
Hybrid periodic mesoporous organosilica designed to improve the properties of immobilized enzymes†
V. Gascón,
I. Díaz,
R. M. Blanco and
C. Márquez-Álvarez*
Molecular Sieves Group. Institute of Catalysis and Petroleum Chemistry (ICP-CSIC), C/ Marie Curie, 2. Cantoblanco 28049, Madrid, Spain. E-mail: cmarquez@icp.csic.es; Fax: +34 91 5854760; Tel: +34 91 5854785
First published on 30th July 2014
Abstract
Two types of highly ordered periodic mesoporous organosilicas have been synthesized as tailor-made supports to immobilize two different enzymes, lipase and laccase. These materials provide an environment where abundant organic groups in close vicinity to the enzyme surface generate a high chemical affinity, which results in high values of enzyme loading, catalytic activity and stabilization. A hydrophobic periodic mesoporous organosilica support (PMO) with a highly ordered hexagonal arrangement of parallel pore channels with a diameter around 7 nm, containing framework hydrocarbon groups (ethylene), was used for lipase immobilization. A novel periodic mesoporous aminosilica (PMA), containing secondary amine groups in its framework and having expanded pores was synthesized and studied for immobilization of a larger enzyme, namely laccase. The synthesis conditions were adjusted, using bis[(3-trimethoxysilyl)propyl]amine and 1,2-bis(trimethoxysilyl)ethane as framework co-precursors and 1,3,5-triisopropylbenzene as micelle expander for producing large pores (>10 nm). The properties of this multifunctional PMA having hydrophilic amino groups and hydrophobic ethylene/propylene groups within the framework were studied. This work compares the confinement of lipase and laccase enzymes in the pores of these hybrid organosilica materials and its effect on immobilization and stabilization parameters. Laccase immobilized on PMA and lipase immobilized on PMO exhibited higher stability in solvents (ethanol and methanol, respectively) compared to enzymes supported on functionalized silica materials with pending organic groups on the surface. High retention of enzymes inside the pores of these materials has been achieved and leaching has been fully prevented. These results can be attributed to the different interactions (hydrophobic, electrostatic and hydrogen bonding) established between the surfaces of the enzyme and the PMO/PMA support, which are enhanced by an optimum pore size adjusted to the enzyme dimensions.
Introduction
Siliceous ordered mesoporous materials (OMM) have burst into the field of enzyme immobilization as excellent supports to obtain nanostructured biocatalysts with improved properties.1 Three main parameters of these materials can be controlled by adjusting the synthesis conditions: (a) pore connectivity through a regular porous network, (b) pore size, which can be fine-tuned to accommodate the dimensions of the enzyme for optimal confinement, and (c) surface chemical functionalization to enhance enzyme-support affinity. This control enables one to obtain biocatalysts with improved properties regarding enzyme loading, catalytic efficiency, stability, and leaching prevention.2The development of hybrid periodic organo-bridged silicas with ordered mesopores and large pore diameter is a breakthrough in the field of ordered mesoporous materials. In these periodic mesoporous organosilicas (PMO), the organic groups are not anchored to the surface but become part of the framework. Using such hybrid supports allows one to avoid the pore size reduction that occurs upon grafting of the organic functionality on the surface of purely inorganic supports, which might hinder diffusion of enzyme molecules.3 However, literature regarding bulky enzyme immobilization on these materials is rather scarce.
The synthesis of PMO materials is carried out using bridged alkoxysilane molecules ((R′O)3Si-R-Si(OR′)3; R: bridging hydrocarbon group, R′: methyl or ethyl) as precursors.4–6 New hybrid organic-inorganic OMM offer more possibilities for application, particularly in enzyme immobilization.4,7,8 The porous structures, surface, and framework properties of PMO materials can be finely tuned by changing the bridging organic groups incorporated and the synthesis conditions employed.9–13 However, the lack of structural rigidity of the organic moiety of the silsesquioxane may result in a disordered material, especially when attempting to synthesize materials with large pore size.
Initially, research on PMOs had been focused on the incorporation of small aliphatic moieties (methylene,14 ethylene,12,15 ethenylene16) into the walls of the channel-like structures, obtaining different morphologies.17,18 Later, organic bridges in the framework were extended to more functional species ranging from hydrocarbons and heteroaromatics to metal complexes. Recent developments in novel PMO involve innovation in their components and structural designs. Multifunctional PMO can be easily obtained by co-condensation of a mixture of bridged organosilane precursors with different surface properties of the pore walls.19–23 For example, Morell et al., 2006 (ref. 24) incorporated aromatic bridging groups (phenylene and thiophene) and obtained PMO materials with pore sizes in the range of 4.8–5.4 nm employing the triblock copolymer Pluronic P123 as structure-directing agent.
The limited pore size of PMO materials may restrict their applications in some areas where large pores are essential for adsorption and immobilization of large biomolecules,25 and only a few works have been published regarding the immobilization of bulky enzymes, for which large pores are required.26,27 In comparison with their purely siliceous counterparts, pore size is more difficult to adjust in PMO.16,28
We have formerly described the immobilization of Candida antarctica lipase B (CaLB) on a PMO material containing ethylene groups.29 Lipases are known to have a hydrophobic domain, which often constitutes a lid responsible for their interfacial activation.30 CaLB lacks this lid,31,32 but still has the hydrophobic domain on its surface. Supports with hydrophobic surfaces can interact with this domain to drive lipase immobilization, therefore, PMO containing ethylene bridges were obtained and tested as supports in our group.
Laccase is a larger enzyme than CaLB, therefore two challenges are to be faced. One is the increase of chemical affinity and the other is obtaining a support with pore size large enough to accommodate the enzyme. We have recently reported33 the immobilization of laccase on OMM with expanded pore size functionalized with amine groups anchored on the silica surface. The large difference between the pI of laccase (around 4) and the pKa value of these groups (close to 10) enabled strong electrostatic interactions in a broad pH range. Based on these results we report here the synthesis of large-pore periodic mesoporous organosilica materials containing amino groups within the framework as supports for immobilization of laccase. Few attempts to introduce nitrogen-containing groups into the walls of hybrid organosilica materials have been reported. Asefa et al., 2003 (ref. 34) prepared periodic mesoporous aminosilica (PMA) materials that contained amine functional groups within the framework via thermal ammonolysis of PMOs under a flow of ammonia gas. The pore diameter of these materials was around 3–4 nm. It has also been reported the synthesis of PMA using silane precursors with amine groups. However, it has to be noticed that the order of the structure is an issue. The increase in concentration of the bridged alkoxysilane with non-rigid organic bridges may lead to a decrease in the order of the material, as reported by Wahab et al., 2004 (ref. 4) even for materials with pore size as small as 3.5 nm. An interesting transition of the mesostructure of this system was observed with increasing content of bis[(3-trimethoxysilyl)propyl]-amine (BTMSPA) in the starting mixture for the co-condensation of BTMSPA and 1,2-bis(trimethoxysilyl)ethane (BTME) in the presence of octadecyltrimethylammonnium chloride.35 When the ratio of BTME–BTMSPA was changed from 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 55:45, a change from a 2D hexagonal (p6mm) to a cubic mesophase took place. Higher BTMSPA concentrations in the reaction mixture led to the collapse of the structure. Tan et al., 2006 (ref. 36) obtained uniform mesopores in a material made with BTMSPA using hexadecyltrimethylammonium bromide (CTAB) as template. When BTMSPA was used, the hydrophilic chain helped to promote co-assembly of the precursor with CTAB micelles, limiting bulk condensation. The reported material had relatively low pore volume and specific surface area, as well as a reduced pore diameter (3.1 nm) but it had a combination of uniform pore size and amine groups integrated in the pore walls. In all these reported works, the pore sizes obtained were always smaller than those attainable for pure silica and did not exceed 6 nm. This factor limits the potential applications of PMAs as supports for the immobilization of large enzymes.
:10 to 55:45, a change from a 2D hexagonal (p6mm) to a cubic mesophase took place. Higher BTMSPA concentrations in the reaction mixture led to the collapse of the structure. Tan et al., 2006 (ref. 36) obtained uniform mesopores in a material made with BTMSPA using hexadecyltrimethylammonium bromide (CTAB) as template. When BTMSPA was used, the hydrophilic chain helped to promote co-assembly of the precursor with CTAB micelles, limiting bulk condensation. The reported material had relatively low pore volume and specific surface area, as well as a reduced pore diameter (3.1 nm) but it had a combination of uniform pore size and amine groups integrated in the pore walls. In all these reported works, the pore sizes obtained were always smaller than those attainable for pure silica and did not exceed 6 nm. This factor limits the potential applications of PMAs as supports for the immobilization of large enzymes.
On the basis of previous works with laccase immobilized on OMM with SBA-15 structure and expanded pores, functionalized with primary amine groups anchored to the siliceous surface, we report here the synthesis of a novel hybrid material: periodic mesoporous aminosilica with large pore size. The aim of this work is to study and compare the behaviour of two model enzymes, laccase and lipase confined within the pores of two different periodic mesoporous organosilica supports.
Experimental
Materials and Reagents
Triblock copolymer PEO20PPO70PEO20 (Pluronic P123) and 1,2-bis(trimethoxysilyl)ethane (BTME) were from Aldrich (USA). 3-Aminopropyltriethoxysilane (APTES) and bis[3-(trimethoxysilyl)propyl]amine (BTMSPA) were purchased from TCI (Belgium). 1,3,5-Tiisopropylbenzene (TIPB) and n-octyltriethoxysilane (OTES) were from Alfa Aesar (Germany). Amorphous silica MS-3030 was kindly donated by Silica PQ Corporation (USA).The extracts of soluble laccase from Myceliophthora thermophila (Suberase) and lipase from Candida antarctica B (Lipozyme CaLB·L) both expressed in Aspergillus oryzae were kindly donated by Novozymes (Denmark). 2′-Aino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), p-nitrophenyl acetate (p-NPA), tributyrin (TB), mercaptoethanol, bromophenol blue and glycerol were from Sigma (USA). Bovine serum albumin (BSA, Sigma-Aldrich, USA) was used as protein standard for protein content determination by the Bradford method.37 The reagents for electrophoresis SDS-PAGE (sodium dodecyl sulfate (SDS), ammonium persulfate, N,N,N′,N′-tetramethylethylenediamine (TEMED), 40% acrylamide/bis solution, 10× Tris base/glycine/SDS buffer and bio-safeTM coomasie G-250 stain) and broad molecular weight standards were from Bio-Rad (USA).
Citric acid, potassium chloride, phosphoric acid, sodium carbonate, sodium bicarbonate and toluene were purchased from Sigma-Aldrich (USA). Acetonitrile and sodium acetate were purchased from Scharlau (Spain). Sodium dihydrogen phosphate 1-hydrate, hydrochloric acid, ethanol, acetic acid, potassium dihydrogen phosphate, disodium hydrogen phosphate, sodium hydroxide, potassium phosphate and acetone were purchased from Panreac (Spain). Trisodium citrate dehydrate was from Analyticals Carlo Erba (Italy). Tris(hydroxymethyl)aminomethane (Tris base) was purchased from Fluka Analytical (USA). Solvents were all analytical or HPLC grade, salts were of high purity and water was Milli-Q grade. All materials were used as obtained without further purification.
Synthesis of hybrid periodic mesoporous materials
Ethylene-bridged periodic mesoporous organosilica (PMO) was synthesized as previously reported29,38 with few modifications. 3.19 g of Pluronic P123 were dissolved at room temperature in 126.84 mL of 0.174 M HCl aqueous solution in a flask with slow stirring. Once the surfactant was dissolved, 9.38 g of KCl were added. Both, the low acid concentration and the presence of inorganic salts play an important role in the formation of highly ordered materials.12,18,38–40 When the resulting solution was homogenized, it was heated in a thermostated water bath to a constant temperature of 40 °C. Then, 3.97 mL of bis-functional alkoxysilane, BTME, were added at once with rapid stirring. The resulting mixture was stirred at 40 °C for 24 h and then aged at 100 °C under static conditions for 24 h. Subsequently, the solid product was recovered by filtration, washed with ethanol and dried at room temperature for 24 h. The surfactant was removed from the material by two successive reflux extractions in ethanol/HCl (1.5 g of as-made PMO in a solution made with 20 mL of 35 wt% HCl and 205 mL of ethanol) for 24 h. The resulting solid was recovered by filtration, washed with ethanol, and dried in air.Aminodipropyl-bridged periodic mesoporous aminosilica (PMA)
Materials synthesized using only the highly flexible aminodipropyl-bridged silane collapsed upon surfactant extraction due to the lack of structural rigidity of the framework.4,34 Therefore, in order to obtain well-ordered pore structures PMA materials were synthesized by co-condensation of ethylene- and aminodipropyl-bridged silanes.For the standard synthesis of periodic mesoporous aminosilica (PMA), 3.19 g of P123 were dissolved in 126.8 mL of a 0.17 M HCl aqueous solution. To this homogeneous mixture, 9.38 g KCl were added. The mixture was slowly stirred for 24 h. Then, 3.49 g of 1,2-bis(trimethoxysilyl)ethane (BTME) and 0.95 g of bis(3-trimethoxysilyl)propylamine (BTMSPA) were added, and the solution was stirred at 40 °C for 24 h. The mixture was aged at 100 °C under static conditions for 24 h. The solution was filtered and the solid product was washed with ethanol and air-dried at room temperature. The surfactant was extracted from the sample by two successive reflux extractions in ethanol/HCl, as indicated above for the PMO sample. The solution was filtered, and the solid product was washed with ethanol and dried under ambient conditions.
The synthesis of pore-expanded PMA (E-PMA) followed similar procedure with some modifications. 1.595 mL of the micelle expander agent TIPB were added to the surfactant solution and the mixture cooled down to 18 °C prior to addition of BTME and BTMSPA. The suspension was kept under stirring at 18 °C for 24 h. The obtained solid was suspended in 200 mL toluene and refluxed for 24 hours before being filtered and washed with ethanol. The solid was then submitted to reflux extraction in ethanol/HCl and dried in the same way as PMA.
Functionalization of mesoporous materials by grafting
For comparative purposes, commercial mesoporous amorphous silica (AS) was functionalized with both amine33 and hydrophobic groups41 to immobilize laccase and lipase, respectively.The parent material (1.1 g) was degassed at 80 °C under vacuum for 18 h and then dispersed in a solution containing the organoalkoxysilane selected for the functionalization process (22 mmol of 3-aminopropyltriethoxysilane (APTES) or 29 mmol of n-octyltriethoxysilane (OTES)) in 100 mL of toluene. The mixture was refluxed under N2 stream for 24 h. The suspension was filtered, washed twice with dry toluene, three times with acetone and finally was dried at room temperature for 24 h. These supports are named NAS (amine-functionalized amorphous silica) and OAS (octyl-functionalized amorphous silica).
Characterization of supports
Mesoscopic order was investigated by low-angle X-ray diffraction (XRD) using a PANalytical X'Pert diffractometer with Cu Kα radiation.Nitrogen adsorption–desorption isotherms were measured at −196 °C using two Micromeritics sorptometers (ASAP 2020 and ASAP 2420) to determine textural properties. Pure silicas were pretreated at 350 °C for 16 h and the functionalized supports, at 120 °C for 16 h. The specific surface area, SBET, was calculated from nitrogen adsorption data in the relative pressure range from 0.04 to 0.2 using the Brunauer–Emmet–Teller (BET) method.42 The total pore volume, Vp, was determined from the amount adsorbed at a relative pressure P/Po of 0.97.42 Pore size distributions were determined from the adsorption branch of the isotherms using the Barrett–Joyner–Halenda (BJH) model with cylindrical geometry of pores. The BJH pore diameter, Dp BJH, is defined as the position of the maximum in the pore size distribution.
Quantitative determination of the nitrogen content of amino-functionalized supports (NAS, PMA and E-PMA) was performed using a LECO CHNS-932 Elemental Analyser with a Perkin Elmer AD-4 autobalance.
Thermogravimetric analyses of the supports were carried out using a Perkin Elmer TGA 7 instrument. Samples were heated under synthetic air flow (60 mL min−1) from 25 to 900 °C at a rate of 20 °C min−1.
Transmission electron micrographs (TEM) were taken using a JEOL 2100 electron microscope operating at 200 kV. The samples for TEM analysis were prepared by suspending a small amount of solid in acetone by sonication in an ultrasonic water bath for 10 min. A drop of this suspension was then poured onto a copper grid coated with a holey carbon film and the solvent allowed to evaporate at room temperature.
Protein determination
The crystal structure of Candida antarctica lipase B, CaLB (PDB: 1TCA)44 was taken from the Protein Data Bank.45 CaLB has a molecular weight of 33 KDa and an isoelectric point (pI) of 6.0. CaLB is a globular α/β type protein with approximate dimensions of 3 nm × 4 nm × 5 nm.Bioinformatic analysis was used to determine the aminoacid sequence and 3D structure of Myceliophthora thermophila laccase (MtL) because the crystal structure is not resolved. The amino acid sequence of MtL was taken from the NCBI Protein Database (accession number AEO 58496.1).46 This amino acid sequence was used as a template to identify homologous sequences of the laccase in BLASTP algorithm.47 The protein BLAST analysis for MtL showed that it shares high identity (73%) and query coverage (99%) with the Melanocarpus albomyces laccase (PDB: 1GWO-A).45,48
The 3D structure of MtL was modelled applying the alignment mode on the Swiss model server47,49–51 using the three-dimensional structure of the Melanocarpus albomyces laccase. The modelled MtL structure has been visualized using Pymol software.52 The molecular analysis of the whole protein using the tools ProtParam,53 UniProt KB (G2QFD0 and G2Q560)54 and Brenda55 showed that it has a molecular weight between 63 and 80 KDa. The predicted isoelectric point (pI) was found to be 4.2. These data are in agreement with those reported by other authors.56,57 MtL has approximate dimensions of 6.3 nm × 7.2 nm × 8.9 nm.
Protein content of solutions was determined with the Bio-Rad Protein Assay (Bio-Rad, USA), based on the Bradford assay,37 using bovine serum albumin (BSA) as protein standard. The commercial extracts of laccase and lipase were found to contain a protein concentration of 3.3 mg mL−1 and 3.0 mg mL−1, respectively.
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed for identification of the enzymes and determination of the purity of the commercial extracts.58
Proteins structural changes by organic solvents were evaluated spectrophotometrically by the changes in the UV-Vis spectra of enzyme solutions with different solvent concentration: laccase in 10% ethanol and 50 mM sodium dihydrogenphosphate/disodium hydrogenphosphate buffer pH 7.0, and lipase in 50% methanol and 50 mM sodium dihydrogenphosphate/disodium hydrogenphosphate buffer pH 7.0 compared to the spectra registered by identical enzyme concentrations in their respective buffers.
Enzymes immobilization
Lipase immobilization was carried out according to a protocol previously reported.41,59,60 Enzyme solutions of different concentrations were prepared in buffered solutions to carry out immobilization at selected pH values. The buffer solutions used and their corresponding pH were: 50 mM glycine/hydrochloric acid (pH 3.5), 50 mM sodium acetate/acetic acid (pH 5.0), 50 mM potassium dihydrogen phosphate/disodium hydrogen phosphate (pH 7.0) and 50 mM sodium carbonate/sodium bicarbonate (pH 9.0). To carry out the immobilization, 100 mg of the support were impregnated with 0.5 mL ethanol (to facilitate its dispersion in aqueous solutions, due to the hydrophobic character of supports), and added to 10 mL enzyme solution. The suspension was kept under gentle stirring at room temperature. Aliquots of the suspension and supernatant were withdrawn at different times and assayed for catalytic activity measurement (pNPA test). The activity of a control enzyme solution was used as reference to evaluate the degree of enzyme immobilization with time. The time at which the residual activity of the supernatant reached a constant value was taken as the end of the immobilization process. The suspensions were then filtered, washed with 200 mM of the respective buffer at the same pH as used for immobilization and subsequently with acetone and allowed to dry at room temperature. High concentration of buffers was preferred for washing in order to preserve hydrophobic interactions and thus preventing enzyme leaching in this step. Dried biocatalysts were stored at 4 °C. Activity of the supported biocatalysts was determined by tributyrin hydrolysis assay. Suspensions with a lipase to support weight ratio between 20 and 600 mg g−1 were used to obtain adsorption isotherms (see Supplementary Information Fig. S1a†) and determine the maximum enzyme loading capacity of the support under the best conditions of immobilization.Laccase immobilization on the different supports was carried out at pH 5.5 (and also pH 6.0 for E-PMA) in 50 mM acetic acid/sodium acetate buffer solutions containing different enzyme concentrations. To the enzyme solution, 50 mg of the support were added and kept in suspension under mild stirring at room temperature. Aliquots were withdrawn at given times and the enzymatic activities of suspension and supernatant were assayed (ABTS assay). The decrease of the supernatant activity to a minimum and constant value indicated the end point of the immobilization process. At this point, the solids were filtered off and washed with acetate buffer (50 mM at the same pH as used for immobilization). The solid samples were first dried under vacuum and then under nitrogen stream and stored at 4 °C for later analysis. Immobilizations were performed using different enzyme to support weight ratios in the range between 25 and 350 mg g−1 in order to determine adsorption isotherms and the maximum loading capacity for all supports (see Fig. S1b† for enzyme adsorption isotherms). To determine the catalytic activity of the supported biocatalysts, 10 mg of the solid were suspended in 1 mL of 50 mM acetate buffer (at the same pH as used for immobilization) and assayed in the ABTS oxidation test.
Enzyme leaching study
The resistance to enzyme leakage from the supports was studied under conditions that presumably favour the release of the protein, namely high dilution and low ionic strength. Lipase catalysts were suspended in 50 mM potassium dihydrogen phosphate/disodium hydrogen phosphate at pH 7.0. The laccase catalysts were incubated in 50 mM acetic acid/sodium acetate buffer at pH 4.5. Suspensions were prepared with 1.25 mg of solid per mL of buffer solution and were incubated at 25 °C. Enzyme leaching was calculated at different incubation times by measuring the amount of enzyme present in the supernatant using the Bradford assay.37 After the leaching treatment, the solids were separated by filtration, suspended in electrophoresis sample buffer (containing SDS, mercaptoethanol, bromophenol blue, Tris buffer pH 6.8 and glycerol) and boiled for 5 min. After this treatment, proteins trapped in the solids would be denatured and the lineal polypeptide chains should be easily released from the pores. The supernatants of these suspensions were withdrawn and analysed by SDS-PAGE electrophoresis.Enzyme activity assays
Routine laccase activity tests were carried out spectrophotometrically by measuring the increase in absorbance at 405 nm caused by the oxidation of ABTS.43 The reaction mixture consisted of a 1.6 mM ABTS solution in 100 mM acetic acid/sodium acetate buffer at pH 4.5. To 1.9 mL of this solution in the cuvette, 50 μL of enzyme solution or suspension were added under stirring and the reaction was monitored continuously at 25 °C for 30 min. One unit of laccase activity (UABTS) was defined as the amount of enzyme required to oxidize 1 μmol of ABTS per minute at 25 °C (the molar absorption coefficient of oxidized ABTS at 405 nm was taken as ε405 nm = 35000 M−1 cm−1).
In order to perform enzyme spectrophotometric assays at different pH values, the isosbestic point of oxidized ABTS (ABTS*) was determined and established at 430 nm, and the molar absorption coefficient of ABTS* was calculated (ε430 nm = 20700 M−1 cm−1). The activities of the free and immobilized laccases (on PMA and NAS) were determined at this wavelength as a function of pH, in 50 mM phosphoric acid/sodium dihydrogenphosphate (pH 2.5) and 50 mM citric acid/trisodium citrate (pH 3.0 to 6.0) buffer solutions at 25 °C (triplicated).
Hydrolysis of p-NPA was used as a routine test to measure the nonspecific esterase activity for monitoring the immobilization of lipase. An aliquot of 50 μL of enzyme solution (control or supernatant) or immobilized enzyme suspension was added to a cuvette with 1.9 mL of 0.4 mM p-NPA aqueous solution (pH 7.0). The activity was determined at 25 °C by measuring the rate of increase of absorbance at 348 nm due to the release of p-nitrophenol (the molar absorption coefficient was taken as ε348 nm = 5150 M−1 cm−1).
Spectrophotometric assays of free enzyme and immobilized enzyme suspensions were performed using an Agilent 8453 UV-Vis spectrophotometer equipped with a stirring device and temperature control.
Lipase biocatalysts stability tests were performed using the hydrolysis of tributyrin as test reaction to evaluate hydrolytic activity of lipase. The activity was determined by titration of the butyric acid released by the hydrolysis of tributyrin. 1.47 mL of tributyrin were added under stirring to 48.5 mL of 10 mM potassium phosphate buffer at pH 7.0 in a thermostated titration vessel at 25 °C. Once equilibrated at pH 7.0, a weighted amount of biocatalyst was added, and the rate of addition of a 0.1 M NaOH solution required to neutralize the acid product and maintain a constant pH of 7.0 was measured. One unit of activity (UTB) was defined as the amount of lipase converting 1 μmol of tributyrin per minute.
Titrimetric determination of lipase activity was performed using a Mettler Toledo DL-50 pH-state.
Stability tests
The thermal stability of free and immobilized enzymes was determined by incubation at 55 °C in 50 mM potassium dihydrogen phosphate/disodium hydrogen phosphate buffer at pH 7.0 (for lipase) and at 60 °C in 50 mM acetic acid/sodium acetate buffer at pH 5.5 (for laccase).Stability of free and immobilized enzymes in organic solvents was evaluated by incubating samples at 25 °C in 50% v/v methanol/water and pure methanol (lipase samples) or 10% v/v ethanol at pH 3.5 (laccase samples).
Incubations were performed in different vials for each aliquot to prevent solvent evaporation during sampling. Aliquots were withdrawn at different times, cooled down and their activities were assayed in the hydrolysis of tributyrin (lipase) or in the oxidation of ABTS (laccase).
Results
Characterization of the supports
TEM images of solvent-extracted hybrid organosilicas (Fig. 1) show large domains with two-dimensional hexagonal arrangements of parallel channels of uniform size. This is consistent with X-ray diffraction (XRD) patterns (see Supplementary Information Fig. S2†), which exhibit an intense reflection and two relatively well resolved weak peaks in the low angle region (0.5–2°), that can be indexed as the 100, 110 and 200 reflections of p6mm hexagonal symmetry. These patterns indicate that the samples possess high degree of mesostructural order. In contrast with the hybrid organosilica supports, the XRD pattern of the commercial amorphous silica (not shown) does not exhibit any diffraction peak at low angle, evidencing the lack of mesoscopic order and, hence, the non-uniform pore structure of this material. | ||
| Fig. 1 TEM micrographs of PMO (a and b), PMA (c and d) and E-PMA (e and f). | ||
Fig. 2a shows nitrogen adsorption–desorption isotherms and the corresponding pore size distributions calculated by the BJH method for the solvent-extracted hybrid aminosilica samples. The isotherms are type IV, with H1 type hysteresis loop at high relative pressure, which is characteristic of mesoporous materials. The calculated pore size distributions (Fig. 2a, inset) indicate that the three samples possess uniform mesopores, in agreement with the regular pore structure shown by XRD and TEM.
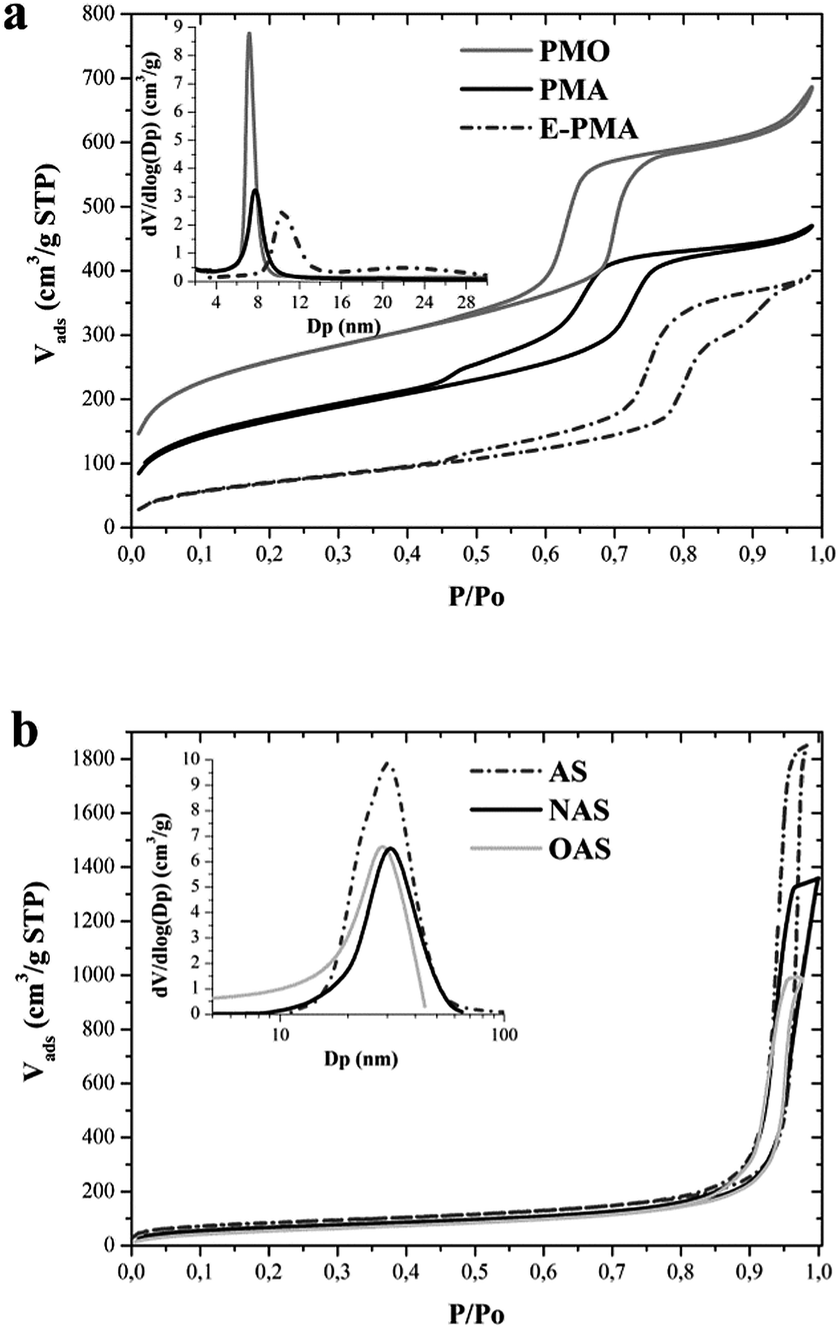 | ||
| Fig. 2 Nitrogen adsorption isotherms and pore size distributions of: (a) hybrid organosilica supports and (b) parent and functionalized amorphous silica supports. | ||
The diameter of pore channels estimated by the BJH method is reported in Table 1. The pore size estimated for PMO and PMA supports is around 7 nm. This value increases up to more than 10 nm when the synthesis of hybrid aminosilica was carried out using the swelling agent (sample E-PMA). In the case of sample E-PMA the nitrogen isotherm (Fig. 2a) shows that, besides the main steep nitrogen uptake at a relative pressure around 0.8, corresponding to the regular mesopore channels, an additional increase of nitrogen adsorption occurs at relative pressures above 0.9. This is also apparent in the pore size distribution (Fig. 2a, inset) that evidences the presence of secondary mesoporosity, with a relatively broad pore size distribution ranging from 15 to 30 nm, which can be attributed to interparticle spaces. The three samples possess high surface area and pore volume (Table 1), although these parameters differ significantly among the three samples. Taking into account the unit cell parameters (a0) calculated from the XRD patterns (11.6, 12.4 and 15.9 for PMO, PMA and E-PMA, respectively) and the diameter of the pore channels determined from the nitrogen isotherms (Table 1), it can be estimated that the pore wall thickness of PMO, PMA and E-PMA is 4.5, 5.2 and 5.5 nm, respectively. Therefore, the differences in textural properties might be attributed to the different pore size and pore wall thickness of the three samples.
| Material | Dp BJHa (nm) | SBETb (m2 g−1) | Vpc (cm3 g−1) | mmol C g−1 SiO2d | mmol N g−1 SiO2e |
|---|---|---|---|---|---|
| a BJH pore diameter (nm).b BET surface area (m2 g−1).c Total pore volume (cm3 g−1).d Hydrophobic (aliphatic carbon) groups content determined from TG analysis (weight loss in the temperature range 150–600 °C).e Concentration of amine groups determined by chemical analysis. | |||||
| AS | 30 | 285 | 2.2 | — | — |
| OAS | 30 | 212 | 1.6 | 4.2 | — |
| NAS | 30 | 236 | 2.1 | — | 1.8 |
| PMO | 7.1 | 882 | 1.0 | 16.1 | — |
| PMA | 7.2 | 594 | 0.7 | — | 1.5 |
| E-PMA | 10.4 | 264 | 0.6 | — | 1 |
Fig. 2b shows the N2 adsorption–desorption isotherms of the parent (AS) and functionalized amorphous silica supports (OAS and NAS). These supports show type IV isotherms corresponding to mesoporous materials.29,41 In contrast with hybrid organosilicas, the commercial amorphous silica has a wide pore size distribution, indicating heterogeneity in the porous structure, and larger pores, with the maximum of the pore size distribution around 30 nm (Fig. 2b, inset). Functionalization of the AS support has a negligible effect on pore size (Fig. 2b, inset and Table 1), as can be expected due to the large pore size. Nevertheless, grafting with aminopropyl or octyl groups on the silica surface produces a small decrease of both surface area and pore volume (Table 1).
Thermogravimetric analysis (TGA) profiles show the incorporation of organic groups in functionalized silica and hybrid organosilica supports (Fig. S3†). All materials show a small weight loss at temperatures below 150 °C that can be attributed to desorption of water or residual organic solvents. The main weight loss occurs in the temperature range 150–600 °C, which corresponds to decomposition and combustion of the organic groups. At temperatures higher than 600 °C a further small weight loss is observed that that might be attributed to dehydroxylation of the silica surface. Therefore, the amount of hydrophobic groups (octyl chains in sample OAS and ethylene bridges in sample PMO) have been calculated from the weight loss in the temperature range 150–600 °C. The results, expressed as mmol C per gram of silica are given in Table 1. It can be seen that the total amount of carbon of the hybrid organosilica material is around four times that of the amorphous silica functionalized with octyl groups. In the case of supports containing amino groups (PMA and E-PMA), the weight loss assigned to removal of organic groups corresponds to both aminodipropyl and ethylene bridges. Therefore, the concentration of amino groups was determined by chemical analysis. All the supports obtained contain a relatively high amount of amine, ranging from 1 to 1.8 mmol per gram of silica (Table 1).
Lipase immobilization on PMO at different pH values
The loading capacity of PMO for lipase immobilization was determined at pH 5.0 by the adsorption isotherm at room temperature (ESI, Fig. S1†). Maximum enzyme loading remained nearly constant (about 90 mg enzyme per gram of support) at a content of enzyme in the liquid phase over 100 mg g−1. This can be attributed to the relatively small pore size that would prevent multilayer adsorption of enzyme. In contrast, on the octyl-functionalized amorphous silica support OAS, lipase loadings up to 400 mg enzyme per gram of support were obtained29 due to its larger pore size. However, this high enzyme loading leads to lower catalytic efficiency (90 UTB per mg of enzyme, compared to 200 UTB/mg for the PMO-supported catalyst29). This is probably due to the intense interaction with the highly hydrophobic octyl groups. Despite each single interaction is mild, the high content in ethylene groups and the high contact surface in PMO due to the confinement of enzyme within the pore, makes the overall interaction intense. But the mild interactions permit the catalyst to preserve high catalytic activity.29Suspensions with 90 mg enzyme per gram of support were prepared to evaluate the effect of pH on lipase immobilization on PMO. The maximum amount of lipase slightly decreased as the pH increased from 3.5 to 9.0 (Table 2). In the whole pH range used for lipase immobilization, silanol groups present on the support should be deprotonated, as the point of zero charge (pzc) of silica is around 2. Taking into account that the isoelectric point (pI) of lipase is around 6.0, electrostatic attractions or repulsions may also be established with the remaining siloxane groups on the surface of PMO22 when the immobilization is performed at pH 3.5–5.0 or pH 7.0–9.0 respectively. Although maximal loading was achieved at pH 3.5 and 5.0, as expected, the values are rather close indicating that the driving forces of the immobilization are hydrophobic interactions.
| pH | tca (h) | Max. loadb (mg g−1) | Biocatalyst activityc (UTB/g) | Cat. eff.d (UTB/mg) |
|---|---|---|---|---|
| a Time at which the maximum loading is achieved.b Maximum enzymatic loading, expressed in milligrams of lipase per gram of support.c Biocatalyst activity expressed in UTB per g of support.d Catalytic efficiency expressed in UTB per mg of lipase. | ||||
| 3.5 | 1.25 | 77.05 | 2377.8 | 30.9 |
| 5 | 1.25 | 70.18 | 4020 | 57.3 |
| 7 | 1.50 | 69.85 | 3958 | 56.7 |
| 9 | 1.75 | 49.14 | 1952.4 | 39.7 |
Laccase immobilization on PMA
The low isoelectric point of laccase (4.2) permits a wide pH range for electrostatic interactions between negatively charged enzyme and positively charged amine groups of the hybrid aminosilica supports (pKa around 11.0). Thus, immobilization was tested at pH 5.5. Because of the similar size between the dimensions of laccase (6.3 × 7.2 × 8.9) and the pore diameter of PMA (7.2 nm), maximal loading in this support is low, as well as activity and efficiency of the biocatalyst (Table 3). The enzyme molecules are probably absorbed only onto the external surface of PMA particles.| Support | pHa | tcb (h) | Max. loadc (mg g−1) | Biocatalyst activityd (UABTS/g) | Cat. eff.e (UABTS/mg) |
|---|---|---|---|---|---|
| a pH of immobilization.b Time at which the maximum loading is achieved.c Maximum enzymatic loading, expressed in milligrams of laccase per gram of supported biocatalyst.d Biocatalyst activity expressed in UABTS per g.e Catalytic efficiency expressed in UABTS per mg of lipase. | |||||
| NAS | 5.5 | 24 | 187 | 170 | 0.91 |
| PMA | 5.5 | 2.0 | 42 | 4.7 | 0.11 |
| E-PMA | 5.5 | 1.5 | 88 | 29 | 0.33 |
| E-PMA | 6.0 | 1.3 | 119 | 19 | 0.16 |
Loading capacity of our expanded pore material E-PMA with 10.2 nm pore diameter was tested. Table 3 shows the loading capacities of the three amine-coated supports. The rise in pore size enabled a twofold increase in the enzyme loading of E-PMA compared to PMA. Also, amine-functionalized amorphous silica was tested for comparative purposes. Similarly to lipase immobilization, the highest laccase loading was achieved in amorphous silica with much larger pore size. However, it is worth noting that adsorption equilibrium was reached more rapidly for the supports with uniform pore size.
It was also observed that increasing the pH of laccase solution to 6.0 led to a faster adsorption and higher enzyme loading on E-PMA: 119 mg g−1 vs. 88.0 mg g−1 at pH 5.5. However the catalytic efficiency was significantly decreased at pH 6.0.
Fig. 3 shows the pH/activity profiles of laccase in soluble and immobilized estates. It can be observed that for laccase supported on NAS, the optimum pH was 0.5 units higher than that of soluble laccase. This result might be explained assuming that amino groups incorporated into materials generate a new environment which may result in a pH gradient from inside the particle towards the external aqueous medium in which catalyst particles are suspended. This trend had been previously found with expanded-pore SBA-15 materials functionalized with primary amine groups, where optimum pH was shifted 0.5 pH units, as well as NAS.33 The shift of optimum pH underwent by laccase immobilized on E-PMA was much more pronounced: from pH 3.0 to pH 5.0. As noted, laccase immobilized in the supports showed higher activity than the free enzyme in the pH range 4.0–6.0. This effect can be attributed to the microenviromental conditions61 in this material, with secondary amines in the close vicinity of enzyme.
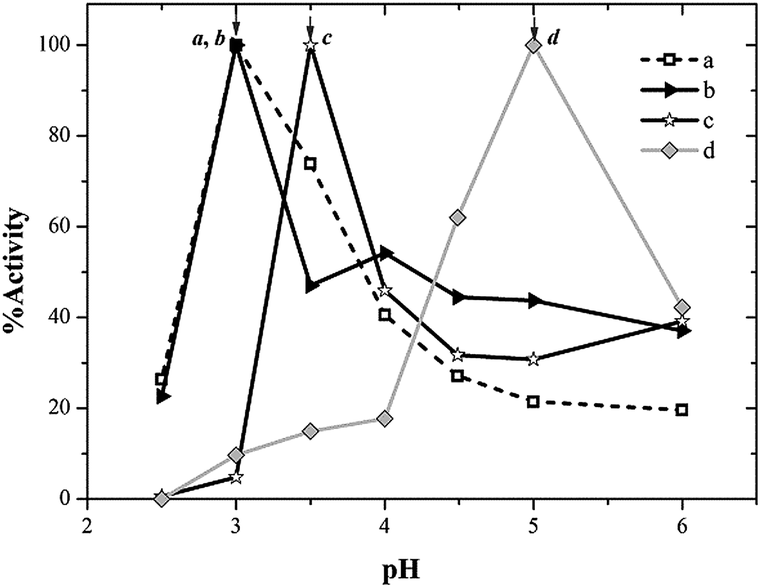 | ||
| Fig. 3 Effect of pH on the activity of (a) free enzyme, (b) AS, (c) NAS and (d) E-PMA. | ||
Leaching and electrophoresis
In former works we had determined the amount of lipase leached from OAS and PMO after two hour incubation under conditions of charge repulsion and high dilution, where the release of the enzyme is favoured and expected.29 We present here a 24 h time course of enzyme leaching (Fig. 4a). Lipase in PMO only undergoes a 15·% initial leaching that may be due to removal of enzyme molecules immobilized on the outer surface of PMO particles, and then no more leaching was detected. Leaching of lipase from OAS is higher and growing with incubation time.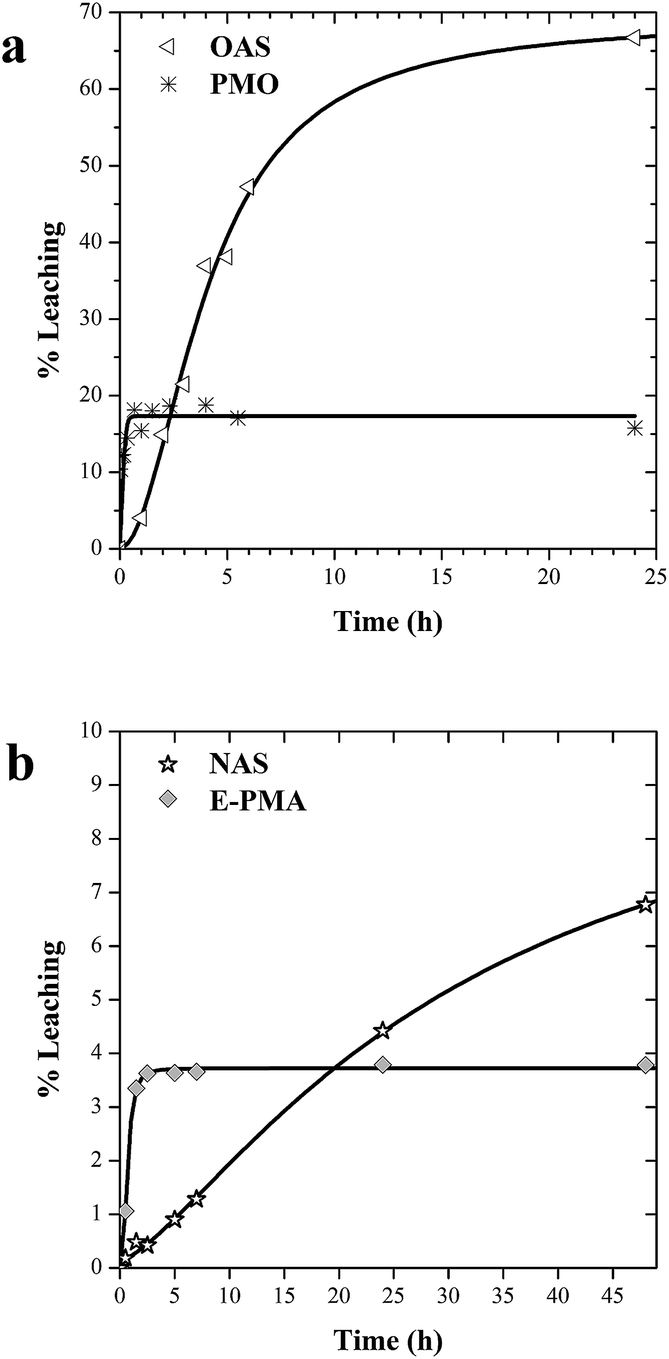 | ||
| Fig. 4 (a) Leaching of lipase from ordered material (PMO) and amorphous mesoporous silica (OAS). (b) Leaching of laccase from ordered material (E-PMA) and amine amorphous silica (NAS). Expressed as percent of the initial enzyme leached on each material as a function of time. | ||
Lipase tightly fitting into pores of PMO and fixed by ethylene groups is highly retained. In contrast, stronger interaction with octyl groups of OAS does not prevent enzyme from leaching through the 30 nm with pores of amorphous silica. These results seem to suggest that it both, chemical affinity and confinement in the pore contribute to minimize enzyme leaching.
The same trend is observed for laccase leaching from catalysts (Fig. 4b). NAS catalyst shows a continuous release of enzyme. Again, the affinity of laccase with amine groups is not enough to prevent leaching from pores with an average diameter of around 30 nm, with no diffusion restrictions. As shown in Fig. 4b, leaching profile in E-PMA showed also initial desorption, probably due to enzyme molecules immobilized on external surfaces. But percent values of laccase leached are much lower than in the case of lipase (4%). The uniform pore size close to the enzyme dimensions along with the increased affinity of the laccase for secondary amine groups make this material more efficient to retain the enzyme inside the inner surfaces.
An attempt to verify the location of enzymes inside the porous network of the materials was made. Since solid samples of the biocatalysts are not suitable for electrophoresis, the enzymes were forced to exit the pores. First, biocatalysts were suspended as described above for leaching tests to desorb as much enzyme as possible, especially to eliminate all protein molecules adsorbed on the external surface of support particles. Then the supernatants were removed and the filtered solids were boiled in sample electrophoresis buffer. In such denaturing conditions including the split of disulphide bonds, the tertiary structure of the protein should be lost and the random coil chain should then be easily released from the pores. However, no protein band could be seen from the E-PMA supernatant electrophoresis (Fig. S4a†). Then, E-PMA samples after the same SDS-PAGE treatment were centrifuged and suspended at pH 11 and 14 respectively and the supernatants were analysed again. Protein band only appeared in the supernatant of the sample suspended at pH 14.0. These results confirm the presence of the enzyme inside the pores, in agreement with previous results based on advanced TEM techniques62 and evidence the strong binding of the enzyme to the inner surface of the support.
The results obtained with lipase catalysts are shown in Fig. S4c.† In this case no further incubation was necessary, and the band corresponding to the immobilized lipase appeared from both, PMO and OAS.
Thermal stability
The thermal stability of enzymes catalysts is important for some industrial applications63 because bioreactors are sometimes operated at elevated temperatures to improve productivity and to avoid microbial contamination.Free and immobilized lipases were incubated in buffer at 55 °C and their inactivation courses are shown in Fig. 5. Lipase on PMO was inactivated at a faster rate than free enzyme, while maximal stabilization was achieved in OAS.
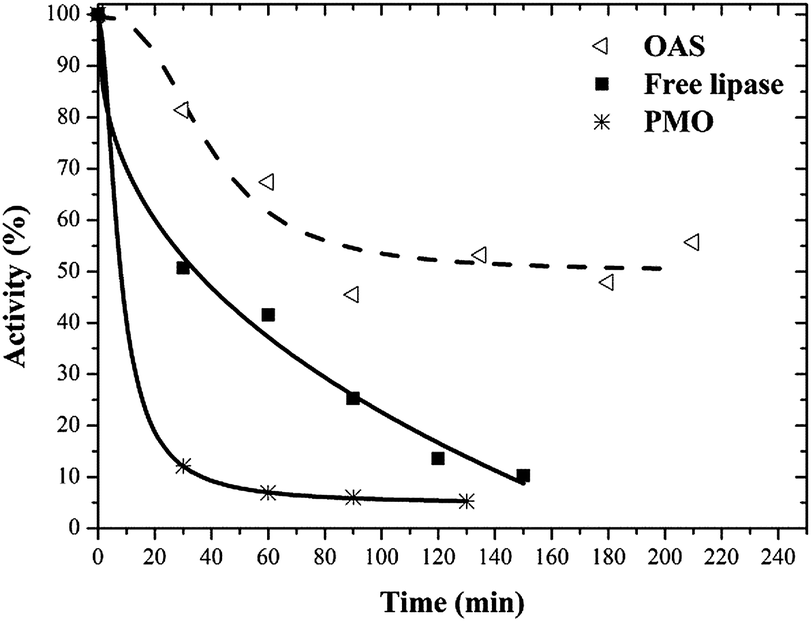 | ||
| Fig. 5 Thermostability profiles at 55 °C of free and immobilized lipase on PMO and octyl amorphous silica (OAS). | ||
Thermal stabilities of free and immobilized laccase on NAS and E-PMA materials were evaluated at 60 °C in the same way. Again, a faster inactivation rate was underwent by laccase in E-PMA and immobilization in NAS resulted in enzyme stabilization (Fig. 6).
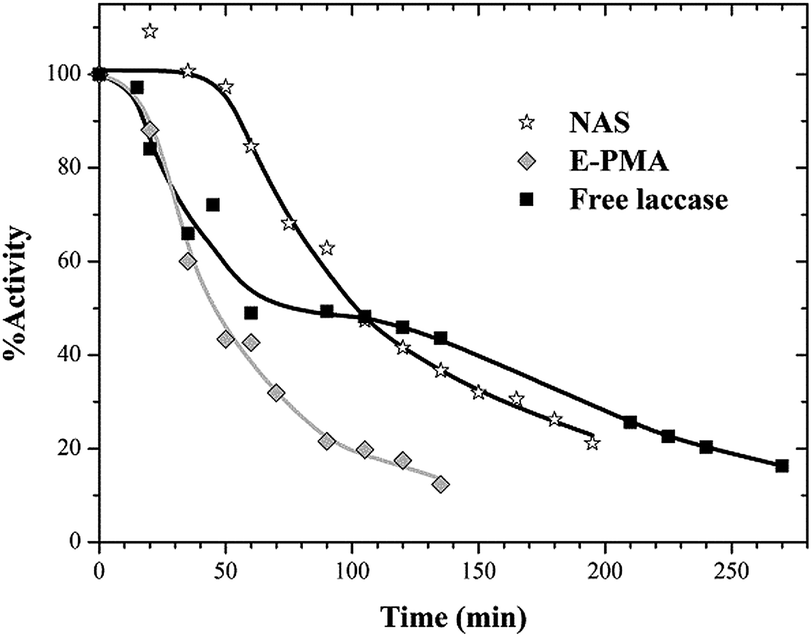 | ||
| Fig. 6 Thermostability profiles at 60 °C of free and immobilized laccase. Runs were performed at pH 5.5. | ||
Stability in organic solvents
Lipase can be used in the synthesis of biodiesel to catalyse both, the hydrolysis of triacylglycerols to release fatty acids and the methanolysis of these fatty acids.64–67 The biocatalytic pathway is attractive due to the easier purification of products and environmental advantages.8,68 However, the necessary presence of methanol involves severe losses of enzymatic activity because of structural and inhibitory damages on the enzyme.66,69 This solvent was chosen to study the structural protection that PMO may provide.PMO-lipase incubated in 50% methanol remained fully active after 24 hours, although the stability of the soluble enzyme was also high, as well as the one on OAS (Fig. 7a). Severe effects on the activity were registered by incubation in 100% methanol, where soluble lipase and OAS-lipase kept only 25% and 32% residual activity after 1 hour respectively. In the same conditions PMO-lipase still kept 75% activity (Fig. 7b). But the activity was almost completely lost at longer incubation time for the three samples, probably because of inhibitory effect of methanol.
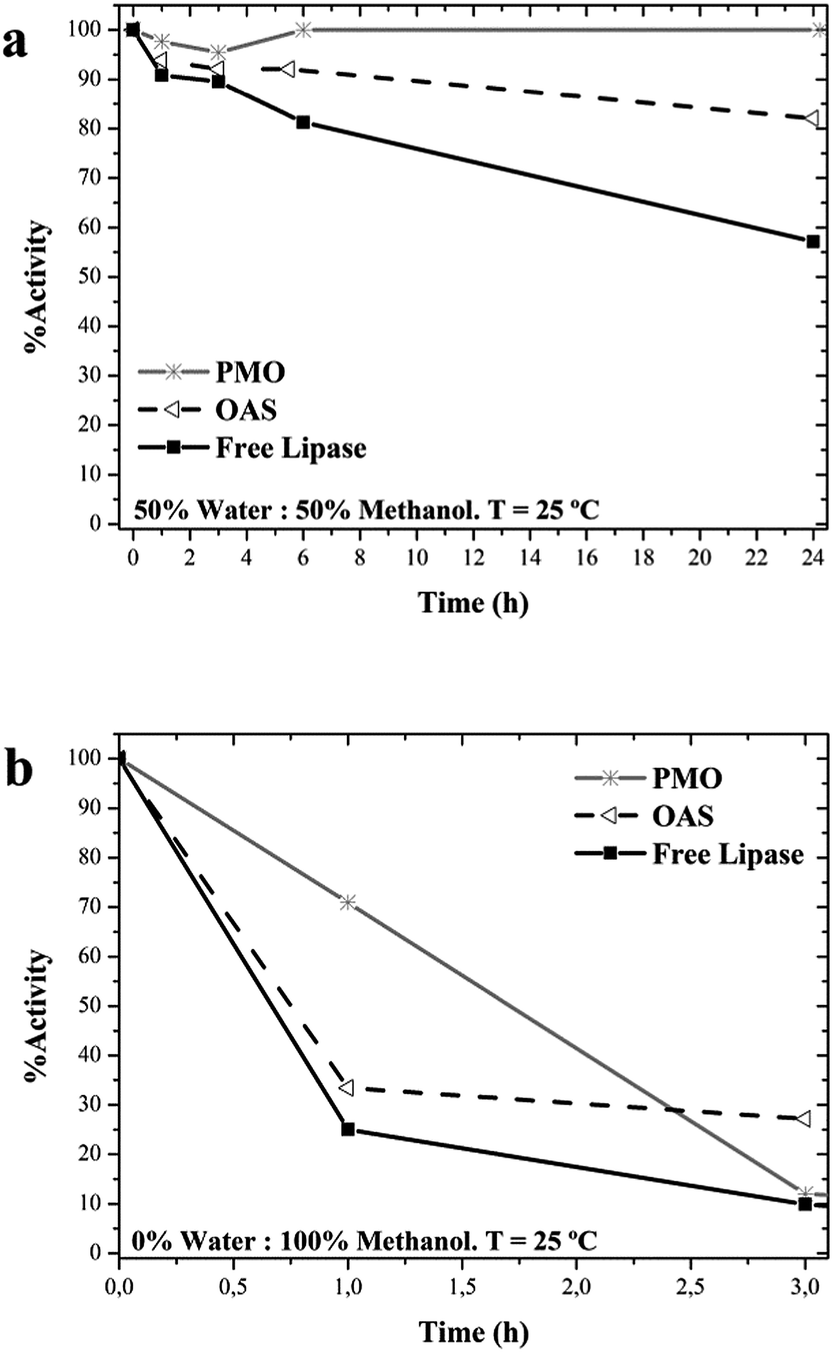 | ||
| Fig. 7 Stability of lipase in solution and supported on OAS and PMO in: (a) 50% aqueous solution: 50% methanol; (b) 0% aqueous solution: 100% methanol. Runs were performed at 25 °C. | ||
Laccase has been used to partially oxidize wine polyphenols.70–72 Therefore, in order to evaluate the potential application of PMA-laccase biocatalysts in wine stabilization, the stability of biocatalysts was tested by incubation in a low ethanol concentration. Free laccase, NAS and E-PMA were incubated in a medium similar to wine: 10% ethanol and acidic pH (Fig. 8). Inactivation was faster in the native enzyme, while NAS preserved around 85% activity after 6 hours incubation and laccase immobilized on PMA remained fully active during the same period of time.
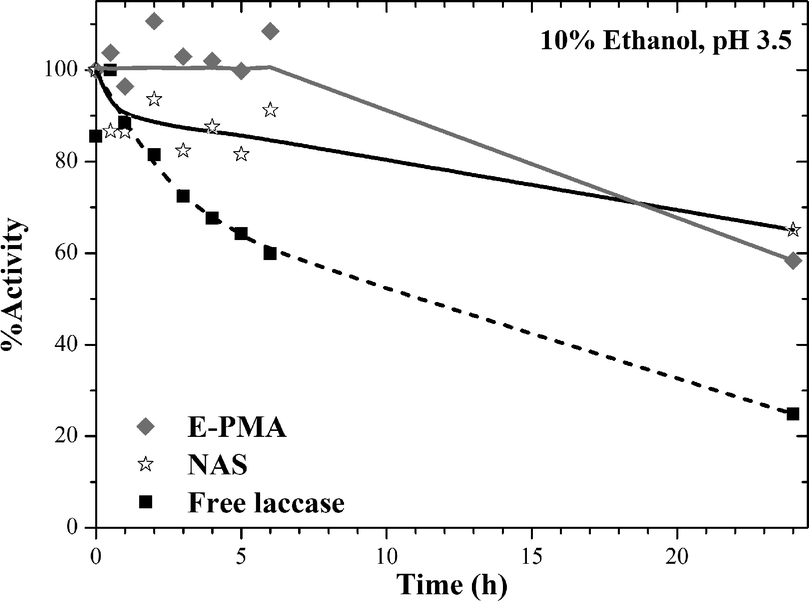 | ||
| Fig. 8 Stability of free and immobilized laccase in ethanol : water solution at acidic pH. Runs were performed at 25 °C. | ||
Fig. 9 shows the UV-Vis spectra of soluble enzymes: lipase in buffer and 50% methanol (a) and laccase in buffer and 10% ethanol (b). Proteins absorbance at 280 nm is due to the presence of aromatic side chain of aminoacids, especially tryptophan. Spectra of the enzyme in the presence of cosolvents show higher peaks of absorbance at 280 nm, which suggests a higher amount of these aromatic aminoacids on the surface of the protein.
 | ||
| Fig. 9 UV-Vis spectra of lipase in aqueous buffer and 50% methanol (a) and laccase in aqueous buffer and 10% ethanol (b). | ||
Discussion
The main characteristics of periodic mesoporous organosilica as support for enzyme immobilization are the confinement and the presence of chemical functionalities. These chemical groups are not anchored on the internal surface of pores but become part of it. These two features mean that there is close vicinity between the side chain groups of the enzyme and the functional groups of the support with no steric hindrances. Apart from the specific organic moieties introduced in the synthesis of the PMO (ethylene bridges) and PMA (dipropylamine and also ethylene bridges), silanol groups are present on the surface of silica that can also play their part.The same loading of lipase on PMO was achieved at pH 5.0 and 7.0 despite repulsion should be established at the higher pH between negative charges on both, enzyme and support. What this probably means is that hydrophobic interactions are the driving forces of the process, over electrostatic ones. The only difference is found in the contact time: immobilization is faster at pH 5.0 than at pH 7.0. Also, the highest values of catalytic efficiency were found at pH 5.0 and 7.0. At pH 3.5 higher loading was achieved as a result of the additional electrostatic attraction (the enzyme should have a net positive charge while the support is negatively charged). Nevertheless, the catalytic efficiency was lower probably because an excessive interaction is established with a distortional effect on the protein structure.
The immobilization of laccase showed higher values of enzyme loading and catalytic efficiency on the amorphous and wide pore support NAS. Although this amine-functionalized amorphous silica has lower surface area than E-PMA, the available surface in E-PMA is much lower because only one molecule of enzyme occupies the whole pore section, as the pore size is close to enzyme dimensions. Thus, the maximal capacity for enzyme immobilization determined for NAS was around four times higher than on E-PMA (Table 3). The intensity of the interaction with laccase is not identical for both supports since there are primary propylamine groups (pKa around 10) in NAS and secondary dipropylamine groups in E-PMA (pKa around 11). The interaction of negative charges of enzyme with these high pKa should be more intense and this is probably the reason why the catalytic efficiency is lower. As discussed for lipase-PMO, the stronger interaction may distort protein structure. This may also explain that immobilization at pH 6.0 displays higher enzyme loading and lower catalytic efficiency than at pH 5.0, because of the higher density of negative charges on the enzyme.
This high pKa in E-PMA is also responsible for a higher density of positive charges at the same pH, and consequently a higher pH gradient between the inner pores of the particles and the external medium. As a consequence, there is a large optimum pH shifting in the activity vs. pH profile, and it is likely that the immobilization inside the particle is occurring at a lower pH than measured in the bulk of the solution.
The absence of enzyme leaching from E-PMA samples can also be explained by this property. The protein unfolded under denaturing conditions does not leave the pores at pH below 11, probably because electrostatic attractions remain even in the random coil configuration. Only at pH extremely high (pH 14.0) all amino groups from support should be deprotonated and repulsive forces would be driving the release of protein from pore channels. These experiments reveal that leaching only occurs in very harsh conditions, showing that this non-covalent immobilization is nearly as irreversible as a covalent one.
Unfolding of the protein is promoted by both, high temperature and organic solvents. These conformational changes may drive the formation of new interactions of side chain amino acids with the support, but the processes do not occur identically.
At high temperature the close vicinity of amines (or ethylenes) and silanol groups promotes abundant new interactions with amino acids exposed upon partial unfolding of the peptide chain. In our experiments, incubation of the catalysts suspensions or enzyme solutions were performed at high temperatures, and cooled down to the assay temperature (25 °C for lipase and laccase). The fact that in both cases: PMO-lipase and E-PMA-laccase resulted the least stable ones may be related to confinement. Probably the interactions established between the enzyme structure, partially unfolded upon heating and the “close-fitting-around” support, are preserved after cooling. Thus, refolding to a more active conformation is prevented and the activity would be irreversibly lost in PMO/E-PMA biocatalysts. Neither enzymes in the wide-pore amorphous silica (OAS, NAS) nor the free enzymes have this restriction from the support, and this absence of steric hindrance would still permit some margin for reversibility. As a consequence the enzymes can partially recover their native conformation at lower temperatures.
Only the lipases naturally contain a hydrophobic domain on their surface. With this exception, hydrophobic side chains of aminoacids are usually placed inside the protein globule, avoiding contact with aqueous medium. Cosolvents miscible in water are known to interfere hydrogen bonds and to unfold proteins, thus forcing some hydrophobic amino acids to arise to the external surface. Spectra in aqueous and organic media of both lipase and laccase (Fig. 9) show a moderate higher absorbance at 280 nm in the organic solvents, which is usually attributed to tryptophan residues (hydrophobic) and confirm this effect of the solvents on both enzymes. PMO and E-PMA surfaces are contain ethylene bridges capable to establish new interactions with these arising hydrophobic groups. The stabilization results confirm that PMO and E-PMA are highly efficient to stabilize lipase and laccase versus methanol and ethanol respectively (Fig. 7 and 8). This higher presence of hydrophobic aminoacids on the external protein surface increases the bonding intensity with the close support surface containing ethylene groups. In the case of E-PMA-laccase, new hydrophobic interactions are added to the former electrostatic ones.
Aminoacids involved in unfolding promoted by high temperature and solvents are different, thus the new interactions and the structure of the partially unfolded proteins are also different. If the change does not lead to inactive conformations (as it happens by heating) the result is the stabilization of an active conformation, which becomes strengthened by abundant non-covalent bonds with the support.
The close match of sizes of enzyme and pore channels is the key to obtain this stabilization since the tertiary structure of the protein is physically prevented to keep on unfolding. However, laccase immobilized on ordered mesoporous materials (OMM) with similar structure and pore size bearing aminopropyl groups anchored on the surface had proven less stable than laccase-E-PMA catalyst in the same conditions.33 The difference between both kinds of materials is that the organic moiety becomes part of the wall of the channel pores in the PMO/E-PMA materials, at the same level as silanols. Aminoacids can easily interact with either the negatively charged siloxanes or the ethylene bridges or dipropylamine groups. It is noticeable that in the case of E-PMA interactions can be established with the three of them. In the OMM materials where organic moieties are anchored to the siliceous surfaces, the silanol groups of the wall are not as accessible, as they are partially hindered by the propylamine or alkyl chains. Thus, additional interactions are not easily established.
Conclusions
We have synthetized a novel periodic mesoporous aminosilica with a pore size that matches with the dimensions of the enzyme laccase. The effects of presence of ethylene bridges and dipropylamine groups becoming part of the surface on laccase immobilization and the behaviour of the catalyst have been studied and compared to lipase immobilized on an ethylene-bridged PMO. The PMO materials display some specific properties which enable the immobilization with high enzyme loading while retaining high catalytic activity and providing protection against inactivation in organic solvents. This is possible because these materials integrate organic and inorganic components in a restricted space where the interaction with the enzyme takes place all around the enzyme molecules and the mobility is prevented. Interactions are also feasible with silanol (or siloxane) groups which are not hindered by organic groups since these are also part of the support framework. Moreover, the number of contact points increase in the presence of an organic cosolvent, where additional hydrophobic interactions are established. The highly ordered structure contributes to keep high catalytic efficiency because it favours pore connectivity.Laccase undergoes an especially strong interaction with secondary amines of E-PMA, which fully prevents leaching at pH below 14. This is a relevant result since the non-covalent nature of the linkage permits to recover the support after enzyme inactivation, namely by suspending it at elevated pH. Lipase immobilization on ethylene-bridged periodic mesoporous organosilica (PMO) appeared to be the most promising approach, since it occurred with high efficiency, maintained enzyme activity, and provided enzyme stability.
Acknowledgements
Authors thank the Spanish MINECO for the financial support through the project MAT 2012-31127. We thank D. Ramiro Martinez (Novozymes, Spain) for his kind help with the supply of enzymes. V. G. acknowledges Ministerio de Educación, Cultura y Deporte for a FPU PhD fellowship (AP2010-2145).Notes and references
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844 RSC.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277–6289 RSC.
- E. Serra, A. Mayoral, Y. Sakamoto, R. M. Blanco and I. Diaz, Microporous Mesoporous Mater., 2008, 114, 201–213 CrossRef CAS PubMed.
- M. A. Wahab, I. Kim and C. S. Ha, J. Solid State Chem., 2004, 177, 3439–3447 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS.
- W. J. Hunks and G. A. Ozin, J. Mater. Chem., 2005, 15, 3716–3724 RSC.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- W. P. Guo, J. Y. Park, M. O. Oh, H. W. Jeong, W. J. Cho, I. Kim and C. S. Ha, Chem. Mater., 2003, 15, 2295–2298 CrossRef CAS.
- P. Van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- X. Y. Bao, X. Li and X. S. Zhao, J. Phys. Chem. B, 2006, 110, 2656–2661 CrossRef CAS PubMed.
- R. M. Grudzien, B. E. Grabicka and M. Jaroniec, Colloids Surf., A, 2007, 300, 235–244 CrossRef CAS PubMed.
- M. Mandal and M. Kruk, J. Mater. Chem., 2010, 20, 7506–7516 RSC.
- C. H. Lee, S. S. Park, S. J. Choe and D. H. Park, Microporous Mesoporous Mater., 2001, 46, 257–264 CrossRef CAS.
- X. Y. Bao, X. S. Zhao, X. Li, P. A. Chia and J. Li, J. Phys. Chem. B, 2004, 108, 4684–4689 CrossRef CAS.
- O. Olkhovyk and M. Jaroniec, Ind. Eng. Chem. Res., 2007, 46, 1745–1751 CrossRef CAS.
- W.-H. Zhang, X. Zhang, L. Zhang, F. Schroeder, P. Harish, S. Hermes, J. Shi and R. A. Fischer, J. Mater. Chem., 2007, 17, 4320–4326 RSC.
- E.-B. Cho, D. Kim and M. Jaroniec, Langmuir, 2009, 25, 13258–13263 CrossRef CAS PubMed.
- F. Goethals, B. Meeus, A. Verberckmoes, P. Van Der Voort and I. Van Driessche, J. Mater. Chem., 2010, 20, 1709–1716 RSC.
- L. Xia, Y. Hu, Y. Wu, M. Zhang and M. Rong, J. Sol-Gel Sci. Technol., 2012, 64, 718–727 CrossRef CAS PubMed.
- J. Morell, M. Gungerich, G. Wolter, J. Jiao, M. Hunger, P. J. Klar and M. Froba, J. Mater. Chem., 2006, 16, 2809–2818 RSC.
- X. Zhou, S. Qiao, N. Hao, X. Wang, C. Yu, L. Wang, D. Zhao and G. Q. Lu, Chem. Mater., 2007, 19, 1870–1876 CrossRef CAS.
- S. Z. Qiao, H. Djojoputro, Q. Hu and G. Q. Lu, Prog. Solid State Chem., 2006, 34, 249–256 CrossRef CAS PubMed.
- Z. Zhou, R. N. Klupp Taylor, S. Kullmann, H. Bao and M. Hartmann, Adv. Mater., 2011, 23, 2627–2632 CrossRef CAS PubMed.
- M. Mandal, A. S. Manchanda, J. Zhuang and M. Kruk, Langmuir, 2012, 28, 8737–8745 CrossRef CAS PubMed.
- E. Serra, E. Diez, I. Diaz and R. M. Blanco, Microporous Mesoporous Mater., 2010, 132, 487–493 CrossRef CAS PubMed.
- S. Rehm, P. Trodler and J. Pleiss, Protein Sci., 2010, 19, 2122–2130 CrossRef CAS PubMed.
- M. Martinelle, M. Holmquist and K. Hult, Biochim. Biophys. Acta, Lipids Lipid Metab., 1995, 1258, 272–276 CrossRef.
- P. Trodler and J. Pleiss, BMC Struct. Biol., 2008, 8, 9 CrossRef PubMed.
- V. Gascón, C. Márquez-Álvarez and R. M. Blanco, Appl. Catal., A, 2014, 482, 116–126 CrossRef PubMed.
- T. Asefa, M. Kruk, N. Coombs, H. Grondey, M. J. MacLachlan, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 11662–11673 CrossRef CAS PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, J. Nanosci. Nanotechnol., 2006, 6, 265–288 CAS.
- B. Tan and S. E. Rankin, J. Non-Cryst. Solids, 2006, 352, 5453–5462 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- S. Z. Qiao, C. Z. Yu, Q. H. Hu, Y. G. Jin, X. F. Zhou, X. S. Zhao and G. Q. Lu, Microporous Mesoporous Mater., 2006, 91, 59–69 CrossRef CAS PubMed.
- X. Y. Bao, X. S. Zhao, S. Z. Qiao and S. K. Bhatia, J. Phys. Chem. B, 2004, 108, 16441–16450 CrossRef CAS.
- S. Urrego, E. Serra, V. Alfredsson, R. M. Blanco and I. Díaz, Microporous Mesoporous Mater., 2010, 129, 173–178 CrossRef CAS PubMed.
- R. M. Blanco, P. Terreros, M. Fernandez-Perez, C. Otero and G. Diaz-Gonzalez, J. Mol. Catal. B: Enzym., 2004, 30, 83–93 CrossRef CAS PubMed.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- O. V. Morozova, G. P. Shumakovich, S. V. Shleev and Y. I. Yaropolov, Appl. Biochem. Microbiol., 2007, 43, 523–535 CrossRef CAS.
- J. Uppenberg, M. T. Hansen, S. Patkar and T. A. Jones, Structure, 1994, 2, 293–308 CrossRef CAS.
- Rutgers and UCSD, RCSB Protein Data Bank, http://www.rcsb.org/pdb/home/home.do, accessed 14 January 2014.
- NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein, accessed 14 January 2014.
- Standard Protein BLAST, http://swissmodel.expasy.org/?pid=smd05, accessed 14 January 2014.
- N. Hakulinen, L. L. Kiiskinen, K. Kruus, M. Saloheimo, A. Koivula and J. Rouvinen, Nat. Struct. Biol., 2002, 9, 601–605 CAS.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed.
- L. Bordoli, F. Kiefer, K. Arnold, P. Benkert, J. Battey and T. Schwede, Nat. Protoc., 2009, 4, 1–13 CrossRef CAS PubMed.
- F. Kiefer, K. Arnold, M. Kunzli, L. Bordoli and T. Schwede, Nucleic Acids Res., 2009, 37, 387–392 CrossRef PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, http://www.pymol.org/, accessed 14 January 2014.
- ExPASy – ProtParamtool, http://web.expasy.org/protparam/, accessed 14 January 2014.
- UniProt KB, http://www.uniprot.org/uniprot/G2QFD0, accessed 14 January 2014.
- B. T. C. E. I. System., http://www.brenda-enzymes.org/php/result_flat.php4?ecno=1.10.3.2, accessed 14 January 2014.
- R. M. Berka, P. Schneider, E. J. Golightly, S. H. Brown, M. Madden, K. M. Brown, T. Halkier, K. Mondorf and F. Xu, Appl. Environ. Microbiol., 1997, 63, 3151–3157 CAS.
- R. M. Berka, I. V. Grigoriev, R. Otillar, A. Salamov, J. Grimwood, I. Reid, N. Ishmael, T. John, C. Darmond, M. C. Moisan, B. Henrissat, P. M. Coutinho, V. Lombard, D. O. Natvig, E. Lindquist, J. Schmutz, S. Lucas, P. Harris, J. Powlowski, A. Bellemare, D. Taylor, G. Butler, R. P. de Vries, I. E. Allijn, J. van den Brink, S. Ushinsky, R. Storms, A. J. Powell, I. T. Paulsen, L. D. Elbourne, S. E. Baker, J. Magnuson, S. Laboissiere, A. J. Clutterbuck, D. Martinez, M. Wogulis, A. L. de Leon, M. W. Rey and A. Tsang, Nat. Biotechnol., 2011, 29, 922–929 CrossRef CAS PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- R. M. Blanco, P. Terreros, N. Munoz and E. Serra, J. Mol. Catal. B: Enzym., 2007, 47, 13–20 CrossRef CAS PubMed.
- E. Serra, V. Alfredsson, R. M. Blanco and I. Diaz, in Zeolites and Related Materials: Trends, Targets and Challenges, Proceedings of the 4th International Feza Conference, ed. A. Gedeon, P. Massiani and F. Babonneau, 2008, pp. 369–372 Search PubMed.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
- A. Mayoral, R. Arenal, V. Gascon, C. Marquez-Alvarez, R. M. Blanco and I. Diaz, ChemCatChem, 2013, 5, 903–909 CrossRef CAS PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- K. Nie, F. Xie, F. Wang and T. Tan, J. Mol. Catal. B: Enzym., 2006, 43, 142–147 CrossRef CAS PubMed.
- C.-H. Kuo, L.-T. Peng, S.-C. Kan, Y.-C. Liu and C.-J. Shieh, Bioresour. Technol., 2013, 145, 229–232 CrossRef CAS PubMed.
- D. T. Tran, Y. J. Lin, C. L. Chen and J. S. Chang, Bioresour. Technol., 2013, 145, 193–203 CrossRef CAS PubMed.
- P. Adlercreutz, Chem. Soc. Rev., 2013, 42, 6406–6436 RSC.
- B. D. Ribeiro, A. M. de Castro, M. A. Coelho and D. M. Freire, Enzyme Res., 2011, 2011, 615803 Search PubMed.
- C. Jose, G. B. Austic, R. D. Bonetto, R. M. Burton and L. E. Briand, Catal. Today, 2013, 213, 73–80 CrossRef CAS PubMed.
- R. C. Minussi, G. M. Pastore and N. Duran, Trends Food Sci. Technol., 2002, 13, 205–216 CrossRef CAS.
- A. Kunamneni, F. J. Plou, A. Ballesteros and M. Alcalde, Recent Pat. Biotechnol., 2008, 2, 10–24 CrossRef CAS.
- J. F. Osma, J. L. Toca-Herrera and S. Rodriguez-Couto, Enzyme Res., 2010, 2010, 918761 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05362a |
| This journal is © The Royal Society of Chemistry 2014 |