with the CLint values of 3.21 and 0.96 μL min−1 mg−1 protein in HLM and HIM, respectively. Furthermore, UGT2B7 (CLint = 0.70 μL min−1 mg−1) and 1A9 (CLint = 0.24 μL min−1 mg−1) were mainly responsible for G2. In addition, G2 was significantly correlated with propofol-glucuronidation (r = 0.681, p = 0.015) and AZT-glucuronidation (r = 0.832, p = 0.0008), respectively. Based on the RAF approach, about 5.98% and 46.16% of G2 in HLM was attributed to UGT1A9 and 2B7. Also, NBKA glucuronidation (G1 and G2) showed marked species differences. Moreover, NBKA displayed a broad-spectrum inhibition against human UGTs. The inhibition of these UGTs followed competitive (UGT1A1), mixed (UGT1A3, 1A6, 1A10, 2B7, 2B15 and 2B17) and non-competitive (UGT1A4, 1A7, 1A8 and 1A9) mechanisms, with Ki values ranging from 2.48 to 20.31 μM. Among them, NBKA exhibited moderate inhibitory effects against UGT1A1, 1A3, 1A4, 1A7, 1A8, 2B15 and 2B17 (both IC50 and Ki values lower than 8 μM). On the contrary, the effect of NBKA was activating for UGT2B10, while UGT2B4 were not altered at three concentrations. Taken altogether, NBKA was primarily metabolized by UGT2B7, and had a potent and broad-spectrum inhibitory effect against most human UGTs which suggested that much caution should be exercised when high-dose BK-included traditional Chinese medicine is co-administered with UGT substrates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Major Project for International Cooperation and Exchange of the National Natural Science Foundation of China (Grant No. 81220108028), the National Major Scientific and Program of Introducing Talents of Discipline to Universities (B13038) and Guangdong Provincial Science and Technology Project (2016B090921005).
References
- X. Zhang, W. Zhao, Y. Wang, J. Lu and X. Chen, Am. J. Chin. Med., 2016, 44, 35–60 CrossRef CAS PubMed.
- Y. Chen, Q. Xiang and Z. Chen, Anal. Methods, 2014, 6, 269–275 RSC.
- S. H. Lim, T. Y. Ha, S. R. Kim, J. Ahn, H. J. Park and S. Kim, Br. J. Nutr., 2009, 101, 1031–1039 CrossRef CAS PubMed.
- J. E. Kim, J. H. Kim, Y. Lee, H. Yang, Y. S. Heo, A. M. Bode, K. W. Lee and Z. Dong, Oncotarget, 2016, 7, 14616–14627 Search PubMed.
- Z. Chen, K. Jin, L. Gao, G. Lou, Y. Jin, Y. Yu and Y. Lou, Eur. J. Pharmacol., 2010, 643, 170–179 CrossRef CAS PubMed.
- H. O. Pae, H. Cho, G. S. Oh, N. Y. Kim, E. K. Song, Y. C. Kim, Y. G. Yun, C. L. Kang, J. D. Kim, J. M. Kim and H. T. Chung, Int. Immunopharmacol., 2001, 1, 1849–1855 CrossRef CAS PubMed.
- P. L. Wang, F. X. Zhang, Z. C. Qiu, Z. H. Yao, M. S. Wong, X. S. Yao and Y. Dai, Fitoterapia, 2016, 109, 31–38 CrossRef CAS PubMed.
- F. Wang, X. Zhou, Q. Wang and B. Zhang, Chin. J. Pharmacol. Toxicol., 2010, 24, 50–58 Search PubMed.
- X. Zhuang, Y. Zhong, M. Yuan and H. Li, J. Pharm. Biomed. Anal., 2013, 75, 18–24 CrossRef CAS PubMed.
- D. M. Yan, Y. X. Chang, Y. F. Wang, E. W. Liu, J. Li, L. Y. Kang and X. M. Gao, J. Ethnopharmacol., 2010, 128, 697–702 CrossRef CAS PubMed.
- S. Q. Zhang and Y. M. Fan, Biomed. Chromatogr., 2014, 28, 433–438 CrossRef CAS PubMed.
- J. Pi, X. Gao, Y. Yu, Y. Zheng, Z. Zhu and Y. Wang, Arch. Pharmacal Res., 2014, 1–11 CAS.
- C. Hu, Q. Liang, X. Tang, Y. Wang, Z. Ma, C. Xiao, H. Tan, Y. Gao and X. Huang, Fitoterapia, 2015, 106, 129–134 CrossRef CAS PubMed.
- P. L. Wang, Z. H. Yao, F. X. Zhang, X. Y. Shen, Y. Dai, L. Qin and X. S. Yao, J. Pharm. Biomed. Anal., 2015, 112, 23–35 CrossRef CAS PubMed.
- X. Zhou, S. Wang, H. Sun and B. Wu, Drug Metab. Pharmacokinet., 2015, 30, 425–433 CrossRef CAS PubMed.
- W. E. Evans and M. V. Relling, Science, 1999, 286, 487–491 CrossRef CAS PubMed.
- P. Mackenzie, K. Bock, B. Burchell, C. Guillemette, S. Ikushiro, T. Iyanagi, J. Miners, I. Owens and D. Nebert, Pharmacogenet. Genomics, 2005, 15, 677–685 CrossRef CAS PubMed.
- P. A. Gregory, R. H. Lewinsky, D. A. Gardner-Stephen and P. I. Mackenzie, Toxicol. Appl. Pharmacol., 2004, 199, 354–363 CrossRef CAS PubMed.
- B. Wu, K. Kulkarni, S. Basu, S. Zhang and M. Hu, J. Pharm. Sci., 2011, 100, 3655–3681 CrossRef CAS PubMed.
- L. Wang, X. Hong, Z. Yao, Y. Dai, G. Zhao, Z. Qin, B. Wu, F. J. Gonzalez and X. Yao, Xenobiotica, 2017, 1–11, DOI:10.1080/00498254.2017.1323139.
- H. Xin, X. Y. Qi, J. J. Wu, X. X. Wang, Y. Li, J. Y. Hong, W. He, W. Xu, G. B. Ge and L. Yang, Food Chem. Toxicol., 2016, 90, 112–122 CrossRef CAS PubMed.
- H. Sun, T. Zhang, Z. Wu and B. Wu, J. Pharm. Sci., 2015, 104, 244–256 CrossRef CAS PubMed.
- H. Sun, Z. Ma, D. Lu and B. Wu, J. Pharm. Sci., 2015, 104, 2369–2377 CrossRef CAS PubMed.
- S. C. Khojasteh, H. Wong and C. E. C. A. Hop, Drug Metabolism and Pharmacokinetics Quick Guide, Springer, New York, 2011 Search PubMed.
- J. Hutzler and T. Tracy, Drug Metab. Dispos., 2002, 30, 355–362 CrossRef CAS.
- L. Zhu, L. Xiao, W. Li, Y. Zhang, W. Han, Y. Zhu, G. Ge and L. Yang, Basic Clin. Pharmacol. Toxicol., 2016, 119, 505–511 CrossRef CAS PubMed.
- A. Cornish-Bowden, Biochem. J., 1974, 137, 143–144 CrossRef CAS PubMed.
- L. Ai, L. Zhu, L. Yang, G. Ge, Y. Cao, Y. Liu, Z. Fang and Y. Zhang, Xenobiotica, 2014, 44, 320–325 CrossRef CAS PubMed.
- L. Zhu, G. Ge, Y. Liu, G. He, S. Liang, Z. Fang, P. Dong, Y. Cao and L. Yang, Xenobiotica, 2012, 42, 1001–1008 CrossRef CAS PubMed.
- V. Uchaipichat, P. I. Mackenzie, D. J. Elliot and J. O. Miners, Drug Metab. Dispos., 2006, 34, 449–456 CAS.
- B. Wu, D. Dong, M. Hu and S. Zhang, Curr. Top. Med. Chem., 2013, 13, 1343–1352 CrossRef CAS PubMed.
- C. R. Bhasker, W. McKinnon, A. Stone, A. C. Lo, T. Kubota, T. IshizaKi and J. O. Miners, Pharmacogenetics, 2000, 10, 679–685 CrossRef CAS PubMed.
- S. Nagar and R. L. Blanchard, Drug Metab. Rev., 2006, 38, 393–409 CrossRef CAS PubMed.
- S. F. Zhou, Z. W. Zhou, C. G. Li, X. Chen, X. Yu, C. C. Xue and A. Herington, Drug Discovery Today, 2007, 12, 664–673 CrossRef CAS PubMed.
- D. G. Dong, Y. Zhang, S. L. Zhang, J. Lv, E. M. Guo, Z. Z. Fang and Y. F. Cao, Pharmazie, 2014, 69, 60–63 CAS.
- Y. Li, Y. Li, H. Chen and W. Sun, Lat. Am. J. Pharm., 2013, 32, 924–926 CAS.
- F. Stickel and D. Shouval, Arch. Toxicol., 2015, 89, 851–865 CrossRef CAS PubMed.
- W. L. Cheung, M. L. Tse, T. Ngan, J. Lin, W. K. Lee, W. T. Poon, T. W. Mak, V. K. S. Leung and T. N. Chau, Clin. Toxicol., 2009, 47, 683–685 CrossRef PubMed.
- R. Teschke and R. Bahre, Ann. Hepatol., 2009, 8, 258–266 Search PubMed.
- J. Wang, Z. Jiang, J. Ji, Y. Li, M. Chen, Y. Wang, Y. Zhang, T. Tai, T. Wang and L. Zhang, J. Ethnopharmacol., 2012, 144, 73–81 CrossRef CAS PubMed.
- X. X. Wang, X. Lv, S. Y. Li, J. Hou, J. Ning, J. Y. Wang, Y. F. Cao, G. B. Ge, B. Guo and L. Yang, Toxicol. Appl. Pharmacol., 2015, 289, 70–78 CrossRef CAS PubMed.
- J. A. Williams, R. Hyland, B. C. Jones, D. A. Smith, S. Hurst, T. C. Goosen, V. PeterKin, J. R. Koup and S. E. Ball, Drug Metab. Dispos., 2004, 32, 1201–1208 CrossRef CAS PubMed.
- A. Rowland, J. O. Miners and P. I. Mackenzie, Int. J. Biochem. Cell Biol., 2013, 45, 1121–1132 CrossRef CAS PubMed.
- S. D. Zucker, X. Qin, S. D. Rouster, F. Yu, R. M. Green, P. Keshavan, J. Feinberg and K. E. Sherman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12671–12676 CrossRef CAS PubMed.
- J. B. Singer, Y. Shou, F. Giles, H. M. Kantarjian, Y. Hsu, A. S. Robeva, P. Rae, A. Weitzman, J. M. Meyer, M. Dugan and O. G. Ottmann, Leukemia, 2007, 21, 2311–2315 CrossRef CAS PubMed.
- B. Y. Bao, B. F. Chuang, Q. Wang, O. Sartor, S. P. Balk, M. Brown, P. W. Kantoff and G. S. Lee, Prostate, 2008, 68, 839–848 CrossRef CAS PubMed.
- W. Jiang and M. Hu, RSC Adv., 2012, 2, 7948–7963 RSC.
- X. Lv, X. X. Wang, J. Hou, Z. Z. Fang, J. J. Wu, Y. F. Cao, S. W. Liu, G. B. Ge and L. Yang, Toxicol. Appl. Pharmacol., 2016, 301, 42–49 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10376j |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Shishi Li‡a,
Zifei Qin
*ab,
Shishi Li‡a,
Zifei Qin