DOI:
10.1039/C6RA02969H
(Paper)
RSC Adv., 2016,
6, 31153-31160
CO2 fixation at atmospheric pressure: porous ZnSnO3 nanocrystals as a highly efficient catalyst for the synthesis of cyclic carbonates†
Received
1st February 2016
, Accepted 16th March 2016
First published on 17th March 2016
Abstract
An efficient methodology for the synthesis of cyclic carbonates from epoxides and CO2 has been developed using self-assembled, ultra small, porous zinc stannate nanocrystals as a catalyst under atmospheric pressure. The catalyst is synthesized by using sodium salicylate as a soft template and the material is thoroughly characterized by powder XRD, HR TEM, N2-sorption and UV-visible spectroscopic tools. Moreover, the catalyst smoothly catalyzes a wide range of reactants to produce a series of cyclic carbonates in excellent yields, under atmospheric pressure of CO2 and in the presence of PEG-600 as a green solvent. The catalyst can be easily recovered from the reaction medium and be reused without significant loss in its catalytic activity and selectivity, suggesting the future potential of this nanocatalyst in the clean-up of the environment.
Introduction
The chemical fixation of carbon dioxide (CO2) to synthesize value added organic fine chemicals is one of the most challenging areas of research today in the context of environmental protection and resource utilization.1 Since CO2 is the most abundant greenhouse gas, cheap, nontoxic and non-flammable its use as the C1 building block for the synthesis of various value added chemicals1–3 is in high demand. Today various methodologies have been developed to transform CO2 into useful organic chemicals.4–8 Among them the synthesis of five-membered cyclic carbonates from CO2 and epoxide is a promising methodology from the standpoint of atom economy and fruitful utilization of resources.9,10 Five-membered cyclic carbonates can be synthesized from the corresponding diols and phosgene, which involve drastic reaction conditions and environmentally corrosive pathways. These cyclic carbonates are extensively used as polycarbonate precursors like dimethyl carbonate (DMC) and diphenyl carbonate (DPC), aprotic polar solvents, electrolytes in rechargeable batteries, intermediates in organic synthesis and fine chemicals etc.11,12 Numerous homogeneous catalysts including alkali metal salts,13,14 metal complexes,15,16 alkali metal halides,17–19 bromine,20 quaternary ammonium/phosphonium salts,21 ionic liquids,22 multi-component peroxometalates,23 etc. have been developed to catalyze this transformation. These homogeneous catalysts suffer from common disadvantages as they can be easily destroyed during the course of the reaction and they cannot be easily recovered after the reaction for reuse. Alternatively, heterogeneous catalytic systems like MOFs,24,25 COFs,26 organic–inorganic hybrid core–shell microspheres27 etc. are developed and successfully employed to overcome these problems. However, most of these catalysts require drastic reaction conditions like very high-pressure of CO2 and addition of hazardous organic solvent in the reaction mixture. From these view points, it is highly desirable to exploit a halogen-free reusable catalytic system, which can work under atmospheric pressure in this CO2 fixation reaction.
On the other hand, zero- and one-dimensional nanostructures of semiconducting oxides, such as TiO2, ZnO, CeO2, ZrO2 and SnO2 have attracted immense interest owing to their unique properties and potential uses in diverse application areas such as catalysis,28 photocatalysis,29 solar cells30 and gas sensors.31 Intensive studies have also been carried out on binary metal oxides nanostructures like CdSnO3,32 ZnSnO3,33 Zn2SnO4,34 Cd2SnO4,35 BaTiO3,36 CuFe–O2,37 and Cd2GeO4 (ref. 38) etc. due to their interesting surface properties. Among these mixed metal oxides, zinc stannate (ZnSnO3) has attracted considerable interest in many frontline areas of chemistry, physics and materials science due to their broad range of applications in sensors, solar cells, transparent conductive oxides and optoelectronic devices.39 Several research groups have reported the synthesis of zinc stannate through different physical and chemical pathways.40 Fan et al. have synthesized nanocrystalline hollow ZnSnO3 microspheres by a hydrothermal method, which showed good sensing property for the hydrocarbons.41 Use of capping agents or templates during the hydrothermal synthesis of the nanocrystals is the most convenient way to generate porosity in the nanostructured materials.42 Thus, design and successful synthesis of ultra small ZnSnO3 nanocrystals with high specific surface area, uniform systems of pores and its utilization as heterogeneous catalyst is highly challenging. Recently, we have developed self-assembled ultra-small zinc stannate nanocrystals (ZnSnO3 and Zn2SnO4) with mesoscopic void spaces through a salicylate templating pathway and it has been employed as an efficient photocatalyst for photodegradation of Rhodamine B under the irradiation of UV/visible light.43 With our continued interest in this material, now we have synthesized self-assembled, tiny zinc stannate nanocrystals with controlled crystalline phases of ZnSnO3 (MZS-1) and explored its catalytic efficiency in the green synthesis of a wide range of five-membered cyclic carbonates from CO2 by using different epoxides in PEG-600 solvent under atmospheric pressure (Scheme 1).
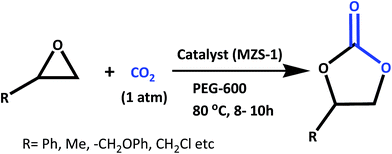 |
| | Scheme 1 Synthesis of cyclic carbonates from terminal epoxides and CO2 at atmospheric pressure. | |
Experimental section
Materials
All the epoxides were purchased from Sigma-Aldrich Chemical Co. Sodium salicylate (GR 99.5%), used as the structure-directing agent, anhydrous zinc chloride (98%) and diethylamine (99.5%, used to maintain the pH of the synthesis gel) were purchased from Merck, India. Tin chloride pentahydrate (98%) was obtained from Loba Chemie, India. All other reagents and solvents were dried and distilled through standard procedure. Thin layer chromatography was done using commercial (MERCK) plates with silica gel 60F254.
Characterization
Powder X-ray diffraction (PXRD) patterns of different MZS-1 samples were recorded with a Bruker D8 Advance X-ray diffractometer operated at a voltage of 40 kV and a current of 40 mA using Ni-filtered Cu Kα (λ = 0.15406 nm) radiation. UV-visible diffuse reflectance spectra were recorded on a Shimadzu UV 2401PC coupled with an integrating sphere attachment. BaSO4 was used as background standard. Transmission electron microscopy (TEM) images of the mesoporous mixed oxide materials were obtained using a JEOL JEM 2010 transmission electron microscope with operating voltage 200 kV. Nitrogen sorption isotherms were obtained using a Quantachrome Autosorb 1C surface area analyzer at 77 K. Prior to the measurement, the samples were degassed at 423 K for approximately 4 h under high vacuum. 1H NMR and 13C NMR spectra of different cyclic carbonates were recorded on the Bruker DPX-400 and DPX-500 instruments.
Synthesis of mesoporous zinc stannate (MZS-1) nanoparticles
The MZS-1 material was synthesized via hydrothermal method using sodium salicylate as a template. In a typical synthesis procedure firstly, 2.4 g (0.015 mol) of sodium salicylate was dissolved into 25 ml distilled water. Then 1.5 ml of diethylamine was added to this solution. The resultant mixture was stirred for 1 h. Then 0.78 g (0.006 mol) anhydrous ZnCl2 and 1.02 g (0.003 mol) of SnCl4, 5H2O, were taken in 10 ml of distilled water separately. Then, both the Zn(II) and Sn(IV) precursors were mixed together under stirring conditions. In the next step, former template solution was slowly dropped into the above mixed acidic salt solution under magnetic stirring. The pH of the resulting gel was raised up to 6.0 by adding additional diethylamine. The above solution was mixed by a magnetic stirrer for 4 h. The obtained white slurry was transferred to Teflon-lined stainless steel autoclave with 100 ml capacity and heated at the temperature of 383 K for 48 h. After the autoclave had cooled naturally to room temperature, the precipitate was filtered and washed with distilled water to remove the residual contaminant ions from the products and dried under vacuum at room temperature. Finally the resultant white solid product was calcined at 550 °C for 5 h to obtain MZS-1 material.
In the similar reaction conditions we have prepared meso-SnO2 and meso-ZnO(1) materials. For meso-ZnO(1) ZnCl2 was used as Zn precursor and we have not added any Sn source. Rest of the reaction conditions were same as that of MZS-1. Similarly meso-SnO2 nanocrystals were prepared.
Typical procedure for the synthesis of cyclic carbonate from epoxide and CO2
In a typical reaction, epoxide (5 mmol), catalyst (15 mg, 0.06 mmol) and PEG-600 (3 ml) were placed in a Schlenk tube equipped with a stirring bar and capped with a rubber cap. After mixing them together, tube was charged with CO2 by balloon for 1 min. The reaction was carried out at 80 °C for 8–10 h and CO2 was provided by a balloon (1 atm). After the reaction the catalyst was filtered and washed with water followed by acetone and dried in oven. The filtrate was extracted three times with ethyl acetate (3 × 10 ml) and washed with water. The organic layers were combined and dried with anhydrous Na2SO4 and the solvent was evaporated to dryness under vacuum. The residue was then purified by column chromatography on silica gel with pet-ether and ethyl acetate to afford the cyclic carbonates. All compounds were characterized on the basis of their spectroscopic data (1H, 13C NMR) and by comparison with those reported in the literature.
Result and discussion
Catalyst characterization
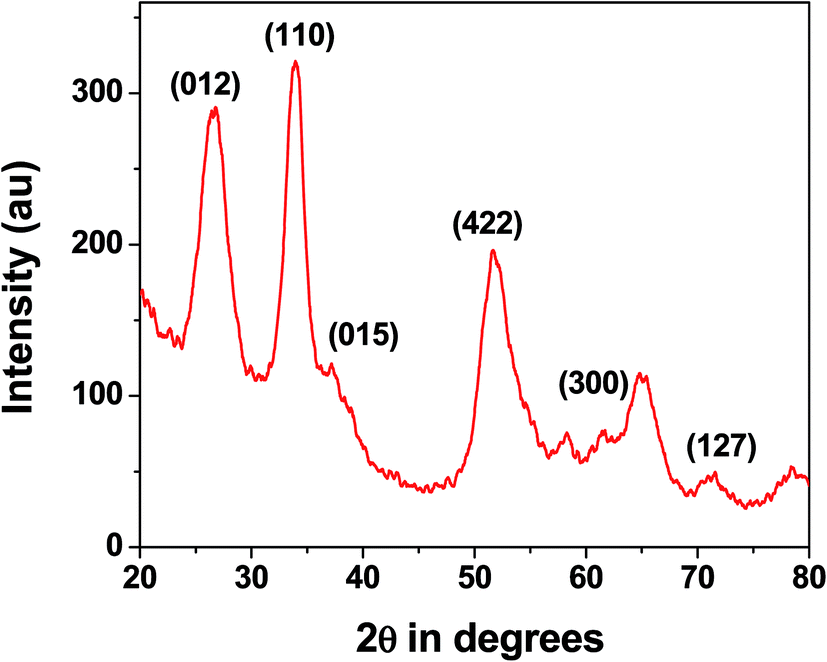
Nanostructure. The wide angle powder X-ray diffraction pattern for the MZS-1 is shown in Fig. 1. The corresponding XRD diffraction peaks of the products can be all indexed to a pure ZnSnO3 (JCPDS: 28-1486). Compared with those of the bulk material, the peaks are relatively broadened, which suggests that the MZS-1 is composed of very tiny nanoparticles. The peaks at 2θ values of 26.58, 34.09, 37.21, 51.8, 61.12 and 71.18 could be assigned to the crystal planes 012, 110, 015, 422, 300 and 127, respectively.44 From XRD pattern it is clear that peaks from other zinc stannate phases are not observed. This result suggested that the product is highly crystalline in nature and composed of perovskite phase with orthorhombic structure. Debye–Scherrer equation for average crystallite size calculation is D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where D is the average crystallite size (nm), λ is the X-ray wavelength (Cu Kα = 0.15406 nm), β is the full width at half maximum (FWHM) in radians and θ is the Bragg diffraction angle of the most intense peak (here 110). Using this equation average crystallite size has been estimated to be 4.5 nm which matches well with the UHR-TEM data. We have performed wide angle PXRD of meso SnO2 and meso ZnO prepared under similar reaction conditions and incorporated the results in ESI (Fig. S1, SnO2 and Fig. S2 for ZnO†). The crystallite sizes using the Debye–Scherrer equation are 4.4 nm and 27.9 nm.
cosθ, where D is the average crystallite size (nm), λ is the X-ray wavelength (Cu Kα = 0.15406 nm), β is the full width at half maximum (FWHM) in radians and θ is the Bragg diffraction angle of the most intense peak (here 110). Using this equation average crystallite size has been estimated to be 4.5 nm which matches well with the UHR-TEM data. We have performed wide angle PXRD of meso SnO2 and meso ZnO prepared under similar reaction conditions and incorporated the results in ESI (Fig. S1, SnO2 and Fig. S2 for ZnO†). The crystallite sizes using the Debye–Scherrer equation are 4.4 nm and 27.9 nm.
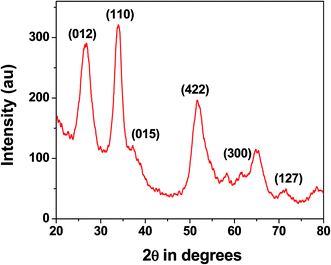 |
| | Fig. 1 Wide angle powder XRD pattern of MZS-1. | |
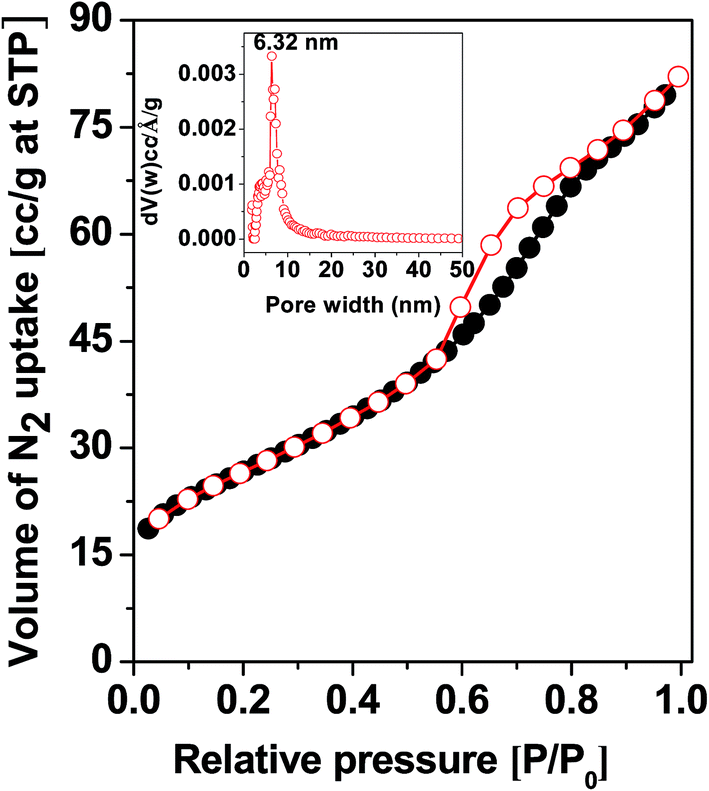
Porosity and BET surface area. In Fig. 2 the N2 adsorption–desorption isotherm of the MZS-1 material is shown. This isotherm can be classified as type IV with an H3 type hysteresis loop which is characteristic for mesoporous materials.45 The BET surface area calculated from the data points in this pressure range P/P0 of 0–0.3 was 96.0 m2 g−1. The hysteresis loop in the P/P0 range from 0.50 to 0.97 is suggesting large uniform mesopores are present in the sample. Pore size distribution of the sample estimated by employing the NLDFT model is shown in the inset of Fig. 2. The observed peak pore width for this sample was 6.32 nm and the narrow distribution suggested uniform assembly of MZS-1 crystallites. In Fig. S3 and S4† we have provided BET analysis data for meso SnO2 and meso-ZnO(1) samples, respectively. All these BET analysis results are tabulated in Table 1.
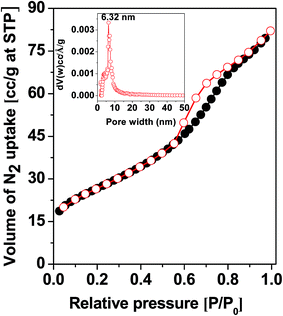 |
| | Fig. 2 N2 adsorption–desorption isotherm of the MZS-1. Inset of the figure PSD employing NLDFT model. | |
Table 1 Physico-chemical properties of different samples
| Sample name |
BET surface area, m2 g−1 |
Pore width (nm) |
Pore volume, cm3 g−1 |
| MZS-1 |
96 |
6.32 |
0.12 |
| Meso-SnO2 |
84 |
6.52 |
0.13 |
| Meso-ZnO |
34 |
2.95 |
0.021 |
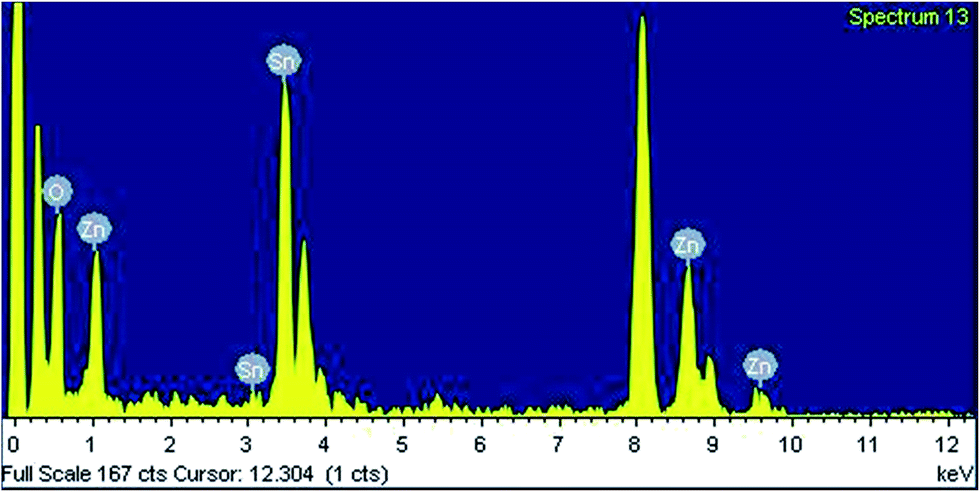
Electron microscopic analysis. In Fig. 3a–d HRTEM images of the mesoporous zinc stannate nanocrystals (MZS-1) are shown. From Fig. 3a spherical nanoparticles having diameter of ca. 2.8–3.7 nm are clearly seen and these are uniformly distributed throughout the specimen grid as self-aggregated nanoparticles. These self-aggregated nanoparticles are responsible for these interparticle voids and mesopores. Fig. 3d represents the lattice fringes pattern for MZS-1. It shows that the distance between two lattice fringes is 0.263 nm, which corresponds to the (110) plane of ZnSnO3 nanocrystals. The selected area electron diffraction (SAED) pattern (upper inset in Fig. 3d) demonstrates the orthorhombic and single-crystalline nature of MZS-1 material. TEM-EDX of MZS-1 (Fig. 4) suggested the presence of all the elements in the framework of MZS-1, namely zinc, tin and oxygen.
 |
| | Fig. 3 HR TEM images of self-assembled zinc stannate nanoparticles (a–c), lattice fringes pattern for the material MZS-1 (d). | |
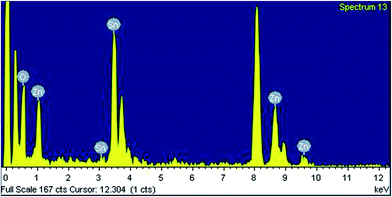 |
| | Fig. 4 EDX spectrum of MZS-1. | |
The thermal stability of the MZS-1 sample is obtained from the thermogravimetric analysis (TGA) under N2 flow. TGA curve of MZS-1 material is shown in Fig. S5.† The TGA profile of MZS-1 showed the first weight loss below 150 °C due to desorption of absorbed water. This was followed by a gradual decrease in the weight upto 390 °C and above this temperature, the weight loss becomes more pronounced and this can be attributed to the thermal decomposition of ZnSnO3 nanoparticles.
Catalytic activity
First, we have examined the reaction conditions including the CO2 pressure, reaction temperature, reaction time, solvent and the catalyst amount to optimize the yield of the cyclic carbonate (Table 2). The coupling of styrene oxide (SO) with CO2 to afford styrene carbonate (SC) is chosen as the model reaction. Among various solvents like toluene, CHCl3, DMF, DMSO, PEG-600, H2O and DCM, PEG-600 showed superior activity over other solvents (Table 2, entries 9–13). Although DMF as solvent showed almost equal activity as compared to PEG-600 in the synthesis of styrene carbonate at atmospheric pressure, but PEG-600 being greener and environment friendly than DMF, we have optimized the reactions with PEG-600. Next, the effect of the reaction temperature and time on the product yield was also examined (Table 2, entries 9–14). The best result in terms of yield (95%) and time was obtained by carrying out the reaction in PEG-600, at 80 °C for 8 h using just 15 mg (0.06 mmol) of catalyst at atmospheric pressure (Table 2, entry 13). On the other hand, with less than 15 mg of the catalyst, the reaction became incomplete and resulted in low yield of the product (Table 2, entry 15).
Table 2 Optimization of reaction conditions for the synthesis of styrene carbonatea
|
| Entry |
Solvent |
CO2 pressure (atm) |
Time (h) |
Temp. (°C) |
Yieldb (%) |
| Reaction conditions: styrene oxide (5 mmol), MZS-1 (15 mg, 0.06 mmol) PEG-600 (3 ml) under 1 atm of CO2. Yields refer to those of isolated pure product. Reactions were performed under refluxing conditions. Catalyst used 12 mg. |
| 1 |
None |
10 |
10 |
100 |
56 |
| 2 |
Toluene |
10 |
10 |
100 |
12 |
| 3c |
CHCl3 |
10 |
10 |
100 |
15 |
| 4c |
DCM |
10 |
10 |
100 |
4 |
| 5c |
H2O |
10 |
10 |
100 |
10 |
| 6 |
DMSO |
10 |
10 |
100 |
83 |
| 7 |
DMF |
5 |
10 |
100 |
95 |
| 8 |
DMF |
1 |
10 |
100 |
95 |
| 9 |
PEG-600 |
1 |
10 |
100 |
95 |
| 10 |
PEG-600 |
1 |
8 |
100 |
95 |
| 11 |
PEG-600 |
1 |
7 |
100 |
81 |
| 12 |
PEG-600 |
1 |
8 |
90 |
95 |
| 13 |
PEG-600 |
1 |
8 |
80 |
95 |
| 14 |
PEG-600 |
1 |
8 |
70 |
85 |
| 15d |
PEG-600 |
1 |
8 |
80 |
82 |
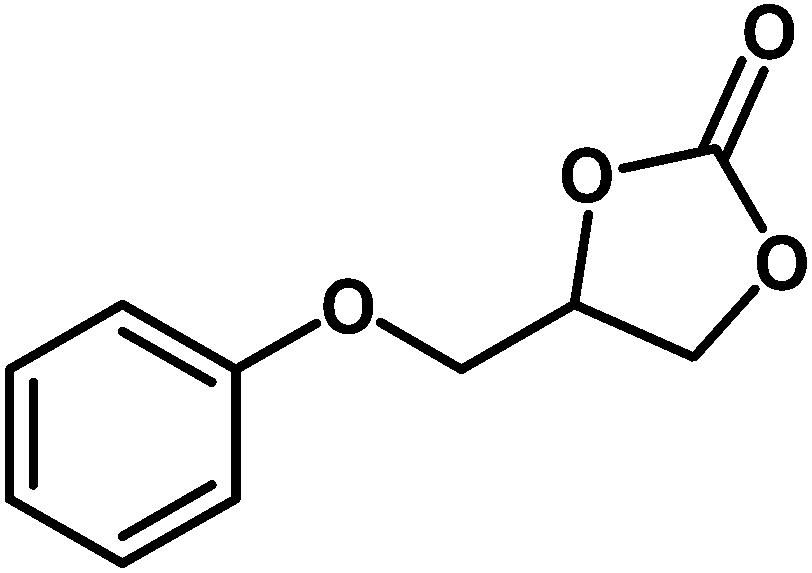
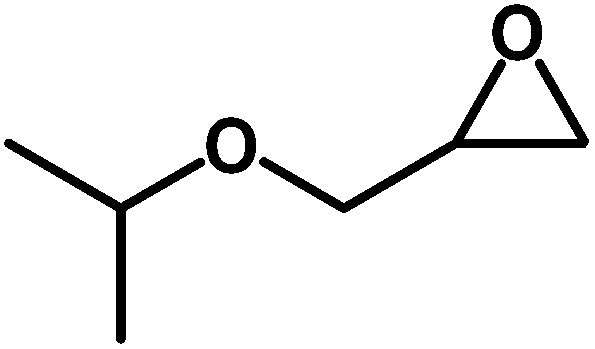
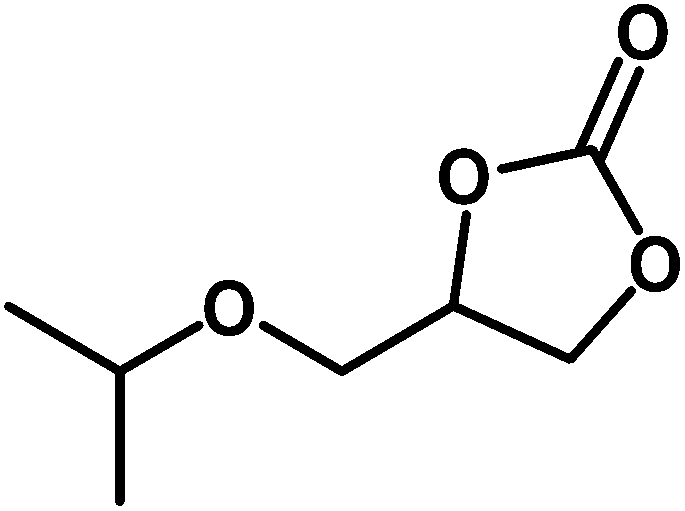
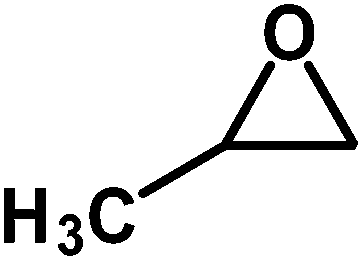
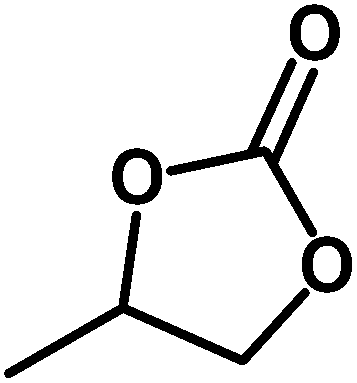
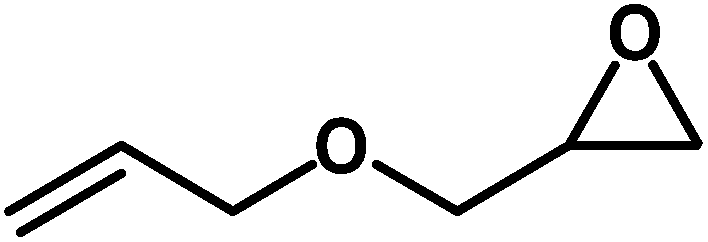
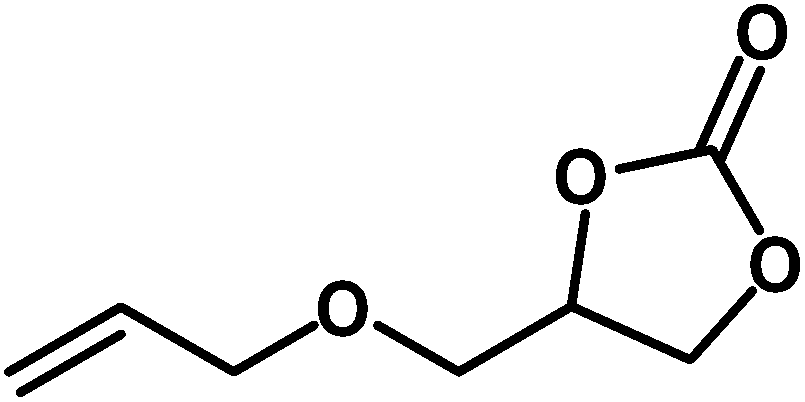
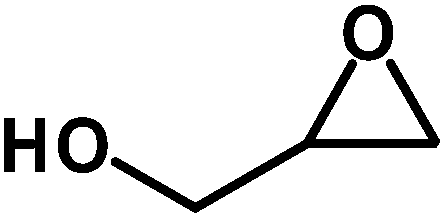
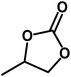
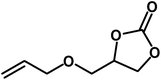
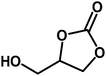
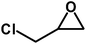
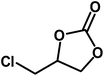
Under the above optimized reaction conditions, we have examined the performances of MZS-1 in chemical fixation of CO2 over different functional group substituted epoxides. Various epoxides, such as styrene oxide (SO), propylene oxide (PO), aryloxy epoxides (1,2-epoxy-3-phenoxy propane), epichlorohydrin (ECH) and allyl glycidyl ether (AGE), glycidol etc. were used as the substrate for the reaction system using MZS-1 as a catalyst. As shown in Table 3, most substrates could be smoothly converted to corresponding cyclic carbonates with high conversion and excellent selectivity (Table 3, entries 1–8). It was demonstrated that terminal epoxides with both electron-donating (Table 3, entries 2–6) and electron-withdrawing groups (Table 3, entry 7) could be transformed to the corresponding cyclic carbonates with almost quantitative yields (91–95%) within 8–10 h under 1 atm CO2 pressure at 80 °C. It is worthy to mention that in the case of propylene oxide, a slightly lower yield of the corresponding cyclic carbonate (Table 3, entry 5, 80%) was observed.
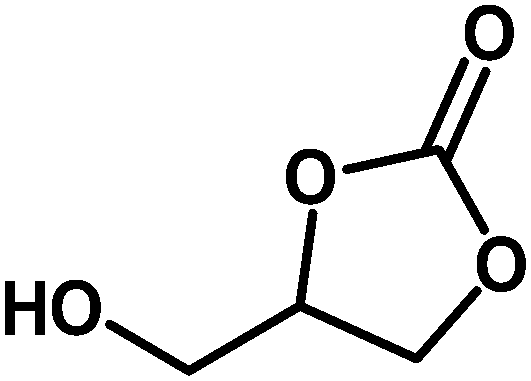
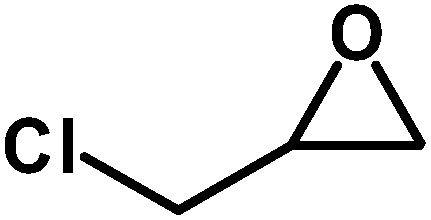
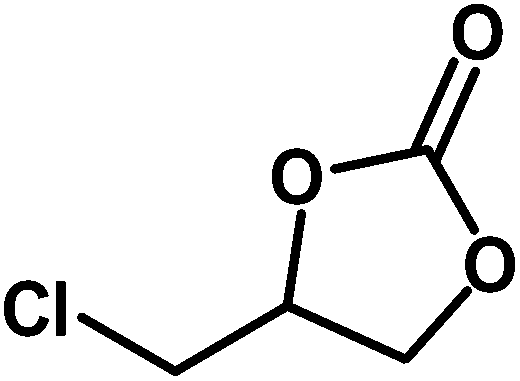
Table 3 Synthesis of different cyclic carbonates by using MZS-1 as catalysta
| Entry |
Epoxide |
Product |
Time (h) |
Yieldb (%) |
| Reaction conditions: epoxide (5 mmol), MZS-1 (15 mg, 0.06 mmol), 80 °C. PEG-600 (3 ml) under 1 atm of CO2. Yields refer to those of isolated pure product. 1H and 13C NMR spectra of the products are shown in the ESI file. |
| 1 |
 |
 |
8 |
95 |
| 2 |
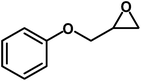 |
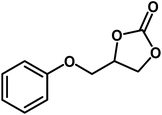 |
9 |
91 |
| 3 |
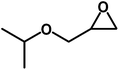 |
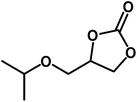 |
10 |
90 |
| 4 |
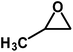 |
 |
8 |
80 |
| 5 |
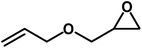 |
 |
8 |
95 |
| 6 |
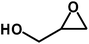 |
 |
10 |
91 |
| 7 |
 |
 |
8 |
94 |
We have also carried out the reaction of styrene oxide with CO2 in the presence of mesoporous zinc oxide (ZnO) and tin oxide (SnO2) nanoparticle as the catalysts and the corresponding results are summarized in Table 4. No product was detected when the reaction was conducted in the absence of any catalyst, and nearly no product was obtained in the presence of mesoporous tin oxide (Meso-SnO2) nanoparticles.46 When we have used mesoporous zinc oxide, Meso-ZnO(1) synthesized by using sodium salicylate as template as the catalyst for the synthesis of styrene carbonate, then the conversions and yield of the desired product is very low compared to our designed mesoporous zinc stannate nanocatalyst MZS-1. We have also performed the reaction with a mixture of free SnO2 and free ZnO as a catalyst and found no such improvement of reaction efficiency as compare to MZS-1 (Table 4, entry 3). We have also used high surface area mesoporous ZnO material Meso-ZnO(2) (BET surface area 456 m2 g−1) by using hexadecyl-2-pyridinylmethylamine as template47 for the CO2 fixation over styrene oxide. The observed yield of styrene carbonate over Meso-ZnO(2) was 36.2%, which is considerably high as compared to Meso-ZnO(1) (Table 4). High surface area of the former could be the reason for higher catalytic activity in this CO2 fixation reaction. The results shown in Table 4 suggested that synergy between the Lewis acidic and the Lewis basic sites of the mixed oxide MZS-1 material could play an important role for the synthesis of cyclic carbonate from epoxide. Moreover, Dai et al. and others have reported that mixed oxide materials shows higher catalytic activity over the individual metal oxides.48,49
Table 4 Synthesis of styrene carbonate in the presence of different catalystsa
| Entry |
Catalyst |
Yieldb (%) |
| Reaction conditions: styrene oxide (5 mmol), MZS-1 (15 mg, 0.06 mmol). PEG-600 (3 ml), CO2 (1 atm), 80 °C, 8 h. Yields refer to those of isolated pure product. |
| 1 |
Meso-SnO2 |
5.0 |
| 2 |
Meso-ZnO(1) |
15.0 |
| 3 |
Meso-ZnO(2) |
36.2 |
| 4 |
Mixture of mesoporous SnO2 and mesoporous ZnO |
30.0 |
| 5 |
MZS-1 |
94.0 |
| 6 |
Without catalyst |
— |
Similarly, we have compared the activity of our present catalytic system with existing state of art heterogeneous catalysts used for synthesis of cyclic carbonate.19,50–52 These results are summarized in Table 5. These results indicate that, other reported reactions involved protic compounds, additives, phase-transfer reagents etc. for the activation of epoxide and often the catalytic systems required high pressure of CO2 (20–50 atm) and high temperature. Whereas, our designed mixed-oxide metal nanocrystals (MZS-1) with mesoporosity and high surface area demonstrates a comparatively better catalytic efficiency for the synthesis of five-membered cyclic carbonates from CO2 and epoxide under 1 atmosphere.
Table 5 Comparison of catalytic activity of MZS-1 over related systems for the synthesis of styrene carbonate
| Entry |
Catalyst |
Reaction conditions |
Yield (%) |
References |
| 1 |
Bifunctional M(salphen) M = Zn, Cu, Ni |
Styrene oxide (10 mmol), catalyst (0.1 mmol), CO2 (2 MPa/19.72 atm), 100 °C, 4 h |
75 |
19 |
| 2 |
Zn(salphen) complex |
Styrene oxide (2 mmol), catalyst (0.05 mmol), CH2Cl2, NBu4I (co-catalyst), CO2 (1 MPa/9.86 atm), 25 °C, 18 h |
66 |
50 |
| 3 |
BIO-1c (Zn porphyrine) |
Styrene oxide (10 mmol), catalyst BIO-1c (0.01 mmol), CO2 (1.7 MPa/16.76 atm), 120 °C, 4 h |
88 |
51 |
| 4 |
MCM-41-IPr-CO2 |
Styrene oxide (50 mmol), catalyst (0.25 mmol), CH2Cl2, CO2 (2.0 MPa/19.72 atm), 120 °C, 48 h |
87 |
52 |
| 5 |
Mesoporous Zn stannate nanocrystal (MZS) |
Styrene oxide (5 mmol), catalyst (0.06 mmol), PEG-600, CO2 (1 atm or 0.1 MPa), 80 °C, 10 h |
95 |
Present study |
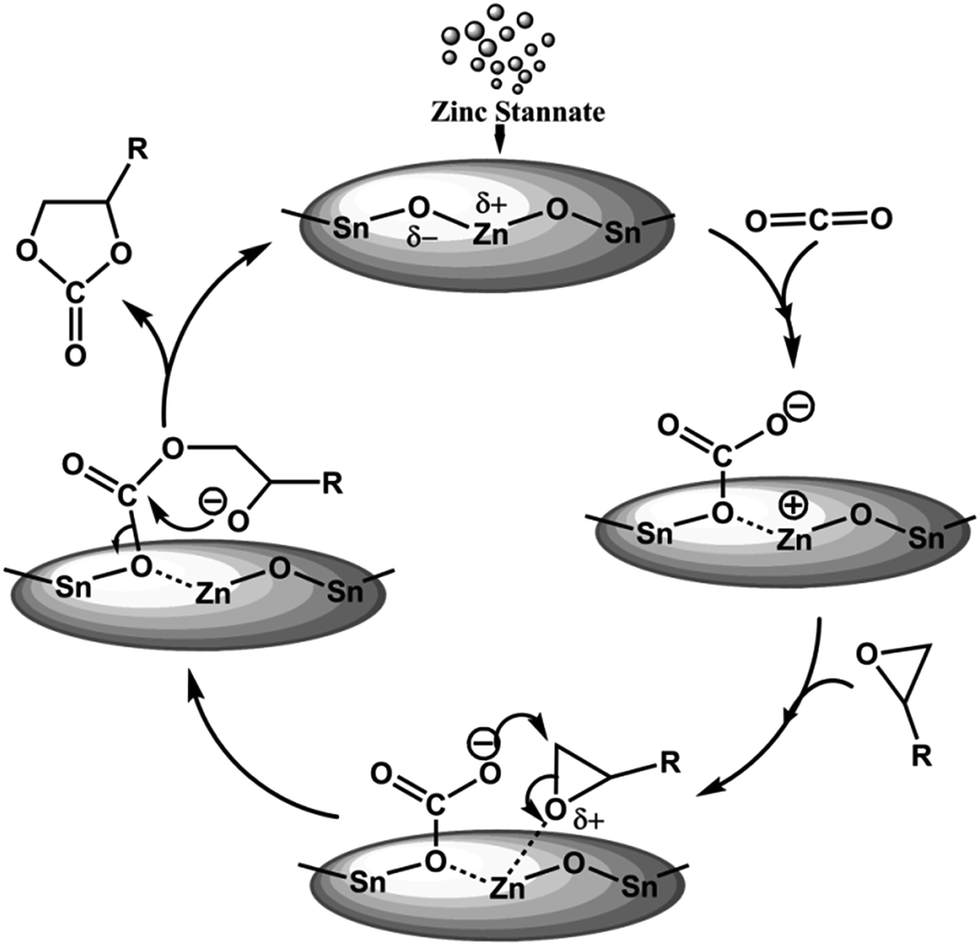
Based on our experimental results we have proposed a plausible bifunctional reaction mechanism in Fig. 5. Considering high Lewis acidic nature of MZS-1 surface the catalyst could facilate the activation of oxiran ring of the epoxide. On the other hand moderate basicity at the Zn(II) surfaces53 (ZnO being amphoteric oxide) in the vicinity of Sn(IV) sites could facilitate the activation/adsorption of CO2 molecules. Thus, the unique cooperative action of acid–base sites derived due to the formation of framework Zn–O–Sn bonds could help the formation of cyclic carbonate from epoxide and CO2 as shown in Fig. 5.
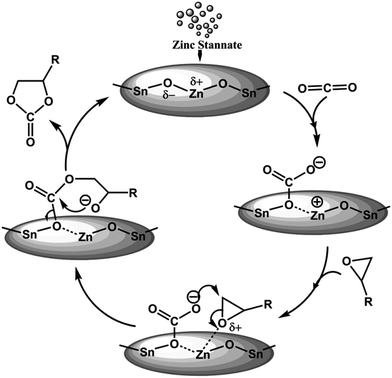 |
| | Fig. 5 Plausible mechanism for the synthesis of cyclic carbonates using MZS-1. | |
Recyclability of MZS-1 material
For a heterogeneous catalyst, it is important to examine its ease of separation, recoverability and reusability.54–57 The reusability of the MZS-1 catalyst was investigated in synthesis of cyclic carbonate for a representative reaction between styrene oxide and CO2. After the completion of the reaction, the contents were centrifuged to separate the solid catalyst from the reaction mixture. The catalyst was then thoroughly washed with distilled water followed by acetone and then dried in an oven at 100 °C for 6 h before reuse.
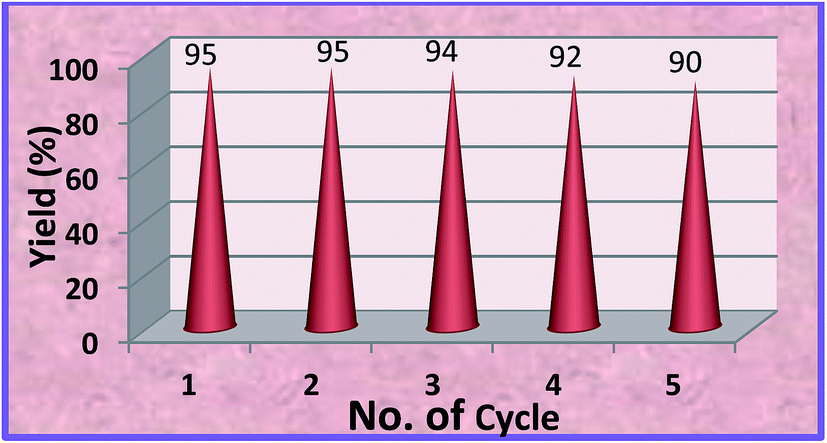
As seen from Fig. 6 that the catalyst can be efficiently recycled and reused for five times without appreciable decrease in product yield.
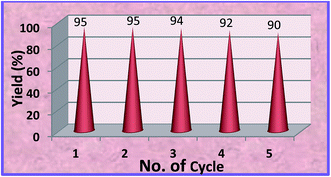 |
| | Fig. 6 Recyclability of MZS-1 catalyst for the synthesis of styrene carbonate. | |
Conclusion
We have designed the synthesis of binary semiconducting metal oxide zinc stannate nanocrystals having mesoporosity and high BET surface area by using sodium salicylate as template, and it is explored for the synthesis of cyclic carbonates from epoxide and carbon dioxide under very mild reaction conditions such as ordinary pressure and in the presence of a green solvent. Tiny ZnSnO3 nanoparticles showed controlled crystalline phase, mesoscopic void spaces, chemical stability and high catalytic activity for the synthesis of a series of cyclic carbonates. The catalyst is also unique as it bears both the acid–base sites derived through the formation of framework Zn–O–Sn bonds. The notable advantages offered by this CO2 fixation protocol are the use of green solvent like PEG-600, reaction at 1 atmosphere CO2 pressure, general applicability, mild reaction conditions, high selectivity, recyclability of the catalyst and high yield of cyclic carbonates. Green and mild catalytic CO2 sequestration pathway displayed by the ZnSnO3 nanocrystals may find huge potential utility as a heterogeneous catalyst for the synthesis of a wide range of value added fine chemicals in future.
Acknowledgements
SMI acknowledges DST-SERB, UGC, New Delhi, India and DST-WB for funding. We acknowledge DST for providing support to the University of Kalyani under FIST, SAP and Purse programme. BB is thankful to CSIR, New Delhi for providing him SPM research fellowship. SR acknowledges University of Kalyani for providing her junior research fellowship (URS). AB wishes to thank DST, New Delhi for extramural project grants through DST-SERB and DST-UKIERI projects.
Notes and references
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed
 .
. - P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281 CAS .
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed .
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092 CAS .
- S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC .
- B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC .
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed .
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed .
- M. Adolph, T. A. Zevaco, C. Altesleben, O. Walter and E. Dinjus, Dalton Trans., 2014, 43, 3285–3296 RSC .
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC .
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554 CrossRef CAS PubMed .
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef .
- Q. Gong, H. Luo, D. Cao, H. Zhang, W. Wang and X. Zhou, Bull. Korean Chem. Soc., 2012, 33, 1945 CrossRef CAS .
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS PubMed .
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC .
- F. W. Li, L. F. Xiao, C. G. Xia and B. Hu, Tetrahedron Lett., 2004, 45, 8307 CrossRef CAS .
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef .
- U. R. Seoa and Y. K. Chung, Adv. Synth. Catal., 2014, 356, 1955 CrossRef .
- Y. Ren, J. Chen, C. Qi and H. Jiang, ChemCatChem, 2015, 7, 1535 CrossRef CAS .
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497 CrossRef CAS PubMed .
- Q. W. Song, L. N. He, J. Q. Wang, H. Yasuda and T. Sakakura, Green Chem., 2013, 15, 110 RSC .
- A. L. Girard, N. Simon, M. Zanatta, S. Marmitt, A. L. Girard, N. Simon, M. Zanatta and S. Marmitt, Green Chem., 2014, 16, 2815 RSC .
- S. M. Chen, Y. Liu, J. P. Guo, P. Z. Li, Z. Y. Huo, P. T. Ma, J. Y. Niu and J. P. Wang, Dalton Trans., 2015, 44, 10152 RSC .
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861 CrossRef CAS PubMed .
- D. J. Darensbourg, W.-C. Chung, K. Wang and H.-C. Zhou, ACS Catal., 2014, 4, 1511 CrossRef CAS .
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793 CrossRef CAS PubMed .
- Q. An, Z. Li, R. Graff, J. Guo, H. Gao and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4969 CAS .
- J. B. Joo, M. Dahl, N. Li, F. Zaera and Y. D. Yin, Energy Environ. Sci., 2013, 6, 2082 CAS .
- A. S. Alshammari, L. N. Chi, X. P. Chen, A. Bagabas, D. Kramer, A. Alromaeh and Z. Jiang, RSC Adv., 2015, 5, 27690 RSC .
- J.-Y. Hong, S.-E. Bae, Y. S. Won and S. J. Huh, J. Colloid Interface Sci., 2015, 448, 467 CrossRef CAS PubMed .
- D. Hu, B. Q. Han, S. J. Deng, Z. P. Feng, Y. Wang, J. Popovic, M. Nuskol, Y. Wang and I. J. Djerdj, J. Phys. Chem. C, 2014, 118, 9832 CAS .
- K. Tang and L. Wang, J. Mater. Chem. A, 2014, 4970 Search PubMed .
-
(a) J. M. Wu, C.-Y. Chen, Y. Zhang, K.-H. Chen, Y. Yang, Y. Hu, J.-H. He and Z. L. Wang, ACS Nano, 2012, 6, 4369 CrossRef CAS PubMed ;
(b) Z. Tian, C. Liang, J. Liu, H. Zhang and L. Zhang, J. Mater. Chem., 2012, 22, 17210 RSC ;
(c) D. Liang, S. Wu, P. Wang, Y. Cai, Z. Tian, J. Liu and C. Liang, RSC Adv., 2014, 4, 26201–26206 RSC .
- T. Lim, H. Kim, M. Meyyappan and S. Ju, ACS Nano, 2012, 6, 4912 CrossRef CAS PubMed .
- S. A. Kelkar, P. A. Shaikh, P. Pachfule and S. B. Ogale, Energy Environ. Sci., 2012, 5, 5681 CAS .
- T.-Y. Ma, H. Li, T.-Z. Ren and Z.-Y. Yuan, RSC Adv., 2012, 2, 2790 RSC .
- C. G. Read, Y. Park and K.-S. Choi, J. Phys. Chem. Lett., 2012, 3, 1872 CrossRef CAS PubMed .
- F. Koffyberg and F. Benko, Appl. Phys. Lett., 2008, 37, 320 CrossRef .
- T. Tharsika, A. S. M. A. Haseeb, S. A. Akbar, M. F. M. Sabri and Y. H. Wong, J. Alloys Compd., 2015, 618, 455 CrossRef CAS .
- C. Liu, R. R. Oder, L. Zhang, Z. Ren, H. Chen, Z. Zhang, C. Ronning and P. X. Gao, J. Mater. Chem. A, 2014, 2, 4157 CAS .
- H. T. Fan, Y. Zeng, X. J. Xu, N. Lv and T. Zhang, Sens. Actuators, B, 2011, 153, 170 CrossRef CAS .
- S. K. Das, M. K. Bhunia, A. K. Sinha and A. Bhaumik, J. Phys. Chem. C, 2009, 113, 8918 CAS .
- V. Kumari, A. K. Patra and A. Bhaumik, RSC Adv., 2014, 4, 13626 RSC .
- Y. Chen, L. Yu, Q. Li, Y. Wu, Q. Li and T. Wang, Nanotechnology, 2012, 23, 415501 CrossRef PubMed .
- M. Pramanik, M. Nandi, H. Uyama and A. Bhaumik, Catal. Sci. Technol., 2012, 2, 613 CAS .
- D. Chandra, N. Mukherjee, A. Mondal and A. Bhaumik, J. Phys. Chem. C, 2008, 112, 8668 CAS .
- D. Chandra, S. Mridha, D. Basak and A. Bhaumik, Chem. Commun., 2009, 2384 RSC .
- W. L. Dai, S. L. Luo, S. F. Yin and C. T. Au, Appl. Catal., A, 2009, 366, 2 CrossRef CAS .
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS .
- A. Decortes, M. Martínez Belmonte, J. B. Buchholza and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC .
- T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485 RSC .
- H. Zhou, Y. M. Wang, W. Z. Zhang, J. P. Qu and X. B. Lu, Green Chem., 2011, 13, 644 RSC .
-
(a) M. Ramin, N. V. Vegten, J. D. Grunwaldt and A. Baiker, J. Mol. Catal. A: Chem., 2006, 258, 165–171 CrossRef CAS ;
(b) B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS ;
(c) W. L. Dai, S. L. Luo, S. F. Yin and C. T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS .
- S. Roy, T. Chatterjee and S. M. Islam, Tetrahedron Lett., 2015, 56, 779 CrossRef CAS .
- S. Roy, T. Chatterjee and S. M. Islam, Green Chem., 2013, 15, 2532 RSC .
- S. Roy, T. Chatterjee, B. Banerjee, N. Salam, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 46075 RSC .
- S. Roy, T. Chatterjee, M. Pramanik, A. Bhaumik and S. M. Islam, J. Mol. Catal. A: Chem., 2014, 386, 78 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available: For 1H NMR and 13C NMR spectral data of all products listed in Table 3. See DOI: 10.1039/c6ra02969h |
| ‡ These two authors have equally contributed in this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.