DOI:
10.1039/C6RA23939K
(Paper)
RSC Adv., 2017,
7, 28979-28986
Preparation and characterization of Fe3O4/gallic acid/graphene oxide magnetic nanocomposites as highly efficient Fenton catalysts
Received
26th September 2016
, Accepted 5th May 2017
First published on 1st June 2017
Abstract
In this study, Fe3O4/gallic acid/graphene oxide magnetic nanocomposites (Fe3O4/GA/GO MNPs) were synthesized as heterogeneous Fenton catalysts to activate H2O2 to degrade methylene blue (MB). The catalysts were characterized by X-ray diffraction (XRD), zeta potential analysis, thermogravimetric analysis (TGA), transmission electron microscopy (TEM) and X-ray Photoelectron Spectroscopy (XPS). The addition of GA resulted in crystal defects on the surface of Fe3O4/GA/GO which improved its catalytic properties. Moreover, Fe3O4/GA/GO had larger surface area, correspondingly exposing more active sites for H2O2 activation, as well as enhancing the adsorption of MB which also promoted its catalytic properties. Complete removal of MB (0.2 mM) and remarkable mineralization of total organic carbon (TOC) (66.7%) were obtained within 420 min at pH 6.0 and 30 °C by using 1.0 g L−1 Fe3O4/GA/GO MNPs and 160.0 mM H2O2. Furthermore, MB could be completely degraded by Fe3O4/GA/GO MNPs in a wide pH value ranging from 3.0 to 10.0. The catalyst showed stable catalytic activity, as well as good mechanical stability, and could be recycled 10 times.
1. Introduction
Advanced Oxidation Processes (AOPs) are promising technologies for the treatment of urban and industrial wastewater due to their high efficiency and versatility.1–4 Among the AOPs, the Fenton reaction has been proven to be effective to degrade organic pollutants due to it generating hydroxyl radicals (˙OH) with a very high oxidation potential (EΘ = +2.8 V vs. NHE in acid environment).5–8 In the Fenton (Fe2+/H2O2) process, the generation of ˙OH and degradation of pollutants are described in eqn (1) and (2). According to eqn (1) and (3), Fe2+ is oxidized to Fe3+ and then regenerated.| | |
Fe2+ + H2O2 → Fe3+ + ˙OH + OH−
| (1) |
| | |
˙OH + P → intermediates → H2O + CO2
| (2) |
| | |
Fe3+ + H2O2 → Fe2+ + ˙HO2 + H+
| (3) |
However, acidic pH (2.5–4.0) is required and a large amount of iron sludge produced from the process needs secondary treatment in the traditional Fenton reaction.9 To circumvent these shortcomings, heterogeneous Fenton processes have been developed that generate ˙OH from the decomposition of H2O2 by solid catalyst recycling in consecutive batch cycles such as nano-zero valence iron,10 iron oxides,11,12 iron-immobilized clays,13 and Fe-containing zeolites.14 Fe3O4 MNPs show efficient catalysis in heterogeneous Fenton system because the Fe(II) in Fe3O4 play an important role in the initiation of the Fenton reaction based on the classical Haber–Weiss mechanism.15 The octahedral sites in the magnetite structure can easily accommodate both Fe(II) and Fe(III), allowing the Fe species to be reversibly oxidized and reduced while keeping the same structure.16 In addition, the magnetite can be easily separated from the reaction medium by an external magnetic field.17
However, Fe3O4 MNPs are apt to aggregate, resulting in reduction of surface/volume ratio and dispersion stability in aqueous solution.18 Moreover, the H2O2-activating ability of Fe3O4 MNPs is generally not strong, and thus the catalytic degradation rate of target substances is low. It is a big challenge to improve the catalytic performance of Fe3O4 MNPs. In order to solve above problems, introducing immobilized iron species on solid supports or modified Fe3O4 with organics as another type of promising heterogeneous catalysts have been considered and is drawing increasing attention recently. Niu et al. prepared humic acid (HA) coated Fe3O4 magnetic nanoparticles (Fe3O4/HA) which exhibited high activity to produce ˙OH by H2O2 decomposition, and the complexed Fe(II)–HA or Fe(III)–HA accelerated electron transfer ability and led to rapid regeneration of Fe(II) species and production of ˙OH.19
Gallic acid (GA) as a kind of flavonoids is capable of binding iron, which can decrease the conversion of Fe(III) to Fe(II) and prevent itself from transforming to quinone.20 Therefore, the addition of GA in Fenton or Fenton-like process can promote the regeneration of Fe(II) which helps the generation of ˙OH.21 Moreover, bidentate enediol ligands containing catechol group have been demonstrated to effectively convert the under-coordinated Fe surface sites back to a bulk-like lattice structure with an octahedral geometry for oxygen-coordinated iron, which may lead to tight binding between ligands and iron oxide.22 Dorniani et al. reported successful preparation of Fe3O4 magnetic nanoparticles coated with gallic acid used for drug delivery.23
Graphene oxide (GO) produced by the oxidation of graphite is heavily oxygenated, bearing hydroxyl and epoxide functional groups on their basal planes, in addition to carbonyl and carboxyl groups located at the sheet edges.24 The presence of these functional groups makes GO sheets strongly hydrophilic, which allows GO to readily swell and disperse in water. These functional properties of GO have been investigated by immobilizing iron oxide on GO sheets to improve the dispersibility of iron oxide. Dong et al. reported that graphene oxide–Fe3O4 magnetic nanocomposites possessed intrinsic peroxidase-like activity and the nanocomposites retained their magnetic properties and could be effectively separated by a magnet.25 Zubir et al. synthesized graphene oxide–iron oxide (GO–Fe3O4) nanocomposites by co-precipitating iron salts onto GO sheets in basic solution and the catalyst behaved higher degradation ability of acid orange 7 than that of Fe3O4 nanoparticles in heterogeneous Fenton-like reaction.26
In this study, we show that GA modified Fe3O4 with the support of GO (Fe3O4/GA/GO MNPs) will greatly increase the catalytic activity of Fe3O4 for H2O2 decomposition. The Fe3O4/GA/GO MNPs were prepared by a facile coprecipitation method and applied in the degradation of methylene blue (MB) in heterogeneous Fenton reaction. MB, as a typical kind of cationic dye, was selected as model compound for the oxidation reaction. The nanocomposites were characterized using X-ray diffractometer (XRD), zeta potentials analysis, thermogravimetric analysis (TGA), transmission electron microscope (TEM) and X-ray photoelectron spectroscopy (XPS). Influences of H2O2 concentration, catalyst dosage, initial MB concentration and solution pH on MB degradation were also assessed. Mechanisms for the formation of Fe3O4/GA/GO MNPs and the heterogeneous Fenton degradation of MB were carefully analyzed.
2. Experimental
2.1. Chemicals
All chemicals were analytical grade and used without further purification. Ferric chloride (FeCl3·6H2O), ferrous sulfate (FeSO4·7H2O), hydrogen peroxide (H2O2, 30 wt%), sulfuric acid (H2SO4, 98%), ammonium hydroxide (NH3·H2O, 25%), methylene blue (MB) and gallic acid (GA) were obtained from Chuandong Chemical Inc., Chengdu, Sichuan, China. 5,5′-Dimethyl-1-pyrroline-N-oxide (DMPO) was purchased from Adamas, Shanghai, China. Graphite powder (99.95%) was purchased from Aladdin, Shanghai, China. All solutions and suspensions were prepared with deionized water.
2.2. Preparation and characterization of catalysts
Graphite oxide was synthesized according to Hummers' method.27 Briefly, 23 mL of concentrated H2SO4 (98%) was added into a 500 mL dry flask containing 0.5 g of NaNO3 placed in an ice-water bath and stirred until the temperature of the mixed liquor is below 5 °C. Then, 1.0 g of flake graphite was added into the flask, and the mixture was stirred for 30 min. Potassium permanganate (3.0 g) was added slowly to the suspension under continuous vigorous stirring and the rate of addition was carefully controlled to keep the reaction temperature below 20 °C. Afterwards, the mixture was stirred at 35 °C for 2 h. Then, 46 mL of H2O was slowly added into the mixture under vigorous stirring, and the reaction temperature was kept around 98 °C for 30 min. After the system cooled to the room temperature, the suspension was centrifuged, washed with HCl (10%) for several times until no SO42− was detected (tested by BaCl2 solution) in the mixture. Then, the suspension was dialyzed until no Cl− was detected in water (tested by AgNO3 solution). Finally, the mixture was subjected to freeze-drying to generate graphite oxide dryness. Graphene oxide solution (GO, 1.0 g L−1) was obtained through ultrasound stripping of graphite oxide in water for reserve.
Fe3O4/GA/GO MNPs were prepared by a facile chemical coprecipitation method.18 In brief, FeCl3·6H2O (10 mmol) and FeSO4·7H2O (5.0 mmol) were dissolved into 15 mL deionized water, mixed with 50 mL 20 mM gallic acid (GA) solution. Then, 100 mL 1.0 g L−1 of GO was added into the mixture under magnetic stirring, after 30 min, the mixture was added into 100 mL of NH3·H2O (3.3 M) rapidly under vigorous mechanical stirring for 120 min. The whole synthesis processes were performed at ambient atmosphere, without protection of inert gases or vacuum. The black magnetic nanoparticles were separated by an external magnetism and washed with deionized water until the pH of mixture was neutral and no iron ions or GA were detected in water, and then dried in a vacuum oven at 50 °C for 24 h. Fe3O4/GA and Fe3O4/GO MNPs were synthesized as above procedures without adding GO and GA, respectively. Fe3O4 MNPs were synthesized as above procedures without adding other reagents. All the products were stored in a desiccator under ambient temperature for further experiments.
The phase structure of the Fe3O4, Fe3O4/GA, Fe3O4/GO and Fe3O4/GA/GO MNPs were determined by powder XRD (Empyrean, Netherlands). The zeta potentials of catalyst suspensions at different pH values were determined by a Malvern 3000 Zetasizer. Thermogravimetric analysis (TGA) was carried out for powder samples using a TGA/DSC 1 thermogravimetric analyzer (TGA) (Mettler-Toledo, Switzerland) under N2 protection at a heating rate of 5 °C min−1. The morphology of the samples were observed by transmission electron microscopy (TEM, JEOL JEM-2100F, Japan). The XPS spectra were recorded on an ESCALAB 250 photoelectron spectrometer (ThermoVG Scientific, USA) with Al Kα (1486.6 eV) as the X-ray source. All XPS spectra were corrected using the C 1s line at 284.6 eV. The Brunauer–Emmett–Teller (BET) surface area was examined by nitrogen adsorption–desorption isotherm measurements at 77 K on a belsorp-max surface area and porosity analyzer (MicrotracBEL, Japan).
2.3. Degradation procedures
The degradation procedures were carried out using 100 mL beaker flask shaken at a speed of 180 rpm. In a typical reaction, 50 mL of MB solution in a certain concentration was prepared with the addition of required amount of Fe3O4/GA/GO MNPs. The pH of the reaction solution was adjusted to a required value by 1.0 M H2SO4 or 1.0 M NaOH. The degradation reactions were initiated by adding H2O2 to the suspension after pre-adsorption equilibrium. At different time intervals, 0.6 mL of suspension sample was collected and immediately quenched with 30 μL pure methanol, then separated by an external magnetism. The supernatant liquid was collected for analysis. Each experiment was run in triplicate. The reported data were the arithmetic mean of three measured values.
2.4. Analytical methods
The concentration of MB was measured by UV-vis spectroscopy at a wavelength of 660 nm, the maximum absorption wavelength of MB.28 Total organic carbon (TOC) concentration was analyzed in an analyticjenaAG MultiN/C3100TOC/TN analyzer to evaluate the mineralization of MB. The total iron leaching of the catalyst was monitored using the orthophenanthroline complexometric method (λ = 510 nm). ˙OH radicals were identified by electron spin resonance (ESR) spectroscopy (Bruker model ESP 300E), operating at 322.767 mT center field and 9055.282 MHz microwave frequency. For electron spin resonance (ESR) assay, 0.5 mL samples were taken instantaneously after heterogeneous Fenton being reacted for 10 min and mixed with 50 μL 500 mM DMPO to form DMPO–OH adduct. The ESR spectra of the reaction filtrate were recorded.
3. Results and discussion
3.1. Formation of Fe3O4/GA/GO MNPs
A hybrid of Fe3O4/GA/GO MNPs was obtained through a two-step procedure. In the first step, graphite was treated with H2SO4, NaNO3 and KMnO4 to obtain GO which contained a variety of functional groups including carboxyl, hydroxy, epoxy and ketone. In the second step, the complexed GA–Fe was well dispersed onto GO sheet and served as nucleation sites. Then, in situ formation of gallic acid modified Fe3O4 nanocomposites started to grow on the surface of GO as NH3·H2O was added. The formation processes of Fe3O4/GA/GO MNPs are shown in Scheme 1.
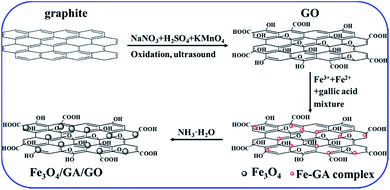 |
| | Scheme 1 The scheme of the formation processes of Fe3O4/GA/GO MNPs. | |
3.2. Characterization of Fe3O4/GA/GO MNPs
The XRD patterns of Fe3O4, Fe3O4/GO, Fe3O4/GA and Fe3O4/GA/GO MNPs are shown in Fig. 1a. The four materials have similar diffraction peaks at 2θ = 29.2°, 34.3°, 41.5°, 51.5°, 54.9°, 60.2°, being assigned to the (220), (311), (400), (422), (511), (440) planes of the cubic spinel structure. The results indicate that the crystal structure of Fe3O4 MNPs is not changed after modification. And no other peaks corresponding to the magnetite are detected in the XRD patterns, indicating that the Fe3O4 MNPs in the composites are pure Fe3O4 with inverse spinel structure.16 Moreover, the peaks of the Fe3O4/GO are narrow and strong, reflecting the fact that the degree of crystallinity of the Fe3O4/GO is higher than that of bare Fe3O4, which indicates that GO promotes the crystallization of Fe3O4. However, Fe3O4/GA peaks are broader and weaker. The reason was that GA as a complexing regent could make the crystallization process of Fe3O4 slower and less regular than that of bare Fe3O4, leading to much more crystal defects benefiting the catalytic activity.29 Therefore, the combination of GA and GO for Fe3O4 modification could retain high crystallinity and also produce some crystal defects which may result in high catalytic property of the Fe3O4/GA/GO MNPs.
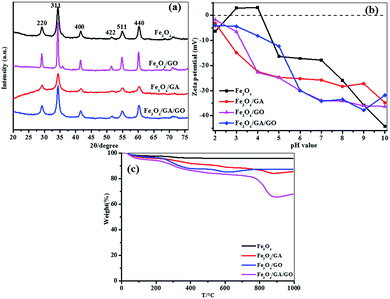 |
| | Fig. 1 (a) XRD patterns (b) zeta potentials (c) TGA curves of Fe3O4, Fe3O4/GO, Fe3O4/GA and Fe3O4/GA/GO MNPs. | |
Zeta potentials are widely used to characterize electronic charges on the surface of particles in aqueous solution. The zeta potentials of Fe3O4, Fe3O4/GA, Fe3O4/GO and Fe3O4/GA/GO MNPs were determined as a function of pH. As presented in Fig. 1b, the zeta potentials of Fe3O4 MNPs are negative when pH value is higher than the point of zero charge (pHPZC: 4.2), while the zeta potentials of Fe3O4/GA, Fe3O4/GO and Fe3O4/GA/GO MNPs are all highly negative in the whole experimental pH range 2.0–10.0 due to the existence of phenolic hydroxyl group in GA and the oxygen-containing functional group in GO on the surface of Fe3O4/GA/GO MNPs, which may be beneficial for cationic sorption.30
The thermal behaviour of Fe3O4, Fe3O4/GA, Fe3O4/GO, and Fe3O4/GA/GO MNPs were further investigated by thermogravimetric analysis (TGA) (Fig. 1c). In the TGA curve of Fe3O4/GA/GO MNPs, the weight loss below 150 °C is due to the loss of physically adsorbed water. The weight loss from 150 °C to 600 °C is ascribed to the loss of oxygen-containing functional groups. The final weight loss from 600 °C to 900 °C is mainly attributed to the burning of carbon.31
The TEM images and the selected area electron diffraction (SAED) patterns of Fe3O4, Fe3O4/GA, Fe3O4/GO and Fe3O4/GA/GO MNPs are shown in Fig. 2. The bare Fe3O4 and Fe3O4/GA MNPs tend to form an agglomerated structure, as shown in Fig. 2a and b. However, in the images of Fe3O4/GO and Fe3O4/GA/GO MNPs, Fe3O4 particles are better dispersed on the surface of GO sheet (Fig. 2c and d), which results from the high specific surface area of GO. The high resolution TEM images and the SAED patterns show the polycrystalline nature of Fe3O4/GO (Fig. 2g) and Fe3O4/GA/GO MNPs (Fig. 2h) compared to the mainly monocrystalline nature of Fe3O4 (Fig. 2e), which indicates that Fe3O4/GO and Fe3O4/GA/GO MNPs having more crystal faces with different surface energy probably benefitting their catalytic properties. Moreover, the lattice distortion presented in Fig. 2f resulted in a lower degree of crystallinity of Fe3O4/GA than that of other catalysts because of the addition of GA, which is in accordance with the XRD patterns (Fig. 1a). However, the weaker crystallinity of Fe3O4/GA led to the crystal defects of Fe3O4, as the green dashed circles shown in Fig. 2f and h, which might also be beneficial to the catalytic activity.29 In conclusion, the GA modified Fe3O4 with the support of GO has more crystal faces and crystal defects, as well as the small particle size, which might all probably enhance the catalytic activity of Fe3O4/GA/GO MNPs, compared to the bare Fe3O4.
 |
| | Fig. 2 TEM images and the SAED patterns (inset) of bare Fe3O4 (a and e), Fe3O4/GA (b and f), Fe3O4/GO (c and g) and Fe3O4/GA/GO MNPs (d and h), respectively. | |
Fig. 3 shows the deconvolution of the O 1s spectra of Fe3O4, Fe3O4/GO and Fe3O4/GA/GO MNPs. The peaks in Fe3O4 are the oxygen in Fe–O bonds belonging to lattice oxygen in Fe3O4 (529.1 eV) and the oxygen in hydroxyl group on the surface of Fe3O4 (–OH, 529.8) ((Fig. 3a)). It consisted of four peaks in Fe3O4/GO: (i) the oxygen in Fe–O bonds in Fe3O4 (529.1 eV), (ii) the oxygen in Fe–O–C bonds suggested possibly coming from the bidentate coordination between the surface Fe and hydroxyl/carbonyl group (529.7 eV), (iii) the oxygen in carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 530.7 eV) and (iv) the oxygen in epoxy group and hydroxyl (C–O–C/C–O, 532.0 eV) (Fig. 3b). The binding energies of Fe–O, Fe–O–C, CO and C–O–C/C–O bonds varied slightly in Fe3O4/GA/GO MNPs, which might be possibly caused by the introduction of GA (Fig. 3c). The oxygen-containing groups on the surface of GO could coordinate with metal ions and served as nucleation sites,32 benefiting the dispersion of metal ions to form well dispersed metal oxides onto GO sheet, which was observed in the TEM images of Fe3O4/GA/GO (Fig. 2d).
O, 530.7 eV) and (iv) the oxygen in epoxy group and hydroxyl (C–O–C/C–O, 532.0 eV) (Fig. 3b). The binding energies of Fe–O, Fe–O–C, CO and C–O–C/C–O bonds varied slightly in Fe3O4/GA/GO MNPs, which might be possibly caused by the introduction of GA (Fig. 3c). The oxygen-containing groups on the surface of GO could coordinate with metal ions and served as nucleation sites,32 benefiting the dispersion of metal ions to form well dispersed metal oxides onto GO sheet, which was observed in the TEM images of Fe3O4/GA/GO (Fig. 2d).
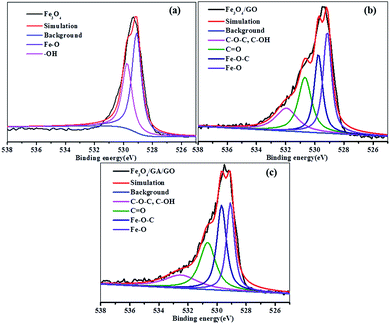 |
| | Fig. 3 O 1s XPS spectra of Fe3O4 (a), Fe3O4/GO (b) and Fe3O4/GA/GO MNPs (c). | |
The specific surface area calculated by using a BET equation was found to be 93.18 m2 g−1, 151.04 m2 g−1, 207.5 m2 g−1 and 226.19 m2 g−1 for Fe3O4, Fe3O4/GA, Fe3O4/GO and Fe3O4/GA/GO MNPs, respectively. The specific surface area of Fe3O4/GA/GO MNPs is larger than the others.
3.3. Heterogeneous Fenton catalytic activity of Fe3O4/PCC MNPs
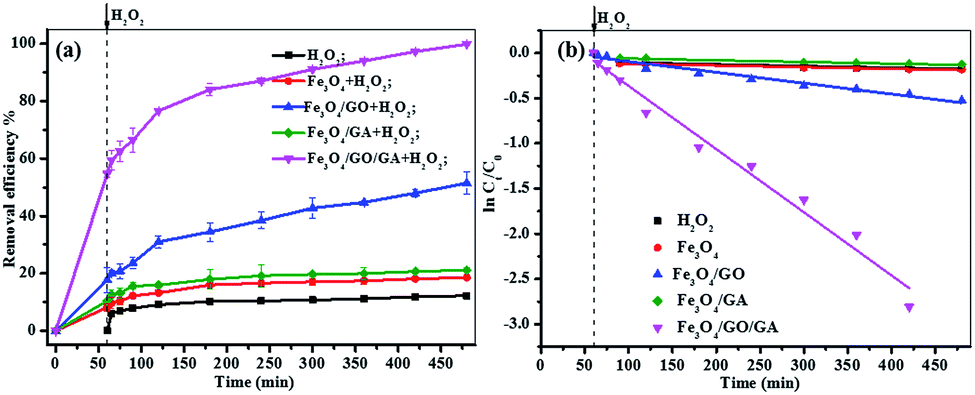
3.3.1 Comparison of catalytic activity. The catalytic activity of both Fe3O4/GO, Fe3O4/GA and Fe3O4/GA/GO MNPs (1.0 g L−1) were evaluated based on the degradation of MB (0.1 mM) with H2O2 (160 mM) at an initial pH of 6.0 (Fig. 4a). As can be seen from the picture, less than 15% of MB degradation was observed in the presence of only 160 mM H2O2 within 420 min. And the degradation efficiency of MB was lower than 20% with Fe3O4 as catalyst. Similarly, less than 21% of MB was removed in the Fe3O4/GA suspension with the addition of H2O2. And there was 51% of MB removal with Fe3O4/GO MNPs as a Fenton catalyst. However, the MB removal efficiency can reach 100% in the presence of H2O2 catalyzed by Fe3O4/GA/GO MNPs within 420 min. As shown in Fig. 4b, the pseudo-first-order kinetics reaction rate constant (kapp) of MB degradation by Fe3O4/GA/GO MNPs was much higher than those of by other catalysts.
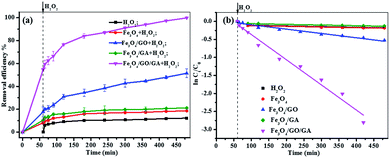 |
| | Fig. 4 (a) Removal curves of MB under different conditions at pH 6.0; (b) pseudo-first-order kinetics of MB degradation ([catalyst] = 1.0 g L−1, [H2O2]0 = 160.0 mM, [MB]0 = 0.2 mM, pH 6.0, T = 30 °C). | |
In the curve of MB degradation by Fe3O4/GA/GO MNPs, the removal of MB was due to the adsorption and degradation by the catalyst. The adsorption of Fe3O4/GA/GO MNPs was due to the electrostatic interaction, thanks to the negative charge of the Fe3O4/GA/GO (Fig. 1b) and its large specific surface area. According to previous studies, adsorption level of organic pollutant on the surface of catalyst strongly influenced their oxidation ability.33 Essentially, the catalyst provided active sites to promote H2O2 decomposition to form ˙OH radicals, then the formed ˙OH radicals diffused into the solution to oxidize organics, while the adsorbed pollutants tend to be more easily attacked by hydroxyl radicals. As Noorjahan et al. indicated, phenol adsorption on zeolite support had positive effect on degradation.34 The similar effect was found by Gu et al. that the sludge derived magnetic porous carbon could be used as an efficient heterogeneous catalyst to adsorb and degrade naphthalene dye (1,2,4-acid) in aqueous solution by Fenton-like reaction.35
Moreover, although Fe3O4/GA or Fe3O4/GO alone had low catalytic activity, their hybrid (Fe3O4/GA/GO) exhibited high catalytic activity in degradation of MB by advanced oxidation technology based on hydroxyl radicals, suggesting the synergistic coupling between GA and GO was indispensable to the high catalytic activity of Fe3O4/GA/GO MNPs.36
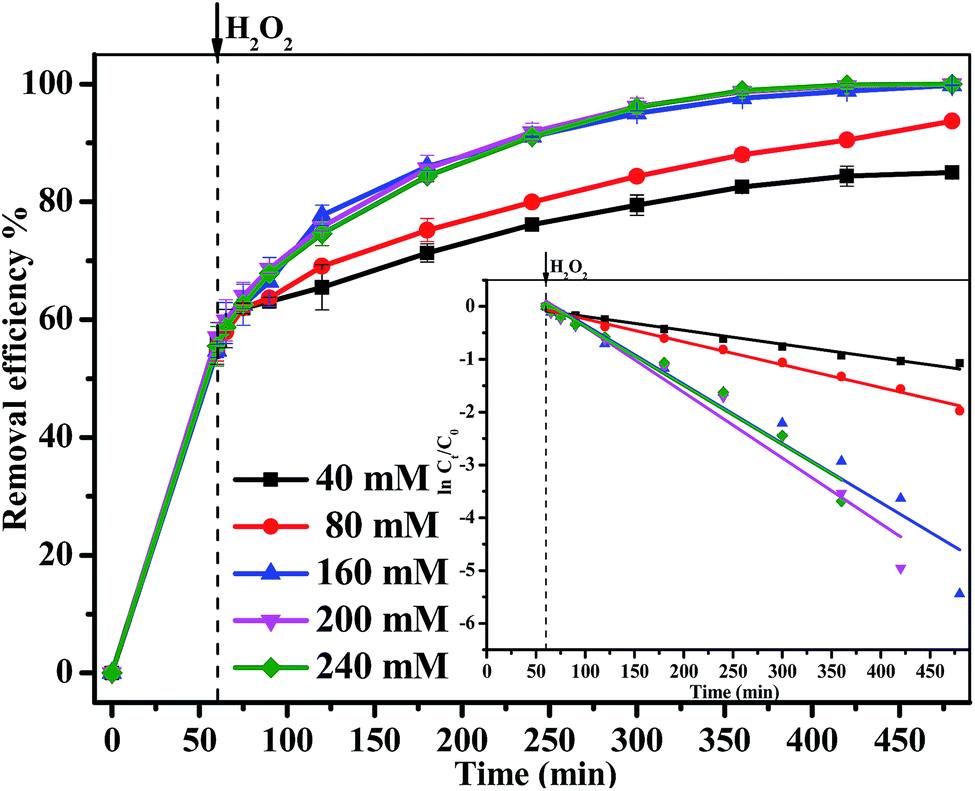
3.3.2 Effect of H2O2 concentration. The degradation of MB over 1.0 g L−1 Fe3O4/GA/GO MNPs at pH 6.0 and 30 °C with different H2O2 concentrations was studied and the results were shown in Fig. 5. It was observed that the degradation percentage of MB increased from 84.3% to 100% when H2O2 concentration increased from 40 mM to 240 mM. The MB degradation approximately followed a pseudo-first-order kinetics with R2 > 0.97, and the pseudo-first-order kinetics reaction rate constant (kapp) raised from 0.003 min−1 to 0.012 min−1 with H2O2 concentration increasing from 40 mM to 200 mM. When H2O2 concentration reached up to 240 mM, the kapp value decreased on the contrary. Since MB removal in the heterogeneous Fenton system was directly related to the concentration of the ˙OH radicals, more MB decomposition was expected with a higher increase of H2O2 concentration. However, there was not a significant improvement on MB removal with a higher H2O2 concentration. This was due to the scavenging effect of ˙OH radicals by H2O2. At a much higher H2O2 concentration more than 160 mM, competitive reactions between the MB and H2O2 may happen. The ˙OH radicals may react with hydrogen peroxide producing hydroperoxy and superoxide anion, of which oxidation potential was much lower than that of ˙OH radicals.37,38
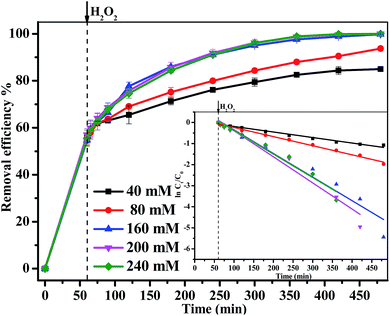 |
| | Fig. 5 The effect of H2O2 concentration on the removal efficiency of MB degraded by Fe3O4/GA/GO MNPs, inset is pseudo-first-order kinetics of MB degradation ([Fe3O4/GA/GO] = 1.0 g L−1, [MB]0 = 0.2 mM, pH 6.0, T = 30 °C). | |
3.3.3 Effect of catalyst dosage. Catalyst dosage was another parameter affecting the heterogeneous Fenton reaction. Fig. 6 shows the effect of Fe3O4/GA/GO MNPs dosages on the degradation of MB. As the initial concentration of catalyst was increased from 0.25 g L−1 to 1.5 g L−1, the degradation efficiency of MB within 420 min at pH 6.0 and 30 °C was significantly increased from 42.0% to 100% with 160 mM H2O2. The increase of Fe3O4/GA/GO concentration enhanced the removal efficiency of MB and the pseudo-first-order kinetics reaction rate constant, which indicated that increasing amount of active sites on catalyst surface accelerated the H2O2 decomposition to form ˙OH radicals. However, the constant rate k was not increased remarkably with the Fe3O4/GA/GO dosage higher than 1.25 g L−1, possibly because of the agglomeration of nano-particles and the scavenging of radicals by the reduced species on the surface of Fe3O4/GA/GO MNPs.17
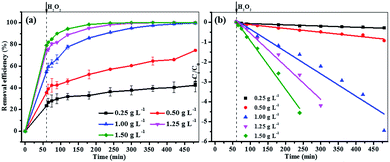 |
| | Fig. 6 The effect of catalyst dosage on MB degradation by Fe3O4/GA/GO (a) removal efficiency of MB (b) pseudo-first-order kinetics of MB degradation ([H2O2]0 = 160 mM, [MB]0 = 0.2 mM, pH 6.0, T = 30 °C). | |
3.3.4 Effect of MB concentration. The results for reaction kinetics as a function of different MB initial concentrations at various reaction times with 160 mM H2O2 and 1.0 g L−1 Fe3O4/GA/GO MNPs at pH 6.0 and 30 °C are displayed in Fig. 7. As can be seen from the figure, the degradation rate of MB was decreased from 100% to 71% with MB concentration increasing from 0.1 mM to 0.4 mM within 420 min. Similarly, the rate constant k declined from 0.057 min−1 to 0.002 min−1 with the increase of MB concentration. This phenomenon could be explained by that more ˙OH radicals were required for the degradation of MB as well as its degradation intermediates at higher initial MB concentration, while the yield of ˙OH radicals were still close under the same concentration of H2O2 and catalyst.38
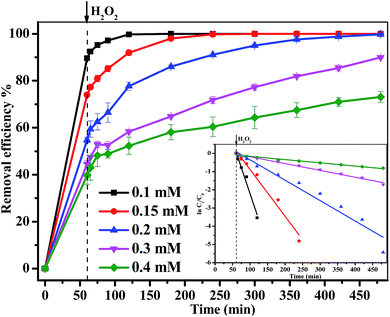 |
| | Fig. 7 The effect of initial MB concentration on the removal efficiency of MB degraded by Fe3O4/GA/GO MNPs, inset is pseudo-first-order kinetics of MB degradation ([H2O2]0 = 160 mM, [Fe3O4/GA/GO] = 1.0 g L−1, pH 6.0, T = 30 °C). | |
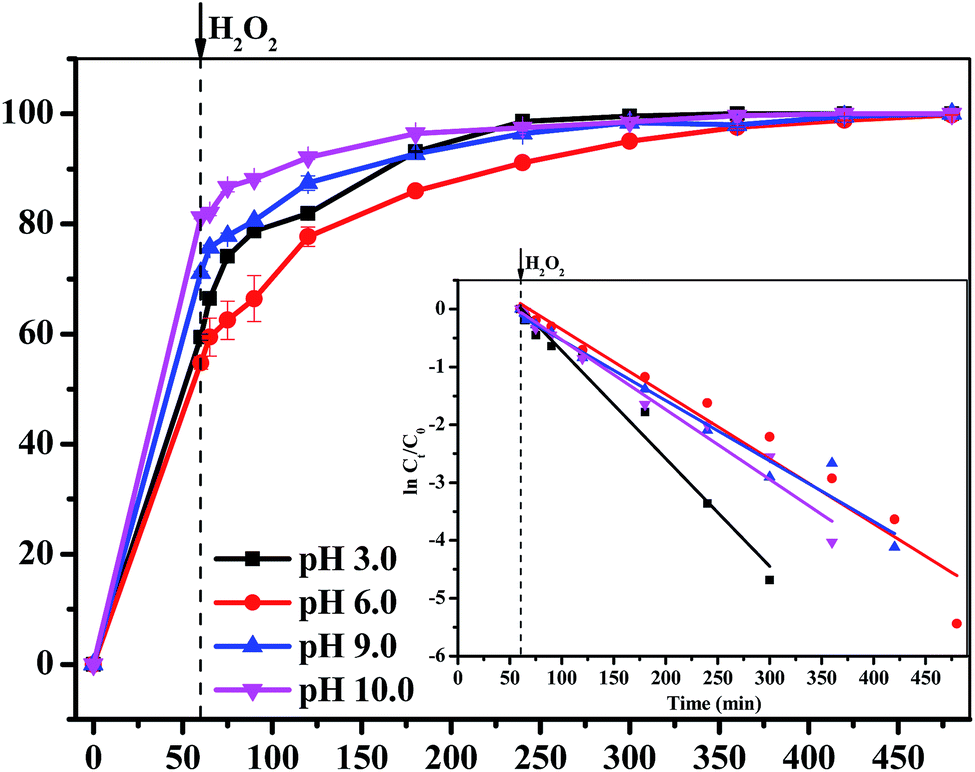
3.3.5 Effect of pH value. The influence of pH value on the degradation efficiency of MB was investigated. As shown in Fig. 8, the removal efficiency of MB all reached as much as 100% with different pH value ranging from 3.0 to 10.0 within 420 min. The rate constant values were 0.019 min−1, 0.011 min−1, 0.010 min−1 and 0.012 min−1 and the total dissolved Fe concentration were 2.70 mg L−1, 1.04 mg L−1, 0.39 mg L−1 and 0.79 mg L−1, corresponding to pH 3.0, pH 6.0, pH 9.0 and pH 10.0, respectively. The results indicated that a lower pH caused a shorter induction time and a higher kinetic rate. This was quite similar to a previous research result of a Fe3O4-based degradation system, in which the removal efficiency sharply decreased when the solution pH was higher than the optimal value.16,39–41 Haber–Weiss circle indicated that the generation of ˙OH from H2O2 was the key step of the whole degradation process. The production of ˙OH on the surface of Fe3O4/GA/GO was gradually restricted with the increase of pH value, which resulted in a slow degradation rate of MB.16
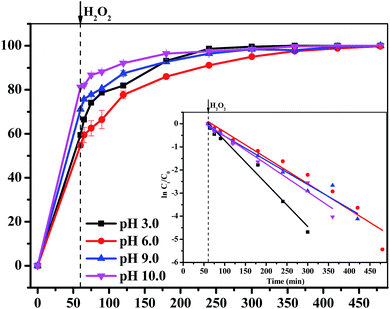 |
| | Fig. 8 The effect of initial pH value of MB solution on the removal efficiency of MB degraded by Fe3O4/GA/GO MNPs, inset is pseudo-first-order kinetics of MB degradation ([Fe3O4/GA/GO] = 1.0 g L−1, [H2O2]0 = 160 mM, [MB]0 = 0.2 mM, T = 30 °C). | |
3.3.6 Stability and reusability of Fe3O4/GA/GO MNPs. In order to evaluate the durability of the Fe3O4/GA/GO MNPs, the recycling experiments of Fe3O4/GA/GO MNPs were studied as shown in Fig. 9. The results demonstrated that the degradation efficiency of MB reached up to 100% even after ten times recycling of the catalyst and dropped to 92% in the eleventh circle. The TOC removal efficiency in the solution increased from 66.7% to 77.9% for the first four cycles, then decreased to 48.0% as the recycling number increased to ten. The decrease in MB mineralization might be caused by the mass loss and aggregation of the catalyst during the recycling process, as well as the adsorption of MB or accumulation of intermediates on the surface of Fe3O4/GA/GO MNPs. The results indicated that Fe3O4/GA/GO MNPs have a good long-term stability and activity for the repeated treatment of MB.
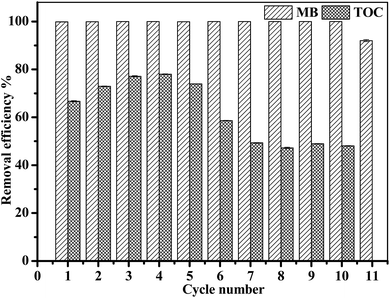 |
| | Fig. 9 Removal efficiency of MB and solution TOC for different cycles of Fe3O4/GA/GO MNPs using identical reaction conditions ([Fe3O4/GA/GO] = 1.0 g L−1, [H2O2] = 160 mM, [MB]0 = 0.2 mM, pH 6.0, T = 30 °C). | |
3.4. Mechanism for H2O2 activation on Fe3O4/GA/GO MNPs
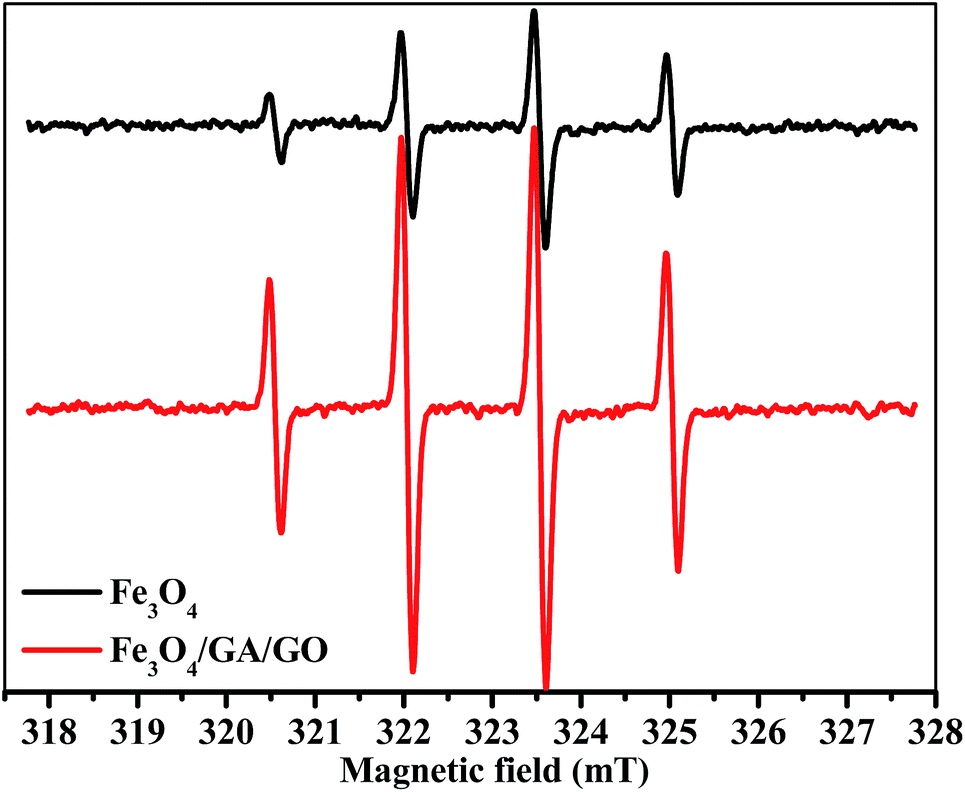
Fig. 10 shows that the ESR spectra in the presence of Fe3O4 MNPs and Fe3O4/GA/GO MNPs displayed a fourfold characteristic peak of the typical DMPO–˙OH adduct with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1, which demonstrated that ˙OH radicals were the main active species involved in the degradation of MB.38,42 The intensity of ˙OH in Fe3O4/GA/GO–H2O2 system was much higher than that of Fe3O4–H2O2 system. The results indicated that the decomposition of H2O2 into ˙OH radicals for MB degradation was indeed enhanced due to the accelerated Fe(II) regeneration rate. Additionally, the crystal defects of Fe3O4/GA/GO MNPs may also contribute to its high catalytic activity for H2O2 decomposition (Fig. 2). The introduction of GA and GO in Fe3O4/GA/GO MNPs could decrease the Fe(III)/Fe(II) redox potential, promote the redox cycle of Fe(III)/Fe(II) and accelerate the Fe(II) regeneration, according to the previous reported organic ligands, greatly promoting the degradation of MB in the heterogeneous Fenton system.43
:2:2:1, which demonstrated that ˙OH radicals were the main active species involved in the degradation of MB.38,42 The intensity of ˙OH in Fe3O4/GA/GO–H2O2 system was much higher than that of Fe3O4–H2O2 system. The results indicated that the decomposition of H2O2 into ˙OH radicals for MB degradation was indeed enhanced due to the accelerated Fe(II) regeneration rate. Additionally, the crystal defects of Fe3O4/GA/GO MNPs may also contribute to its high catalytic activity for H2O2 decomposition (Fig. 2). The introduction of GA and GO in Fe3O4/GA/GO MNPs could decrease the Fe(III)/Fe(II) redox potential, promote the redox cycle of Fe(III)/Fe(II) and accelerate the Fe(II) regeneration, according to the previous reported organic ligands, greatly promoting the degradation of MB in the heterogeneous Fenton system.43
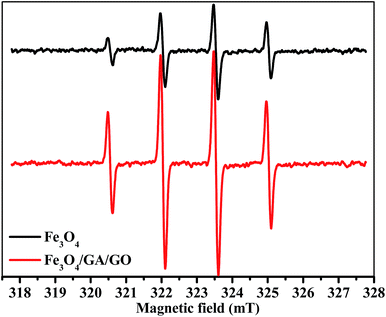 |
| | Fig. 10 DMPO spin-trapping ESR spectra of ˙OH radicals in heterogeneous Fenton reaction with Fe3O4 and Fe3O4/GA/GO MNPs as catalyst ([catalyst] = 1.0 g L−1, [H2O2]0 = 160 mM, [DMPO] = 500 mM, pH 6.0, T = 30 °C, t = 10 min). | |
Based on above results, a mechanism for the Fenton-like oxidation of MB in this system was illustrated as follows. Firstly, MB was adsorbed on Fe3O4/GA/GO MNPs. Then, the surface Fe(II) of Fe3O4/GA/GO MNPs catalyzed H2O2 decomposition to form ˙OH radicals (eqn (4)); ˙OH radicals oxidized MB, mineralizing MB to carbon dioxide and water (eqn (5)); H2O2 also reacted with surface Fe(III) to produce Fe(II) and ˙O2− (eqn (6));18 the produced ˙O2− was rapidly consumed in the effective reduction of Fe(III) (eqn (7)); in addition, this study had confirmed that the addition of GA could decrease the Fe(III)/Fe(II) redox potential as the previous reported organic ligands did.19 Therefore, the accelerated reduction of Fe(III) to Fe(II) continuously supplied Fe(II) for the decomposition of H2O2 into ˙OH. Finally, the adsorbed MB was readily degraded by ˙OH generated on the catalyst surface. In conclusion, Fe3O4/GA/GO MNPs exhibited a high catalytic capacity for H2O2 decomposition.
| |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe(II) + H2O2 → Fe(III) + ˙OH Fe(II) + H2O2 → Fe(III) + ˙OH
| (4) |
| | |
˙OH + MB → intermediates → H2O + CO2
| (5) |
| | |
Fe(III) + H2O2 → Fe(II) + ˙O2−
| (6) |
| | |
Fe(III) + ˙O2− → Fe(II) + O2
| (7) |
4. Conclusion
Fe3O4/GA/GO MNPs were successfully fabricated by a simple coprecipitation method and used as heterogeneous Fenton catalyst. The catalyst exhibited high catalytic activity for the Fenton oxidation of MB in aqueous solution within a wide pH range. Characterization results and catalytic tests confirmed that the high catalytic activity of the Fe3O4/GA/GO could be attributed to the addition of GA and the support of GO. On one hand, the addition of GA make the crystallization process of Fe3O4 slower and less regular than that of bare Fe3O4, leading to much more crystal defects on the surface of Fe3O4/GA/GO MNPs. On the other hand, GO could improve the dispersibility of Fe3O4, and increase the specific surface area of Fe3O4/GA/GO, benefiting the adsorption of MB onto catalysts. The large surface area, added surface crystal defects and low Fe(III)/Fe(II) redox potential of Fe3O4/GA/GO MNPs jointly contribute to a good performance of removing the MB. The adsorption of MB facilitated the oxidation reaction by ˙OH produced on the catalyst surface. In conclusion, this study provides a new pathway for preparation of heterogeneous Fenton catalysts with potential applications in water treatment.
Acknowledgements
The present research supported by the Natural Science Foundation of China (No. 51378494, No. 51208539 and No. 51678554).
Notes and references
- A. Machulek Jr, J. E. F. Moraes, C. Vautier-Giongo, C. A. Silverio, L. C. Friedrich, C. A. O. Nascimento, M. C. Gonzalez and F. H. Quina, Environ. Sci. Technol., 2007, 41, 8459–8463 CrossRef.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- J. H. Ma, W. H. Ma, W. J. Song, C. C. Chen, Y. L. Tang, J. C. Zhao, Y. P. Huang, Y. M. Xu and L. Zang, Environ. Sci. Technol., 2006, 40, 618–624 CrossRef CAS PubMed.
- C. L. P. S. Zanta, L. C. Friedrich, A. Machulek Jr, K. M. Higa and F. H. Quina, J. Hazard. Mater., 2010, 178, 258–263 CrossRef CAS PubMed.
- P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, J. Chem. Technol. Biotechnol., 2008, 83, 1323–1338 CrossRef CAS.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176, 249–265 CrossRef.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- A. Georgi, A. Schierz, U. Trommler, C. P. Horwitz, T. J. Collins and F. D. Kopinke, Appl. Catal., B, 2007, 72, 26–36 CrossRef CAS.
- L. Xu and J. Wang, J. Hazard. Mater., 2011, 186, 256–264 CrossRef CAS PubMed.
- S. S. Lin and M. D. Gurol, Environ. Sci. Technol., 1998, 32, 1417–1423 CrossRef CAS.
- W. P. Kwan and B. M. Voelker, Environ. Sci. Technol., 2003, 37, 1150–1158 CrossRef CAS PubMed.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- R. Gonzalez-Olmos, M. J. Martin, A. Georgi, F.-D. Kopinke, I. Oller and S. Malato, Appl. Catal., B, 2012, 125, 51–58 CrossRef CAS.
- R. C. C. Costa, M. F. F. Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. Rios, C. N. Silva and R. M. Lago, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed.
- X. B. Hu, B. Z. Liu, Y. H. Deng, H. Z. Chen, S. Luo, C. Sun, P. Yang and S. G. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef CAS.
- L. Xu and J. Wang, Appl. Catal., B, 2012, 123, 117–126 CrossRef.
- W. Wang, Y. Liu, T. Li and M. Zhou, Chem. Eng. J., 2014, 242, 1–9 CrossRef CAS.
- H. Niu, D. Zhang, S. Zhang, X. Zhang, Z. Meng and Y. Cai, J. Hazard. Mater., 2011, 190, 559–565 CrossRef CAS PubMed.
- M. J. Hynes and M. O. Coinceanainn, J. Inorg. Biochem., 2001, 85, 131–142 CrossRef CAS PubMed.
- H. K. J. Powell and M. C. Taylor, Aust. J. Chem., 1982, 35, 739–756 CrossRef.
- C. J. Xu, K. M. Xu, H. W. Gu, R. K. Zheng, H. Liu, X. X. Zhang, Z. H. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- D. Dorniani, M. Z. Bin Hussein, A. U. Kura, S. Fakurazi, A. H. Shaari and Z. Ahmad, Int. J. Nanomed., 2012, 7, 5745–5756 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- Y. L. Dong, H. G. Zhang, Z. U. Rahman, L. Su, X. J. Chen, J. Hu and X. G. Chen, Nanoscale, 2012, 4, 3969–3976 RSC.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. D. da Costa, Sci. Rep., 2014, 4, 4594 CrossRef PubMed.
- P. H. Shi, R. J. Su, F. Z. Wan, M. C. Zhu, D. X. Li and S. H. Xu, Appl. Catal., B, 2012, 123, 265–272 CrossRef.
- M. R. Malekbala, M. A. Khan, S. Hosseini, L. C. Abdullah and T. S. Y. Choong, J. Ind. Eng. Chem., 2015, 21, 369–377 CrossRef CAS.
- J. W. Fang, H. Q. Fan, Y. Ma, Z. Wang and Q. Chang, Appl. Surf. Sci., 2015, 332, 47–54 CrossRef CAS.
- L. Bai, Z. Li, Y. Zhang, T. Wang, R. Lu, W. Zhou, H. Gao and S. Zhang, Chem. Eng. J., 2015, 279, 757–766 CrossRef CAS.
- J. W. Zhang, M. S. Azam, C. Shi, J. Huang, B. Bin, Q. X. Liu and H. B. Zeng, RSC Adv., 2015, 5, 32272–32282 RSC.
- P. H. Shi, R. J. Su, F. Z. Wan, M. C. Zhu, D. X. Li and S. H. Xu, Appl. Catal., B, 2012, 123, 265–272 CrossRef.
- X. Xue, K. Hanna, M. Abdelmoula and N. Deng, Appl. Catal., B, 2009, 89, 432–440 CrossRef CAS.
- A. Noorjahan, V. D. Kumari, A. Subrahmanyam and L. Panda, Appl. Catal., B, 2005, 57, 291–298 CrossRef.
- L. Gu, N. Zhu, H. Guo, S. Huang, Z. Lou and H. Yuan, J. Hazard. Mater., 2013, 246, 145–153 CrossRef PubMed.
- P. H. Shi, R. J. Su, S. B. Zhu, M. C. Zhu, D. X. Li and S. H. Xu, J. Hazard. Mater., 2012, 229, 331–339 CrossRef PubMed.
- L. Wang, Y. Yao, Z. Zhang, L. Sun, W. Lu, W. Chen and H. Chen, Chem. Eng. J., 2014, 251, 348–354 CrossRef CAS.
- S.-P. Sun, X. Zeng, C. Li and A. T. Lemley, Chem. Eng. J., 2014, 244, 44–49 CrossRef CAS.
- X. Zhang, M. He, J. H. Liu, R. Liao, L. Zhao, J. Xie, R. Wang, S. T. Yang, H. Wang and Y. Liu, Chin. Sci. Bull., 2014, 59, 3406–3412 CrossRef CAS.
- X. Zhang, Y. Ding, H. Tang, X. Han, L. Zhu and N. Wang, Chem. Eng. J., 2014, 236, 251–262 CrossRef CAS.
- W. Wang, Y. Wang, Y. Liu and T. Li, Chem. Lett., 2012, 41, 897–899 CrossRef CAS.
- N. Wang, L. Zhu, D. Wang, M. Wang, Z. Lin and H. Tang, Ultrason. Sonochem., 2010, 17, 526–533 CrossRef CAS PubMed.
- Z. Ma, L. Ren, S. Xing, Y. Wu and Y. Gao, J. Phys. Chem. C, 2015, 119, 23068–23074 CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Sha Wanga,
Juan Xiaoc,
Chang Cuiab and
Chuan Wang*a
ab,
Sha Wanga,
Juan Xiaoc,
Chang Cuiab and
Chuan Wang*a