Synthesis of [1]benzothieno[3,2-b][1]benzothiophene pendant and norbornene random co-polymers via ring opening metathesis†
Craig M. S.
Combe
*a,
Laure
Biniek
b,
Bob C.
Schroeder
a and
Iain
McCulloch
*a
aDepartment of Chemistry, Imperial College, London, SW7 2AZ, UK. E-mail: imcculloch@imperial.ac.uk
bInstitut Charles Sadron, CNRS-Université de Strasbourg, 23 rue du Loess, 67034 Strasbourg, France. E-mail: laure.biniek@ics-cnrs.unistra.fr
First published on 19th November 2013
Abstract
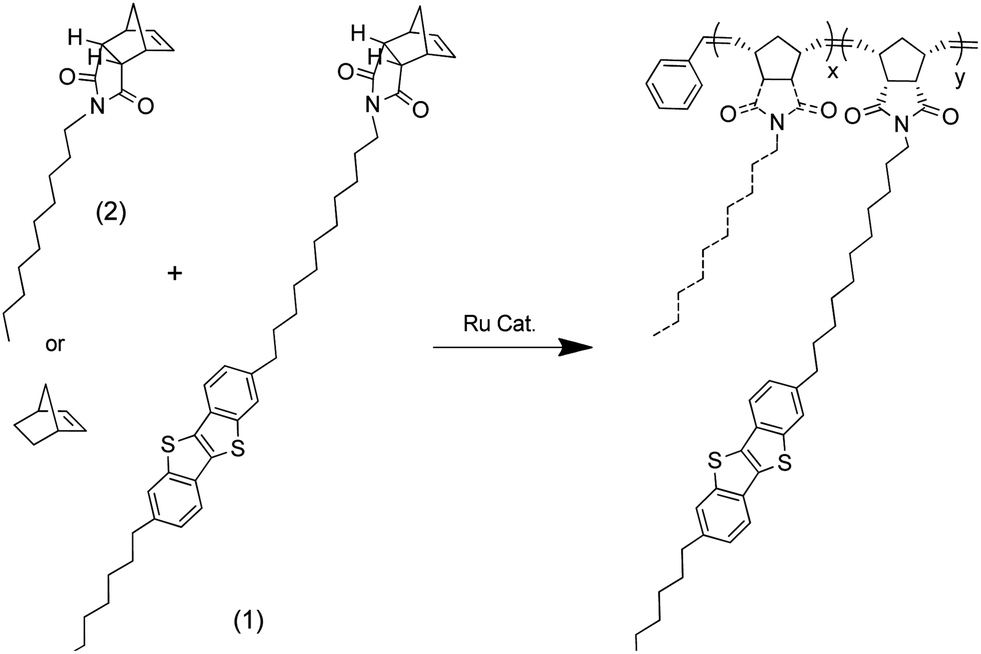
2-Octyl-7-(11-(cis-5-norbornene-endo-2,3-dicarboxylicimide)undecane)[1]benzothienopheno[3,2-b] benzothienophene (1) was synthesised and co-polymerised with varying amounts of 1-decyl(cis-5-norbornene-endo-2,3-dicarboxylic) imide (2) or norbornene, using a ruthenium catalyst, to produce a series of random co-polymers.
Introduction
Alkyl [1]benzothieno[3,2-b][1]benzothiophenes (BTBT) have attracted attention in recent years as a high performing organic transistor material,1–3 with 2-tridecyl[1]benzothieno[3,2-b][1]-benzothiophene (C13-BTBT) displaying hole mobilities as high as 17.2 cm2 V−1 s−1 in polycrystalline thin films grown from solution.4 Although there have been examples of polymers utilising this unit, coupled along a conjugated backbone,5,6 there have been no examples of its incorporation as a side-chain, or “comb-shaped” polymer, where the unit is attached as a pendant group, via a flexible spacer chain to a polymer backbone. This would potentially allow enhanced processability and possible facile alignment of the BTBT units in a controlled way, as it is decoupled from the backbone where molecular motion is usually very restricted.7–10Ring-opening metathesis polymerisation (ROMP)11–14 was chosen as the preferred mechanism as it has proven tolerance with electron rich aromatic groups, due to the absence of radicals, and polynorbornene materials being used in plastic electronics for OLEDs15,16 and OPVs17 devices. The fast initiation rate of catalyst dichloro[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene](benzylidene)bis(3-bromopyridine) ruthenium(II)18 resulted in a lack of selectivity between the different norbornene monomers facilitating the synthesis of a random copolymer.19
Synthesis
BTBT was synthesised in a one step process from 2-chlorobenzaldehyde and sodium hydrosulfide hydrate, following literature procedures,20 to produce 15 g of material in a yield typical for the reaction. Friedel–Crafts acylation of BTBT (3 g) with octanoyl chloride (Scheme 1) at low temperature produced the mono-addition product, [1]benzothieno[3,2-b][1]benzothiophene-2-octan-1-one (3), in very high yields.21 The ketone was reduced using two and a half equivalents of aluminium chloride and lithium aluminium hydride to generate 2-octyl-[1]benzothieno[3,2-b][1]benzothiophene (4) quantitatively. These steps were repeated with 11-bromoundecanoic acid chloride, synthesised from 11-bromoundecanoic acid and oxalyl chloride, to produce 2-octyl-7-(11-bromoundecan-1-one)[1]benzothienopheno[3,2-b] benzothienophene (5) then 2-octyl-7-(11-bromoundecane) [1]benzothieno[3,2-b][1]benzothiophene (alkylbromo-BTBT) (6) in high yields.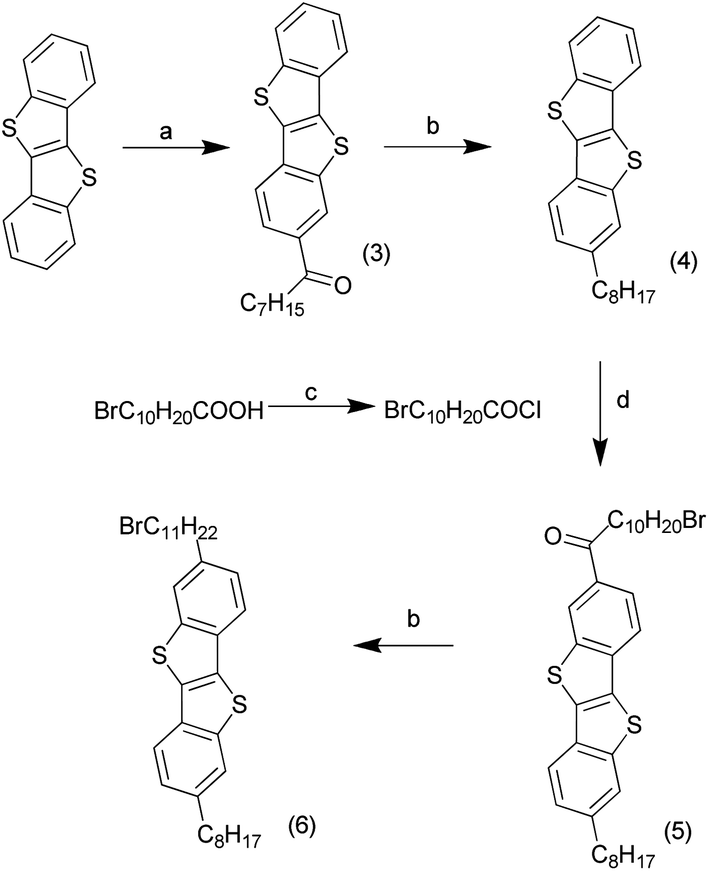 | ||
| Scheme 1 Synthetic route to molecule 6. (a) 1.1 eq. C7H15COCl, 2.5 eq. AlCl3, dry DCM, Ar, −78 °C, 4 h, 98%. (b) 2.5 eq. AlCl3, 2.5 eq. 1 M LiAlH4 in Et2O, Ar, dry Et2O and DCM, rt, O/N, 100/98%. (c) 1.4 eq. oxalyl chloride, cat. DMF, Ar, dry DCM, 1 h, 100%. (d) 2.5 eq. AlCl3, dry DCM, Ar, −78 °C, 4 h, 88%. | ||
The alkylbromo-BTBT underwent nucleophilic substitution with cis-5-norbornene-endo-2,3-dicarboxylic imide, synthesised from the anhydride and ammonium acetate,22 to produce ROMP monomer 2-octyl-7-(11-(cis-5-norbornene-endo-2,3-dicarboxylicimide)undecane)[1]benzothienopheno[3,2-b] benzothienophene (BTBT norbornene imide) (1) (Scheme 2). 1-Decyl(cis-5-norbornene-endo-2,3-dicarboxylic) imide (alkyl norbornene imide) (2) was also synthesised from the anhydride and 1-decylamine, using a Dean–Stark apparatus, to be used as a co-monomer.23
 | ||
Scheme 2 Synthesis of molecules 1 and 2. (a) 1 eq. NH4OAc, AcOH, 140 °C, 16 h, 100%. (b) 1 eq. C8BTBTC11Br (0.5 g), 1.5 eq. K2CO3, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:THF, reflux, 16 h, 96%. (c) 1 eq. 1-decylamine, 1 eq. triethylamine, Ar, dry toluene, reflux, 16 h, Dean–Stark app., 86%. :1 DMF:THF, reflux, 16 h, 96%. (c) 1 eq. 1-decylamine, 1 eq. triethylamine, Ar, dry toluene, reflux, 16 h, Dean–Stark app., 86%. | ||
Norbornene-BTBT underwent ROMP polymerisation with bicyclo[2.2.1]hept-2-ene (norbornene) or the alkyl norbornene imide as co-monomers in different ratios using dichloro[1,3-bis(2,4,6-trimethylphenyl)-2-imidasolidinylidene](benzylidene) bis(3-bromopyridine)ruthenium(II) as catalyst (Scheme 3) (Table 1). Attempts to synthesise a norbornene-BTBT homo-polymer produced a material too insoluble to handle and was not further pursued.
 | ||
| Scheme 3 Polymerisation of norbornene-BTBT with either norbornene or alkyl norbornene imide. | ||
| Polymer | Co-monomer & equivalents | Catalyst (eq.) | M n (kg mol−1) | M w (kg mol−1) | tMn (kg mol−1) | PDI | Ratiob |
|---|---|---|---|---|---|---|---|
| a Determined by gel permeation chromatography in chlorobenzene relative to monodispersed polystyrene standards. b Ratio of co-monomers in polymer measured by 1H NMR – (norbornene/alkyl-norbornene2): norbornene-BTBT. | |||||||
| p-BTBT1 | 5 eq. norbornene | 0.03 | 13 | 49 | 25 | 3.7 | 2.5:1 |
| p-BTBT2 | 7.5 eq. norbornene | 0.03 | 9.7 | 61 | 46 | 6.3 | 7:1 |
| p-BTBT3 | 10 eq. norbornene | 0.03 | 8.6 | 44 | 54 | 5.1 | 10:1 |
| p-BTBT4 | 1 eq. alkyl-norbornene | 0.015 | 69 | 89 | 65 | 1.3 | 1:1 |
| p-BTBT5 | 2 eq. alkyl-norbornene | 0.01 | 104 | 160 | 127 | 1.5 | 2:1 |
Analysis
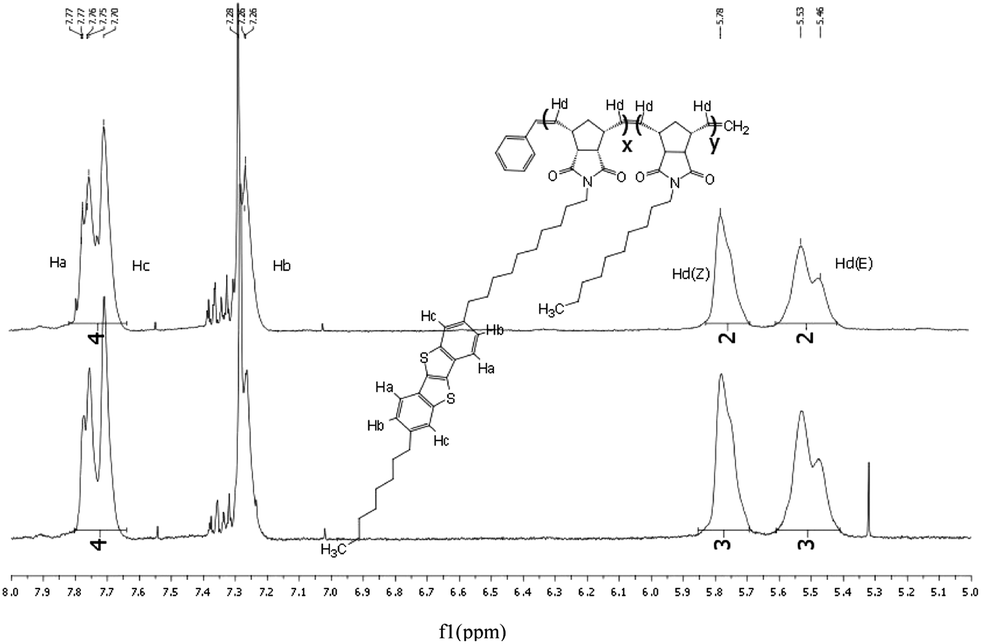
The table in Table 1 displays the number-averaged molecular weight (Mn), average molecular weight (Mw), theoretical average molecular weight (tMn, see ESI for calculation), and the polydispersity index (PDI) of the corresponding polymers. Polymers with the norbornene co-monomer produced low Mn and large PDI, while polymers with the alkyl norbornene imide co-monomer produced a Mn very close to the tMn and a small PDI. It is hypothesised that the rigidity of the norbornene unit compromises the polymer solubility, causing aggregation, and precipitation preventing high molecular weights from being formed while the copolymers incorporating alkyl chains on the norbornene imide had sufficient solubility to allow for propagation of the polymer chain.NMR analysis24 of the polymers show key hydrogen peaks that quantify the ratio of co-polymerisation between the monomers. Peaks at 7.7 ppm correspond to the aromatic hydrogens in the 3, 4, 8 and 9 positions of BTBT, integrating at 4 for every BTBT monomer, and give the most distinct signals for the monomer. p-BTBT1, 2 and 3 have distinct peaks at 5.2 and 5.3 corresponding to the respective Z and E polymer backbone alkenes formed from norbornene ring opening and peaks at 5.5 and 5.8 corresponding to the respective Z and E alkenes formed from norbornene-BTBT (Fig. 1). Due to the broadness of polymer 1H NMR signals, accurate integration of the individual peaks is not possible so all were integrated together, from 5.1 to 5.8, to give the total number of vinyl backbone hydrogens per monomer. The ratio of co-monomers can then be measured; p-BTBT1 showed a poor incorporation of norbornene relative to the BTBT, roughly half of the expected amount, while p-BTBT2 and 3 had approximately the calculated ratio. It is suspected that this is the result of the same factors that caused the polymers to have a poor molecular weight, aggregation that prevented the polymerisation to proceed sufficiently.
 | ||
| Fig. 1 Key proton peaks of p-BTBT1, 2, and 3. | ||
Similarly, p-BTBT4 and 5 only have peaks at 5.5 and 5.8 as the co-monomers have the same polymerisable group (Fig. 2). The ratio between the aromatic and vinyl hydrogen peaks corresponds well with the predicted values given the ratio of co-monomers polymerised. Combined with the molecular weight data, the strategy of using co-monomers with an alkyl chain has been successful in producing polymers of the desired size and form.
 | ||
| Fig. 2 Key proton peaks of p-BTBT4, and 5. | ||
Ultraviolet and visible spectroscopic (UV-vis) analysis spectra of BTBT ROMP polymers displayed a λmax of 322 nm in solution and 322 nm, with a broadening of the signal, in the drop cast thin film typical for all five polymers. This red shift is too small to be head-to-tail, J-aggregation of the BTBT pendants25–27 that would also create a narrowing of the signal. An absence of solvent–solute interactions is a likely explanation for this effect (Fig. 3).
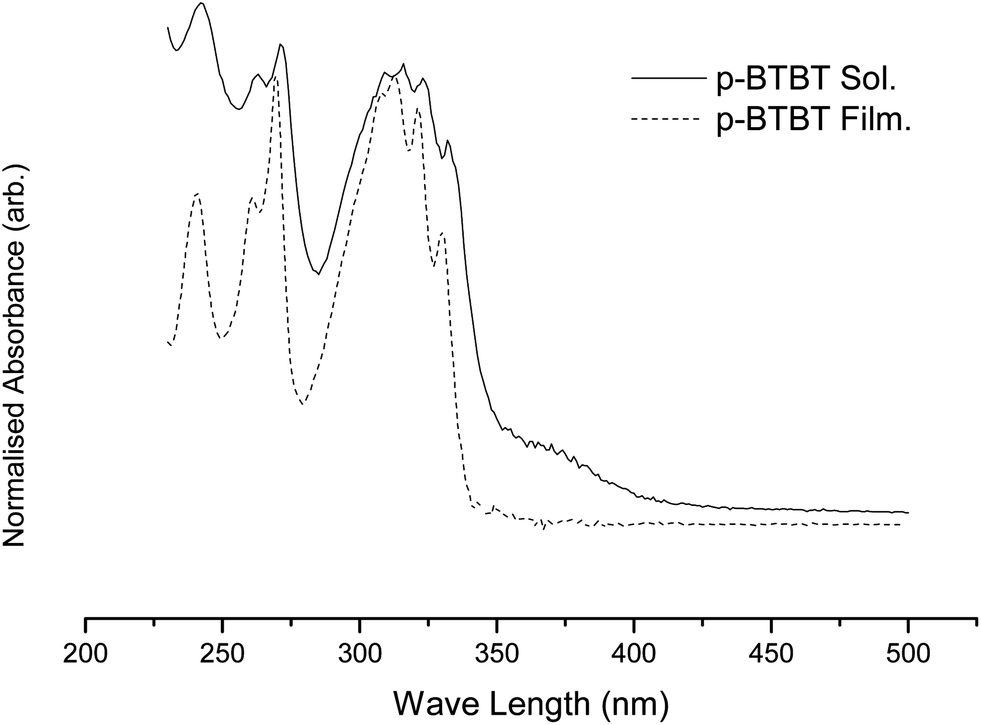 | ||
| Fig. 3 Typical UV-vis of ROMP polymers in solution and thin film. | ||
Devices made using these materials produced no transistor performance. It is hypothesised that there is too much crowding of the BTBT by the aliphatic poly-norbornene and alkyl chains, preventing a clear channel for hole transport through the material. This is corroborated with the UV-vis data that exhibits no indication of π-orbital overlap.
Conclusions
Pendant polymers of BTBT were synthesised utilising ROMP with varying success. Co-polymerisation with solubilising alkyl-pendants produced materials very close to theoretical predictions. However, the materials proved unsuccessful as transistor devices due to a lack of extended π-stacking. Further exploration of the ratio of aliphatic to aromatic is needed to ensure there is sufficient aromatic interactions for charge transport, while maintaining solubility. This could be achieved with a fine-tuning of the alkyl chain length and possible removal from one side of the BTBT pendant. ROMP materials have enormous potential for the controlled polymerisation of radical sensitive materials and provide interesting morphologies due polynorbornene's backbone, justifying further interest.Notes and references
- H. Minemawari, T. Yamada, H. Matsui, J. y. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS PubMed
.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed
- A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548–16550 CrossRef CAS PubMed
- L. Biniek, B. C. Schroeder, J. E. Donaghey, N. Yaacobi-Gross, R. S. Ashraf, Y. W. Soon, C. B. Nielsen, J. R. Durrant, T. D. Anthopoulos and I. McCulloch, Macromolecules, 2013, 46, 727–735 CrossRef CAS
- W. Shin, M. Y. Jo, D. S. You, Y. S. Jeong, D. Y. Yoon, J.-W. Kang, J. H. Cho, G. D. Lee, S.-S. Hong and J. H. Kim, Synth. Met., 2012, 162, 768–774 CrossRef CAS PubMed
- M. Wind, R. Graf, S. Renker and H. W. Spiess, Macromol. Chem. Phys., 2005, 206, 142–156 CrossRef CAS
- B. R. Maughon, M. Weck, B. Mohr and R. H. Grubbs, Macromolecules, 1997, 30, 257–265 CrossRef CAS
- H. Finkelmann, M. Happ, M. Portugal and H. Ringsdorf, Die Makromolekulare Chemie, 1978, 179, 2541–2544 CrossRef CAS
- H. Finkelmann, H. Ringsdorf and J. H. Wendorff, Die Makromolekulare Chemie, 1978, 179, 273–276 CrossRef CAS
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2000, 34, 18–29 CrossRef PubMed
- T.-L. Choi and R. H. Grubbs, Angew. Chem., 2003, 115, 1785–1788 CrossRef
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed
- A. Meyers, A. Kimyonok and M. Weck, Macromolecules, 2005, 38, 8671–8678 CrossRef CAS
- E. Bellmann, S. E. Shaheen, S. Thayumanavan, S. Barlow, R. H. Grubbs, S. R. Marder, B. Kippelen and N. Peyghambarian, Chem. Mater., 1998, 10, 1668–1676 CrossRef CAS
- A. de la Escosura, M. V. Martínez-Díaz, T. Torres, R. H. Grubbs, D. M. Guldi, H. Neugebauer, C. Winder, M. Drees and N. S. Sariciftci, Chem.– Asian J., 2006, 1, 148–154 CrossRef CAS PubMed
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS
- F. Niedermair, M. Sandholzer, G. Kremser and C. Slugovc, Organometallics, 2009, 28, 2888–2896 CrossRef CAS
- M. Saito, I. Osaka, E. Miyazaki, K. Takimiya, H. Kuwabara and M. Ikeda, Tetrahedron Lett., 2011, 52, 285–288 CrossRef CAS PubMed
- B. Kosata, V. Kozmik, J. í. Svoboda, V. Novotná, P. Vanek and M. Glogarová, Liq. Cryst., 2003, 30, 603–610 CrossRef CAS
- F. M. Mansfeld, G. Feng and S. Otto, Org. Biomol. Chem., 2009, 7, 4289–4295 CAS
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 118–126 CrossRef CAS
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS
- S. Kim, T. K. An, J. Chen, I. Kang, S. H. Kang, D. S. Chung, C. E. Park, Y.-H. Kim and S.-K. Kwon, Adv. Funct. Mater., 2011, 21, 1616–1623 CrossRef CAS
- H. Menzel, B. Weichart, A. Schmidt, S. Paul, W. Knoll, J. Stumpe and T. Fischer, Langmuir, 1994, 10, 1926–1933 CrossRef CAS
- J.-J. Aaron, Z. Mechbal, A. Adenier, C. Parkanyi, V. Kozmik and J. Svoboda, J. Fluoresc., 2002, 12, 231–239 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tc32058h |
| This journal is © The Royal Society of Chemistry 2014 |